
The Role of AhR in the Hallmarks of Brain Aging: Friend and Foe

Abstract
:
{kind=link}
{kind=link}
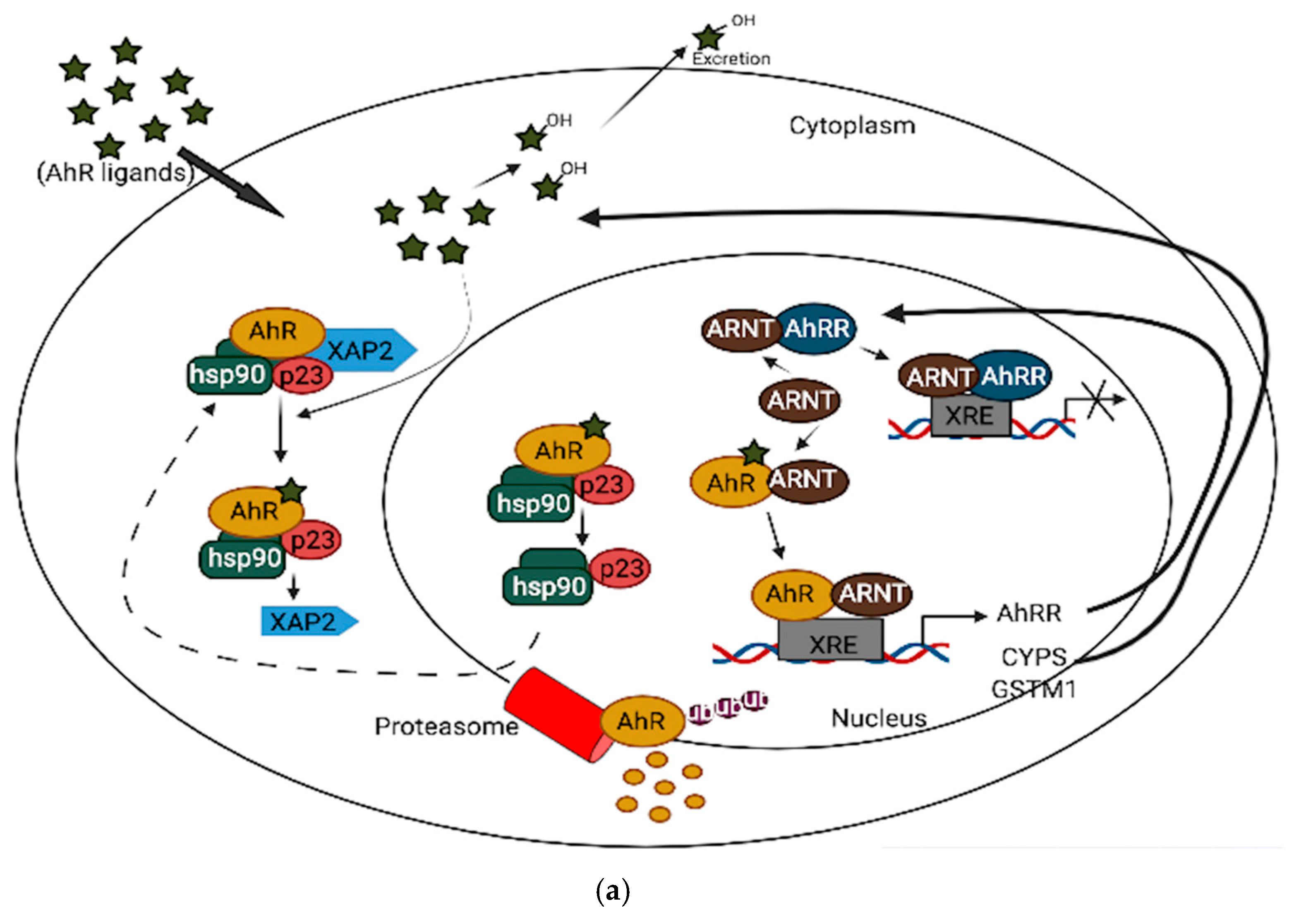
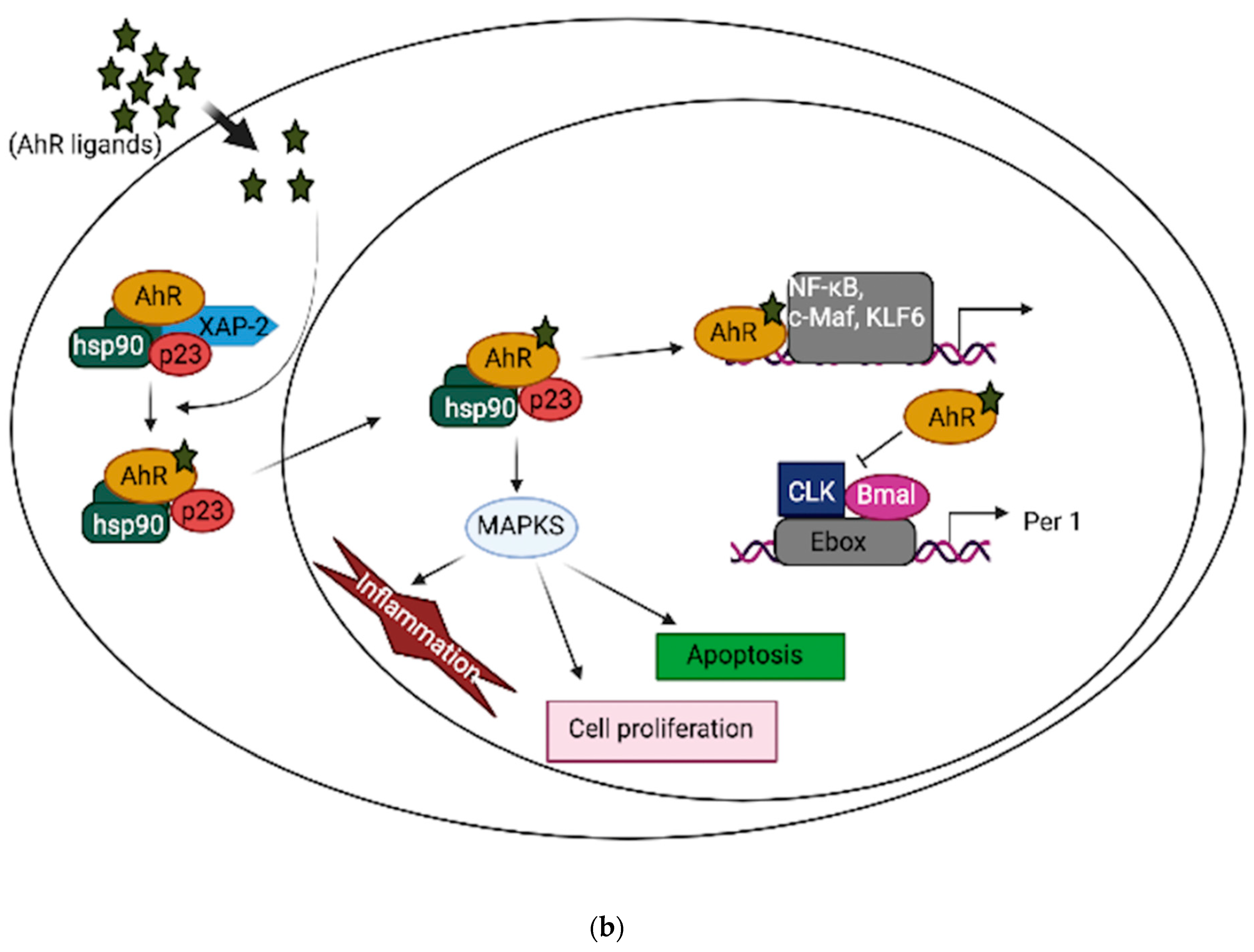
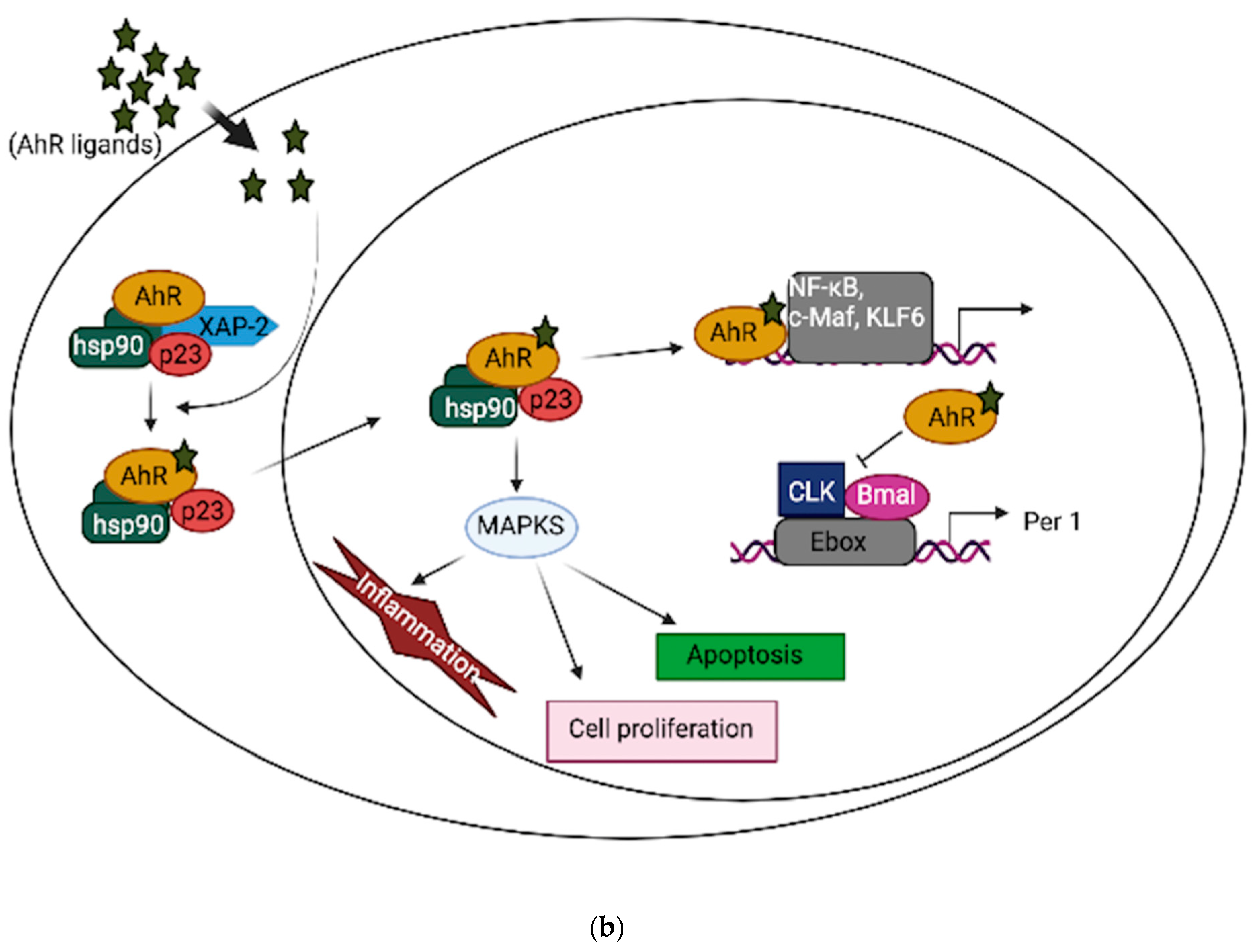{kind=link}
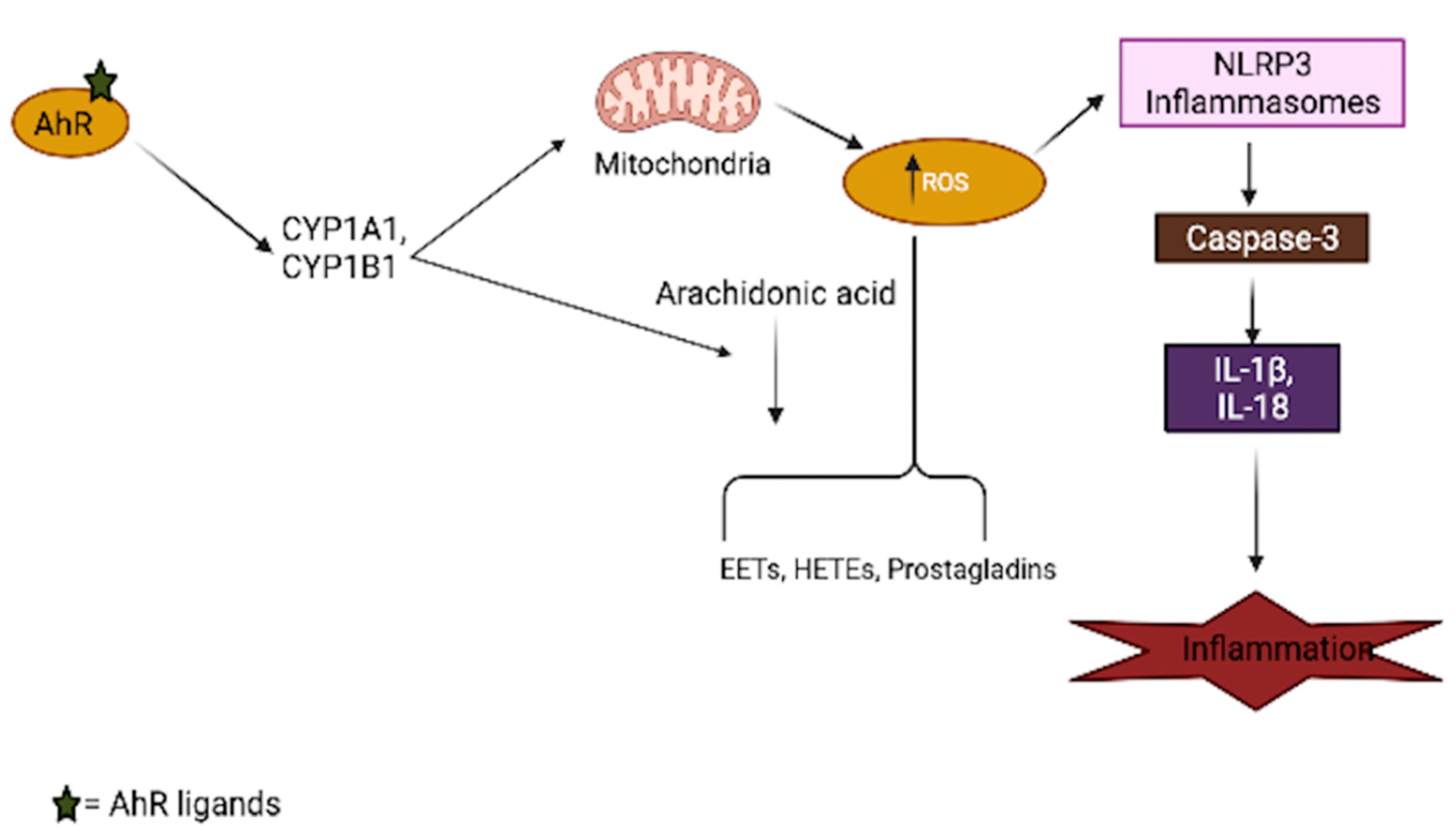{kind=link}
{kind=link}
1. Introduction
2. AhR Expression, Functions, and Signaling in the Brain
3. AhR and Aging Hallmarks in the Brain
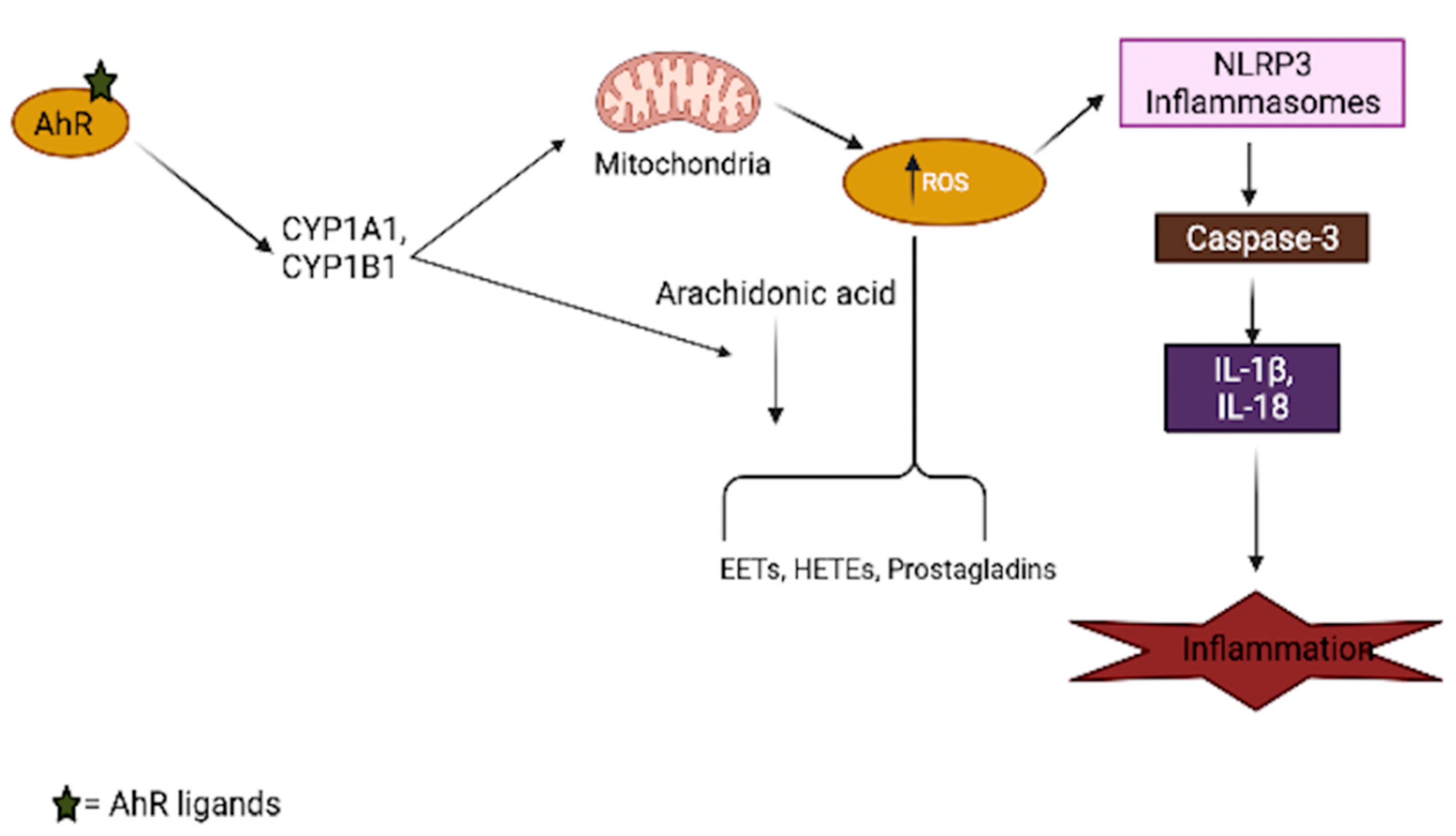
3.1. Oxidative Stress
3.2. Stress Response
3.3. Neurogenesis and Neuronal Plasticity
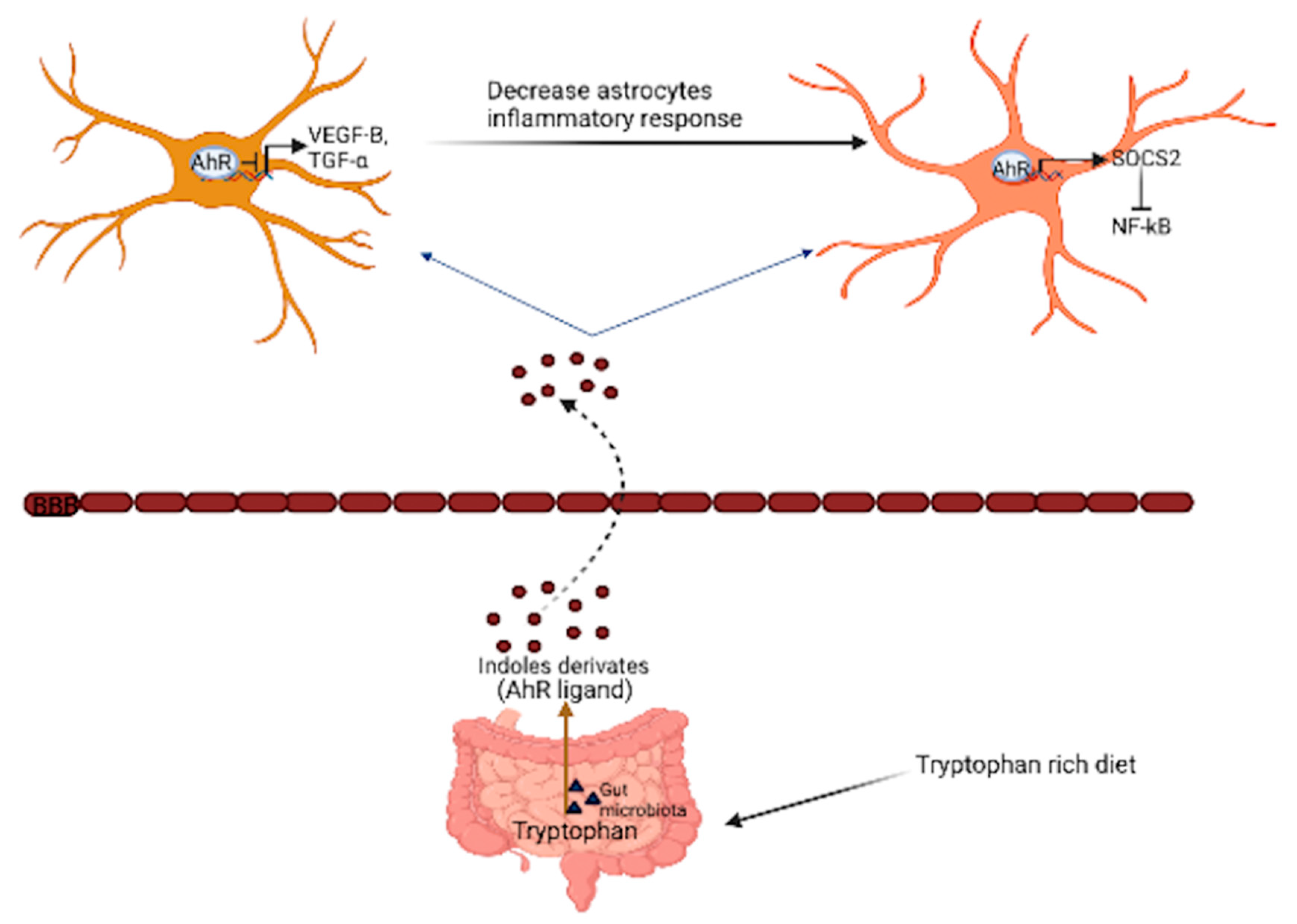
3.4. Inflammation and Glial Cell Activation
4. AhR Signaling Mechanism in Aging-Related Brain Diseases
4.1. Parkinson’s Disease (PD)
4.2. Alzheimer’s and Huntington’s
4.3. Multiple Sclerosis and Amyotrophic Lateral Sclerosis
5. Concluding Remarks
Funding
Conflicts of Interest
References
- Bryson, D. Anatomical and physiological changes with age. In Textile-Led Design for the Active Ageing Population; Woodhead Publishing: Cambridge, UK, 2015; pp. 107–116. [Google Scholar]
- Yankner, B.A.; Lu, T.; Loerch, P. The aging brain. Annu. Rev. Pathol. 2008, 3, 41–66. [Google Scholar] [CrossRef]
- Franceschi, C.; Garagnani, P.; Morsiani, C.; Conte, M.; Santoro, A.; Grignolio, A.; Monti, D.; Capri, M.; Salvioli, S. The Continuum of Aging and Age-Related Diseases: Common Mechanisms but Different Rates. Front. Med. 2018, 5, 61. [Google Scholar] [CrossRef] [Green Version]
- Ortman, J.M.; Velkoff, V.A.; Hogan, H. An Aging Nation: The Older Population in the United States; United States Census Bureau: Sutland, MD, USA, 2014; pp. 1–28.
- Aunan, J.R.; Cho, W.C.; Soreide, K. The Biology of Aging and Cancer: A Brief Overview of Shared and Divergent Molecular Hallmarks. Aging Dis. 2017, 8, 628–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacNee, W.; Rabinovich, R.A.; Choudhury, G. Ageing and the border between health and disease. Eur. Respir. J. 2014, 44, 1332–1352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Longo, M.; Bellastella, G.; Maiorino, M.I.; Meier, J.J.; Esposito, K.; Giugliano, D. Diabetes and Aging: From Treatment Goals to Pharmacologic Therapy. Front. Endocrinol. 2019, 10, 45. [Google Scholar] [CrossRef] [PubMed]
- Fotenos, A.F.; Mintun, M.A.; Snyder, A.Z.; Morris, J.C.; Buckner, R.L. Brain volume decline in aging: Evidence for a relation between socioeconomic status, preclinical Alzheimer disease, and reserve. Arch. Neurol. 2008, 65, 113–120. [Google Scholar] [CrossRef]
- Seidler, R.D.; Bernard, J.A.; Burutolu, T.B.; Fling, B.W.; Gordon, M.T.; Gwin, J.T.; Kwak, Y.; Lipps, D.B. Motor control and aging: Links to age-related brain structural, functional, and biochemical effects. Neurosci. Biobehav. Rev. 2010, 34, 721–733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murman, D.L. The Impact of Age on Cognition. Semin. Hear. 2015, 36, 111–121. [Google Scholar] [CrossRef]
- Hou, Y.; Dan, X.; Babbar, M.; Wei, Y.; Hasselbalch, S.G.; Croteau, D.L.; Bohr, V.A. Ageing as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 2019, 15, 565–581. [Google Scholar] [CrossRef]
- Cole, J.H.; Marioni, R.E.; Harris, S.E.; Deary, I.J. Brain age and other bodily ‘ages’: Implications for neuropsychiatry. Mol. Psychiatry 2019, 24, 266–281. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guerville, F.; De Souto Barreto, P.; Ader, I.; Andrieu, S.; Casteilla, L.; Dray, C.; Fazilleau, N.; Guyonnet, S.; Langin, D.; Liblau, R.; et al. Revisiting the Hallmarks of Aging to Identify Markers of Biological Age. J. Prev. Alzheimers Dis. 2020, 7, 56–64. [Google Scholar] [CrossRef]
- Eckers, A.; Jakob, S.; Heiss, C.; Haarmann-Stemmann, T.; Goy, C.; Brinkmann, V.; Cortese-Krott, M.M.; Sansone, R.; Esser, C.; Ale-Agha, N.; et al. The aryl hydrocarbon receptor promotes aging phenotypes across species. Sci. Rep. 2016, 6, 19618. [Google Scholar] [CrossRef] [Green Version]
- Gao, D.; Wu, M.; Wang, C.; Wang, Y.; Zuo, Z. Chronic exposure to low benzo[a]pyrene level causes neurodegenerative disease-like syndromes in zebrafish (Danio rerio). Aquat. Toxicol. 2015, 167, 200–208. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, V.; Schiavi, A.; Shaik, A.; Puchta, D.R.; Ventura, N. Ahr-bacterial diet interaction modulates aging and associated pathologies in C. elegans. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Blagosklonny, M.V.; Hall, M.N. Growth and aging: A common molecular mechanism. Aging 2009, 1, 357–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchionni, S.; Sell, C.; Lorenzini, A. Development and Longevity: Cellular and Molecular Determinants—A Mini-Review. Gerontology 2020, 66, 223–230. [Google Scholar] [CrossRef]
- Herculano-Houzel, S. Longevity and sexual maturity vary across species with number of cortical neurons, and humans are no exception. J. Comp. Neurol. 2019, 527, 1689–1705. [Google Scholar] [CrossRef]
- Esser, C.; Rannug, A. The aryl hydrocarbon receptor in barrier organ physiology, immunology, and toxicology. Pharmacol. Rev. 2015, 67, 259–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hahn, M.E. Aryl hydrocarbon receptors: Diversity and evolution. Rev. Chem. Biol. Interact. 2002, 141, 131–160. [Google Scholar] [CrossRef]
- Kimura, E.; Tohyama, C. Embryonic and Postnatal Expression of Aryl Hydrocarbon Receptor mRNA in Mouse Brain. Front. Neuroanat. 2017, 11, 4. [Google Scholar] [CrossRef] [Green Version]
- Qin, H.; Powell-Coffman, J.A. The Caenorhabditis elegans aryl hydrocarbon receptor, AHR-1, regulates neuronal development. Dev. Biol. 2004, 270, 64–75. [Google Scholar] [CrossRef]
- Gassmann, K.; Abel, J.; Bothe, H.; Haarmann-Stemmann, T.; Merk, H.F.; Quasthoff, K.N.; Rockel, T.D.; Schreiber, T.; Fritsche, E. Species-specific differential AhR expression protects human neural progenitor cells against developmental neurotoxicity of PAHs. Environ. Health Perspect. 2010, 118, 1571–1577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Latchney, S.E.; Hein, A.M.; O’Banion, M.K.; DiCicco-Bloom, E.; Opanashuk, L.A. Deletion or activation of the aryl hydrocarbon receptor alters adult hippocampal neurogenesis and contextual fear memory. J. Neurochem. 2013, 125, 430–445. [Google Scholar] [CrossRef] [Green Version]
- Abbot, B.D.; Birnbaum, L.S.; Perdew, G.H. Developmental Expression of Two Members of a New Class of Transcription Factors: I. Expression of Aryl Hydrocarbon Receptor in the C57BL/6N Mouse Embryo. Dev. Dyn. 1995, 204, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Byers, J.P.; Masters, K.; Sarver, J.G.; Hassoun, E.A. Association between the Levels of Biogenic Amines and Superoxide Anion Production in Brain Regions of Rats after Subchronic Exposure to TCDD. Toxicology 2006, 228, 291–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Hawkins, B.T.; Miller, D.S. Aryl hydrocarbon receptor-mediated up-regulation of ATP-driven xenobiotic efflux transporters at the blood-brain barrier. FASEB J. 2011, 25, 644–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.C.; Chang, L.H.; Huang, S.S.; Huang, Y.J.; Chih, C.L.; Kuo, H.C.; Lee, Y.H.; Lee, I.H. Aryl hydrocarbon receptor modulates stroke-induced astrogliosis and neurogenesis in the adult mouse brain. J. Neuroinflamm. 2019, 16, 187. [Google Scholar] [CrossRef] [PubMed]
- Juricek, L.; Coumoul, X. The Aryl Hydrocarbon Receptor and the Nervous System. Int. J. Mol. Sci. 2018, 19, 2504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.H.; Lin, C.H.; Hsu, P.C.; Sun, Y.Y.; Huang, Y.J.; Zhuo, J.H.; Wang, C.Y.; Gan, Y.L.; Hung, C.C.; Kuan, C.Y.; et al. Aryl hydrocarbon receptor mediates both proinflammatory and anti-inflammatory effects in lipopolysaccharide-activated microglia. Glia 2015, 63, 1138–1154. [Google Scholar] [CrossRef] [PubMed]
- Moon, B.H.; Hong, C.G.; Kim, S.Y.; Kim, H.J.; Shin, S.K.; Kang, S.; Lee, K.J.; Kim, Y.K.; Lee, M.S.; Shin, K.H. A single administration of 2,3,7,8-tetrachlorodibenzo-p-dioxin that produces reduced food and water intake induces long-lasting expression of corticotropin-releasing factor, arginine vasopressin, and proopiomelanocortin in rat brain. Toxicol. Appl. Pharmacol. 2008, 233, 314–322. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Mizuyachi, K.; Terranova, P.F.; Rozman, K.K. 2,3,7,8-tetrachlorodibenzo-p-dioxin decreases responsiveness of the hypothalamus to estradiol as a feedback inducer of preovulatory gonadotropin secretion in the immature gonadotropin-primed rat. Toxicol. Appl. Pharmacol. 2001, 170, 181–190. [Google Scholar] [CrossRef]
- Desaulniers, D.; Xiao, G.H.; Leingartner, K.; Chu, I.; Musicki, B.; Tsang, B.K. Comparisons of brain, uterus, and liver mRNA expression for cytochrome p450s, DNA methyltransferase-1, and catechol-o-methyltransferase in prepubertal female Sprague-Dawley rats exposed to a mixture of aryl hydrocarbon receptor agonists. Toxicol. Sci. 2005, 86, 175–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wendling, J.M.; Orth, R.G.; Poiger, H. Determination of [3H]-2,3,7,8-Tetrach10rodibenz~-p-dioxin in Human Feces to Ascertain Its Relative Metabolism in Man. Anal. Chem. 1990, 62, 796–800. [Google Scholar] [CrossRef] [PubMed]
- Bock, K.W. Aryl hydrocarbon receptor (AHR): From selected human target genes and crosstalk with transcription factors to multiple AHR functions. Biochem. Pharmacol. 2019, 168, 65–70. [Google Scholar] [CrossRef]
- Vogel, C.F.; Sciullo, E.; Li, W.; Wong, P.; Lazennec, G.; Matsumura, F. RelB, a new partner of aryl hydrocarbon receptor-mediated transcription. Mol. Endocrinol. 2007, 21, 2941–2955. [Google Scholar] [CrossRef] [Green Version]
- Hayes, J.D.; Dinkova-Kostova, A.T.; McMahon, M. Cross-talk between transcription factors AhR and Nrf2: Lessons for cancer chemoprevention from dioxin. Toxicol. Sci. 2009, 111, 199–201. [Google Scholar] [CrossRef]
- Yeager, R.L.; Reisman, S.A.; Aleksunes, L.M.; Klaassen, C.D. Introducing the “TCDD-inducible AhR-Nrf2 gene battery”. Toxicol. Sci. 2009, 111, 238–246. [Google Scholar] [CrossRef] [Green Version]
- Kaminska, B.; Gozdz, A.; Zawadzka, M.; Ellert-Miklaszewska, A.; Lipko, M. MAPK signal transduction underlying brain inflammation and gliosis as therapeutic target. Anat. Rec. 2009, 292, 1902–1913. [Google Scholar] [CrossRef]
- Sun, Y.; Liu, W.Z.; Liu, T.; Feng, X.; Yang, N.; Zhou, H.F. Signaling pathway of MAPK/ERK in cell proliferation, differentiation, migration, senescence and apoptosis. J. Recept. Signal. Transduct. Res. 2015, 35, 600–604. [Google Scholar] [CrossRef]
- Tischkau, S.A. Mechanisms of circadian clock interactions with aryl hydrocarbon receptor signalling. Eur. J. Neurosci. 2020, 51, 379–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, A.R.; Jeong, Y.J.; Hwang, C.Y.; Yoon, K.S.; Choe, W.; Ha, J.; Kim, S.S.; Pak, Y.K.; Yeo, E.J.; Kang, I. Alpha-naphthoflavone induces apoptosis through endoplasmic reticulum stress via c-Src-, ROS-, MAPKs-, and arylhydrocarbon receptor-dependent pathways in HT22 hippocampal neuronal cells. Neurotoxicology 2019, 71, 39–51. [Google Scholar] [CrossRef]
- Drutel, G.; Heron, A.; Kathmann, M.; Gros, C.; Mace, S.; Plotkine, M.; Schwartz, J.C.; Arrang, J.M. ARNT2, a transcription factor for brain neuron survival? Eur. J. Neurosci. 1999, 11, 1545–1553. [Google Scholar] [CrossRef]
- Dougherty, E.J.; Pollenz, R.S. Analysis of Ah Receptor-ARNT and Ah Receptor-ARNT2 Complexes In Vitro and in Cell Culture. Toxicol. Sci. 2008, 103, 191–206. [Google Scholar] [CrossRef] [Green Version]
- Cuartero, M.I.; Ballesteros, I.; de la Parra, J.; Harkin, A.L.; Abautret-Daly, A.; Sherwin, E.; Fernandez-Salguero, P.; Corbi, A.L.; Lizasoain, I.; Moro, M.A. L-kynurenine/aryl hydrocarbon receptor pathway mediates brain damage after experimental stroke. Circulation 2014, 130, 2040–2051. [Google Scholar] [CrossRef] [Green Version]
- Ma, N.; He, T.; Johnston, L.J.; Ma, X. Host-microbiome interactions: The aryl hydrocarbon receptor as a critical node in tryptophan metabolites to brain signaling. Gut Microbes 2020, 11, 1203–1219. [Google Scholar] [CrossRef] [PubMed]
- Schwarcz, R.; Stone, T.W. The kynurenine pathway and the brain: Challenges, controversies and promises. Neuropharmacology 2017, 112, 237–247. [Google Scholar] [CrossRef] [Green Version]
- Dougherty, D.M.; Marsh-Richard, D.M.; Mathias, C.W.; Hood, A.J.; Addicott, M.A.; Moeller, F.G.; Morgan, C.J.; Badawy, A.A. Comparison of 50- and 100-g L -tryptophan depletion and loading formulations for altering 5-HT synthesis: Pharmacokinetics, side effects, and mood states. Psychopharmacology 2008, 198, 431–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rothhammer, V.; Mascanfroni, I.D.; Bunse, L.; Takenaka, M.C.; Kenison, J.E.; Mayo, L.; Chao, C.C.; Patel, B.; Yan, R.; Blain, M.; et al. Type I interferons and microbial metabolites of tryptophan modulate astrocyte activity and central nervous system inflammation via the aryl hydrocarbon receptor. Nat. Med. 2016, 22, 586–597. [Google Scholar] [CrossRef] [PubMed]
- Rothhammer, V.; Borucki, D.M.; Tjon, E.C.; Takenaka, M.C.; Chao, C.C.; Ardura-Fabregat, A.; de Lima, K.A.; Gutierrez-Vazquez, C.; Hewson, P.; Staszewski, O.; et al. Microglial control of astrocytes in response to microbial metabolites. Nature 2018, 557, 724–728. [Google Scholar] [CrossRef] [PubMed]
- Maitre, M.; Klein, C.; Patte-Mensah, C.; Mensah-Nyagan, A.G. Tryptophan metabolites modify brain Abeta peptide degradation: A role in Alzheimer’s disease? Prog. Neurobiol. 2020, 190, 101800. [Google Scholar] [CrossRef]
- Gramatzki, D.; Pantazis, G.; Schittenhelm, J.; Tabatabai, G.; Kohle, C.; Wick, W.; Schwarz, M.; Weller, M.; Tritschler, I. Aryl hydrocarbon receptor inhibition downregulates the TGF-beta/Smad pathway in human glioblastoma cells. Oncogene 2009, 28, 2593–2605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silginer, M.; Burghardt, I.; Gramatzki, D.; Bunse, L.; Leske, H.; Rushing, E.J.; Hao, N.; Platten, M.; Weller, M.; Roth, P. The aryl hydrocarbon receptor links integrin signaling to the TGF-beta pathway. Oncogene 2016, 35, 3260–3271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harman, D. Aging: A theory based on free radical and radiation chemistry. J. Gerontol. 1956, 11, 298–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gladyshev, V.N. The free radical theory of aging is dead. Long live the damage theory! Antioxid. Redox Signal. 2014, 20, 727–731. [Google Scholar] [CrossRef]
- Liguori, I.; Russo, G.; Curcio, F.; Bulli, G.; Aran, L.; Della-Morte, D.; Gargiulo, G.; Testa, G.; Cacciatore, F.; Bonaduce, D.; et al. Oxidative stress, aging, and diseases. Clin. Interv. Aging 2018, 13, 757–772. [Google Scholar] [CrossRef] [Green Version]
- Tan, B.L.; Norhaizan, M.E.; Liew, W.P.; Sulaiman Rahman, H. Antioxidant and Oxidative Stress: A Mutual Interplay in Age-Related Diseases. Front. Pharmacol. 2018, 9, 1162. [Google Scholar] [CrossRef] [Green Version]
- Zheng, F.; Goncalves, F.M.; Abiko, Y.; Li, H.; Kumagai, Y.; Aschner, M. Redox toxicology of environmental chemicals causing oxidative stress. Redox Biol. 2020, 34, 101475. [Google Scholar] [CrossRef]
- Gemma, C.; Vila, J.; Bachstetter, A.; Bickford, P.C. Oxidative Stress and the Aging Brain: From Theory to Prevention. In Brain Aging: Models, Methods, and Mechanisms; Riddle, D.R., Ed.; CRC Press/Taylor & Francis: Boca Raton, FL, USA, 2007; pp. 353–374. [Google Scholar] [CrossRef]
- Lenaz, G. Role of mitochondria in oxidative stress and ageing. Biochim. Biophys. Acta 1998, 1366, 53–67. [Google Scholar] [CrossRef] [Green Version]
- Dai, D.F.; Chiao, Y.A.; Marcinek, D.J.; Szeto, H.H.; Rabinovitch, P.S. Mitochondrial oxidative stress in aging and healthspan. Longev. Healthspan 2014, 3, 6. [Google Scholar] [CrossRef] [Green Version]
- Demers-Lamarche, J.; Guillebaud, G.; Tlili, M.; Todkar, K.; Belanger, N.; Grondin, M.; Nguyen, A.P.; Michel, J.; Germain, M. Loss of Mitochondrial Function Impairs Lysosomes. J. Biol. Chem. 2016, 291, 10263–10276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lefaki, M.; Papaevgeniou, N.; Chondrogianni, N. Redox regulation of proteasome function. Redox Biol. 2017, 13, 452–458. [Google Scholar] [CrossRef] [PubMed]
- Dalton, T.P.; Puga, A.; Shertzer, H.G. Induction of cellular oxidative stress by aryl hydrocarbon receptor activation. Chem.Biol. Interact. 2002, 141, 77–95. [Google Scholar] [CrossRef]
- Liu, H.; Shi, L.; Giesy, J.P.; Yu, H. Polychlorinated diphenyl sulfides can induce ROS and genotoxicity via the AhR-CYP1A1 pathway. Chemosphere 2019, 223, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Vogel, C.F.A.; Van Winkle, L.S.; Esser, C.; Haarmann-Stemmann, T. The aryl hydrocarbon receptor as a target of environmental stressors—Implications for pollution mediated stress and inflammatory responses. Redox Biol. 2020, 34, 101530. [Google Scholar] [CrossRef]
- Zhang, Y.; Nie, X.; Tao, T.; Qian, W.; Jiang, S.; Jiang, J.; Li, A.; Guo, A.; Xu, G.; Wu, Q. 2,3,7,8-Tetrachlorodibenzo-p-dioxin promotes astrocyte activation and the secretion of tumor necrosis factor-alpha via PKC/SSeCKS-dependent mechanisms. J. Neurochem. 2014, 129, 839–849. [Google Scholar] [CrossRef] [Green Version]
- Hassoun, E.A.; Li, F.; Abushaban, A.; Stohs, S.J. The relative abilities of TCDD and its congeners to induce oxidative stress in the hepatic and brain tissues of rats after subchronic exposure. Toxicology 2000, 145, 103–113. [Google Scholar] [CrossRef]
- Lin, J.; Zhao, H.S.; Qin, L.; Li, X.N.; Zhang, C.; Xia, J.; Li, J.L. Atrazine Triggers Mitochondrial Dysfunction and Oxidative Stress in Quail (Coturnix C. coturnix) Cerebrum via Activating Xenobiotic-Sensing Nuclear Receptors and Modulating Cytochrome P450 Systems. J. Agric. Food Chem. 2018, 66, 6402–6413. [Google Scholar] [CrossRef]
- Huang, P.; Rannug, A.; Ahlbom, E.; Hakansson, H.; Ceccatelli, S. Effect of 2,3,7,8-tetrachlorodibenzo-p-dioxin on the expression of cytochrome P450 1A1, the aryl hydrocarbon receptor, and the aryl hydrocarbon receptor nuclear translocator in rat brain and pituitary. Toxicol. Appl. Pharmacol. 2000, 169, 159–167. [Google Scholar] [CrossRef]
- Bansal, S.; Leu, A.N.; Gonzalez, F.J.; Guengerich, F.P.; Chowdhury, A.R.; Anandatheerthavarada, H.K.; Avadhani, N.G. Mitochondrial targeting of cytochrome P450 (CYP) 1B1 and its role in polycyclic aromatic hydrocarbon-induced mitochondrial dysfunction. J. Biol. Chem. 2014, 289, 9936–9951. [Google Scholar] [CrossRef] [Green Version]
- Albertolle, M.E.; Peter Guengerich, F. The relationships between cytochromes P450 and H2O2: Production, reaction, and inhibition. J. Inorg. Biochem. 2018, 186, 228–234. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Tardivel, A.; Thorens, B.; Choi, I.; Tschopp, J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol. 2010, 11, 136–140. [Google Scholar] [CrossRef]
- Larigot, L.; Juricek, L.; Dairou, J.; Coumoul, X. AhR signaling pathways and regulatory functions. Biochim. Open 2018, 7, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Nebert, D.W.; Karp, C.L. Endogenous functions of the aryl hydrocarbon receptor (AHR): Intersection of cytochrome P450 1 (CYP1)-metabolized eicosanoids and AHR biology. J. Biol. Chem. 2008, 283, 36061–36065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nannelli, A.; Rossignolo, F.; Tolando, R.; Rossato, P.; Longo, V.; Gervasi, P.G. Effect of beta-naphthoflavone on AhR-regulated genes (CYP1A1, 1A2, 1B1, 2S1, Nrf2, and GST) and antioxidant enzymes in various brain regions of pig. Toxicology 2009, 265, 69–79. [Google Scholar] [CrossRef]
- Garcia-Lara, L.; Perez-Severiano, F.; Gonzalez-Esquivel, D.; Elizondo, G.; Segovia, J. Absence of aryl hydrocarbon receptors increases endogenous kynurenic acid levels and protects mouse brain against excitotoxic insult and oxidative stress. J. Neurosci. Res. 2015, 93, 1423–1433. [Google Scholar] [CrossRef] [PubMed]
- Kultz, D. Molecular and evolutionary basis of the cellular stress response. Annu. Rev. Physiol. 2005, 67, 225–257. [Google Scholar] [CrossRef]
- Beere, H.M. Death versus survival: Functional interaction between the apoptotic and stress-inducible heat shock protein pathways. J. Clin. Investg. 2005, 115, 2633–2639. [Google Scholar] [CrossRef] [Green Version]
- Miller, D.J.; Fort, P.E. Heat Shock Proteins Regulatory Role in Neurodevelopment. Front. Neurosci. 2018, 12, 821. [Google Scholar] [CrossRef] [Green Version]
- Lupien, S.J.; de Leon, M.; de Santi, S.; Convit, A.; Tarshish, C.; Nair, N.P.V.; Thakur, M.; McEwen, B.S.; Hauger, R.L.; Meaney, M.J. Cortisol levels during human aging predict hippocampal atrophy and memory deficits. Nat. Neurosci. 1998, 1, 69–73. [Google Scholar] [CrossRef]
- Kourtis, N.; Tavernarakis, N. Cellular stress response pathways and ageing: Intricate molecular relationships. EMBO J. 2011, 30, 2520–2531. [Google Scholar] [CrossRef] [Green Version]
- Calderwood, S.K.; Murshid, A.; Prince, T. The shock of aging: Molecular chaperones and the heat shock response in longevity and aging—A mini-review. Gerontology 2009, 55, 550–558. [Google Scholar] [CrossRef] [Green Version]
- Lindholm, D.; Wootz, H.; Korhonen, L. ER stress and neurodegenerative diseases. Cell Death Differ. 2006, 13, 385–392. [Google Scholar] [CrossRef]
- Aluru, N.; Vijayan, M.M. Brain transcriptomics in response to beta-naphthoflavone treatment in rainbow trout: The role of aryl hydrocarbon receptor signaling. Aquat. Toxicol. 2008, 87, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Gesto, M.; Soengas, J.L.; Miguez, J.M. Acute and prolonged stress responses of brain monoaminergic activity and plasma cortisol levels in rainbow trout are modified by PAHs (naphthalene, beta-naphthoflavone and benzo(a)pyrene) treatment. Aquat. Toxicol. 2008, 86, 341–351. [Google Scholar] [CrossRef]
- Guerrina, N.; Aloufi, N.; Shi, F.; Prasade, K.; Mehrotra, C.; Traboulsi, H.; Matthews, J.; Eidelman, D.H.; Hamid, Q.; Baglole, C.J. The aryl hydrocarbon receptor reduces LC3II expression and controls endoplasmic reticulum stress. Am. J. Physiol. Lung Cell Mol. Physiol. 2021, 320, L339–L355. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.C.; Zhou, Y.; Huang, S.K. SHP-2 phosphatase controls aryl hydrocarbon receptor-mediated ER stress response in mast cells. Arch. Toxicol. 2017, 91, 1739–1748. [Google Scholar] [CrossRef] [PubMed]
- Pearson-Leary, J.; Osborne, D.M.; McNay, E.C. Role of Glia in Stress-Induced Enhancement and Impairment of Memory. Front. Integr. Neurosci. 2015, 9, 63. [Google Scholar] [CrossRef] [Green Version]
- Jauregui-Huerta, F.; Delgadillo, Y.R.; Gonzalez-Perez, O.; Gonzalez-Castañeda, R.; Garcia-Estrada, J.; Luqui, S. Responses of Glial Cells to Stress and Glucocorticoids. Curr. Immunol. Rev. 2010, 6, 195–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gage, F.H. Mammalian neural stem cells. Science 2000, 287, 1433–1438. [Google Scholar] [CrossRef] [PubMed]
- Shors, T.J.; Miesegaes, G.; Beylin, A.; Zhao, M.; Rydel, T.; Gould, E. Neurogenesis in the adult is involved in the formation of trace memories. Nature 2001, 410, 372–376. [Google Scholar] [CrossRef]
- Roy, N.S.; Benraiss, A.; Wang, S.; Richard, A.R.; Goodman, R.; Couldwell, W.T.; Nedergaard, M.; Kawaguchi, A.; Okano, H.; Goldman, S.A. Promoter-Targeted Selection and Isolation of Neural Progenitor Cells from the Adult Human Ventricular Zone. J. Neurosci. Res. 2001, 59, 321–331. [Google Scholar] [CrossRef]
- Curtis, M.A.; Kam, M.; Nannmark, U.; Anderson, M.F.; Axell, M.Z.; Wikkelso, C. Human neuroblasts migrate to the olfactory bulb via a lateral ventricular extension. Science 2007, 315, 1243–1249. [Google Scholar] [CrossRef]
- Donovan, M.H.; Yazdani, U.; Norris, R.D.; Games, D.; German, D.C.; Eisch, A.J. Decreased adult hippocampal neurogenesis in the PDAPP mouse model of Alzheimer’s disease. J. Comp. Neurol. 2006, 495, 70–83. [Google Scholar] [CrossRef] [PubMed]
- Bouab, M.; Paliouras, G.N.; Aumont, A.; Forest-Berard, K.; Fernandes, K.J. Aging of the subventricular zone neural stem cell niche: Evidence for quiescence-associated changes between early and mid-adulthood. Neuroscience 2011, 173, 135–149. [Google Scholar] [CrossRef]
- Carmichael, O.; Lockhart, S. The role of diffusion tensor imaging in the study of cognitive aging. Curr. Top. Behav. Neurosci. 2012, 11, 289–320. [Google Scholar] [CrossRef]
- Bjornsson, C.S.; Apostolopoulou, M.; Tian, Y.; Temple, S. It takes a village: Constructing the neurogenic niche. Dev. Cell 2015, 32, 435–446. [Google Scholar] [CrossRef] [Green Version]
- Garber, C.; Vasek, M.J.; Vollmer, L.L.; Sun, T.; Jiang, X.; Klein, R.S. Astrocytes decrease adult neurogenesis during virus-induced memory dysfunction via IL-1. Nat. Immunol. 2018, 19, 151–161. [Google Scholar] [CrossRef]
- Chen, Y.; Xu, L.; Xie, H.Q.H.; Xu, T.; Fu, H.; Zhang, S.; Sha, R.; Xia, Y.; Zhao, B. Identification of differentially expressed genes response to TCDD in rat brain after long-term low-dose exposure. J. Environ. Sci. 2017, 62, 92–99. [Google Scholar] [CrossRef] [PubMed]
- De la Parra, J.; Cuartero, M.I.; Perez-Ruiz, A.; Garcia-Culebras, A.; Martin, R.; Sanchez-Prieto, J.; Garcia-Segura, J.M.; Lizasoain, I.; Moro, M.A. AhR Deletion Promotes Aberrant Morphogenesis and Synaptic Activity of Adult-Generated Granule Neurons and Impairs Hippocampus-Dependent Memory. eNeuro 2018, 5. [Google Scholar] [CrossRef] [PubMed]
- Di Giaimo, R.; Durovic, T.; Barquin, P.; Kociaj, A.; Lepko, T.; Aschenbroich, S.; Breunig, C.T.; Irmler, M.; Cernilogar, F.M.; Schotta, G.; et al. The Aryl Hydrocarbon Receptor Pathway Defines the Time Frame for Restorative Neurogenesis. Cell Rep. 2018, 25, 3241–3251.e3245. [Google Scholar] [CrossRef] [Green Version]
- Keshavarzi, M.; Khoshnoud, M.J.; Ghaffarian Bahraman, A.; Mohammadi-Bardbori, A. An Endogenous Ligand of Aryl Hydrocarbon Receptor 6-Formylindolo[3,2-b]Carbazole (FICZ) Is a Signaling Molecule in Neurogenesis of Adult Hippocampal Neurons. J. Mol. Neurosci. 2020, 70, 806–817. [Google Scholar] [CrossRef]
- Grabert, K.; Michoel, T.; Karavolos, M.H.; Clohisey, S.; Baillie, J.K.; Stevens, M.P.; Freeman, T.C.; Summers, K.M.; McColl, B.W. Microglial brain region-dependent diversity and selective regional sensitivities to aging. Nat. Neurosci. 2016, 19, 504–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calabrese, V.; Santoro, A.; Monti, D.; Crupi, R.; Di Paola, R.; Latteri, S.; Cuzzocrea, S.; Zappia, M.; Giordano, J.; Calabrese, E.J.; et al. Aging and Parkinson’s Disease: Inflammaging, neuroinflammation and biological remodeling as key factors in pathogenesis. Free Radic. Biol. Med. 2018, 115, 80–91. [Google Scholar] [CrossRef] [PubMed]
- Zuo, L.; Prather, E.R.; Stetskiv, M.; Garrison, D.E.; Meade, J.R.; Peace, T.I.; Zhou, T. Inflammaging and Oxidative Stress in Human Diseases: From Molecular Mechanisms to Novel Treatments. Int. J. Mol. Sci. 2019, 20, 4472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bektas, A.; Schurman, S.H.; Sen, R.; Ferrucci, L. Aging, inflammation and the environment. Exp. Gerontol. 2018, 105, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Xia, S.; Zhang, X.; Zheng, S.; Khanabdali, R.; Kalionis, B.; Wu, J.; Wan, W.; Tai, X. An Update on Inflamm-Aging: Mechanisms, Prevention, and Treatment. J. Immunol. Res. 2016, 2016, 8426874. [Google Scholar] [CrossRef]
- Clarke, L.E.; Liddelow, S.A.; Chakraborty, C.; Munch, A.E.; Heiman, M.; Barres, B.A. Normal aging induces A1-like astrocyte reactivity. Proc. Natl. Acad. Sci. USA 2018, 115, E1896–E1905. [Google Scholar] [CrossRef] [Green Version]
- Godbout, J.P.; Chen, J.; Abraham, J.; Richwine, A.F.; Berg, B.M.; Kelley, K.W.; Johnson, R.W. Exaggerated neuroinflammation and sickness behavior in aged mice following activation of the peripheral innate immune system. FASEB J. 2005, 19, 1329–1331. [Google Scholar] [CrossRef]
- Vogel, C.F.; Khan, E.M.; Leung, P.S.; Gershwin, M.E.; Chang, W.L.; Wu, D.; Haarmann-Stemmann, T.; Hoffmann, A.; Denison, M.S. Cross-talk between aryl hydrocarbon receptor and the inflammatory response: A role for nuclear factor-kappaB. J. Biol. Chem. 2014, 289, 1866–1875. [Google Scholar] [CrossRef] [Green Version]
- Gunther, J.; Fallarino, F.; Fuchs, D.; Wirthgen, E. Editorial: Immunomodulatory Roles of Tryptophan Metabolites in Inflammation and Cancer. Front. Immunol. 2020, 11, 1497. [Google Scholar] [CrossRef] [PubMed]
- Gulaj, E.; Pawlak, K.; Bien, B.; Pawlak, D. Kynurenine and its metabolites in Alzheimer’s disease patients. Adv. Med. Sci. 2010, 55, 204–211. [Google Scholar] [CrossRef]
- Capuron, L.; Schroecksnadel, S.; Feart, C.; Aubert, A.; Higueret, D.; Barberger-Gateau, P.; Laye, S.; Fuchs, D. Chronic low-grade inflammation in elderly persons is associated with altered tryptophan and tyrosine metabolism: Role in neuropsychiatric symptoms. Biol. Psychiatry 2011, 70, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Mezrich, J.D.; Fechner, J.H.; Zhang, X.; Johnson, B.P.; Burlingham, W.J.; Bradfield, C.A. An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. J. Immunol. 2010, 185, 3190–3198. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, N.T.; Kimura, A.; Nakahama, T.; Chinen, I.; Masuda, K.; Nohara, K.; Fujii-Kuriyama, Y.; Kishimoto, T. Aryl hydrocarbon receptor negatively regulates dendritic cell immunogenicity via a kynurenine-dependent mechanism. Proc. Natl. Acad. Sci. USA 2010, 107, 19961–19966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, A.S.; Langmann, T. Indole-3-carbinol regulates microglia homeostasis and protects the retina from degeneration. J. Neuroinflamm. 2020, 17, 327. [Google Scholar] [CrossRef]
- Barroso, A.; Mahler, J.V.; Fonseca-Castro, P.H.; Quintana, F.J. The aryl hydrocarbon receptor and the gut-brain axis. Cell Mol. Immunol. 2021, 18, 259–268. [Google Scholar] [CrossRef]
- Marsland, B.J. Regulating inflammation with microbial metabolites. Nat. Med. 2016, 22, 581–583. [Google Scholar] [CrossRef]
- Bruck, W.; Pfortner, R.; Pham, T.; Zhang, J.; Hayardeny, L.; Piryatinsky, V.; Hanisch, U.K.; Regen, T.; van Rossum, D.; Brakelmann, L.; et al. Reduced astrocytic NF-kappaB activation by laquinimod protects from cuprizone-induced demyelination. Acta Neuropathol. 2012, 124, 411–424. [Google Scholar] [CrossRef] [Green Version]
- Rothhammer, V.; Borucki, D.M.; Kenison, J.E.; Hewson, P.; Wang, Z.; Bakshi, R.; Sherr, D.H.; Quintana, F.J. Detection of aryl hydrocarbon receptor agonists in human samples. Sci. Rep. 2018, 8, 4970. [Google Scholar] [CrossRef] [Green Version]
- Lowery, R.L.; Latchney, S.E.; Peer, R.P.; Lamantia, C.E.; Opanashuk, L.; McCall, M.; Majewska, A.K. Acute 2,3,7,8-Tetrachlorodibenzo-p-dioxin exposure in adult mice does not alter the morphology or inflammatory response of cortical microglia. Neurosci. Lett. 2021, 742, 135516. [Google Scholar] [CrossRef] [PubMed]
- Bravo-Ferrer, I.; Cuartero, M.I.; Medina, V.; Ahedo-Quero, D.; Pena-Martinez, C.; Perez-Ruiz, A.; Fernandez-Valle, M.E.; Hernandez-Sanchez, C.; Fernandez-Salguero, P.M.; Lizasoain, I.; et al. Lack of the aryl hydrocarbon receptor accelerates aging in mice. FASEB J. 2019, 33, 12644–12654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jayaramayya, K.; Iyer, M.; Venkatesan, D.; Balasubramanian, V.; Narayanasamy, A.; Subramaniam, M.D.; Cho, S.G.; Vellingiri, B. Unraveling correlative roles of dopamine transporter (DAT) and Parkin in Parkinson’s disease (PD)—A road to discovery? Brain Res. Bull. 2020, 157, 169–179. [Google Scholar] [CrossRef]
- Gonzalez-Barbosa, E.; Garcia-Aguilar, R.; Vega, L.; Cabanas-Cortes, M.A.; Gonzalez, F.J.; Segovia, J.; Morales-Lazaro, S.L.; Cisneros, B.; Elizondo, G. Parkin is transcriptionally regulated by the aryl hydrocarbon receptor: Impact on alpha-synuclein protein levels. Biochem. Pharmacol. 2019, 168, 429–437. [Google Scholar] [CrossRef]
- Rath, S.N.; Jena, L.; Patri, M. Understanding ligands driven mechanism of wild and mutant aryl hydrocarbon receptor in presence of phytochemicals combating Parkinson’s disease: An in silico and in vivo study. J. Biomol. Struct. Dyn. 2020, 38, 807–826. [Google Scholar] [CrossRef] [PubMed]
- Datla, K.P.; Christidou, M.; Widmer, W.W.; Rooprai, H.K.; Dexter, D.T. Tissue distribution and neuroprotective effects of citrus flavonoid tangeretin in a rat model of Parkinson’s disease. Neuroreport 2001, 12, 3871–3875. [Google Scholar] [CrossRef] [PubMed]
- Akahoshi, E.; Yoshimura, S.; Uruno, S.; Ishihara-Sugano, M. Effect of dioxins on regulation of tyrosine hydroxylase gene expression by aryl hydrocarbon receptor: A neurotoxicology study. Environ. Health 2009, 8, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramos-Garcia, N.A.; Orozco-Ibarra, M.; Estudillo, E.; Elizondo, G.; Gomez Apo, E.; Chavez Macias, L.G.; Sosa-Ortiz, A.L.; Torres-Ramos, M.A. Aryl Hydrocarbon Receptor in Post-Mortem Hippocampus and in Serum from Young, Elder, and Alzheimer’s Patients. Int. J. Mol. Sci. 2020, 21, 1983. [Google Scholar] [CrossRef] [Green Version]
- Duan, Z.; Zhang, S.; Liang, H.; Xing, Z.; Guo, L.; Shi, L.; Du, L.; Kuang, C.; Takikawa, O.; Yang, Q. Amyloid beta neurotoxicity is IDO1-Kyn-AhR dependent and blocked by IDO1 inhibitor. Signal Transduct. Target. Ther. 2020, 5, 96. [Google Scholar] [CrossRef]
- Finch, C.E.; Kulminski, A.M. The Alzheimer’s Disease Exposome. Alzheimers Dement. 2019, 15, 1123–1132. [Google Scholar] [CrossRef]
- Margaret, G.; Reynolds, C.A.; Fratiglioni, L.; Johansson, B.; Mortimer, J.A.; Berg, S.; Fiske, A.; Pedersen, N.L. Role of genes and environments for explaining Alzheimer disease. Arch. Gen. Psychiatry 2006, 63, 168–174. [Google Scholar]
- Angeles-Lopez, Q.D.; Garcia-Lara, L.; Aguirre-Pineda, N.; Castaneda-Arellano, R.; Elizondo-Azuela, G.; Perez-Severiano, F.; Segovia, J. The absence of the aryl hydrocarbon receptor in the R6/1 transgenic mouse model of Huntington’s disease improves the neurological phenotype. Behav. Brain Res. 2021, 408, 113230. [Google Scholar] [CrossRef]
- Jauch, D.; Urbariska, E.M.; Guidetti, P.; Bird, E.D.; Vonsattel, J.P.; Whetsell, W.O., Jr.; Schwarcz, R. Dysfunction of brain kynurenic acid metabolism in Huntington’s disease: Focus on kynurenine aminotransferases. J. Neurol. Sci. 1995, 130, 39–47. [Google Scholar] [CrossRef]
- Bondulich, M.K.; Fan, Y.; Song, Y.; Giorgini, F.; Bates, G.P. Ablation of kynurenine 3-monooxygenase rescues plasma inflammatory cytokine levels in the R6/2 mouse model of Huntington’s disease. Sci. Rep. 2021, 11, 5484. [Google Scholar] [CrossRef] [PubMed]
- Jangi, S.; Gandhi, R.; Cox, L.M.; Li, N.; von Glehn, F.; Yan, R.; Patel, B.; Mazzola, M.A.; Liu, S.; Glanz, B.L.; et al. Alterations of the human gut microbiome in multiple sclerosis. Nat. Commun. 2016, 7, 12015. [Google Scholar] [CrossRef] [PubMed]
- Zorlu, N.; Hoffjan, S.; Haghikia, A.; Deyneko, I.V.; Epplen, J.T. Evaluation of variation in genes of the arylhydrocarbon receptor pathway for an association with multiple sclerosis. J. Neuroimmunol. 2019, 334, 576979. [Google Scholar] [CrossRef]
- Wheeler, M.A.; Quintana, F.J. Regulation of Astrocyte Functions in Multiple Sclerosis. Cold Spring Harb. Perspect. Med. 2019, 9, a029009. [Google Scholar] [CrossRef]
- Kaye, J.; Piryatinsky, V.; Birnberg, T.; Hingaly, T.; Raymond, E.; Kashi, R.; Amit-Romach, E.; Caballero, I.S.; Towfic, F.; Ator, M.A.; et al. Laquinimod arrests experimental autoimmune encephalomyelitis by activating the aryl hydrocarbon receptor. Proc. Natl. Acad. Sci. USA 2016, 113, E6145–E6152. [Google Scholar] [CrossRef] [Green Version]
- Wingerchuk, D.M. Smoking: Effects on multiple sclerosis susceptibility and disease progression. Ther. Adv. Neurol. Disord. 2012, 5, 13–22. [Google Scholar] [CrossRef] [Green Version]
- Zeilinger, S.; Kuhnel, B.; Klopp, N.; Baurecht, H.; Kleinschmidt, A.; Gieger, C.; Weidinger, S.; Lattka, E.; Adamski, J.; Peters, A.; et al. Tobacco smoking leads to extensive genome-wide changes in DNA methylation. PLoS ONE 2013, 8, e63812. [Google Scholar] [CrossRef]
- Mackenzie, I.R.; Rademakers, R. The role of transactive response DNA-binding protein-43 in amyotrophic lateral sclerosis and frontotemporal dementia. Curr. Opin. Neurol. 2008, 21, 693–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ash, P.E.A.; Stanford, E.A.; Al Abdulatif, A.; Ramirez-Cardenas, A.; Ballance, H.I.; Boudeau, S.; Jeh, A.; Murithi, J.M.; Tripodis, Y.; Murphy, G.J.; et al. Dioxins and related environmental contaminants increase TDP-43 levels. Mol. Neurodegener. 2017, 12, 35. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ojo, E.S.; Tischkau, S.A. The Role of AhR in the Hallmarks of Brain Aging: Friend and Foe. Cells 2021, 10, 2729. https://doi.org/10.3390/cells10102729
Ojo ES, Tischkau SA. The Role of AhR in the Hallmarks of Brain Aging: Friend and Foe. Cells. 2021; 10(10):2729. https://doi.org/10.3390/cells10102729
Chicago/Turabian StyleOjo, Emmanuel S., and Shelley A. Tischkau. 2021. "The Role of AhR in the Hallmarks of Brain Aging: Friend and Foe" Cells 10, no. 10: 2729. https://doi.org/10.3390/cells10102729
APA StyleOjo, E. S., & Tischkau, S. A. (2021). The Role of AhR in the Hallmarks of Brain Aging: Friend and Foe. Cells, 10(10), 2729. https://doi.org/10.3390/cells10102729