
The Role of Glucocorticoids in Inflammatory Diseases

 , and
, and
Abstract
:1. Introduction
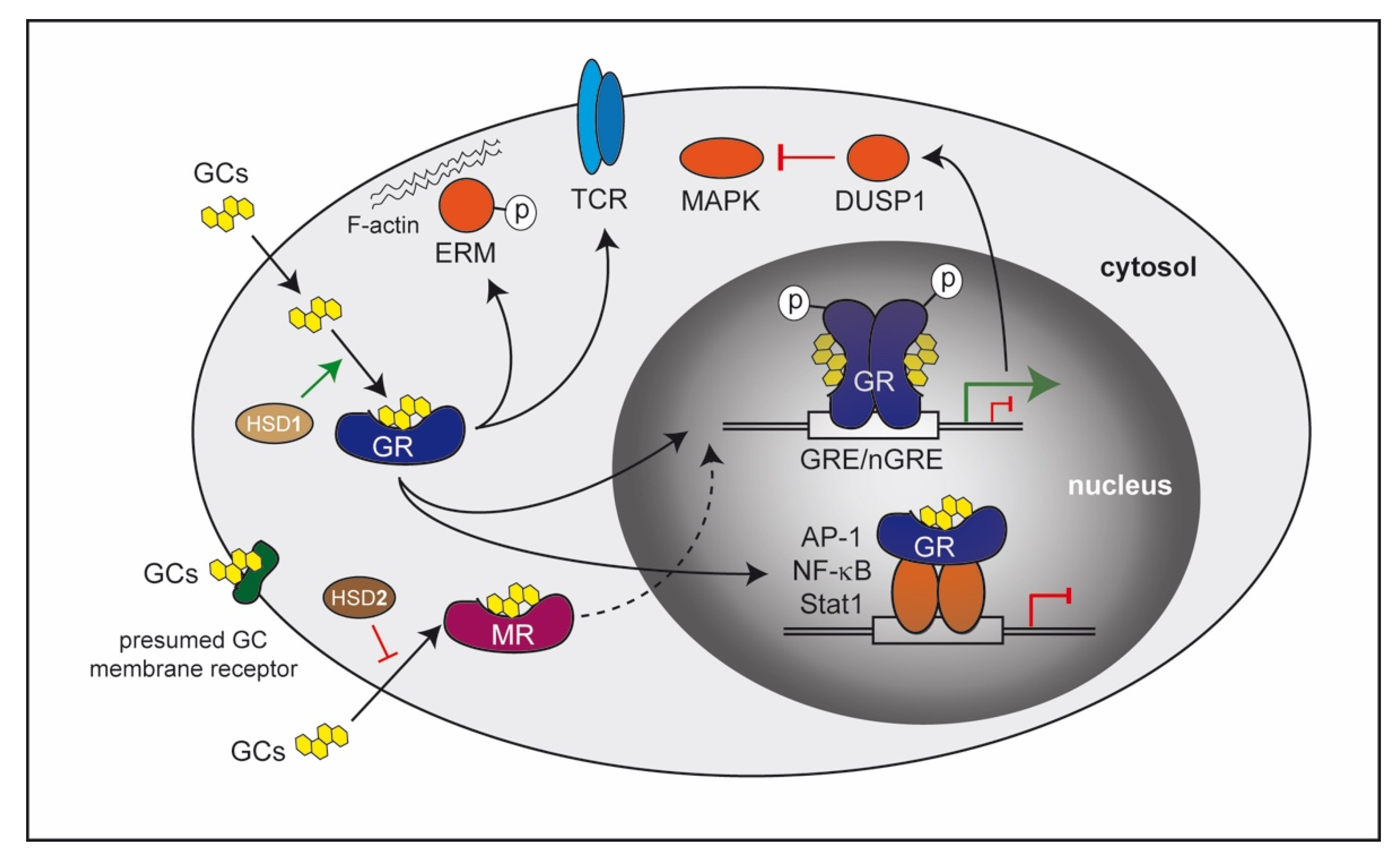
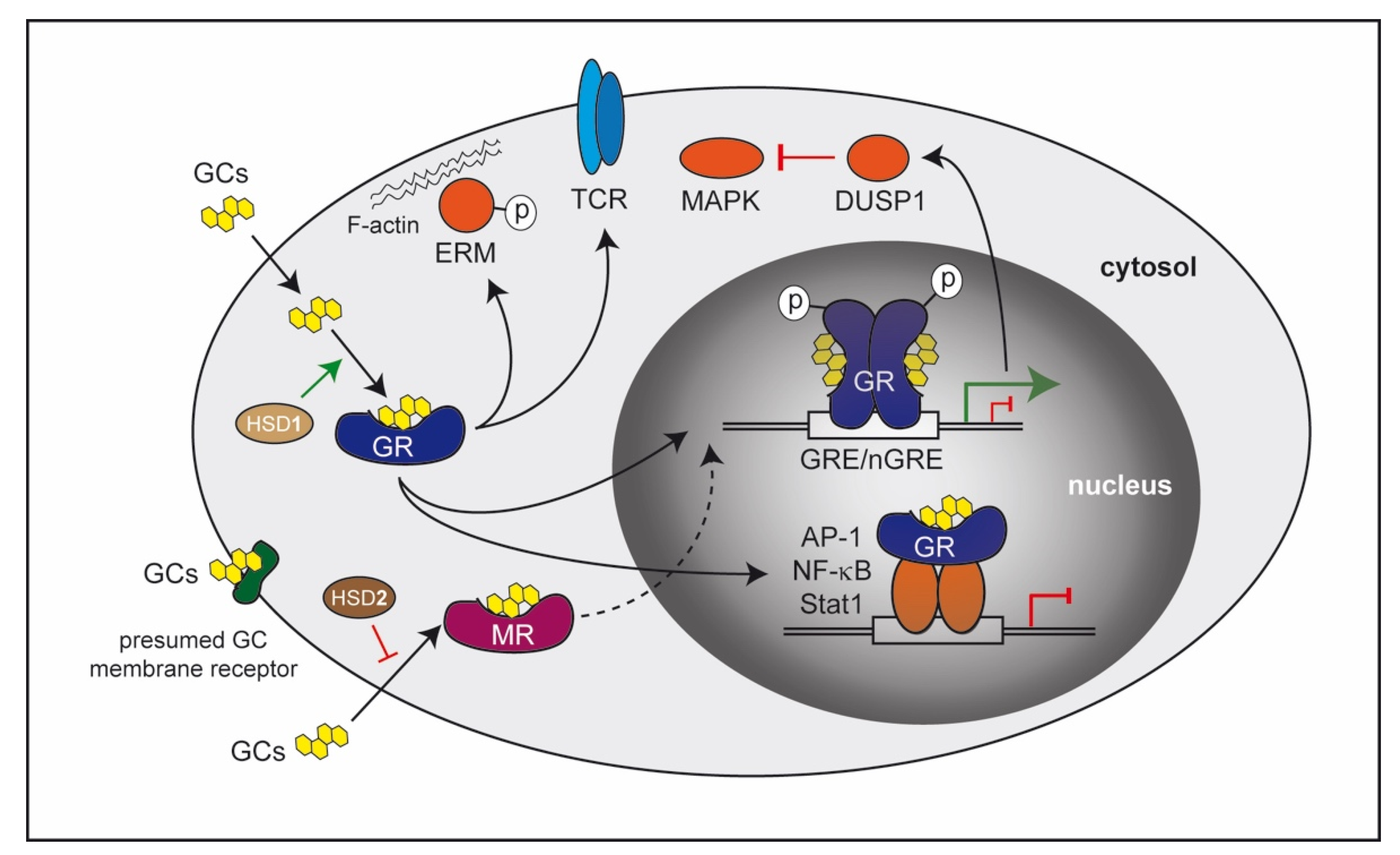
2. Molecular Mechanisms of GC Action
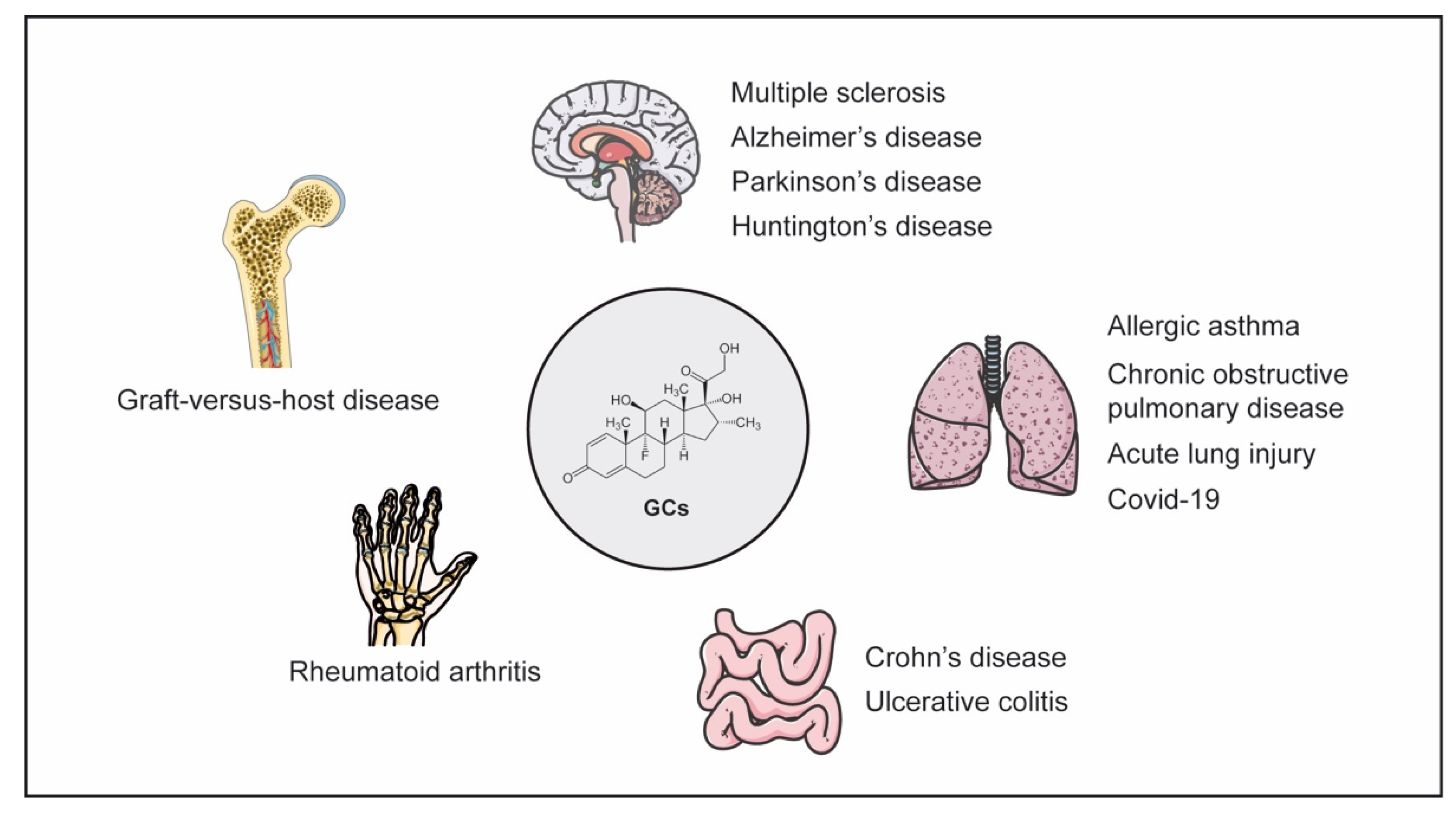
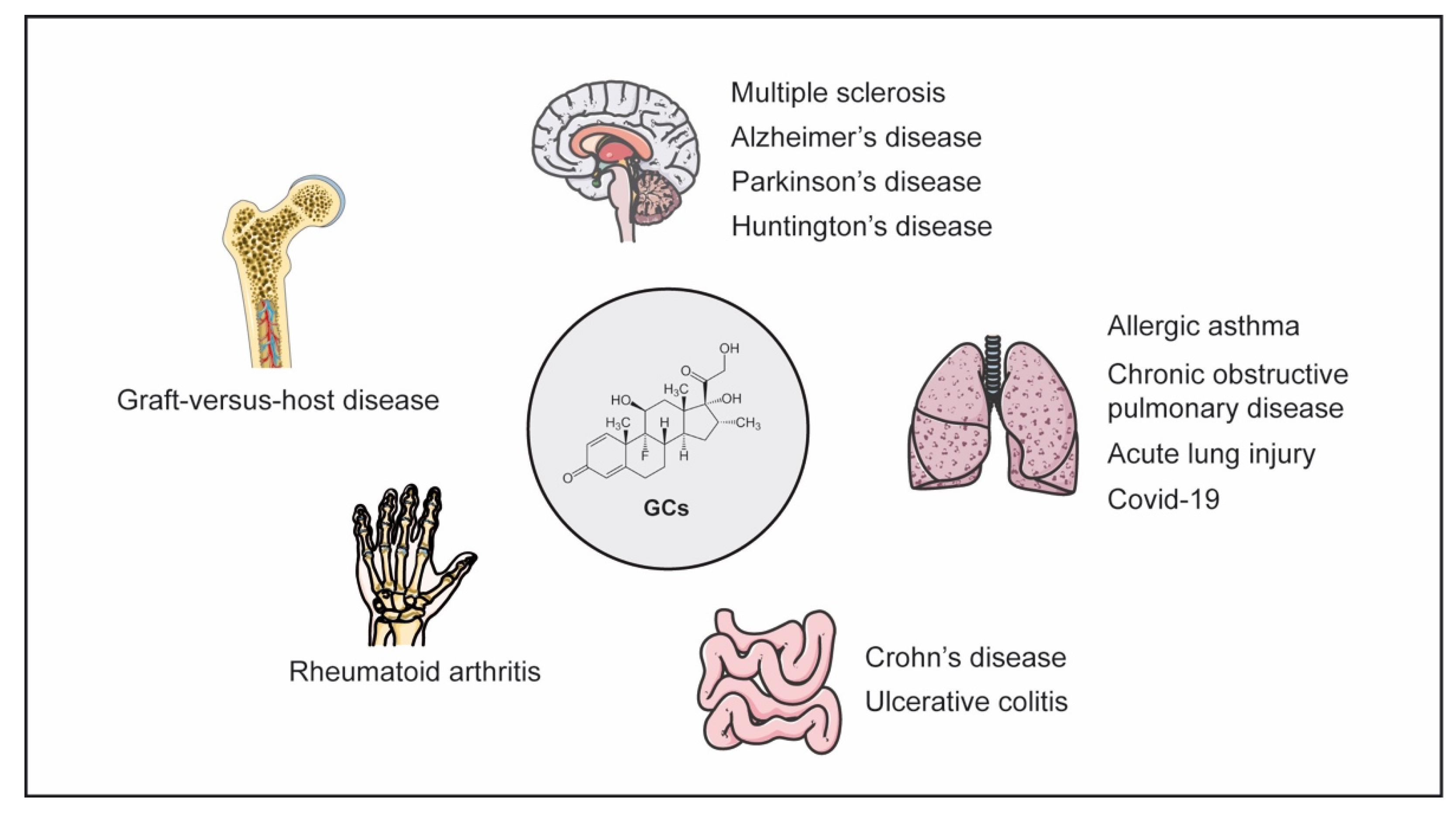
3. GCs in the Pathogenesis and Treatment of Inflammatory Diseases
3.1. Neuroinflammatory Diseases
3.2. Inflammatory Bowel Disease

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Type of Mutation | Model | Disease | Therapy (Free GCs) | Reference |
|---|---|---|---|---|---|
| GR | myeloid cell-specific deletion | DSS-colitis | aggravated | n.d. | [83] |
| GR | IEC-specific deletion, inducible | DSS-colitis | aggravated | n.d. | [7] |
| GR | Treg cell-specific deletion | T-cell transfer colitis | aggravated | n.d. | [84] |
| GR | impaired dimerization, ubiquitous | TNFα-induced intestinal inflammation | aggravated | abrogated | [27] |
| GILZ | neutrophil-specific deletion | DNBS-colitis | aggravated | n.d. | [90] |
| GILZ | T cell-specific deletion | DNBS-colitis | aggravated | n.d. | [91] |
3.3. Pulmonary Diseases
| Gene | Type of Mutation | Model | Disease | Therapy (Free GCs) | Reference |
|---|---|---|---|---|---|
| GR | deletion in immune cells | Ova/Alum immunization | unaltered | unaltered | [8] |
| GR | deletion in stromal cells | Ova/Alum immunization | unaltered | abrogated | [8] |
| GR | impaired dimerization, ubiquitous | Ova/Alum immunization | unaltered | abrogated | [8] |
| GR | AT2-specific deletion, inducible | Ova/Alum immunization | unaltered | partially abrogated | [8] |
| GR | myeloid cell-specific deletion | LPS/Oleic acidtreatment | unaltered | abrogated | [123] |
| GR | impaired dimerization, ubiquitous | LPS/Oleic acid treatment | unaltered | abrogated | [123] |
3.4. Rheumatoid Arthritis
| Gene | Type of Mutation | Model | Disease | Therapy (Free GCs) | Reference |
|---|---|---|---|---|---|
| GR | T cell-specific deletion | AIA | unaltered | abrogated | [141] |
| GR | myeloid cell-specific deletion | AIA | unaltered | unaltered | [141] |
| GR | B cell-specific deletion | AIA | unaltered | unaltered | [141] |
| GR | impaired dimerization, ubiquitous | AIA | unaltered | abrogated | [141] |
| GR | deletion in immune cells | STIA | unaltered | unaltered | [143] |
| GR | deletion in stromal cells | STIA | unaltered | abrogated | [143] |
| GR | FLS-specific deletion, inducible | STIA | unaltered | partially abrogated | [143] |
| 11β-HSD1 | ubiquitous deletion | STIA | aggravated | partially abrogated | [144,147] |
| AnX1 | ubiquitous deletion | STIA | unaltered | abrogated | [156] |
| GILZ | ubiquitous deletion | CIA | unaltered | unaltered | [162] |
| DUSP1 | ubiquitous deletion | CIA | aggravated | n.d. | [163] |
3.5. Graft-Versus-Host Disease
4. Novel Therapeutic Approaches
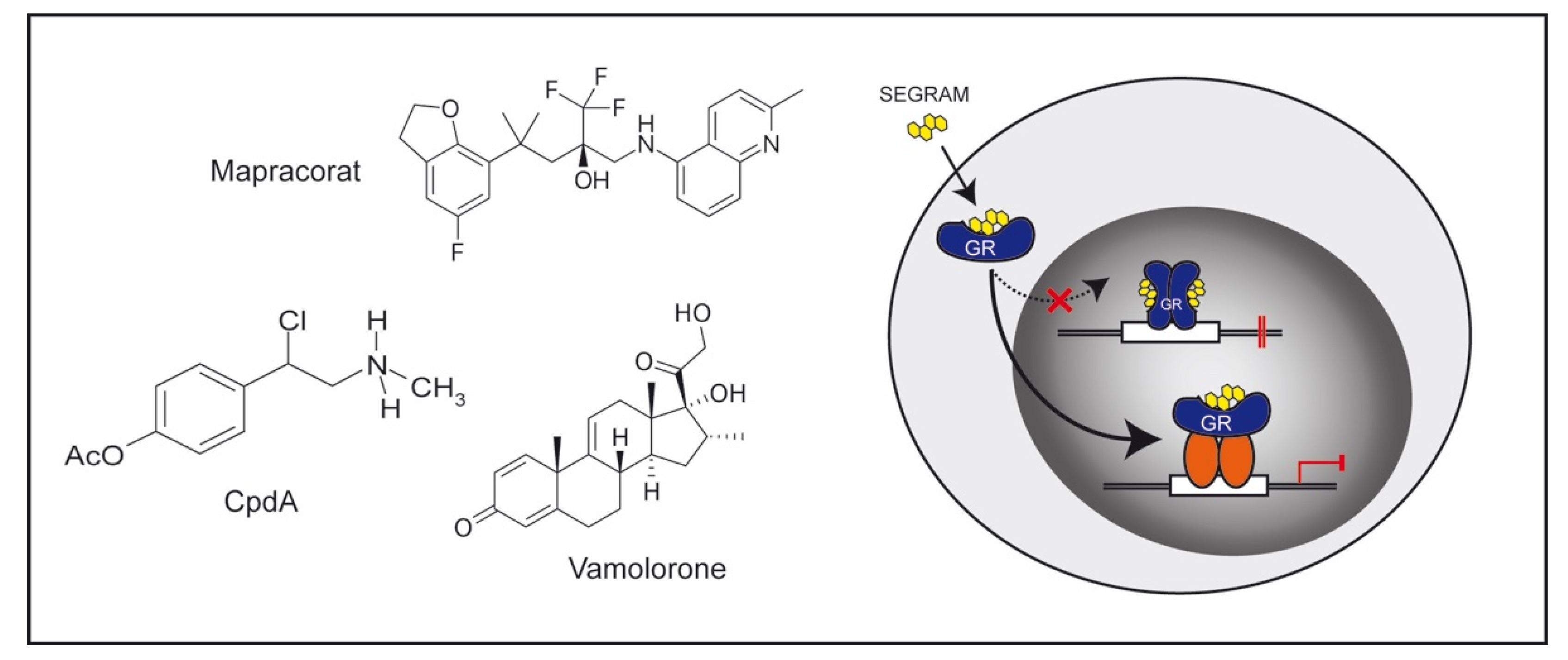
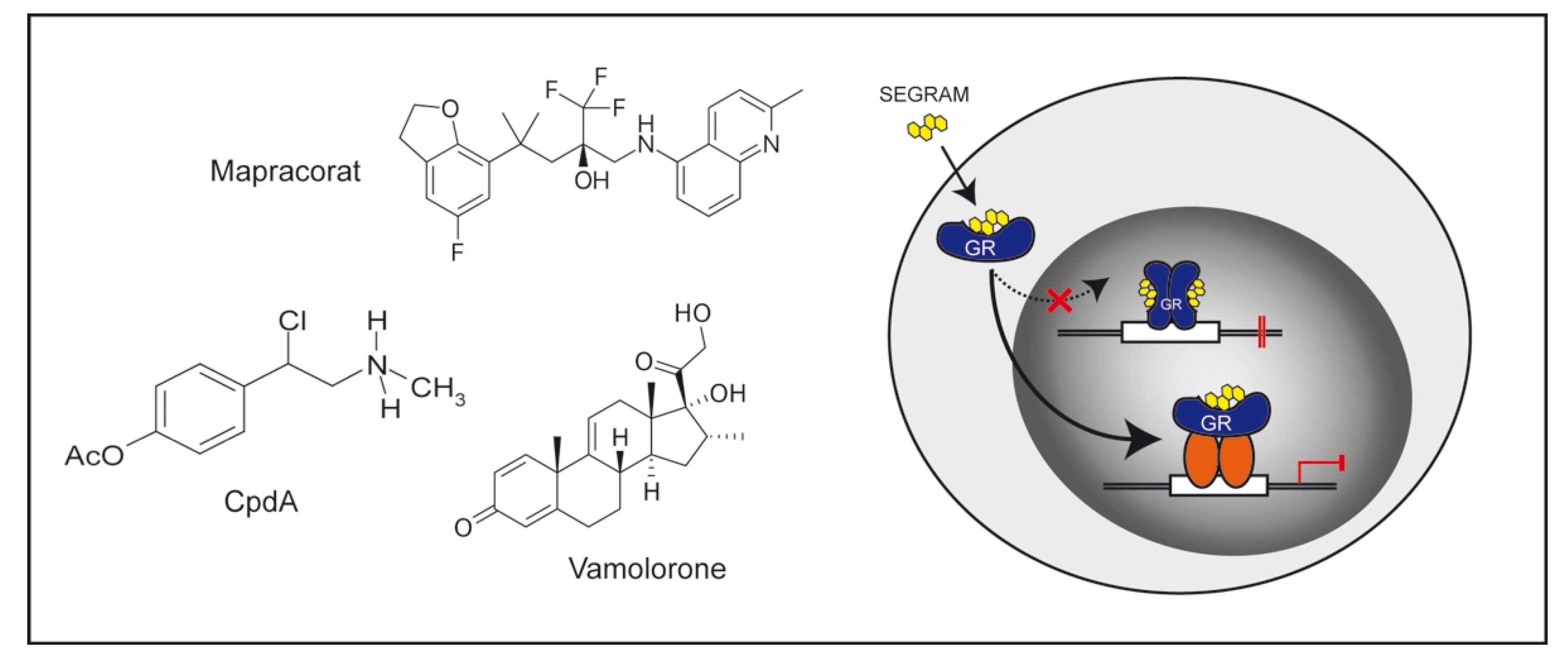
4.1. GC Derivatives
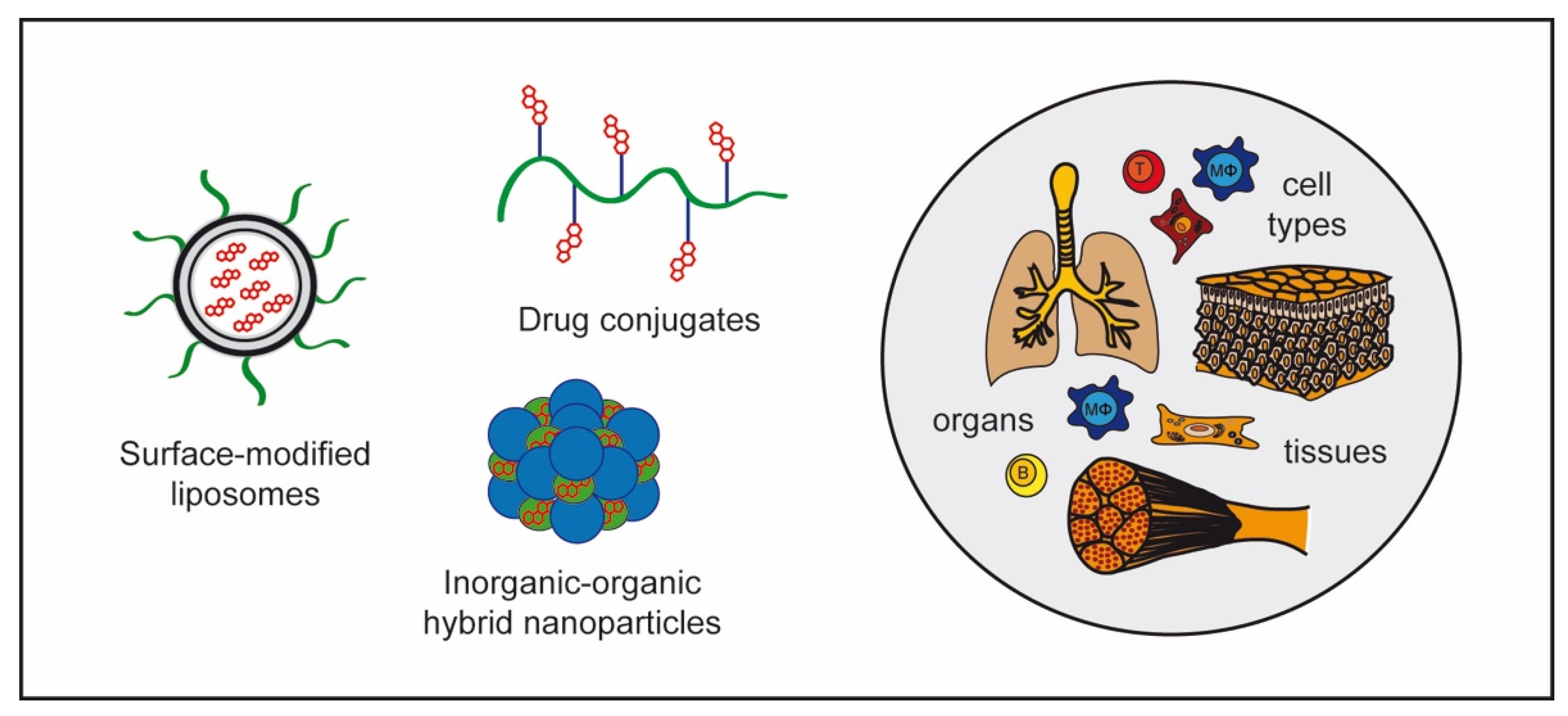
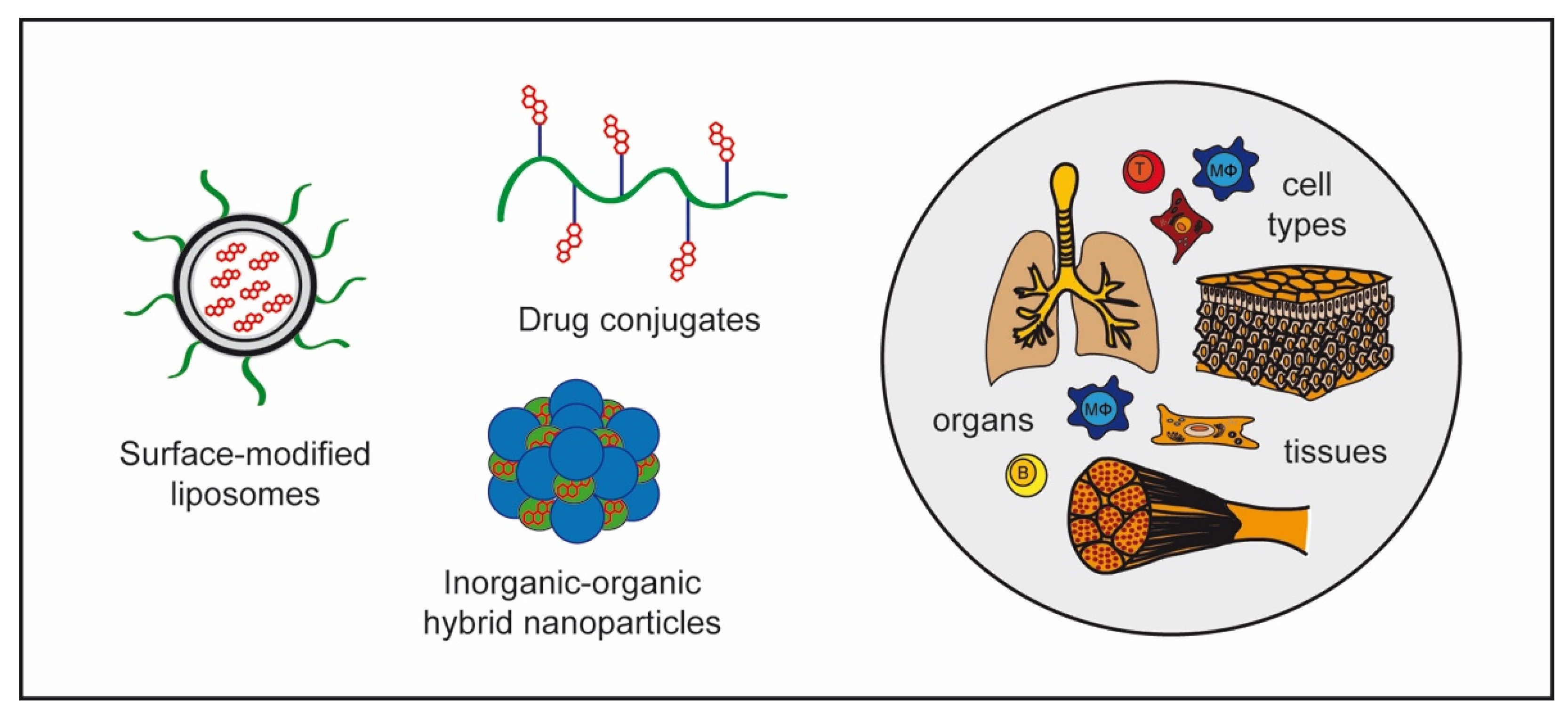
4.2. GC Nanoformulations
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bach, J.F. The effect of infections on susceptibility to autoimmune and allergic diseases. N. Engl. J. Med. 2002, 347, 911–920. [Google Scholar] [CrossRef]
- Kotsovilis, S.; Andreakos, E. Therapeutic human monoclonal antibodies in inflammatory diseases. Methods Mol. Biol. 2014, 1060, 37–59. [Google Scholar] [CrossRef] [PubMed]
- Escoter-Torres, L.; Caratti, G.; Mechtidou, A.; Tuckermann, J.; Uhlenhaut, N.H.; Vettorazzi, S. Fighting the Fire: Mechanisms of Inflammatory Gene Regulation by the Glucocorticoid Receptor. Front. Immunol. 2019, 10, 1859. [Google Scholar] [CrossRef]
- Vandewalle, J.; Luypaert, A.; De Bosscher, K.; Libert, C. Therapeutic Mechanisms of Glucocorticoids. Trends Endocrinol. Metab. 2018, 29, 42–54. [Google Scholar] [CrossRef]
- Schäcke, H.; Docke, W.D.; Asadullah, K. Mechanisms involved in the side effects of glucocorticoids. Pharmacol. Ther. 2002, 96, 23–43. [Google Scholar] [CrossRef]
- Saad, M.J.; Folli, F.; Kahn, J.A.; Kahn, C.R. Modulation of insulin receptor, insulin receptor substrate-1, and phosphatidylinositol 3-kinase in liver and muscle of dexamethasone-treated rats. J. Clin. Investig. 1993, 92, 2065–2072. [Google Scholar] [CrossRef]
- Muzzi, C.; Watanabe, N.; Twomey, E.; Meers, G.K.; Reichardt, H.M.; Bohnenberger, H.; Reichardt, S.D. The Glucocorticoid Receptor in Intestinal Epithelial Cells Alleviates Colitis and Associated Colorectal Cancer in Mice. Cell Mol. Gastroenterol. Hepatol. 2021, 11, 1505–1518. [Google Scholar] [CrossRef]
- Klassen, C.; Karabinskaya, A.; Dejager, L.; Vettorazzi, S.; Van Moorleghem, J.; Lühder, F.; Meijsing, S.H.; Tuckermann, J.P.; Bohnenberger, H.; Libert, C.; et al. Airway Epithelial Cells Are Crucial Targets of Glucocorticoids in a Mouse Model of Allergic Asthma. J. Immunol. 2017, 199, 48–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lloyd, C.M.; Saglani, S. Epithelial cytokines and pulmonary allergic inflammation. Curr. Opin. Immunol. 2015, 34, 52–58. [Google Scholar] [CrossRef]
- Reichardt, S.D.; Lühder, F.; Wiegers, G.J.; Reichardt, H.M. A flow cytometric approach to study glucocorticoid receptor expression in immune cell subpopulations of genetically engineered mice. Immunol. Lett. 2021, 233, 68–79. [Google Scholar] [CrossRef] [PubMed]
- Sundahl, N.; Bridelance, J.; Libert, C.; De Bosscher, K.; Beck, I.M. Selective glucocorticoid receptor modulation: New directions with non-steroidal scaffolds. Pharmacol. Ther. 2015, 152, 28–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lühder, F.; Reichardt, H.M. Novel Drug Delivery Systems Tailored for Improved Administration of Glucocorticoids. Int. J. Mol. Sci. 2017, 18, 1836. [Google Scholar] [CrossRef]
- Hollenberg, S.M.; Weinberger, C.; Ong, E.S.; Cerelli, G.; Oro, A.; Lebo, R.; Thompson, E.B.; Rosenfeld, M.G.; Evans, R.M. Primary structure and expression of a functional human glucocorticoid receptor cDNA. Nature 1985, 318, 635–641. [Google Scholar] [CrossRef]
- Arriza, J.L.; Weinberger, C.; Cerelli, G.; Glaser, T.M.; Handelin, B.L.; Housman, D.E.; Evans, R.M. Cloning of human mineralocorticoid receptor complementary DNA: Structural and functional kinship with the glucocorticoid receptor. Science 1987, 237, 268–275. [Google Scholar] [CrossRef]
- Gomez-Sanchez, E.P.; Gomez-Sanchez, C.E. 11beta-hydroxysteroid dehydrogenases: A growing multi-tasking family. Mol. Cell. Endocrinol. 2021, 526, 111210. [Google Scholar] [CrossRef]
- Lim, H.Y.; Müller, N.; Herold, M.J.; van den Brandt, J.; Reichardt, H.M. Glucocorticoids exert opposing effects on macrophage function dependent on their concentration. Immunology 2007, 122, 47–53. [Google Scholar] [CrossRef]
- Panettieri, R.A.; Schaafsma, D.; Amrani, Y.; Koziol-White, C.; Ostrom, R.; Tliba, O. Non-genomic Effects of Glucocorticoids: An Updated View. Trends Pharmacol. Sci. 2019, 40, 38–49. [Google Scholar] [CrossRef]
- Abraham, S.M.; Lawrence, T.; Kleiman, A.; Warden, P.; Medghalchi, M.; Tuckermann, J.; Saklatvala, J.; Clark, A.R. Antiinflammatory effects of dexamethasone are partly dependent on induction of dual specificity phosphatase 1. J. Exp. Med. 2006, 203, 1883–1889. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, M.C.; Baatar, D.; Collins, G.; Carter, A.; Indig, F.; Biragyn, A.; Taub, D.D. Dexamethasone augments CXCR4-mediated signaling in resting human T cells via the activation of the Src kinase Lck. Blood 2009, 113, 575–584. [Google Scholar] [CrossRef] [Green Version]
- Müller, N.; Fischer, H.J.; Tischner, D.; van den Brandt, J.; Reichardt, H.M. Glucocorticoids induce effector T cell depolarization via ERM proteins, thereby impeding migration and APC conjugation. J. Immunol. 2013, 190, 4360–4370. [Google Scholar] [CrossRef] [Green Version]
- Lim, H.W.; Uhlenhaut, N.H.; Rauch, A.; Weiner, J.; Hübner, S.; Hübner, N.; Won, K.J.; Lazar, M.A.; Tuckermann, J.; Steger, D.J. Genomic redistribution of GR monomers and dimers mediates transcriptional response to exogenous glucocorticoid in vivo. Genome Res. 2015, 25, 836–844. [Google Scholar] [CrossRef] [Green Version]
- Surjit, M.; Ganti, K.P.; Mukherji, A.; Ye, T.; Hua, G.; Metzger, D.; Li, M.; Chambon, P. Widespread negative response elements mediate direct repression by agonist-liganded glucocorticoid receptor. Cell 2011, 145, 224–241. [Google Scholar] [CrossRef] [Green Version]
- Schiller, B.J.; Chodankar, R.; Watson, L.C.; Stallcup, M.R.; Yamamoto, K.R. Glucocorticoid receptor binds half sites as a monomer and regulates specific target genes. Genome Biol. 2014, 15, 418. [Google Scholar] [CrossRef]
- Chen, W.; Dang, T.; Blind, R.D.; Wang, Z.; Cavasotto, C.N.; Hittelman, A.B.; Rogatsky, I.; Logan, S.K.; Garabedian, M.J. Glucocorticoid receptor phosphorylation differentially affects target gene expression. Mol. Endocrinol. 2008, 22, 1754–1766. [Google Scholar] [CrossRef] [Green Version]
- Yang Yen, H.F.; Chambard, J.C.; Sun, Y.L.; Smeal, T.; Schmidt, T.J.; Drouin, J.; Karin, M. Transcriptional interference between c-Jun and the glucocorticoid receptor: Mutual inhibition of DNA binding due to direct protein-protein interaction. Cell 1990, 62, 1205–1215. [Google Scholar] [CrossRef]
- Ray, A.; Prefontaine, K.E. Physical association and functional antagonism between the p65 subunit of transcription factor NF-kappa B and the glucocorticoid receptor. Proc. Natl. Acad. Sci. USA 1994, 91, 752–756. [Google Scholar] [CrossRef] [Green Version]
- Ballegeer, M.; Van Looveren, K.; Timmermans, S.; Eggermont, M.; Vandevyver, S.; Thery, F.; Dendoncker, K.; Souffriau, J.; Vandewalle, J.; Van Wyngene, L.; et al. Glucocorticoid receptor dimers control intestinal STAT1 and TNF-induced inflammation in mice. J. Clin. Investig. 2018, 128, 3265–3279. [Google Scholar] [CrossRef]
- Uhlenhaut, N.H.; Barish, G.D.; Yu, R.T.; Downes, M.; Karunasiri, M.; Liddle, C.; Schwalie, P.; Hubner, N.; Evans, R.M. Insights into Negative Regulation by the Glucocorticoid Receptor from Genome-wide Profiling of Inflammatory Cistromes. Mol. Cell. 2013, 49, 158–171. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.S.; Choi, H.; Han, B.S.; Kim, W.K.; Lee, S.C.; Oh, K.J.; Bae, K.H. c-Jun regulates adipocyte differentiation via the KLF15-mediated mode. Biochem. Biophys. Res. Commun. 2016, 469, 552–558. [Google Scholar] [CrossRef]
- Vettorazzi, S.; Nalbantoglu, D.; Gebhardt, J.C.M.; Tuckermann, J. A guide to changing paradigms of glucocorticoid receptor function-a model system for genome regulation and physiology. FEBS J. 2021. online ahead of print. [Google Scholar] [CrossRef]
- Oh, K.S.; Patel, H.; Gottschalk, R.A.; Lee, W.S.; Baek, S.; Fraser, I.D.C.; Hager, G.L.; Sung, M.H. Anti-Inflammatory Chromatinscape Suggests Alternative Mechanisms of Glucocorticoid Receptor Action. Immunity 2017, 47, 298–309.e5. [Google Scholar] [CrossRef] [PubMed]
- Escoter-Torres, L.; Greulich, F.; Quagliarini, F.; Wierer, M.; Uhlenhaut, N.H. Anti-inflammatory functions of the glucocorticoid receptor require DNA binding. Nucleic Acids Res. 2020, 48, 8393–8407. [Google Scholar] [CrossRef]
- Schweingruber, N.; Fischer, H.J.; Fischer, L.; van den Brandt, J.; Karabinskaya, A.; Labi, V.; Villunger, A.; Kretzschmar, B.; Huppke, P.; Simons, M.; et al. Chemokine-mediated redirection of T cells constitutes a critical mechanism of glucocorticoid therapy in autoimmune CNS responses. Acta Neuropathol. 2014, 127, 713–729. [Google Scholar] [CrossRef] [Green Version]
- Gebert, L.F.R.; MacRae, I.J. Regulation of microRNA function in animals. Nat. Rev. Mol. Cell Biol. 2019, 20, 21–37. [Google Scholar] [CrossRef] [PubMed]
- Clayton, S.A.; Jones, S.W.; Kurowska-Stolarska, M.; Clark, A.R. The role of microRNAs in glucocorticoid action. J. Biol. Chem. 2018, 293, 1865–1874. [Google Scholar] [CrossRef] [Green Version]
- Puimege, L.; Van Hauwermeiren, F.; Steeland, S.; Van Ryckeghem, S.; Vandewalle, J.; Lodens, S.; Dejager, L.; Vandevyver, S.; Staelens, J.; Timmermans, S.; et al. Glucocorticoid-induced microRNA-511 protects against TNF by down-regulating TNFR1. EMBO Mol. Med. 2015, 7, 1004–1017. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Nguyen, Q.T.; Lee, J.; Lee, S.H.; Janocha, A.; Kim, S.; Le, H.T.; Dvorina, N.; Weiss, K.; Cameron, M.J.; et al. Anti-inflammatory Roles of Glucocorticoids Are Mediated by Foxp3(+) Regulatory T Cells via a miR-342-Dependent Mechanism. Immunity 2020, 53, 581–596.e5. [Google Scholar] [CrossRef]
- Robertson, S.; Diver, L.A.; Alvarez-Madrazo, S.; Livie, C.; Ejaz, A.; Fraser, R.; Connell, J.M.; MacKenzie, S.M.; Davies, E. Regulation of Corticosteroidogenic Genes by MicroRNAs. Int. J. Endocrinol. 2017, 2017, 2021903. [Google Scholar] [CrossRef] [Green Version]
- Louveau, A.; Harris, T.H.; Kipnis, J. Revisiting the Mechanisms of CNS Immune Privilege. Trends Immunol. 2015, 36, 569–577. [Google Scholar] [CrossRef] [Green Version]
- Forrester, J.V.; McMenamin, P.G.; Dando, S.J. CNS infection and immune privilege. Nat. Rev. Neurosci. 2018, 19, 655–671. [Google Scholar] [CrossRef]
- Aspelund, A.; Antila, S.; Proulx, S.T.; Karlsen, T.V.; Karaman, S.; Detmar, M.; Wiig, H.; Alitalo, K. A dural lymphatic vascular system that drains brain interstitial fluid and macromolecules. J. Exp. Med. 2015, 212, 991–999. [Google Scholar] [CrossRef]
- Louveau, A.; Herz, J.; Alme, M.N.; Salvador, A.F.; Dong, M.Q.; Viar, K.E.; Herod, S.G.; Knopp, J.; Setliff, J.C.; Lupi, A.L.; et al. CNS lymphatic drainage and neuroinflammation are regulated by meningeal lymphatic vasculature. Nat. Neurosci. 2018, 21, 1380–1391. [Google Scholar] [CrossRef]
- Perry, V.H. Microglia. MicroBiol. Spectr. 2016, 4, 3. [Google Scholar] [CrossRef] [Green Version]
- Milligan, N.M.; Newcombe, R.; Compston, D.A. A double-blind controlled trial of high dose methylprednisolone in patients with multiple sclerosis: 1. Clinical effects. J. Neurol. Neurosurg Psychiatry 1987, 50, 511–516. [Google Scholar] [CrossRef] [Green Version]
- Gold, R.; Linington, C.; Lassmann, H. Understanding pathogenesis and therapy of multiple sclerosis via animal models: 70 years of merits and culprits in experimental autoimmune encephalomyelitis research. Brain 2006, 129, 1953–1971. [Google Scholar] [CrossRef]
- Schweingruber, N.; Haine, A.; Tiede, K.; Karabinskaya, A.; van den Brandt, J.; Wüst, S.; Metselaar, J.M.; Gold, R.; Tuckermann, J.P.; Reichardt, H.M.; et al. Liposomal encapsulation of glucocorticoids alters their mode of action in the treatment of experimental autoimmune encephalomyelitis. J. Immunol. 2011, 187, 4310–4318. [Google Scholar] [CrossRef]
- Wüst, S.; van den Brandt, J.; Tischner, D.; Kleiman, A.; Tuckermann, J.P.; Gold, R.; Lühder, F.; Reichardt, H.M. Peripheral T cells are the therapeutic targets of glucocorticoids in experimental autoimmune encephalomyelitis. J. Immunol. 2008, 180, 8434–8443. [Google Scholar] [CrossRef] [Green Version]
- Montes-Cobos, E.; Ring, S.; Fischer, H.J.; Heck, J.; Strauss, J.; Schwaninger, M.; Reichardt, S.D.; Feldmann, C.; Lühder, F.; Reichardt, H.M. Targeted delivery of glucocorticoids to macrophages in a mouse model of multiple sclerosis using inorganic-organic hybrid nanoparticles. J. Control. Release 2017, 245, 157–169. [Google Scholar] [CrossRef]
- Bier, J.; Steiger, S.M.; Reichardt, H.M.; Lühder, F. Protection of Antigen-Primed Effector T Cells From Glucocorticoid-Induced Apoptosis in Cell Culture and in a Mouse Model of Multiple Sclerosis. Front. Immunol. 2021, 12, 671258. [Google Scholar] [CrossRef]
- Tischner, D.; van den Brandt, J.; Weishaupt, A.; Lühder, F.; Herold, M.J.; Reichardt, H.M. Stable silencing of the glucocorticoid receptor in myelin-specific T effector cells by retroviral delivery of shRNA: Insight into neuroinflammatory disease. Eur. J. Immunol. 2009, 39, 2361–2370. [Google Scholar] [CrossRef]
- Elovaara, I.; Lalla, M.; Spare, E.; Lehtimaki, T.; Dastidar, P. Methylprednisolone reduces adhesion molecules in blood and cerebrospinal fluid in patients with MS. Neurology 1998, 51, 1703–1708. [Google Scholar] [CrossRef]
- Kiefer, R.; Kreutzberg, G.W. Effects of dexamethasone on microglial activation in vivo: Selective downregulation of major histocompatibility complex class II expression in regenerating facial nucleus. J. NeuroImmunol. 1991, 34, 99–108. [Google Scholar] [CrossRef]
- Montes-Cobos, E.; Schweingruber, N.; Li, X.; Fischer, H.J.; Reichardt, H.M.; Lühder, F. Deletion of the Mineralocorticoid Receptor in Myeloid Cells Attenuates Central Nervous System Autoimmunity. Front. Immunol. 2017, 8, 1319. [Google Scholar] [CrossRef] [Green Version]
- Reder, A.T.; Thapar, M.; Jensen, M.A. A reduction in serum glucocorticoids provokes experimental allergic encephalomyelitis: Implications for treatment of inflammatory brain disease. Neurology 1994, 44, 2289–2294. [Google Scholar] [CrossRef]
- Nerius, M.; Haenisch, B.; Gomm, W.; Doblhammer, G.; Schneider, A. Glucocorticoid Therapy is Associated with a Lower Risk of Dementia. J. Alzheimers Dis. 2020, 73, 175–183. [Google Scholar] [CrossRef]
- Hui, Z.; Zhijun, Y.; Yushan, Y.; Liping, C.; Yiying, Z.; Difan, Z.; Chunglit, C.T.; Wei, C. The combination of acyclovir and dexamethasone protects against Alzheimer’s disease-related cognitive impairments in mice. Psychopharmacology 2020, 237, 1851–1860. [Google Scholar] [CrossRef]
- Castano, A.; Herrera, A.J.; Cano, J.; Machado, A. The degenerative effect of a single intranigral injection of LPS on the dopaminergic system is prevented by dexamethasone, and not mimicked by rh-TNF-alpha, IL-1beta and IFN-gamma. J. Neurochem. 2002, 81, 150–157. [Google Scholar] [CrossRef]
- Carrillo-de Sauvage, M.A.; Maatouk, L.; Arnoux, I.; Pasco, M.; Sanz Diez, A.; Delahaye, M.; Herrero, M.T.; Newman, T.A.; Calvo, C.F.; Audinat, E.; et al. Potent and multiple regulatory actions of microglial glucocorticoid receptors during CNS inflammation. Cell Death Differ. 2013, 20, 1546–1557. [Google Scholar] [CrossRef]
- Maatouk, L.; Compagnion, A.C.; Sauvage, M.C.; Bemelmans, A.P.; Leclere-Turbant, S.; Cirotteau, V.; Tohme, M.; Beke, A.; Trichet, M.; Bazin, V.; et al. TLR9 activation via microglial glucocorticoid receptors contributes to degeneration of midbrain dopamine neurons. Nat. Commun. 2018, 9, 2450. [Google Scholar] [CrossRef]
- Qi, Y.; Klyubin, I.; Ondrejcak, T.; Hu, N.W.; Rowan, M.J. Enduring glucocorticoid-evoked exacerbation of synaptic plasticity disruption in male rats modelling early Alzheimer’s disease amyloidosis. Neuropsychopharmacology 2021, 46, 2170–2179. [Google Scholar] [CrossRef]
- Bolshakov, A.P.; Tret’yakova, L.V.; Kvichansky, A.A.; Gulyaeva, N.V. Glucocorticoids: Dr. Jekyll and Mr. Hyde of Hippocampal Neuroinflammation. Biochemistry (Mosc.) 2021, 86, 156–167. [Google Scholar] [CrossRef] [PubMed]
- Harry, G.J. Microglia in Neurodegenerative Events-An Initiator or a Significant Other? Int. J. Mol. Sci. 2021, 22, 5818. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Jimenez, D.; Kolb, J.P.; Cidlowski, J.A. Glucocorticoids as Regulators of Macrophage-Mediated Tissue Homeostasis. Front. Immunol. 2021, 12, 669891. [Google Scholar] [CrossRef] [PubMed]
- Espinosa-Oliva, A.M.; de Pablos, R.M.; Villaran, R.F.; Arguelles, S.; Venero, J.L.; Machado, A.; Cano, J. Stress is critical for LPS-induced activation of microglia and damage in the rat hippocampus. Neurobiol. Aging 2011, 32, 85–102. [Google Scholar] [CrossRef]
- Frank, M.G.; Miguel, Z.D.; Watkins, L.R.; Maier, S.F. Prior exposure to glucocorticoids sensitizes the neuroinflammatory and peripheral inflammatory responses to E. coli lipopolysaccharide. Brain Behav. Immun. 2010, 24, 19–30. [Google Scholar] [CrossRef]
- Gass, P.; Kretz, O.; Wolfer, D.P.; Berger, S.; Tronche, F.; Reichardt, H.M.; Kellendonk, C.; Lipp, H.P.; Schmid, W.; Schütz, G. Genetic disruption of mineralocorticoid receptor leads to impaired neurogenesis and granule cell degeneration in the hippocampus of adult mice. EMBO Rep. 2000, 1, 447–451. [Google Scholar] [CrossRef] [Green Version]
- Karst, H.; Karten, Y.J.; Reichardt, H.M.; de Kloet, E.R.; Schütz, G.; Joels, M. Corticosteroid actions in hippocampus require DNA binding of glucocorticoid receptor homodimers. Nat. Neurosci. 2000, 3, 977–978. [Google Scholar] [CrossRef]
- Hill, A.R.; Spencer-Segal, J.L. Glucocorticoids and the Brain after Critical Illness. Endocrinology 2021, 162, bqaa242. [Google Scholar] [CrossRef]
- Feng, X.; Zhao, Y.; Yang, T.; Song, M.; Wang, C.; Yao, Y.; Fan, H. Glucocorticoid-Driven NLRP3 Inflammasome Activation in Hippocampal Microglia Mediates Chronic Stress-Induced Depressive-Like Behaviors. Front. Mol. Neurosci. 2019, 12, 210. [Google Scholar] [CrossRef]
- Gass, P.; Reichardt, H.M.; Strekalova, T.; Henn, F.; Tronche, F. Mice with targeted mutations of glucocorticoid and mineralocorticoid receptors: Models for depression and anxiety? Physiol. Behav. 2001, 73, 811–825. [Google Scholar] [CrossRef]
- Saeedi, M.; Rashidy-Pour, A. Association between chronic stress and Alzheimer’s disease: Therapeutic effects of Saffron. Biomed Pharmacother 2021, 133, 110995. [Google Scholar] [CrossRef]
- Kline, S.A.; Mega, M.S. Stress-Induced Neurodegeneration: The Potential for Coping as Neuroprotective Therapy. Am. J. Alzheimers Dis. Other Demen 2020, 35, 1533317520960873. [Google Scholar] [CrossRef]
- Vyas, S.; Rodrigues, A.J.; Silva, J.M.; Tronche, F.; Almeida, O.F.; Sousa, N.; Sotiropoulos, I. Chronic Stress and Glucocorticoids: From Neuronal Plasticity to Neurodegeneration. Neural Plast. 2016, 2016, 6391686. [Google Scholar] [CrossRef] [Green Version]
- Watermeyer, T.; Robb, C.; Gregory, S.; Udeh-Momoh, C. Therapeutic implications of hypothalamic-pituitaryadrenal-axis modulation in Alzheimer’s disease: A narrative review of pharmacological and lifestyle interventions. Front. Neuroendocrinol. 2021, 60, 100877. [Google Scholar] [CrossRef]
- Ubeda, C.; Djukovic, A.; Isaac, S. Roles of the intestinal microbiota in pathogen protection. Clin. Transl. Immunol. 2017, 6, e128. [Google Scholar] [CrossRef]
- Okumura, R.; Takeda, K. Maintenance of gut homeostasis by the mucosal immune system. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2016, 92, 423–435. [Google Scholar] [CrossRef] [Green Version]
- Peterson, L.W.; Artis, D. Intestinal epithelial cells: Regulators of barrier function and immune homeostasis. Nat. Rev. Immunol. 2014, 14, 141–153. [Google Scholar] [CrossRef]
- Ordas, I.; Eckmann, L.; Talamini, M.; Baumgart, D.C.; Sandborn, W.J. Ulcerative colitis. Lancet 2012, 380, 1606–1619. [Google Scholar] [CrossRef] [Green Version]
- Fakhoury, M.; Negrulj, R.; Mooranian, A.; Al-Salami, H. Inflammatory bowel disease: Clinical aspects and treatments. J. Inflamm. Res. 2014, 7, 113–120. [Google Scholar] [CrossRef] [Green Version]
- Seyedian, S.S.; Nokhostin, F.; Malamir, M.D. A review of the diagnosis, prevention, and treatment methods of inflammatory bowel disease. J. Med. Life 2019, 12, 113–122. [Google Scholar] [CrossRef]
- Hazel, K.; O’Connor, A. Emerging treatments for inflammatory bowel disease. Ther. Adv. Chronic. Dis. 2020, 11, 2040622319899297. [Google Scholar] [CrossRef]
- Dubois-Camacho, K.; Ottum, P.A.; Franco-Munoz, D.; De la Fuente, M.; Torres-Riquelme, A.; Diaz-Jimenez, D.; Olivares-Morales, M.; Astudillo, G.; Quera, R.; Hermoso, M.A. Glucocorticosteroid therapy in inflammatory bowel diseases: From clinical practice to molecular biology. World J. Gastroenterol. 2017, 23, 6628–6638. [Google Scholar] [CrossRef]
- Meers, G.K.; Bohnenberger, H.; Reichardt, H.M.; Lühder, F.; Reichardt, S.D. Impaired resolution of DSS-induced colitis in mice lacking the glucocorticoid receptor in myeloid cells. PLoS ONE 2018, 13, e0190846. [Google Scholar] [CrossRef]
- Rocamora-Reverte, L.; Tuzlak, S.; von Raffay, L.; Tisch, M.; Fiegl, H.; Drach, M.; Reichardt, H.M.; Villunger, A.; Tischner, D.; Wiegers, G.J. Glucocorticoid Receptor-Deficient Foxp3(+) Regulatory T Cells Fail to Control Experimental Inflammatory Bowel Disease. Front. Immunol. 2019, 10, 472. [Google Scholar] [CrossRef]
- Zhang, Z.; Dong, L.; Jia, A.; Chen, X.; Yang, Q.; Wang, Y.; Wang, Y.; Liu, R.; Cao, Y.; He, Y.; et al. Glucocorticoids Promote the Onset of Acute Experimental Colitis and Cancer by Upregulating mTOR Signaling in Intestinal Epithelial Cells. Cancers 2020, 12, 945. [Google Scholar] [CrossRef] [Green Version]
- Crielaard, B.J.; Lammers, T.; Morgan, M.E.; Chaabane, L.; Carboni, S.; Greco, B.; Zaratin, P.; Kraneveld, A.D.; Storm, G. Macrophages and liposomes in inflammatory disease: Friends or foes? Int. J. Pharm. 2011, 416, 499–506. [Google Scholar] [CrossRef]
- Yang, M.; Jia, W.; Wang, D.; Han, F.; Niu, W.; Zhang, H.; Shih, D.Q.; Zhang, X. Effects and Mechanism of Constitutive TL1A Expression on Intestinal Mucosal Barrier in DSS-Induced Colitis. Dig. Dis. Sci. 2019, 64, 1844–1856. [Google Scholar] [CrossRef]
- Xu, P.; Elizalde, M.; Masclee, A.; Pierik, M.; Jonkers, D. Corticosteroid enhances epithelial barrier function in intestinal organoids derived from patients with Crohn’s disease. J. Mol. Med. 2021, 99, 805–815. [Google Scholar] [CrossRef]
- Ronchetti, S.; Migliorati, G.; Riccardi, C. GILZ as a Mediator of the Anti-Inflammatory Effects of Glucocorticoids. Front. Endocrinol. (Lausanne) 2015, 6, 170. [Google Scholar] [CrossRef] [Green Version]
- Ricci, E.; Ronchetti, S.; Gabrielli, E.; Pericolini, E.; Gentili, M.; Roselletti, E.; Vecchiarelli, A.; Riccardi, C. GILZ restrains neutrophil activation by inhibiting the MAPK pathway. J. Leukoc. Biol. 2019, 105, 187–194. [Google Scholar] [CrossRef] [Green Version]
- Bereshchenko, O.; Coppo, M.; Bruscoli, S.; Biagioli, M.; Cimino, M.; Frammartino, T.; Sorcini, D.; Venanzi, A.; Di Sante, M.; Riccardi, C. GILZ promotes production of peripherally induced Treg cells and mediates the crosstalk between glucocorticoids and TGF-beta signaling. Cell Rep. 2014, 7, 464–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- You, P.; Chen, N.; Su, L.; Peng, T.; Chen, G.; Liu, Y. Local level of TGF-beta1 determines the effectiveness of dexamethasone through regulating the balance of Treg/Th17 cells in TNBS-induced mouse colitis. Exp. Ther. Med. 2018, 15, 3639–3649. [Google Scholar] [CrossRef] [PubMed]
- Franzin, M.; Stefancic, K.; Lucafo, M.; Decorti, G.; Stocco, G. Microbiota and Drug Response in Inflammatory Bowel Disease. Pathogens 2021, 10, 211. [Google Scholar] [CrossRef]
- Pigneur, B.; Lepage, P.; Mondot, S.; Schmitz, J.; Goulet, O.; Dore, J.; Ruemmele, F.M. Mucosal Healing and Bacterial Composition in Response to Enteral Nutrition Vs Steroid-based Induction Therapy-A Randomised Prospective Clinical Trial in Children With Crohn’s Disease. J. Crohns Colitis 2019, 13, 846–855. [Google Scholar] [CrossRef] [Green Version]
- Sood, A.; Mahajan, R.; Juyal, G.; Midha, V.; Grewal, C.S.; Mehta, V.; Singh, A.; Joshi, M.C.; Narang, V.; Kaur, K.; et al. Efficacy of fecal microbiota therapy in steroid dependent ulcerative colitis: A real world intention-to-treat analysis. Intest. Res. 2019, 17, 78–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Dwyer, D.N.; Gurczynski, S.J.; Moore, B.B. Pulmonary immunity and extracellular matrix interactions. Matrix Biol. 2018, 73, 122–134. [Google Scholar] [CrossRef]
- Invernizzi, R.; Lloyd, C.M.; Molyneaux, P.L. Respiratory microbiome and epithelial interactions shape immunity in the lungs. Immunology 2020, 160, 171–182. [Google Scholar] [CrossRef] [Green Version]
- Maselli, D.J.; Hardin, M.; Christenson, S.A.; Hanania, N.A.; Hersh, C.P.; Adams, S.G.; Anzueto, A.; Peters, J.I.; Han, M.K.; Martinez, F.J. Clinical Approach to the Therapy of Asthma-COPD Overlap. Chest 2019, 155, 168–177. [Google Scholar] [CrossRef] [PubMed]
- Grayson, M.H.; Feldman, S.; Prince, B.T.; Patel, P.J.; Matsui, E.C.; Apter, A.J. Advances in asthma in 2017: Mechanisms, biologics, and genetics. J. Allergy Clin. Immunol. 2018, 142, 1423–1436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wenzel, S.E. Asthma phenotypes: The evolution from clinical to molecular approaches. Nat. Med. 2012, 18, 716–725. [Google Scholar] [CrossRef]
- Lemanske, R.F., Jr.; Busse, W.W. Asthma: Clinical expression and molecular mechanisms. J. Allergy Clin. Immunol. 2010, 125, S95–S102. [Google Scholar] [CrossRef]
- Lambrecht, B.N.; Hammad, H. The immunology of asthma. Nat. Immunol. 2015, 16, 45–56. [Google Scholar] [CrossRef]
- Celli, B.R.; Wedzicha, J.A. Update on Clinical Aspects of Chronic Obstructive Pulmonary Disease. N. Engl. J. Med. 2019, 381, 1257–1266. [Google Scholar] [CrossRef] [Green Version]
- Holgate, S.T.; Polosa, R. Treatment strategies for allergy and asthma. Nat. Rev. Immunol. 2008, 8, 218–230. [Google Scholar] [CrossRef]
- Barnes, P.J. Inhaled corticosteroids are not beneficial in chronic obstructive pulmonary disease. Am. J. Respir. Crit Care Med. 2000, 161, 342–344; discussion 344. [Google Scholar] [CrossRef] [PubMed]
- Calverley, P.M. Inhaled corticosteroids are beneficial in chronic obstructive pulmonary disease. Am. J. Respir. Crit Care Med. 2000, 161, 341–342; discussion 344. [Google Scholar] [CrossRef] [PubMed]
- Ronchetti, S.; Ricci, E.; Migliorati, G.; Gentili, M.; Riccardi, C. How Glucocorticoids Affect the Neutrophil Life. Int. J. Mol. Sci. 2018, 19, 4090. [Google Scholar] [CrossRef] [Green Version]
- Meagher, L.C.; Cousin, J.M.; Seckl, J.R.; Haslett, C. Opposing effects of glucocorticoids on the rate of apoptosis in neutrophilic and eosinophilic granulocytes. J. Immunol. 1996, 156, 4422–4428. [Google Scholar] [PubMed]
- Irusen, E.; Matthews, J.G.; Takahashi, A.; Barnes, P.J.; Chung, K.F.; Adcock, I.M. p38 Mitogen-activated protein kinase-induced glucocorticoid receptor phosphorylation reduces its activity: Role in steroid-insensitive asthma. J. Allergy Clin. Immunol. 2002, 109, 649–657. [Google Scholar] [CrossRef]
- Trevor, J.L.; Deshane, J.S. Refractory asthma: Mechanisms, targets, and therapy. Allergy 2014, 69, 817–827. [Google Scholar] [CrossRef]
- Dejager, L.; Dendoncker, K.; Eggermont, M.; Souffriau, J.; Van Hauwermeiren, F.; Willart, M.; Van Wonterghem, E.; Naessens, T.; Ballegeer, M.; Vandevyver, S.; et al. Neutralizing TNFalpha restoRes. glucocorticoid sensitivity in a mouse model of neutrophilic airway inflammation. Mucosal Immunol. 2015, 8, 1212–1225. [Google Scholar] [CrossRef] [Green Version]
- Ware, L.B.; Matthay, M.A. The acute respiratory distress syndrome. N. Engl. J. Med. 2000, 342, 1334–1349. [Google Scholar] [CrossRef]
- Englert, J.A.; Bobba, C.; Baron, R.M. Integrating molecular pathogenesis and clinical translation in sepsis-induced acute respiratory distress syndrome. JCI Insight 2019, 4, e124061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubenfeld, G.D.; Caldwell, E.; Peabody, E.; Weaver, J.; Martin, D.P.; Neff, M.; Stern, E.J.; Hudson, L.D. Incidence and outcomes of acute lung injury. N. Engl. J. Med. 2005, 353, 1685–1693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimmerman, J.J.; Akhtar, S.R.; Caldwell, E.; Rubenfeld, G.D. Incidence and outcomes of pediatric acute lung injury. Pediatrics 2009, 124, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Huppert, L.A.; Matthay, M.A.; Ware, L.B. Pathogenesis of Acute Respiratory Distress Syndrome. Semin. Respir. Crit. Care Med. 2019, 40, 31–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, C.Y.; Chen, C.S.; Yiang, G.T.; Cheng, Y.L.; Yong, S.B.; Wu, M.Y.; Li, C.J. New Insights into the Immune Molecular Regulation of the Pathogenesis of Acute Respiratory Distress Syndrome. Int. J. Mol. Sci. 2018, 19, 588. [Google Scholar] [CrossRef] [Green Version]
- Tang, B.M.; Craig, J.C.; Eslick, G.D.; Seppelt, I.; McLean, A.S. Use of corticosteroids in acute lung injury and acute respiratory distress syndrome: A systematic review and meta-analysis. Crit. Care Med. 2009, 37, 1594–1603. [Google Scholar] [CrossRef] [PubMed]
- Levitt, J.E.; Matthay, M.A. Treatment of acute lung injury: Historical perspective and potential future therapies. Semin. Respir. Crit. Care Med. 2006, 27, 426–437. [Google Scholar] [CrossRef]
- Khilnani, G.C.; Hadda, V. Corticosteroids and ARDS: A review of treatment and prevention evidence. Lung India 2011, 28, 114–119. [Google Scholar] [CrossRef] [PubMed]
- Diaz, J.V.; Brower, R.; Calfee, C.S.; Matthay, M.A. Therapeutic strategies for severe acute lung injury. Crit Care Med. 2010, 38, 1644–1650. [Google Scholar] [CrossRef] [Green Version]
- Tu, G.W.; Shi, Y.; Zheng, Y.J.; Ju, M.J.; He, H.Y.; Ma, G.G.; Hao, G.W.; Luo, Z. Glucocorticoid attenuates acute lung injury through induction of type 2 macrophage. J. Transl. Med. 2017, 15, 181. [Google Scholar] [CrossRef]
- Vettorazzi, S.; Bode, C.; Dejager, L.; Frappart, L.; Shelest, E.; Klassen, C.; Tasdogan, A.; Reichardt, H.M.; Libert, C.; Schneider, M.; et al. Glucocorticoids limit acute lung inflammation in concert with inflammatory stimuli by induction of SphK1. Nat. Commun. 2015, 6, 7796. [Google Scholar] [CrossRef] [Green Version]
- Wepler, M.; Preuss, J.M.; Merz, T.; Hartmann, C.; Wachter, U.; McCook, O.; Vogt, J.; Kress, S.; Groger, M.; Fink, M.; et al. Impaired Glucocorticoid Receptor Dimerization Aggravates LPS-Induced Circulatory and Pulmonary Dysfunction. Front. Immunol. 2019, 10, 3152. [Google Scholar] [CrossRef] [PubMed]
- Alexaki, V.I.; Henneicke, H. The Role of Glucocorticoids in the Management of COVID-19. Horm. Metab. Res. 2021, 53, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Z.; Liu, J.; Shi, D.; Chen, W.; Li, J.; Yan, R.; Bi, Y.; Hu, W.; Zhu, Z.; Yu, Y.; et al. Glucocorticoids improve severe or critical COVID-19 by activating ACE2 and reducing IL-6 levels. Int. J. Biol. Sci. 2020, 16, 2382–2391. [Google Scholar] [CrossRef] [PubMed]
- Group, R.C.; Horby, P.; Lim, W.S.; Emberson, J.R.; Mafham, M.; Bell, J.L.; Linsell, L.; Staplin, N.; Brightling, C.; Ustianowski, A.; et al. Dexamethasone in Hospitalized Patients with Covid-19. N. Engl. J. Med. 2021, 384, 693–704. [Google Scholar] [CrossRef]
- Ramiro, S.; Mostard, R.L.M.; Magro-Checa, C.; van Dongen, C.M.P.; Dormans, T.; Buijs, J.; Gronenschild, M.; de Kruif, M.D.; van Haren, E.H.J.; van Kraaij, T.; et al. Historically controlled comparison of glucocorticoids with or without tocilizumab versus supportive care only in patients with COVID-19-associated cytokine storm syndrome: Results of the CHIC study. Ann. Rheum. Dis. 2020, 79, 1143–1151. [Google Scholar] [CrossRef]
- Group, W.H.O.R.E.A.f.C.-T.W.; Sterne, J.A.C.; Murthy, S.; Diaz, J.V.; Slutsky, A.S.; Villar, J.; Angus, D.C.; Annane, D.; Azevedo, L.C.P.; Berwanger, O.; et al. Association Between Administration of Systemic Corticosteroids and Mortality Among Critically Ill Patients With COVID-19: A Meta-analysis. JAMA 2020, 324, 1330–1341. [Google Scholar] [CrossRef]
- Edalatifard, M.; Akhtari, M.; Salehi, M.; Naderi, Z.; Jamshidi, A.; Mostafaei, S.; Najafizadeh, S.R.; Farhadi, E.; Jalili, N.; Esfahani, M.; et al. Intravenous methylprednisolone pulse as a treatment for hospitalised severe COVID-19 patients: Results from a randomised controlled clinical trial. Eur. Respir. J. 2020, 56, 2002808. [Google Scholar] [CrossRef]
- Ye, Z.W.; Yuan, S.; Chan, J.F.; Zhang, A.J.; Yu, C.Y.; Ong, C.P.; Yang, D.; Chan, C.C.; Tang, K.; Cao, J.; et al. Beneficial effect of combinational methylprednisolone and remdesivir in hamster model of SARS-CoV-2 infection. Emerg. Microbes Infect. 2021, 10, 291–304. [Google Scholar] [CrossRef] [PubMed]
- Rochwerg, B.; Agarwal, A.; Siemieniuk, R.A.; Agoritsas, T.; Lamontagne, F.; Askie, L.; Lytvyn, L.; Leo, Y.S.; Macdonald, H.; Zeng, L.; et al. A living WHO guideline on drugs for covid-19. BMJ 2020, 370, m3379. [Google Scholar] [CrossRef]
- Hench, P.S.; Kendall, E.C.; Slocumb, C.H.; Polley, H.F. The effect of a hormone of the adrenal cortex (17-hydroxy-11-dehydrocorticosterone; compound E) and of pituitary adrenocorticotropic hormone on rheumatoid arthritis. Proc. Staff. Meet Mayo Clin. 1949, 24, 181–197. [Google Scholar] [PubMed]
- Kvien, T.K. Epidemiology and burden of illness of rheumatoid arthritis. Pharmacoeconomics 2004, 22, 1–12. [Google Scholar] [CrossRef]
- Scott, D.L.; Wolfe, F.; Huizinga, T.W. Rheumatoid arthritis. Lancet 2010, 376, 1094–1108. [Google Scholar] [CrossRef]
- Alamanos, Y.; Drosos, A.A. Epidemiology of adult rheumatoid arthritis. Autoimmun. Rev. 2005, 4, 130–136. [Google Scholar] [CrossRef]
- Silman, A.J.; Pearson, J.E. Epidemiology and genetics of rheumatoid arthritis. Arthritis Res. 2002, 4 (Suppl. S3), S265–S272. [Google Scholar] [CrossRef] [Green Version]
- Karami, J.; Aslani, S.; Jamshidi, A.; Garshasbi, M.; Mahmoudi, M. Genetic implications in the pathogenesis of rheumatoid arthritis; an updated review. Gene 2019, 702, 8–16. [Google Scholar] [CrossRef]
- Yap, H.Y.; Tee, S.Z.; Wong, M.M.; Chow, S.K.; Peh, S.C.; Teow, S.Y. Pathogenic Role of Immune Cells in Rheumatoid Arthritis: Implications in Clinical Treatment and Biomarker Development. Cells 2018, 7, 161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bevaart, L.; Vervoordeldonk, M.J.; Tak, P.P. Evaluation of therapeutic targets in animal models of arthritis: How does it relate to rheumatoid arthritis? Arthritis Rheum. 2010, 62, 2192–2205. [Google Scholar] [CrossRef] [PubMed]
- Baschant, U.; Frappart, L.; Rauchhaus, U.; Bruns, L.; Reichardt, H.M.; Kamradt, T.; Brauer, R.; Tuckermann, J.P. Glucocorticoid therapy of antigen-induced arthritis depends on the dimerized glucocorticoid receptor in T cells. Proc. Natl. Acad. Sci. USA 2011, 108, 19317–19322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Misharin, A.V.; Cuda, C.M.; Saber, R.; Turner, J.D.; Gierut, A.K.; Haines, G.K., 3rd; Berdnikovs, S.; Filer, A.; Clark, A.R.; Buckley, C.D.; et al. Nonclassical Ly6C(-) monocytes drive the development of inflammatory arthritis in mice. Cell Rep. 2014, 9, 591–604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koenen, M.; Culemann, S.; Vettorazzi, S.; Caratti, G.; Frappart, L.; Baum, W.; Kronke, G.; Baschant, U.; Tuckermann, J.P. Glucocorticoid receptor in stromal cells is essential for glucocorticoid-mediated suppression of inflammation in arthritis. Ann. Rheum. Dis. 2018, 77, 1610–1618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fenton, C.; Martin, C.; Jones, R.; Croft, A.; Campos, J.; Naylor, A.J.; Taylor, A.E.; Chimen, M.; Cooper, M.; Lavery, G.G.; et al. Local steroid activation is a critical mediator of the anti-inflammatory actions of therapeutic glucocorticoids. Ann. Rheum. Dis. 2021, 80, 250–260. [Google Scholar] [CrossRef] [PubMed]
- Croft, A.P.; Campos, J.; Jansen, K.; Turner, J.D.; Marshall, J.; Attar, M.; Savary, L.; Wehmeyer, C.; Naylor, A.J.; Kemble, S.; et al. Distinct fibroblast subsets drive inflammation and damage in arthritis. Nature 2019, 570, 246–251. [Google Scholar] [CrossRef] [PubMed]
- Hardy, R.S.; Fenton, C.; Croft, A.P.; Naylor, A.J.; Begum, R.; Desanti, G.; Buckley, C.D.; Lavery, G.; Cooper, M.S.; Raza, K. 11 Beta-hydroxysteroid dehydrogenase type 1 regulates synovitis, joInt. destruction, and systemic bone loss in chronic polyarthritis. J. Autoimmun. 2018, 92, 104–113. [Google Scholar] [CrossRef]
- Coutinho, A.E.; Gray, M.; Brownstein, D.G.; Salter, D.M.; Sawatzky, D.A.; Clay, S.; Gilmour, J.S.; Seckl, J.R.; Savill, J.S.; Chapman, K.E. 11beta-Hydroxysteroid dehydrogenase type 1, but not type 2, deficiency worsens acute inflammation and experimental arthritis in mice. Endocrinology 2012, 153, 234–240. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Dong, Y.; Zou, F.; Wu, M.; Fan, C.; Ding, Y. 11beta-Hydroxysteroid dehydrogenase 1 inhibition attenuates collagen-induced arthritis. Int. Immunopharmacol. 2013, 17, 489–494. [Google Scholar] [CrossRef]
- Macfarlane, E.; Seibel, M.J.; Zhou, H. Arthritis and the role of endogenous glucocorticoids. Bone Res. 2020, 8, 33. [Google Scholar] [CrossRef]
- Buttgereit, F.; Zhou, H.; Kalak, R.; Gaber, T.; Spies, C.M.; Huscher, D.; Straub, R.H.; Modzelewski, J.; Dunstan, C.R.; Seibel, M.J. Transgenic disruption of glucocorticoid signaling in mature osteoblasts and osteocytes attenuates K/BxN mouse serum-induced arthritis in vivo. Arthritis Rheum. 2009, 60, 1998–2007. [Google Scholar] [CrossRef]
- Tu, J.; Stoner, S.; Fromm, P.D.; Wang, T.; Chen, D.; Tuckermann, J.; Cooper, M.S.; Seibel, M.J.; Zhou, H. Endogenous glucocorticoid signaling in chondrocytes attenuates joInt. inflammation and damage. FASEB J. 2018, 32, 478–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ehrchen, J.; Steinmuller, L.; Barczyk, K.; Tenbrock, K.; Nacken, W.; Eisenacher, M.; Nordhues, U.; Sorg, C.; Sunderkotter, C.; Roth, J. Glucocorticoids induce differentiation of a specifically activated, anti-inflammatory subtype of human monocytes. Blood 2007, 109, 1265–1274. [Google Scholar] [CrossRef] [Green Version]
- Goulding, N.J.; Godolphin, J.L.; Sharland, P.R.; Peers, S.H.; Sampson, M.; Maddison, P.J.; Flower, R.J. Anti-inflammatory lipocortin 1 production by peripheral blood leucocytes in response to hydrocortisone. Lancet 1990, 335, 1416–1418. [Google Scholar] [CrossRef]
- Perretti, M.; D’Acquisto, F. Annexin A1 and glucocorticoids as effectors of the resolution of inflammation. Nat. Rev. Immunol. 2009, 9, 62–70. [Google Scholar] [CrossRef]
- Purvis, G.S.D.; Solito, E.; Thiemermann, C. Annexin-A1: Therapeutic Potential in Microvascular Disease. Front. Immunol. 2019, 10, 938. [Google Scholar] [CrossRef] [Green Version]
- Patel, H.B.; Kornerup, K.N.; Sampaio, A.L.; D’Acquisto, F.; Seed, M.P.; Girol, A.P.; Gray, M.; Pitzalis, C.; Oliani, S.M.; Perretti, M. The impact of endogenous annexin A1 on glucocorticoid control of inflammatory arthritis. Ann. Rheum. Dis. 2012, 71, 1872–1880. [Google Scholar] [CrossRef] [Green Version]
- Solito, E.; Kamal, A.; Russo-Marie, F.; Buckingham, J.C.; Marullo, S.; Perretti, M. A novel calcium-dependent proapoptotic effect of annexin 1 on human neutrophils. FASEB J. 2003, 17, 1544–1546. [Google Scholar] [CrossRef]
- Vago, J.P.; Nogueira, C.R.; Tavares, L.P.; Soriani, F.M.; Lopes, F.; Russo, R.C.; Pinho, V.; Teixeira, M.M.; Sousa, L.P. Annexin A1 modulates natural and glucocorticoid-induced resolution of inflammation by enhancing neutrophil apoptosis. J. Leukoc. Biol. 2012, 92, 249–258. [Google Scholar] [CrossRef]
- Maderna, P.; Yona, S.; Perretti, M.; Godson, C. Modulation of phagocytosis of apoptotic neutrophils by supernatant from dexamethasone-treated macrophages and annexin-derived peptide Ac(2-26). J. Immunol. 2005, 174, 3727–3733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalli, J.; Jones, C.P.; Cavalcanti, D.M.; Farsky, S.H.; Perretti, M.; Rankin, S.M. Annexin A1 regulates neutrophil clearance by macrophages in the mouse bone marrow. FASEB J. 2012, 26, 387–396. [Google Scholar] [CrossRef] [PubMed]
- Beaulieu, E.; Ngo, D.; Santos, L.; Yang, Y.H.; Smith, M.; Jorgensen, C.; Escriou, V.; Scherman, D.; Courties, G.; Apparailly, F.; et al. Glucocorticoid-induced leucine zipper is an endogenous antiinflammatory mediator in arthritis. Arthritis Rheum. 2010, 62, 2651–2661. [Google Scholar] [CrossRef] [PubMed]
- Ngo, D.; Beaulieu, E.; Gu, R.; Leaney, A.; Santos, L.; Fan, H.; Yang, Y.; Kao, W.; Xu, J.; Escriou, V.; et al. Divergent effects of endogenous and exogenous glucocorticoid-induced leucine zipper in animal models of inflammation and arthritis. Arthritis Rheum. 2013, 65, 1203–1212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vattakuzhi, Y.; Abraham, S.M.; Freidin, A.; Clark, A.R.; Horwood, N.J. Dual-specificity phosphatase 1-null mice exhibit spontaneous osteolytic disease and enhanced inflammatory osteolysis in experimental arthritis. Arthritis Rheum. 2012, 64, 2201–2210. [Google Scholar] [CrossRef] [PubMed]
- Hoppstadter, J.; Ammit, A.J. Role of Dual-Specificity Phosphatase 1 in Glucocorticoid-Driven Anti-inflammatory Responses. Front. Immunol. 2019, 10, 1446. [Google Scholar] [CrossRef]
- Mathe, G.; Amiel, J.L.; Schwarzenberg, L.; Cattan, A.; Schneider, M. Adoptive immunotherapy of acute leukemia: Experimental and clinical results. Cancer Res. 1965, 25, 1525–1531. [Google Scholar]
- Biernacki, M.A.; Sheth, V.S.; Bleakley, M. T cell optimization for graft-versus-leukemia responses. JCI Insight 2020, 5. [Google Scholar] [CrossRef] [PubMed]
- Shlomchik, W.D. Graft-versus-host disease. Nat. Rev. Immunol. 2007, 7, 340–352. [Google Scholar] [CrossRef] [PubMed]
- Van Lint, M.T.; Uderzo, C.; Locasciulli, A.; Majolino, I.; Scime, R.; Locatelli, F.; Giorgiani, G.; Arcese, W.; Iori, A.P.; Falda, M.; et al. Early treatment of acute graft-versus-host disease with high- or low-dose 6-methylprednisolone: A multicenter randomized trial from the Italian Group for Bone Marrow Transplantation. Blood 1998, 92, 2288–2293. [Google Scholar]
- Fuji, S.; Byrne, M.; Nagler, A.; Mohty, M.; Savani, B.N. How we can mitigate the side effects associated with systemic glucocorticoid after allogeneic hematopoietic cell transplantation. Bone Marrow Transplant. 2021, 56, 1248–1256. [Google Scholar] [CrossRef]
- Pidala, J.; Hamadani, M.; Dawson, P.; Martens, M.; Alousi, A.M.; Jagasia, M.; Efebera, Y.A.; Chhabra, S.; Pusic, I.; Holtan, S.G.; et al. Randomized multicenter trial of sirolimus vs prednisone as initial therapy for standard-risk acute GVHD: The BMT CTN 1501 trial. Blood 2020, 135, 97–107. [Google Scholar] [CrossRef]
- Bacigalupo, A.; Milone, G.; Cupri, A.; Severino, A.; Fagioli, F.; Berger, M.; Santarone, S.; Chiusolo, P.; Sica, S.; Mammoliti, S.; et al. Steroid treatment of acute graft-versus-host disease grade I: A randomized trial. Haematologica 2017, 102, 2125–2133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDonald, G.B.; Tabellini, L.; Storer, B.E.; Martin, P.J.; Lawler, R.L.; Rosinski, S.L.; Schoch, H.G.; Hansen, J.A. Predictive Value of Clinical Findings and Plasma Biomarkers after Fourteen Days of Prednisone Treatment for Acute Graft-versus-host Disease. Biol. Blood Marrow Transplant. 2017, 23, 1257–1263. [Google Scholar] [CrossRef]
- Hill, L.; Alousi, A.; Kebriaei, P.; Mehta, R.; Rezvani, K.; Shpall, E. New and emerging therapies for acute and chronic graft versus host disease. Ther. Adv. Hematol. 2018, 9, 21–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braun, L.M.; Zeiser, R. Immunomodulatory Therapies for the Treatment of Graft-versus-host Disease. Hemasphere 2021, 5, e581. [Google Scholar] [CrossRef] [PubMed]
- Bouazzaoui, A.; Spacenko, E.; Mueller, G.; Huber, E.; Schubert, T.; Holler, E.; Andreesen, R.; Hildebrandt, G.C. Steroid treatment alters adhesion molecule and chemokine expression in experimental acute graft-vs.-host disease of the intestinal tract. Exp. Hematol. 2011, 39, 238–249.e231. [Google Scholar] [CrossRef] [PubMed]
- Theiss-Suennemann, J.; Jorss, K.; Messmann, J.J.; Reichardt, S.D.; Montes-Cobos, E.; Lühder, F.; Tuckermann, J.P.; Wolff, H.A.; Dressel, R.; Gröne, H.J.; et al. Glucocorticoids attenuate acute graft-versus-host disease by suppressing the cytotoxic capacity of CD8(+) T cells. J. Pathol. 2015, 235, 646–655. [Google Scholar] [CrossRef]
- Menger, L.; Gouble, A.; Marzolini, M.A.; Pachnio, A.; Bergerhoff, K.; Henry, J.Y.; Smith, J.; Pule, M.; Moss, P.; Riddell, S.R.; et al. TALEN-mediated genetic inactivation of the glucocorticoid receptor in cytomegalovirus-specific T cells. Blood 2015, 126, 2781–2789. [Google Scholar] [CrossRef] [Green Version]
- Basar, R.; Daher, M.; Uprety, N.; Gokdemir, E.; Alsuliman, A.; Ensley, E.; Ozcan, G.; Mendt, M.; Hernandez Sanabria, M.; Kerbauy, L.N.; et al. Large-scale GMP-compliant CRISPR-Cas9-mediated deletion of the glucocorticoid receptor in multivirus-specific T cells. Blood Adv. 2020, 4, 3357–3367. [Google Scholar] [CrossRef] [PubMed]
- Baake, T.; Jorss, K.; Suennemann, J.; Rossmann, L.; Bohnenberger, H.; Tuckermann, J.P.; Reichardt, H.M.; Fischer, H.J.; Reichardt, S.D. The glucocorticoid receptor in recipient cells keeps cytokine secretion in acute graft-versus-host disease at bay. Oncotarget 2018, 9, 15437–15450. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Kaiser, T.K.; Borschiwer, M.; Bohnenberger, H.; Reichardt, S.D.; Lühder, F.; Walter, L.; Dressel, R.; Meijsing, S.H.; Reichardt, H.M. Glucocorticoid resistance of allogeneic T cells alters the gene expression profile in the inflamed small intestine of mice suffering from acute graft-versus-host disease. J. Steroid Biochem. Mol. Biol. 2019, 195, 105485. [Google Scholar] [CrossRef]
- Macintyre, A.N.; Gerriets, V.A.; Nichols, A.G.; Michalek, R.D.; Rudolph, M.C.; Deoliveira, D.; Anderson, S.M.; Abel, E.D.; Chen, B.J.; Hale, L.P.; et al. The glucose transporter Glut1 is selectively essential for CD4 T cell activation and effector function. Cell Metab. 2014, 20, 61–72. [Google Scholar] [CrossRef] [Green Version]
- Uhl, F.M.; Chen, S.; O’Sullivan, D.; Edwards-Hicks, J.; Richter, G.; Haring, E.; Andrieux, G.; Halbach, S.; Apostolova, P.; Buscher, J.; et al. Metabolic reprogramming of donor T cells enhances graft-versus-leukemia effects in mice and humans. Sci. Transl. Med. 2020, 12, eabb8969. [Google Scholar] [CrossRef]
- Chountoulesi, M.; Demetzos, C. Promising Nanotechnology Approaches in Treatment of Autoimmune Diseases of Central Nervous System. Brain Sci. 2020, 10, 338. [Google Scholar] [CrossRef]
- Hao, Y.; Zhang, F.; Mo, S.; Zhao, J.; Wang, X.; Zhao, Y.; Zhang, L. Biomedical Applications of Supramolecular Materials in the Controllable Delivery of Steroids. Front. Mol. BioSci. 2021, 8, 700712. [Google Scholar] [CrossRef]
- Chapman, K.E.; Coutinho, A.E.; Zhang, Z.; Kipari, T.; Savill, J.S.; Seckl, J.R. Changing glucocorticoid action: 11beta-hydroxysteroid dehydrogenase type 1 in acute and chronic inflammation. J. Steroid Biochem. Mol. Biol. 2013, 137, 82–92. [Google Scholar] [CrossRef] [Green Version]
- Pereira, C.D.; Azevedo, I.; Monteiro, R.; Martins, M.J. 11beta-Hydroxysteroid dehydrogenase type 1: Relevance of its modulation in the pathophysiology of obesity, the metabolic syndrome and type 2 diabetes mellitus. Diabetes Obes. Metab. 2012, 14, 869–881. [Google Scholar] [CrossRef] [PubMed]
- Heck, S.; Bender, K.; Kullmann, M.; Göttlicher, M.; Herrlich, P.; Cato, A.C. I kappaB alpha-independent downregulation of NF-kappaB activity by glucocorticoid receptor. EMBO J. 1997, 16, 4698–4707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heck, S.; Kullmann, M.; Gast, A.; Ponta, H.; Rahmsdorf, H.J.; Herrlich, P.; Cato, A.C. A distinct modulating domain in glucocorticoid receptor monomers in the repression of activity of the transcription factor AP-1. EMBO J. 1994, 13, 4087–4095. [Google Scholar] [CrossRef] [PubMed]
- Reichardt, H.M.; Kaestner, K.H.; Tuckermann, J.; Kretz, O.; Wessely, O.; Bock, R.; Gass, P.; Schmid, W.; Herrlich, P.; Angel, P.; et al. DNA binding of the glucocorticoid receptor is not essential for survival. Cell 1998, 93, 531–541. [Google Scholar] [CrossRef] [Green Version]
- Reichardt, H.M.; Tuckermann, J.P.; Göttlicher, M.; Vujic, M.; Weih, F.; Angel, P.; Herrlich, P.; Schütz, G. Repression of inflammatory responses in the absence of DNA binding by the glucocorticoid receptor. EMBO J. 2001, 20, 7168–7173. [Google Scholar] [CrossRef] [Green Version]
- Tuckermann, J.P.; Kleiman, A.; Moriggl, R.; Spanbroek, R.; Neumann, A.; Illing, A.; Clausen, B.E.; Stride, B.; Förster, I.; Habenicht, A.J.; et al. Macrophages and neutrophils are the targets for immune suppression by glucocorticoids in contact allergy. J. Clin. Investig. 2007, 117, 1381–1390. [Google Scholar] [CrossRef]
- Frijters, R.; Fleuren, W.; Toonen, E.J.; Tuckermann, J.P.; Reichardt, H.M.; van der Maaden, H.; van Elsas, A.; van Lierop, M.J.; Dokter, W.; de Vlieg, J.; et al. Prednisolone-induced differential gene expression in mouse liver carrying wild type or a dimerization-defective glucocorticoid receptor. BMC Genom. 2010, 11, 359. [Google Scholar] [CrossRef] [Green Version]
- Rauch, A.; Seitz, S.; Baschant, U.; Schilling, A.F.; Illing, A.; Stride, B.; Kirilov, M.; Mandic, V.; Takacz, A.; Schmidt-Ullrich, R.; et al. Glucocorticoids suppress bone formation by attenuating osteoblast differentiation via the monomeric glucocorticoid receptor. Cell Metab. 2010, 11, 517–531. [Google Scholar] [CrossRef] [Green Version]
- Waddell, D.S.; Baehr, L.M.; van den Brandt, J.; Johnsen, S.A.; Reichardt, H.M.; Furlow, J.D.; Bodine, S.C. The glucocorticoid receptor and FOXO1 synergistically activate the skeletal muscle atrophy-associated MuRF1 gene. Am. J. Physiol. 2008, 295, E785–E797. [Google Scholar] [CrossRef] [PubMed]
- Johnson, T.A.; Paakinaho, V.; Kim, S.; Hager, G.L.; Presman, D.M. Genome-wide binding potential and regulatory activity of the glucocorticoid receptor’s monomeric and dimeric forms. Nat. Commun. 2021, 12, 1987. [Google Scholar] [CrossRef]
- Hübner, S.; Dejager, L.; Libert, C.; Tuckermann, J.P. The glucocorticoid receptor in inflammatory processes: Transrepression is not enough. Biol. Chem. 2015, 396, 1223–1231. [Google Scholar] [CrossRef] [PubMed]
- Vayssiere, B.M.; Dupont, S.; Choquart, A.; Petit, F.; Garcia, T.; Marchandeau, C.; Gronemeyer, H.; Resche-Rigon, M. Synthetic glucocorticoids that dissociate transactivation and AP-1 transrepression exhibit antiinflammatory activity in vivo. Mol. Endocrinol. 1997, 11, 1245–1255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coghlan, M.J.; Jacobson, P.B.; Lane, B.; Nakane, M.; Lin, C.W.; Elmore, S.W.; Kym, P.R.; Luly, J.R.; Carter, G.W.; Turner, R.; et al. A novel antiinflammatory maintains glucocorticoid efficacy with reduced side effects. Mol. Endocrinol. 2003, 17, 860–869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Lierop, M.J.; Alkema, W.; Laskewitz, A.J.; Dijkema, R.; van der Maaden, H.M.; Smit, M.J.; Plate, R.; Conti, P.G.; Jans, C.G.; Timmers, C.M.; et al. Org 214007-0: A novel non-steroidal selective glucocorticoid receptor modulator with full anti-inflammatory properties and improved therapeutic index. PLoS ONE 2012, 7, e48385. [Google Scholar] [CrossRef] [Green Version]
- Baiula, M.; Spampinato, S. Mapracorat, a novel non-steroidal selective glucocorticoid receptor agonist for the treatment of allergic conjunctivitis. Inflamm. Allergy Drug Targets 2014, 13, 289–298. [Google Scholar] [CrossRef]
- Cavet, M.E.; Volhejn, S.; Harrington, K.L.; Zhang, J.Z. Anti-allergic effects of mapracorat, a novel selective glucocorticoid receptor agonist, in human conjunctival fibroblasts and epithelial cells. Mol. Vis. 2013, 19, 1515–1525. [Google Scholar]
- De Bosscher, K.; Vanden Berghe, W.; Beck, I.M.; Van Molle, W.; Hennuyer, N.; Hapgood, J.; Libert, C.; Staels, B.; Louw, A.; Haegeman, G. A fully dissociated compound of plant origin for inflammatory gene repression. Proc. Natl. Acad. Sci. USA 2005, 102, 15827–15832. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Zhang, Z.Y.; Schluesener, H.J. Compound A, a plant origin ligand of glucocorticoid receptors, increases regulatory T cells and M2 macrophages to attenuate experimental autoimmune neuritis with reduced side effects. J. Immunol. 2009, 183, 3081–3091. [Google Scholar] [CrossRef]
- Wüst, S.; Tischner, D.; John, M.; Tuckermann, J.P.; Menzfeld, C.; Hanisch, U.K.; van den Brandt, J.; Lühder, F.; Reichardt, H.M. Therapeutic and adverse effects of a non-steroidal glucocorticoid receptor ligand in a mouse model of multiple sclerosis. PLoS ONE 2009, 4, e8202. [Google Scholar] [CrossRef] [Green Version]
- Dewint, P.; Gossye, V.; De Bosscher, K.; Vanden Berghe, W.; Van Beneden, K.; Deforce, D.; Van Calenbergh, S.; Muller-Ladner, U.; Vander Cruyssen, B.; Verbruggen, G.; et al. A plant-derived ligand favoring monomeric glucocorticoid receptor conformation with impaired transactivation potential attenuates collagen-induced arthritis. J. Immunol. 2008, 180, 2608–2615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reber, L.L.; Daubeuf, F.; Plantinga, M.; De Cauwer, L.; Gerlo, S.; Waelput, W.; Van Calenbergh, S.; Tavernier, J.; Haegeman, G.; Lambrecht, B.N.; et al. A dissociated glucocorticoid receptor modulator reduces airway hyperresponsiveness and inflammation in a mouse model of asthma. J. Immunol. 2012, 188, 3478–3487. [Google Scholar] [CrossRef] [Green Version]
- Hua, G.; Zein, N.; Daubeuf, F.; Chambon, P. Glucocorticoid receptor modulators CpdX and CpdX-D3 exhibit the same in vivo antiinflammatory activities as synthetic glucocorticoids. Proc. Natl. Acad. Sci. USA 2019, 116, 14191–14199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rauch, A.; Gossye, V.; Bracke, D.; Gevaert, E.; Jacques, P.; Van Beneden, K.; Vandooren, B.; Rauner, M.; Hofbauer, L.C.; Haegeman, G.; et al. An anti-inflammatory selective glucocorticoid receptor modulator preserves osteoblast differentiation. FASEB J. 2011. [Google Scholar] [CrossRef]
- Rauner, M.; Goettsch, C.; Stein, N.; Thiele, S.; Bornhaeuser, M.; De Bosscher, K.; Haegeman, G.; Tuckermann, J.; Hofbauer, L.C. Dissociation of osteogenic and immunological effects by the selective glucocorticoid receptor agonist, compound A, in human bone marrow stromal cells. Endocrinology 2011, 152, 103–112. [Google Scholar] [CrossRef]
- Hua, G.; Zein, N.; Paulen, L.; Chambon, P. The glucocorticoid receptor agonistic modulators CpdX and CpdX-D3 do not generate the debilitating effects of synthetic glucocorticoids. Proc. Natl. Acad. Sci. USA 2019, 116, 14200–14209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reeves, E.K.M.; Hoffman, E.P.; Nagaraju, K.; Damsker, J.M.; McCall, J.M. VBP15: Preclinical characterization of a novel anti-inflammatory delta 9,11 steroid. Bioorg. Med. Chem. 2013, 21, 2241–2249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Damsker, J.M.; Dillingham, B.C.; Rose, M.C.; Balsley, M.A.; Heier, C.R.; Watson, A.M.; Stemmy, E.J.; Jurjus, R.A.; Huynh, T.; Tatem, K.; et al. VBP15, a glucocorticoid analogue, is effective at reducing allergic lung inflammation in mice. PLoS ONE 2013, 8, e63871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Damsker, J.M.; Conklin, L.S.; Sadri, S.; Dillingham, B.C.; Panchapakesan, K.; Heier, C.R.; McCall, J.M.; Sandler, A.D. VBP15, a novel dissociative steroid compound, reduces NFkappaB-induced expression of inflammatory cytokines in vitro and symptoms of murine trinitrobenzene sulfonic acid-induced colitis. Inflamm. Res. 2016, 65, 737–743. [Google Scholar] [CrossRef]
- Akkad, H.; Cacciani, N.; Llano-Diez, M.; Corpeno Kalamgi, R.; Tchkonia, T.; Kirkland, J.L.; Larsson, L. Vamorolone treatment improves skeletal muscle outcome in a critical illness myopathy rat model. Acta Physiol. 2019, 225, e13172. [Google Scholar] [CrossRef] [PubMed]
- Beenakker, E.A.; Fock, J.M.; Van Tol, M.J.; Maurits, N.M.; Koopman, H.M.; Brouwer, O.F.; Van der Hoeven, J.H. Intermittent prednisone therapy in Duchenne muscular dystrophy: A randomized controlled trial. Arch. Neurol. 2005, 62, 128–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matthews, E.; Brassington, R.; Kuntzer, T.; Jichi, F.; Manzur, A.Y. Corticosteroids for the treatment of Duchenne muscular dystrophy. Cochrane Database Syst. Rev. 2016, CD003725. [Google Scholar] [CrossRef] [Green Version]
- Hoffman, E.P.; Riddle, V.; Siegler, M.A.; Dickerson, D.; Backonja, M.; Kramer, W.G.; Nagaraju, K.; Gordish-Dressman, H.; Damsker, J.M.; McCall, J.M. Phase 1 trial of vamorolone, a first-in-class steroid, shows improvements in side effects via biomarkers bridged to clinical outcomes. Steroids 2018, 134, 43–52. [Google Scholar] [CrossRef]
- Conklin, L.S.; Damsker, J.M.; Hoffman, E.P.; Jusko, W.J.; Mavroudis, P.D.; Schwartz, B.D.; Mengle-Gaw, L.J.; Smith, E.C.; Mah, J.K.; Guglieri, M.; et al. Phase IIa trial in Duchenne muscular dystrophy shows vamorolone is a first-in-class dissociative steroidal anti-inflammatory drug. Pharmacol. Res. 2018, 136, 140–150. [Google Scholar] [CrossRef]
- Hoffman, E.P.; Schwartz, B.D.; Mengle-Gaw, L.J.; Smith, E.C.; Castro, D.; Mah, J.K.; McDonald, C.M.; Kuntz, N.L.; Finkel, R.S.; Guglieri, M.; et al. Vamorolone trial in Duchenne muscular dystrophy shows dose-related improvement of muscle function. Neurology 2019, 93, e1312–e1323. [Google Scholar] [CrossRef] [Green Version]
- Avnir, Y.; Turjeman, K.; Tulchinsky, D.; Sigal, A.; Kizelsztein, P.; Tzemach, D.; Gabizon, A.; Barenholz, Y. Fabrication principles and their contribution to the superior in vivo therapeutic efficacy of nano-liposomes remote loaded with glucocorticoids. PLoS ONE 2011, 6, e25721. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, J.; Metselaar, J.M.; Gold, R. Intravenous liposomal prednisolone downregulates in situ TNF-alpha production by T-cells in experimental autoimmune encephalomyelitis. J. Histochem. Cytochem. 2003, 51, 1241–1244. [Google Scholar] [CrossRef]
- Turjeman, K.; Bavli, Y.; Kizelsztein, P.; Schilt, Y.; Allon, N.; Katzir, T.B.; Sasson, E.; Raviv, U.; Ovadia, H.; Barenholz, Y. Nano-Drugs Based on Nano Sterically Stabilized Liposomes for the Treatment of Inflammatory Neurodegenerative Diseases. PLoS ONE 2015, 10, e0130442. [Google Scholar] [CrossRef] [Green Version]
- Linker, R.A.; Weller, C.; Lühder, F.; Mohr, A.; Schmidt, J.; Knauth, M.; Metselaar, J.M.; Gold, R. Liposomal glucocorticosteroids in treatment of chronic autoimmune demyelination: Long-term protective effects and enhanced efficacy of methylprednisolone formulations. Exp. Neurol. 2008, 211, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Tiebosch, I.A.; Crielaard, B.J.; Bouts, M.J.; Zwartbol, R.; Salas-Perdomo, A.; Lammers, T.; Planas, A.M.; Storm, G.; Dijkhuizen, R.M. Combined treatment with recombinant tissue plasminogen activator and dexamethasone phosphate-containing liposomes improves neurological outcome and restricts lesion progression after embolic stroke in rats. J. Neurochem. 2012, 123 (Suppl. S2), 65–74. [Google Scholar] [CrossRef]
- Gaillard, P.J.; Appeldoorn, C.C.; Rip, J.; Dorland, R.; van der Pol, S.M.; Kooij, G.; de Vries, H.E.; Reijerkerk, A. Enhanced brain delivery of liposomal methylprednisolone improved therapeutic efficacy in a model of neuroinflammation. J. Control. Release 2012, 164, 364–369. [Google Scholar] [CrossRef] [PubMed]
- Verma, A.; Jain, A.; Tiwari, A.; Saraf, S.; Panda, P.K.; Agrawal, G.P.; Jain, S.K. Folate Conjugated Double Liposomes Bearing Prednisolone and Methotrexate for Targeting Rheumatoid Arthritis. Pharm. Res. 2019, 36, 123. [Google Scholar] [CrossRef]
- Wang, S.; Yang, S.; Lai, X.; Song, Y.; Hu, L.; Li, C.; Shi, T.; Liu, X.; Deng, Y.; Chen, G. Sialic Acid Conjugate-Modified Liposomal Dexamethasone Palmitate Targeting Neutrophils for Rheumatoid Arthritis Therapy: Influence of Particle Size. AAPS PharmSciTech 2021, 22, 16. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.C.; Chiang, P.F.; Kuo, Y.J.; Peng, C.L.; Chen, K.Y.; Chiang, Y.C. Hyaluronan-Loaded Liposomal Dexamethasone-Diclofenac Nanoparticles for Local Osteoarthritis Treatment. Int. J. Mol. Sci. 2021, 22, 665. [Google Scholar] [CrossRef]
- Nishiwaki, S.; Nakayama, T.; Murata, M.; Nishida, T.; Terakura, S.; Saito, S.; Kato, T.; Mizuno, H.; Imahashi, N.; Seto, A.; et al. Dexamethasone palmitate ameliorates macrophages-rich graft-versus-host disease by inhibiting macrophage functions. PLoS ONE 2014, 9, e96252. [Google Scholar] [CrossRef]
- Heck, J.G.; Napp, J.; Simonato, S.; Mollmer, J.; Lange, M.; Reichardt, H.M.; Staudt, R.; Alves, F.; Feldmann, C. Multifunctional phosphate-based inorganic-organic hybrid nanoparticles. J. Am. Chem. Soc. 2015, 137, 7329–7336. [Google Scholar] [CrossRef]
- Kaiser, T.K.; Khorenko, M.; Moussavi, A.; Engelke, M.; Boretius, S.; Feldmann, C.; Reichardt, H.M. Highly selective organ distribution and cellular uptake of inorganic-organic hybrid nanoparticles customized for the targeted delivery of glucocorticoids. J. Control. Release 2020, 319, 360–370. [Google Scholar] [CrossRef]
- Napp, J.; Markus, M.A.; Heck, J.G.; Dullin, C.; Mobius, W.; Gorpas, D.; Feldmann, C.; Alves, F. Therapeutic Fluorescent Hybrid Nanoparticles for Traceable Delivery of Glucocorticoids to Inflammatory Sites. Theranostics 2018, 8, 6367–6383. [Google Scholar] [CrossRef]
- Kaiser, T.K.; Li, H.; Rossmann, L.; Reichardt, S.D.; Bohnenberger, H.; Feldmann, C.; Reichardt, H.M. Glucocorticoids delivered by inorganic-organic hybrid nanoparticles mitigate acute graft-versus-host disease and sustain graft-versus-leukemia activity. Eur. J. Immunol. 2020, 50, 1220–1233. [Google Scholar] [CrossRef] [Green Version]
- Abou-ElNour, M.; Ishak, R.A.H.; Tiboni, M.; Bonacucina, G.; Cespi, M.; Casettari, L.; Soliman, M.E.; Geneidi, A.S. Triamcinolone acetonide-loaded PLA/PEG-PDL microparticles for effective intra-articular delivery: Synthesis, optimization, in vitro and in vivo evaluation. J. Control. Release 2019, 309, 125–144. [Google Scholar] [CrossRef] [PubMed]
- Jadhav, D.; Vavia, P. Dexamethasone Sodium Phosphate Loaded Modified Cyclodextrin Based Nanoparticles: An Efficient Treatment for Rheumatoid Arthritis. J. Pharm. Sci. 2021, 110, 1206–1218. [Google Scholar] [CrossRef] [PubMed]
- Gadade, D.D.; Pekamwar, S.S. Cyclodextrin Based Nanoparticles for Drug Delivery and Theranostics. Adv. Pharm. Bull. 2020, 10, 166–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Mu, J.; Xu, Z.; Zhong, H.; Chen, Z.; Ni, Q.; Liang, X.J.; Guo, S. Modular Acid-Activatable Acetone-Based Ketal-Linked Nanomedicine by Dexamethasone Prodrugs for Enhanced Anti-Rheumatoid Arthritis with Low Side Effects. Nano Lett. 2020, 20, 2558–2568. [Google Scholar] [CrossRef]
- Perni, S.; Prokopovich, P. Optimisation and feature selection of poly-beta-amino-ester as a drug delivery system for cartilage. J. Mater. Chem. B 2020, 8, 5096–5108. [Google Scholar] [CrossRef] [PubMed]
- Yan, F.; Zhong, Z.; Wang, Y.; Feng, Y.; Mei, Z.; Li, H.; Chen, X.; Cai, L.; Li, C. Exosome-based biomimetic nanoparticles targeted to inflamed joints for enhanced treatment of rheumatoid arthritis. J. Nanobiotechnol. 2020, 18, 115. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.H.; Jung, W.; Keum, H.; Kim, T.W.; Jon, S. Nanoparticles Derived from the Natural Antioxidant Rosmarinic Acid Ameliorate Acute Inflammatory Bowel Disease. ACS Nano 2020, 14, 6887–6896. [Google Scholar] [CrossRef]
- Chen, S.Q.; Song, Y.Q.; Wang, C.; Tao, S.; Yu, F.Y.; Lou, H.Y.; Hu, F.Q.; Yuan, H. Chitosan-modified lipid nanodrug delivery system for the targeted and responsive treatment of ulcerative colitis. Carbohydr. Polym. 2020, 230, 115613. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.Y.; Cheon, J.H.; Lee, S.H.; Min, J.Y.; Back, S.Y.; Song, J.G.; Kim, D.H.; Lim, S.J.; Han, H.K. Ternary nanocomposite carriers based on organic clay-lipid vesicles as an effective colon-targeted drug delivery system: Preparation and in vitro/in vivo characterization. J. Nanobiotechnol. 2020, 18, 17. [Google Scholar] [CrossRef] [PubMed]
- Lazaro, C.M.; de Oliveira, C.C.; Gambero, A.; Rocha, T.; Cereda, C.M.S.; de Araujo, D.R.; Tofoli, G.R. Evaluation of Budesonide-Hydroxypropyl-beta-Cyclodextrin Inclusion Complex in Thermoreversible Gels for Ulcerative Colitis. Dig Dis. Sci. 2020, 65, 3297–3304. [Google Scholar] [CrossRef] [PubMed]
- Nizic Nodilo, L.; Ugrina, I.; Spoljaric, D.; Amidzic Klaric, D.; Jakobusic Brala, C.; Perkusic, M.; Pepic, I.; Lovric, J.; Sarson, V.; Safundzic Kucuk, M.; et al. A Dry Powder Platform for Nose-to-Brain Delivery of Dexamethasone: Formulation Development and Nasal Deposition Studies. Pharmaceutics 2021, 13, 795. [Google Scholar] [CrossRef] [PubMed]
- Tada, Y.; Iversen, L.; Koo, J. Early efficacy and safety data with fixed-dose combination calcipotriol/betamethasone dipropionate foam attributed to mechanism of absorption and steroid potency. J. Eur. Acad. Dermatol. Venereol. 2021, 35 (Suppl. S1), 5–9. [Google Scholar] [CrossRef]
- Formica, M.L.; Ullio Gamboa, G.V.; Tartara, L.I.; Luna, J.D.; Benoit, J.P.; Palma, S.D. Triamcinolone acetonide-loaded lipid nanocapsules for ophthalmic applications. Int. J. Pharm. 2020, 573, 118795. [Google Scholar] [CrossRef] [PubMed]
- Formica, M.L.; Legeay, S.; Bejaud, J.; Montich, G.G.; Ullio Gamboa, G.V.; Benoit, J.P.; Palma, S.D. Novel hybrid lipid nanocapsules loaded with a therapeutic monoclonal antibody—Bevacizumab-and Triamcinolone acetonide for combined therapy in neovascular ocular pathologies. Mater Sci. Eng. C Mater Biol. Appl. 2021, 119, 111398. [Google Scholar] [CrossRef] [PubMed]
- Quarta, C.; Clemmensen, C.; Zhu, Z.; Yang, B.; Joseph, S.S.; Lutter, D.; Yi, C.X.; Graf, E.; Garcia-Caceres, C.; Legutko, B.; et al. Molecular Integration of Incretin and Glucocorticoid Action Reverses Immunometabolic Dysfunction and Obesity. Cell Metab. 2017, 26, 620–632.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Valk, F.M.; van Wijk, D.F.; Lobatto, M.E.; Verberne, H.J.; Storm, G.; Willems, M.C.; Legemate, D.A.; Nederveen, A.J.; Calcagno, C.; Mani, V.; et al. Prednisolone-containing liposomes accumulate in human atherosclerotic macrophages upon intravenous administration. Nanomedicine 2015, 11, 1039–1046. [Google Scholar] [CrossRef] [Green Version]
- Kanhai, K.M.S.; Zuiker, R.; Stavrakaki, I.; Gladdines, W.; Gaillard, P.J.; Klaassen, E.S.; Groeneveld, G.J. Glutathione-PEGylated liposomal methylprednisolone in comparison to free methylprednisolone: Slow release characteristics and prolonged lymphocyte depression in a first-in-human study. Br J. Clin. Pharmacol. 2018, 84, 1020–1028. [Google Scholar] [CrossRef]
| Gene | Type of Mutation | Model | Disease | Therapy (Free GCs) | Reference |
|---|---|---|---|---|---|
| GR | T cell-specific deletion | MOG-EAE | aggravated | abrogated | [47] |
| GR | myeloid cell-specific deletion | MOG-EAE | aggravated | unaltered | [47] |
| GR | impaired dimerization, ubiquitous | MOG-EAE | unaltered | unaltered | [33] |
| MR | myeloid cell-specific deletion | MOG-EAE | ameliorated | n.d. | [53] |
| Gene | Type of Mutation | Model | Disease | Therapy (Free GCs) | Reference |
|---|---|---|---|---|---|
| GR | T cell-specific deletion | fully MHC-mismatched | aggravated | partially abrogated | [176] |
| GR | T cell-specific deletion | MHC class I- disparate | aggravated | n.d. | [176] |
| GR | myeloid cell-specific deletion | fully MHC-mismatched | aggravated | n.d. | [179] |
| GR | impaired dimerization, ubiquitous | fully MHC-mismatched | aggravated | n.d. | [179] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reichardt, S.D.; Amouret, A.; Muzzi, C.; Vettorazzi, S.; Tuckermann, J.P.; Lühder, F.; Reichardt, H.M. The Role of Glucocorticoids in Inflammatory Diseases. Cells 2021, 10, 2921. https://doi.org/10.3390/cells10112921
Reichardt SD, Amouret A, Muzzi C, Vettorazzi S, Tuckermann JP, Lühder F, Reichardt HM. The Role of Glucocorticoids in Inflammatory Diseases. Cells. 2021; 10(11):2921. https://doi.org/10.3390/cells10112921
Chicago/Turabian StyleReichardt, Sybille D., Agathe Amouret, Chiara Muzzi, Sabine Vettorazzi, Jan P. Tuckermann, Fred Lühder, and Holger M. Reichardt. 2021. "The Role of Glucocorticoids in Inflammatory Diseases" Cells 10, no. 11: 2921. https://doi.org/10.3390/cells10112921
APA StyleReichardt, S. D., Amouret, A., Muzzi, C., Vettorazzi, S., Tuckermann, J. P., Lühder, F., & Reichardt, H. M. (2021). The Role of Glucocorticoids in Inflammatory Diseases. Cells, 10(11), 2921. https://doi.org/10.3390/cells10112921