
Antihistamines Potentiate Dexamethasone Anti-Inflammatory Effects. Impact on Glucocorticoid Receptor-Mediated Expression of Inflammation-Related Genes

 ,
,
Abstract
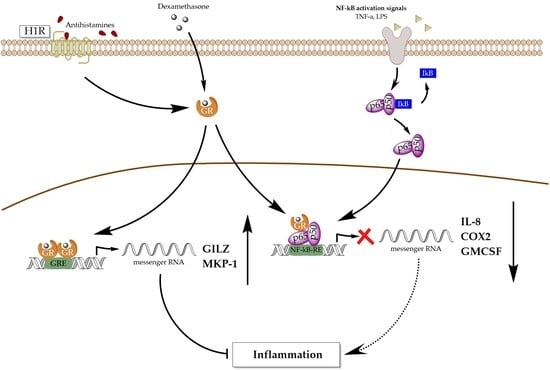
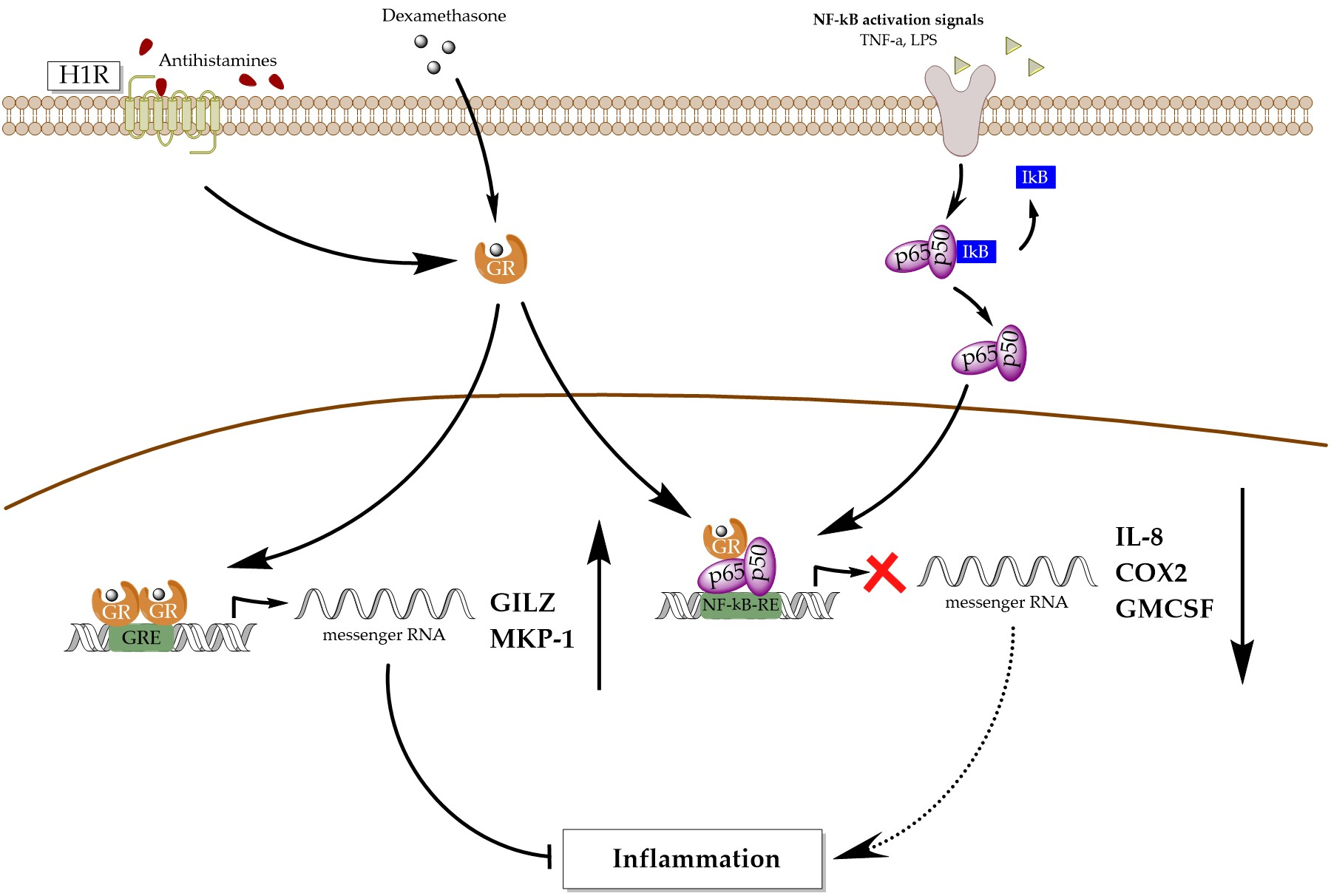
1. Introduction
2. Materials & Methods
2.1. Materials
2.2. Plasmid Constructions
2.3. Cell Culture
Transfection and Reporter Gene Assays
2.4. RT-PCR and Quantitative Real-Time PCR
2.5. MTS Assay
2.6. Data Analysis
3. Results
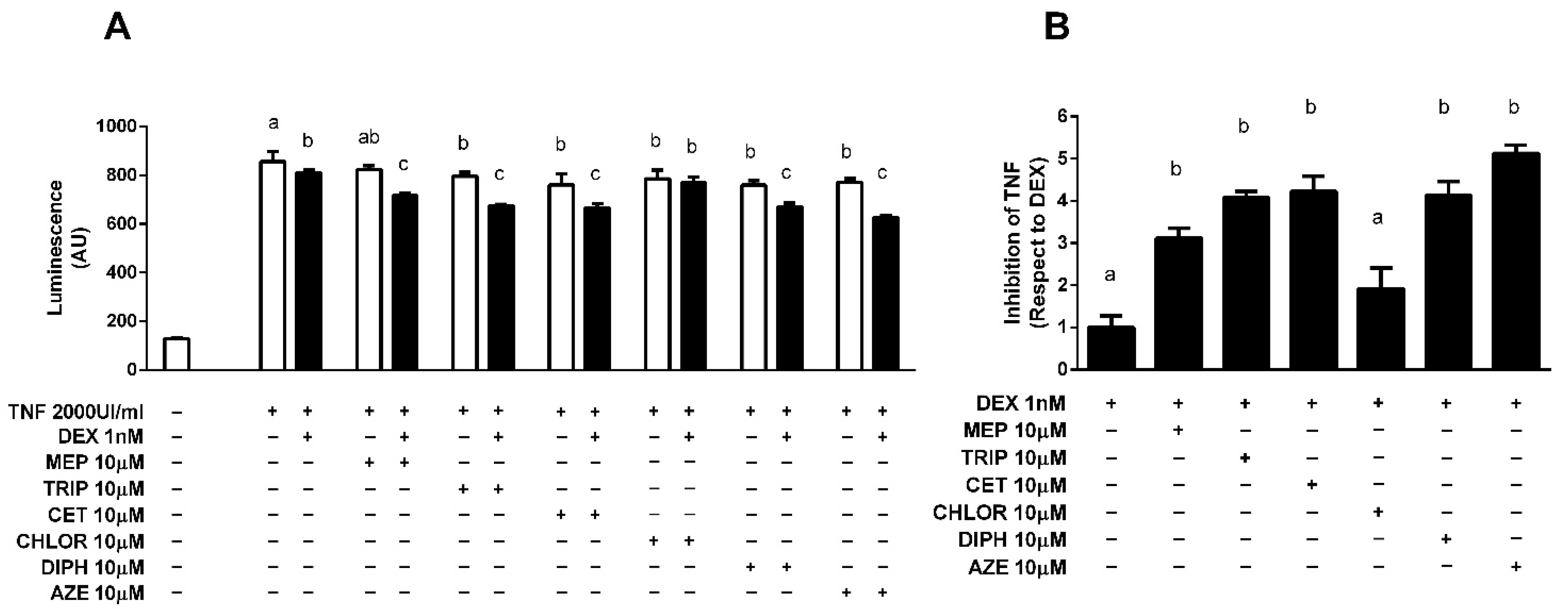
3.1. Antihistamines Enhance Dexamethasone-Mediated Inhibition of NF-κB Activity Induced by TNF-α
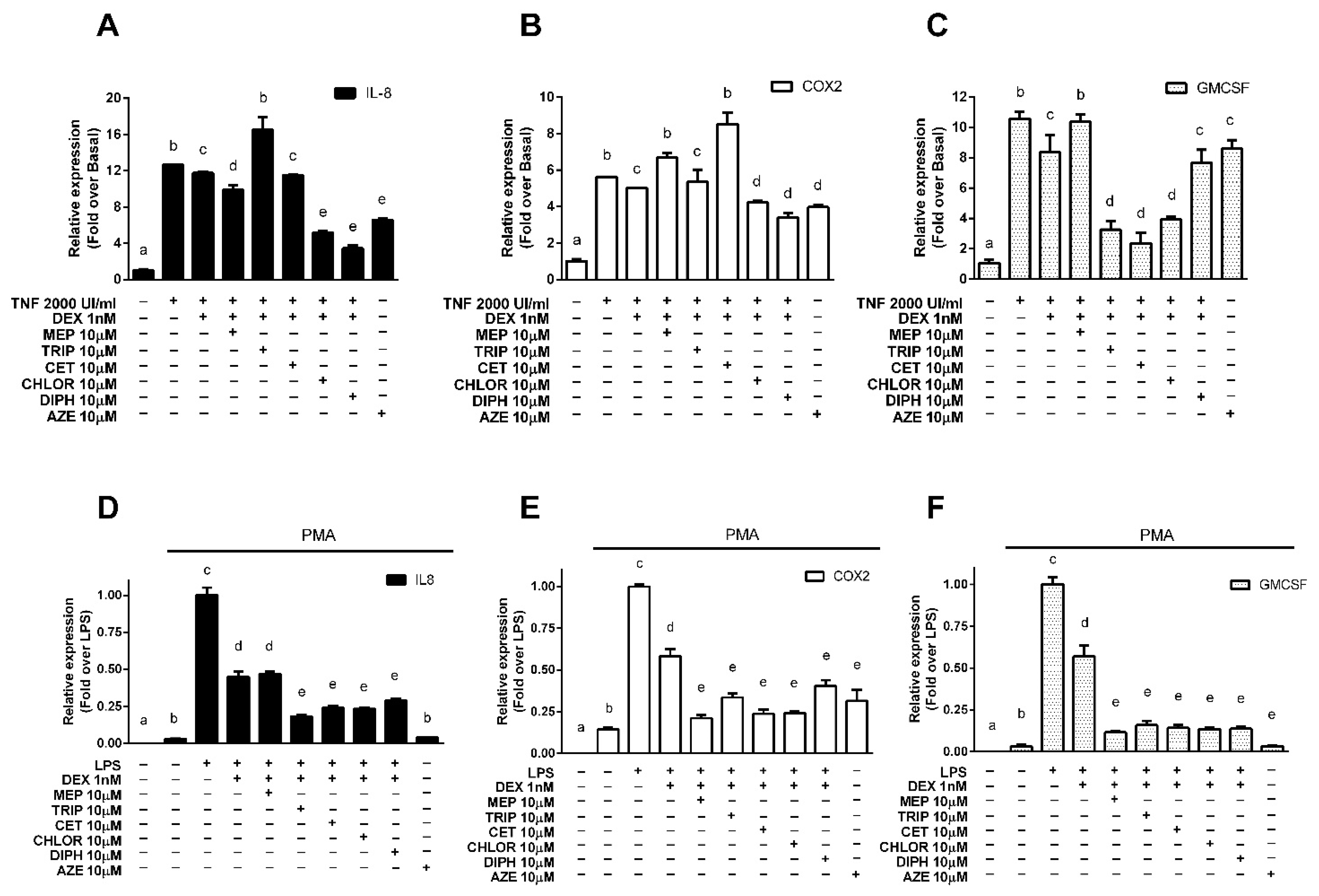
3.2. Modulation of Endogenous Pro-Inflammatory Gene Expression
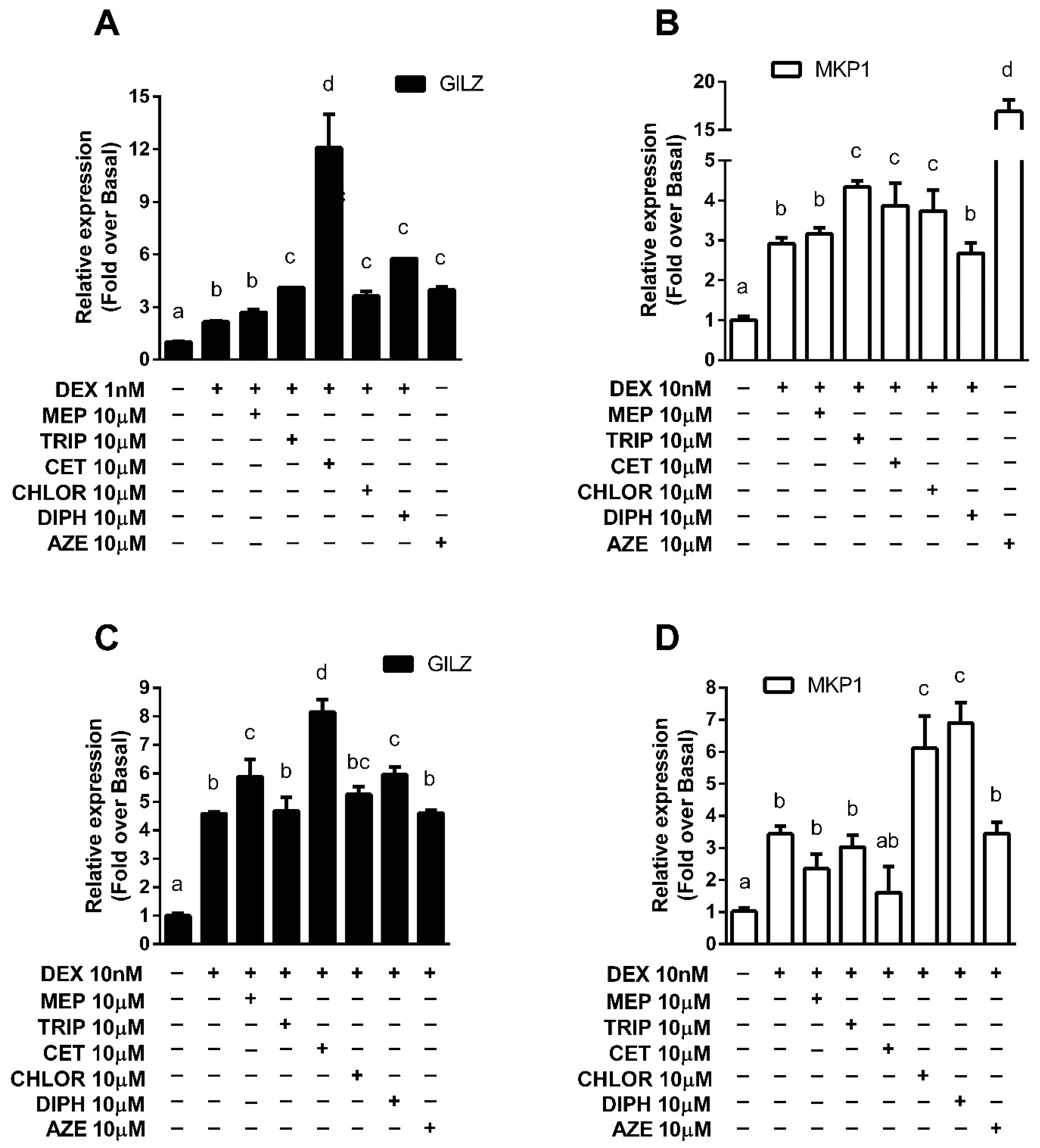
3.3. Modulation of Endogenous Anti-Inflammatory Gene Expression
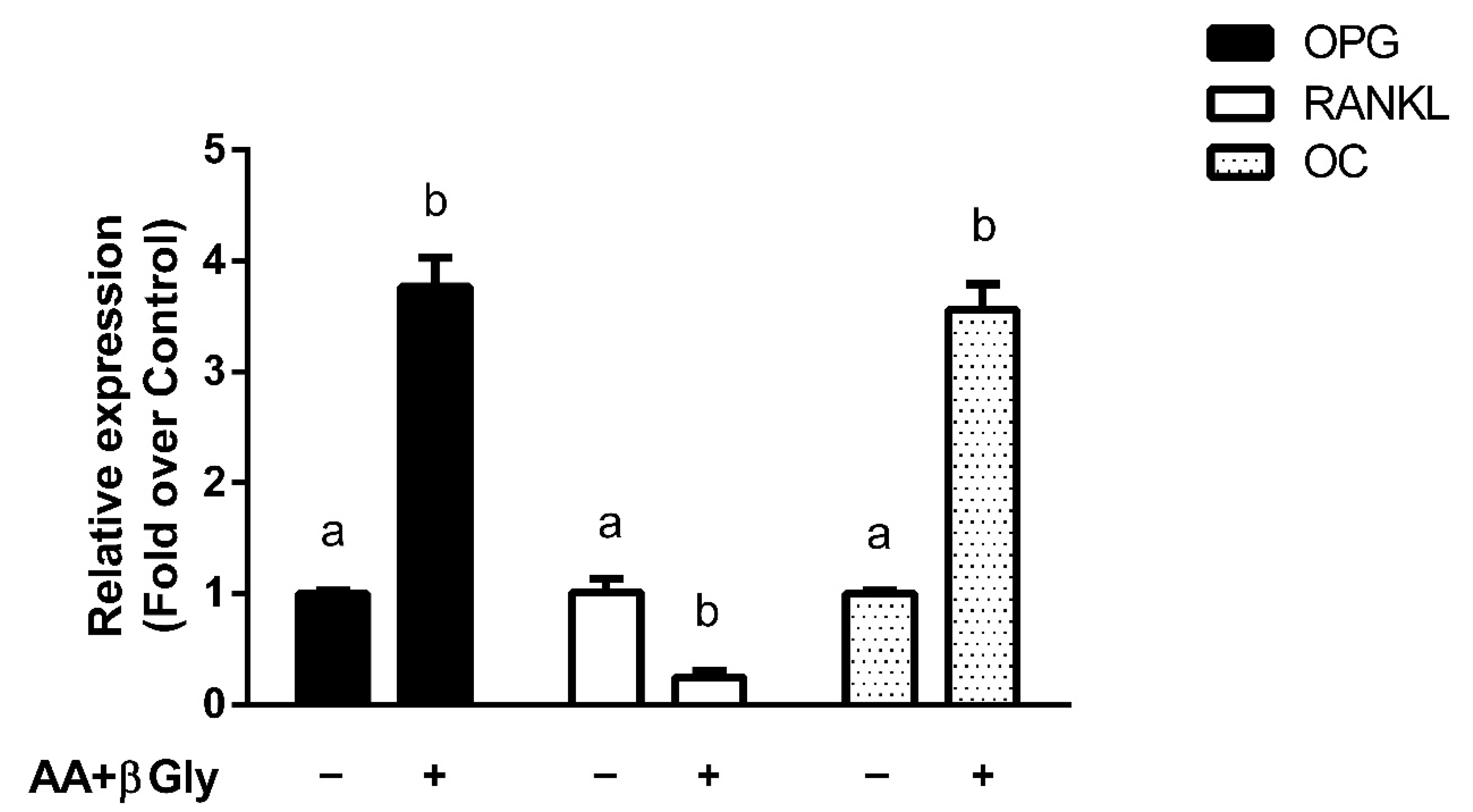
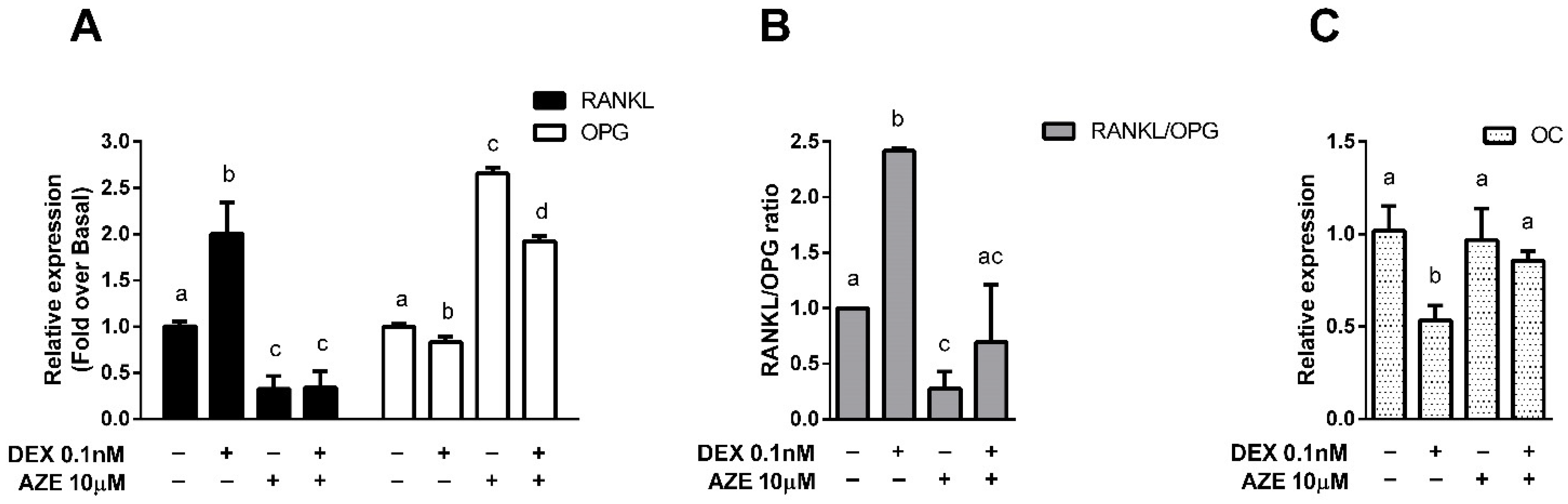
3.4. Modulation of Bone Metabolism Markers Expression
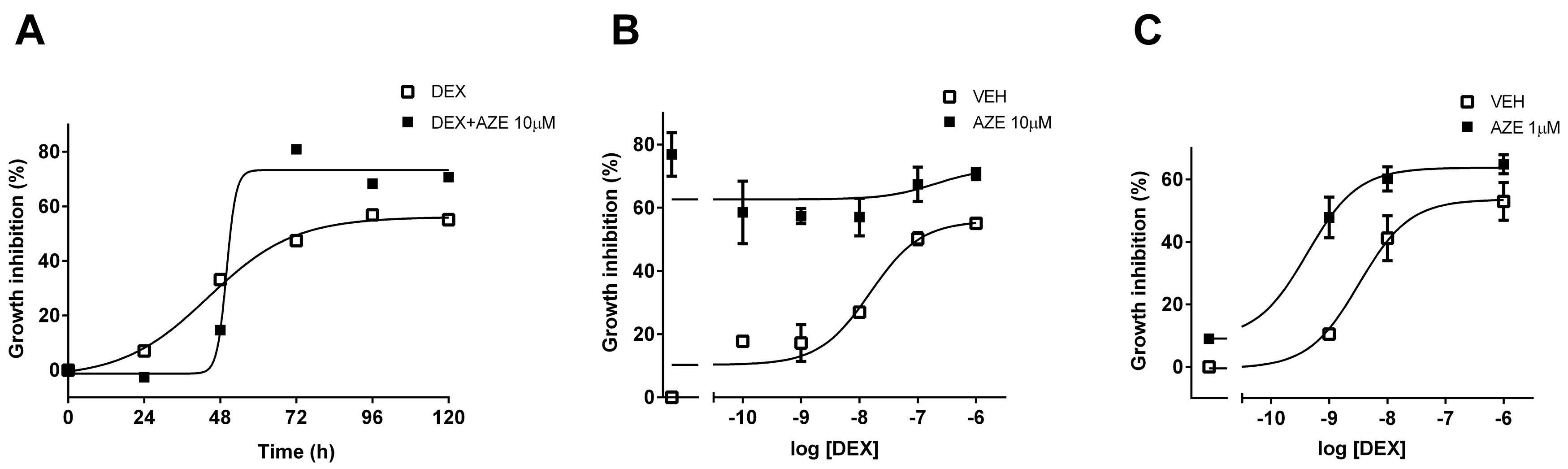
3.5. Effect of AZE on Differentiated MC3T3-E1 Cell Proliferation
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Furman, D.; Campisi, J.; Verdin, E.; Carrera-Bastos, P.; Targ, S.; Franceschi, C.; Ferrucci, L.; Gilroy, D.W.; Fasano, A.; Miller, G.W.; et al. Chronic Inflammation in the Etiology of Disease across the Life Span. Nat. Med. 2019, 25, 1822–1832. [Google Scholar] [CrossRef]
- Buttorff, C.; Ruder, T.; Bauman, M. Multiple Chronic Conditions in the United States; RAND Corporation: Santa Monica, CA, USA, 2017. [Google Scholar]
- Barnes, P.J. How corticosteroids control inflammation: Quintiles prize lecture 2005. Br. J. Pharmacol. 2006, 148, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Clark, A.R.; Belvisi, M.G. Maps and legends: The quest for dissociated ligands of the glucocorticoid receptor. Pharmacol. Ther. 2012, 134, 54–67. [Google Scholar] [CrossRef] [PubMed]
- Nicolaides, N.C.; Galata, Z.; Kino, T.; Chrousos, G.P.; Charmandari, E. The human glucocorticoid receptor: Molecular basis of biologic function. Steroids 2010, 75, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Rhen, T.; Cidlowski, J.A. Antiinflammatory action of glucocorticoids—New mechanisms for old drugs. N. Engl. J. Med. 2005, 353, 1711–1723. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, S.; Lozach, J.; Benner, C.; Pascual, G.; Tangirala, R.K.; Westin, S.; Hoffmann, A.; Subramaniam, S.; David, M.; Rosenfeld, M.G.; et al. Molecular determinants of crosstalk between nuclear receptors and toll-like receptors. Cell 2005, 122, 707–721. [Google Scholar] [CrossRef] [PubMed]
- Reily, M.M.; Pantoja, C.; Hu, X.; Chinenov, Y.; Rogatsky, I. The GRIP1:IRF3 Interaction as a target for glucocorticoid receptor-mediated immunosuppression. EMBO J. 2006, 25, 108–117. [Google Scholar] [CrossRef]
- De Bosscher, K.; Haegeman, G.; Elewaut, D. Targeting inflammation using selective glucocorticoid receptor modulators. Curr. Opin. Pharmacol. 2010, 10, 497–504. [Google Scholar] [CrossRef]
- Glass, C.K.; Saijo, K. Nuclear receptor transrepression pathways that regulate inflammation in macrophages and T cells. Nat. Rev. Immunol. 2010, 10, 365–376. [Google Scholar] [CrossRef]
- Newton, R.; Holden, N.S. Separating transrepression and transactivation: A distressing divorce for the glucocorticoid receptor? Mol. Pharmacol. 2007, 72, 799–809. [Google Scholar] [CrossRef]
- Schäcke, H.; Rehwinkel, H.; Asadullah, K. Dissociated glucocorticoid receptor ligands: Compounds with an improved therapeutic index. Curr. Opin. Investig. Drugs 2005, 6, 503–507. [Google Scholar] [PubMed]
- Kleiman, A.; Hübner, S.; Rodriguez Parkitna, J.M.; Neumann, A.; Hofer, S.; Weigand, M.A.; Bauer, M.; Schmid, W.; Schütz, G.; Libert, C.; et al. Glucocorticoid Receptor dimerization is required for survival in septic shock via suppression of interleukin-1 in macrophages. FASEB J. 2012, 26, 722–729. [Google Scholar] [CrossRef] [PubMed]
- Tuckermann, J.P.; Kleiman, A.; Moriggl, R.; Spanbroek, R.; Neumann, A.; Illing, A.; Clausen, B.E.; Stride, B.; Förster, I.; Habenicht, A.J.R.; et al. Macrophages and neutrophils are the targets for immune suppression by glucocorticoids in contact allergy. J. Clin. Invest. 2007, 117, 1381–1390. [Google Scholar] [CrossRef] [PubMed]
- Vandevyver, S.; Dejager, L.; Van Bogaert, T.; Kleyman, A.; Liu, Y.; Tuckermann, J.; Libert, C. Glucocorticoid receptor dimerization induces MKP1 to protect against TNF-induced inflammation. J. Clin. Invest. 2012, 122, 2130–2140. [Google Scholar] [CrossRef] [PubMed]
- Chi, H.; Flavell, R.A. Acetylation of MKP-1 and the control of inflammation. Sci. Signal 2008, 1, pe44. [Google Scholar] [CrossRef]
- Ayroldi, E.; Cannarile, L.; Migliorati, G.; Nocentini, G.; Delfino, D.V.; Riccardi, C. Mechanisms of the anti-inflammatory effects of glucocorticoids: Genomic and nongenomic interference with MAPK signaling pathways. FASEB J. 2012, 26, 4805–4820. [Google Scholar] [CrossRef]
- Vandevyver, S.; Dejager, L.; Tuckermann, J.; Libert, C. New insights into the anti-inflammatory mechanisms of glucocorticoids: An emerging role for glucocorticoid-receptor-mediated transactivation. Endocrinology 2013, 154, 993–1007. [Google Scholar] [CrossRef]
- Quax, R.A.; Manenschijn, L.; Koper, J.W.; Hazes, J.M.; Lamberts, S.W.J.; van Rossum, E.F.C.; Feelders, R.A. Glucocorticoid sensitivity in health and disease. Nat. Rev. Endocrinol. 2013, 9, 670–686. [Google Scholar] [CrossRef]
- Thiele, S.; Ziegler, N.; Tsourdi, E.; De Bosscher, K.; Tuckermann, J.; Hofbauer, L.; Rauner, M. Selective Glucocorticoid receptor modulation maintains bone mineral density in mice. J. Bone Miner. Res. 2012, 27, 2242–2250. [Google Scholar] [CrossRef]
- Boyce, B.F.; Xing, L. The RANKL/RANK/OPG pathway. Curr. Osteoporos. Rep. 2007, 5, 98–104. [Google Scholar] [CrossRef]
- McLaughlin, F.; Mackintosh, J.; Hayes, B.P.; McLaren, A.; Uings, I.J.; Salmon, P.; Humphreys, J.; Meldrum, E.; Farrow, S.N. Glucocorticoid-Induced osteopenia in the mouse as assessed by histomorphometry, microcomputed tomography, and biochemical markers. Bone 2002, 30, 924–930. [Google Scholar] [CrossRef]
- Hofbauer, L.C.; Gori, F.; Riggs, B.L.; Lacey, D.L.; Dunstan, C.R.; Spelsberg, T.C.; Khosla, S. Stimulation of osteoprotegerin ligand and inhibition of osteoprotegerin production by glucocorticoids in human osteoblastic lineage cells: Potential paracrine mechanisms of glucocorticoid-induced osteoporosis. Endocrinology 1999, 140, 4382–4389. [Google Scholar] [CrossRef]
- Zappia, C.D.; Granja-Galeano, G.; Fernández, N.; Shayo, C.; Davio, C.; Fitzsimons, C.P.; Monczor, F. Effects of histamine H1 receptor signaling on glucocorticoid receptor activity. Role of canonical and non-canonical pathways. Sci. Rep. 2015, 5, 17476. [Google Scholar] [CrossRef] [PubMed]
- Zappia, C.D.; Soto, A.; Granja-Galeano, G.; Fenoy, I.; Fernandez, N.; Davio, C.A.; Shayo, C.; Fitzsimons, C.P.; Goldman, A.; Monczor, F. Azelastine potentiates antiasthmatic dexamethasone effect on a murine asthma model. Pharmacol. Res. Perspect. 2019, 7, e00531. [Google Scholar] [CrossRef] [PubMed]
- Zappia, C.D.; Monczor, F. Therapeutic utility of glucocorticoids and antihistamines cotreatment. Rationale and perspectives. Pharmacol. Res. Perspect. 2019, 7, e00530. [Google Scholar] [CrossRef] [PubMed]
- Godowski, P.J.; Rusconi, S.; Miesfeld, R.; Yamamoto, K.R. Glucocorticoid receptor mutants that are constitutive activators of transcriptional enhancement. Nature 1987, 325, 365–368. [Google Scholar] [CrossRef] [PubMed]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef]
- Ghosh, C.C.; Ramaswami, S.; Juvekar, A.; Vu, H.-Y.; Galdieri, L.; Davidson, D.; Vancurova, I. Gene-specific repression of proinflammatory cytokines in stimulated human macrophages by nuclear IκBα. J. Immunol. 2010, 185, 3685–3693. [Google Scholar] [CrossRef]
- Grkovich, A.; Johnson, C.A.; Buczynski, M.W.; Dennis, E.A. Lipopolysaccharide-induced Cyclooxygenase-2 expression in human U937 macrophages is phosphatidic acid Phosphohydrolase-1-Dependent. J. Biol. Chem. 2006, 281, 32978–32987. [Google Scholar] [CrossRef]
- Nishida, T.; Takano, M.; Kawakami, T.; Nishino, N.; Nakai, S.; Hirai, Y. The transcription of the interleukin 1 Beta Gene is induced with PMA and inhibited with dexamethasone in U937 cells. Biochem. Biophys. Res. Commun. 1988, 156, 269–274. [Google Scholar] [CrossRef]
- Schreiber, J.; Jenner, R.G.; Murray, H.L.; Gerber, G.K.; Gifford, D.K.; Young, R.A. Coordinated binding of NF-KappaB family members in the response of human cells to lipopolysaccharide. Proc. Natl. Acad. Sci. USA 2006, 103, 5899–5904. [Google Scholar] [CrossRef] [PubMed]
- Sharif, O.; Bolshakov, V.N.; Raines, S.; Newham, P.; Perkins, N.D. Transcriptional profiling of the LPS induced NF-KappaB response in macrophages. BMC Immunol. 2007, 8, 1. [Google Scholar] [CrossRef] [PubMed]
- Czekanska, E.M.; Stoddart, M.J.; Richards, R.G.; Hayes, J.S. In search of an osteoblast cell model for in vitro research. Eur. Cell. Mater. 2012, 24, 1–17. [Google Scholar] [CrossRef]
- Fitzsimons, C.P.; Monczor, F.; Fernández, N.; Shayo, C.; Davio, C. Mepyramine, a histamine H1 receptor inverse agonist, binds preferentially to a G protein-coupled form of the receptor and sequesters G protein. J. Biol. Chem. 2004, 279, 34431–34439. [Google Scholar] [CrossRef] [PubMed]
- Bakker, R.A.; Schoonus, S.B.; Smit, M.J.; Timmerman, H.; Leurs, R. Histamine H(1)-receptor activation of nuclear factor-Kappa B: Roles for G beta gamma- and G alpha(q/11)-subunits in constitutive and agonist-mediated signaling. Mol. Pharmacol. 2001, 60, 1133–1142. [Google Scholar] [CrossRef]
- Bakker, R.A.; Nicholas, M.W.; Smith, T.T.; Burstein, E.S.; Hacksell, U.; Timmerman, H.; Leurs, R.; Brann, M.R.; Weiner, D.M. In vitro pharmacology of clinically used central nervous system-active drugs as inverse H(1) receptor agonists. J. Pharmacol. Exp. Ther. 2007, 322, 172–179. [Google Scholar] [CrossRef] [PubMed]
- Burghi, V.; Echeverría, E.B.; Zappia, C.D.; Díaz Nebreda, A.; Ripoll, S.; Gómez, N.; Shayo, C.; Davio, C.A.; Monczor, F.; Fernández, N.C. Biased agonism at histamine H1 receptor: Desensitization, internalization and MAPK activation triggered by antihistamines. Eur. J. Pharmacol. 2021, 896, 173913. [Google Scholar] [CrossRef] [PubMed]
- Monczor, F.; Fernandez, N.; Fitzsimons, C.P.; Shayo, C.; Davio, C. Antihistaminergics and inverse agonism: Potential therapeutic applications. Eur. J. Pharmacol. 2013, 715, 26–32. [Google Scholar] [CrossRef]
- John, S.; Johnson, T.A.; Sung, M.-H.; Biddie, S.C.; Trump, S.; Koch-Paiz, C.A.; Davis, S.R.; Walker, R.; Meltzer, P.S.; Hager, G.L. Kinetic complexity of the global response to glucocorticoid receptor action. Endocrinology 2009, 150, 1766–1774. [Google Scholar] [CrossRef]
- Bolton, E.C.; So, A.Y.; Chaivorapol, C.; Haqq, C.M.; Li, H.; Yamamoto, K.R. Cell- and gene-specific regulation of primary target genes by the androgen receptor. Genes Dev. 2007, 21, 2005–2017. [Google Scholar] [CrossRef]
- Gertz, J.; Savic, D.; Varley, K.E.; Partridge, E.C.; Safi, A.; Jain, P.; Cooper, G.M.; Reddy, T.E.; Crawford, G.E.; Myers, R.M. Distinct properties of cell-type-specific and shared transcription factor binding sites. Mol. Cell 2013, 52, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Rogatsky, I.; Wang, J.-C.; Derynck, M.K.; Nonaka, D.F.; Khodabakhsh, D.B.; Haqq, C.M.; Darimont, B.D.; Garabedian, M.J.; Yamamoto, K.R. Target-specific utilization of transcriptional regulatory surfaces by the glucocorticoid receptor. Proc. Natl. Acad. Sci. USA 2003, 100, 13845–13850. [Google Scholar] [CrossRef]
- So, A.Y.-L.; Chaivorapol, C.; Bolton, E.C.; Li, H.; Yamamoto, K.R. Determinants of cell- and gene-specific transcriptional regulation by the glucocorticoid receptor. PLoS Genet. 2007, 3, e94. [Google Scholar] [CrossRef]
- Collingwood, T.N.; Urnov, F.D.; Wolffe, A.P. Nuclear receptors: Coactivators, Corepressors and chromatin remodeling in the control of transcription. J. Mol. Endocrinol. 1999, 23, 255–275. [Google Scholar] [CrossRef] [PubMed]
- Monczor, F.; Chatzopoulou, A.; Zappia, C.D.; Houtman, R.; Meijer, O.C.; Fitzsimons, C.P. A model of glucocorticoid receptor interaction with coregulators predicts transcriptional regulation of target genes. Front. Pharmacol. 2019, 10, 214. [Google Scholar] [CrossRef] [PubMed]
- Smoak, K.A.; Cidlowski, J.A. Mechanisms of glucocorticoid receptor signaling during inflammation. Mech. Ageing Dev. 2004, 125, 697–706. [Google Scholar] [CrossRef] [PubMed]
- Tsaprouni, L.G.; Ito, K.; Adcock, I.M.; Punchard, N. Suppression of lipopolysaccharide- and tumour necrosis factor-alpha-induced interleukin (IL)-8 expression by glucocorticoids involves changes in IL-8 promoter acetylation. Clin. Exp. Immunol. 2007, 150, 151–157. [Google Scholar] [CrossRef]
- Nissen, R.; Yamamoto, K. The glucocorticoid receptor inhibits NFκB by interfering with serine-2 phosphorylation of the RNA polymerase II carboxy-terminal domain. Genes Dev. 2000, 14, 2314–2329. [Google Scholar] [CrossRef]
- Ito, K.; Barnes, P.J.; Adcock, I.M. Glucocorticoid receptor recruitment of histone deacetylase 2 inhibits interleukin-1beta-induced histone H4 acetylation on lysines 8 and 12. Mol. Cell. Biol. 2000, 20, 6891–6903. [Google Scholar] [CrossRef]
- Cho, I.J.; Kim, S.G. A novel mitogen-activated protein kinase phosphatase-1 and glucocorticoid receptor (GR) interacting protein-1-dependent combinatorial mechanism of gene transrepression by GR. Mol. Endocrinol. 2009, 23, 86–99. [Google Scholar] [CrossRef]
- Barrios-Rodiles, M.; Tiraloche, G.; Chadee, K. Lipopolysaccharide modulates cyclooxygenase-2 Transcriptionally and posttranscriptionally in human macrophages independently from endogenous IL-1 Beta and TNF-Alpha. J. Immunol. 1999, 163, 963–969. [Google Scholar]
- Schäcke, H.; Döcke, W.D.; Asadullah, K. Mechanisms involved in the side effects of glucocorticoids. Pharmacol. Ther. 2002, 96, 23–43. [Google Scholar] [CrossRef]
- Whittier, X.; Saag, K.G. Glucocorticoid-induced osteoporosis. Rheum. Dis. Clin. N. Am. 2016, 42, 177–189. [Google Scholar] [CrossRef] [PubMed]
- Hardy, R.S.; Zhou, H.; Seibel, M.J.; Cooper, M.S. Glucocorticoids and bone: Consequences of Endogenous and exogenous excess and replacement therapy. Endocr. Rev. 2018, 39, 519–548. [Google Scholar] [CrossRef] [PubMed]
- Biosse-Duplan, M.; Baroukh, B.; Dy, M.; de Vernejoul, M.-C.; Saffar, J.-L. Histamine promotes osteoclastogenesis through the differential expression of histamine receptors on osteoclasts and osteoblasts. Am. J. Pathol. 2009, 174, 1426–1434. [Google Scholar] [CrossRef] [PubMed]
- Ikawa, Y.; Yonekawa, T.; Ohkuni, Y.; Kuribayashi, M.; Fukino, K.; Ueno, K. A comparative study of histamine activities on differentiation of osteoblasts and osteoclasts. J. Toxicol. Sci. 2007, 32, 555–564. [Google Scholar] [CrossRef]
- Kinjo, M.; Setoguchi, S.; Solomon, D.H. Antihistamine therapy and bone mineral density: Analysis in a population-based US sample. Am. J. Med. 2008, 121, 1085–1091. [Google Scholar] [CrossRef][Green Version]
- Wiercigroch, M.; Folwarczna, J. Histamine in regulation of bone remodeling processes. Postepy Hig. Med. Dosw. 2013, 67, 887–895. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| A549 | U937 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| IL-8 | COX2 | GMCSF | GILZ | MKP1 | IL-8 | COX2 | GMCSF | GILZ | MKP1 | |
| Mepyramine | 3.12 ± 0.95 * | −2.71 ± 1.05 * | 0.08 ± 0.38 * | 1.26 ± 0.11 | 1.09 ± 0.08 | 0.97 ± 0.06 | 1.88 ± 0.09 * | 2.05 ± 0.03 * | 1.28 ± 0.23 * | 0.68 ± 0.30 |
| Trans-triprolidine | −4.41 ± 2.71 * | 0.71 ± 2.96 | 3.32 ± 0.37 * | 1.91 ± 0.01 * | 1.49 ± 0.09 * | 1.48 ± 0.04 * | 1.59 ± 0.10 * | 1.95 ± 0.10 * | 1.02 ± 0.18 | 0.88 ± 0.24 |
| Cetirizine | 1.28 ± 0.07 | −7.54 ± 4.00 * | 3.73 ± 0.44 * | 5.63 ± 1.26 * | 1.33 ± 0.33 * | 1.37 ± 0.03 * | 1.82 ± 0.11 * | 1.99 ± 0.08 * | 1.78 ± 0.17 * | 0.47 ± 0.57 |
| Chlorpheniramine | 8.47 ± 0.29 * | 3.60 ± 0.39 * | 3.01 ± 0.12 * | 1.69 ± 0.23 * | 1.28 ± 0.31 * | 1.39 ± 0.03 * | 1.81 ± 0.04 * | 2.00 ± 0.04 * | 1.15 ± 0.10 | 1.78 ± 0.41 * |
| Diphenhydramine | 10.47 ± 0.64 * | 5.69 ± 1.03 * | 1.31 ± 0.55 | 2.69 ± 0.01 * | 0.92 ± 0.15 | 1.28 ± 0.03 * | 1.42 ± 0.14 * | 2.00 ± 0.05 * | 1.30 ± 0.10 * | 2.01 ± 0.26 * |
| Azelastine | 6.90 ± 0.34 * | 2.83 ± 2.47 * | 0.89 ± 0.37 | 1.85 ± 0.15 * | 5.81 ± 0.73 * | 1.74 ± 0.01 * | 1.63 ± 0.27 * | 2.25 ± 0.03 * | 1.01 ± 0.04 | 1.01 ± 0.15 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zappia, C.D.; Torralba-Agu, V.; Echeverria, E.; Fitzsimons, C.P.; Fernández, N.; Monczor, F. Antihistamines Potentiate Dexamethasone Anti-Inflammatory Effects. Impact on Glucocorticoid Receptor-Mediated Expression of Inflammation-Related Genes. Cells 2021, 10, 3026. https://doi.org/10.3390/cells10113026
Zappia CD, Torralba-Agu V, Echeverria E, Fitzsimons CP, Fernández N, Monczor F. Antihistamines Potentiate Dexamethasone Anti-Inflammatory Effects. Impact on Glucocorticoid Receptor-Mediated Expression of Inflammation-Related Genes. Cells. 2021; 10(11):3026. https://doi.org/10.3390/cells10113026
Chicago/Turabian StyleZappia, Carlos Daniel, Valeria Torralba-Agu, Emiliana Echeverria, Carlos P. Fitzsimons, Natalia Fernández, and Federico Monczor. 2021. "Antihistamines Potentiate Dexamethasone Anti-Inflammatory Effects. Impact on Glucocorticoid Receptor-Mediated Expression of Inflammation-Related Genes" Cells 10, no. 11: 3026. https://doi.org/10.3390/cells10113026
APA StyleZappia, C. D., Torralba-Agu, V., Echeverria, E., Fitzsimons, C. P., Fernández, N., & Monczor, F. (2021). Antihistamines Potentiate Dexamethasone Anti-Inflammatory Effects. Impact on Glucocorticoid Receptor-Mediated Expression of Inflammation-Related Genes. Cells, 10(11), 3026. https://doi.org/10.3390/cells10113026