
Transcriptome Analysis of Testicular Aging in Mice


Abstract
1. Introduction
2. Materials and Methods
2.1. Animals and RNA Preparation
2.2. Total RNA Sequencing
2.3. Tissue Expression Estimation
2.4. Investigating the Potential Features of Aging-Related Transcripts
3. Results
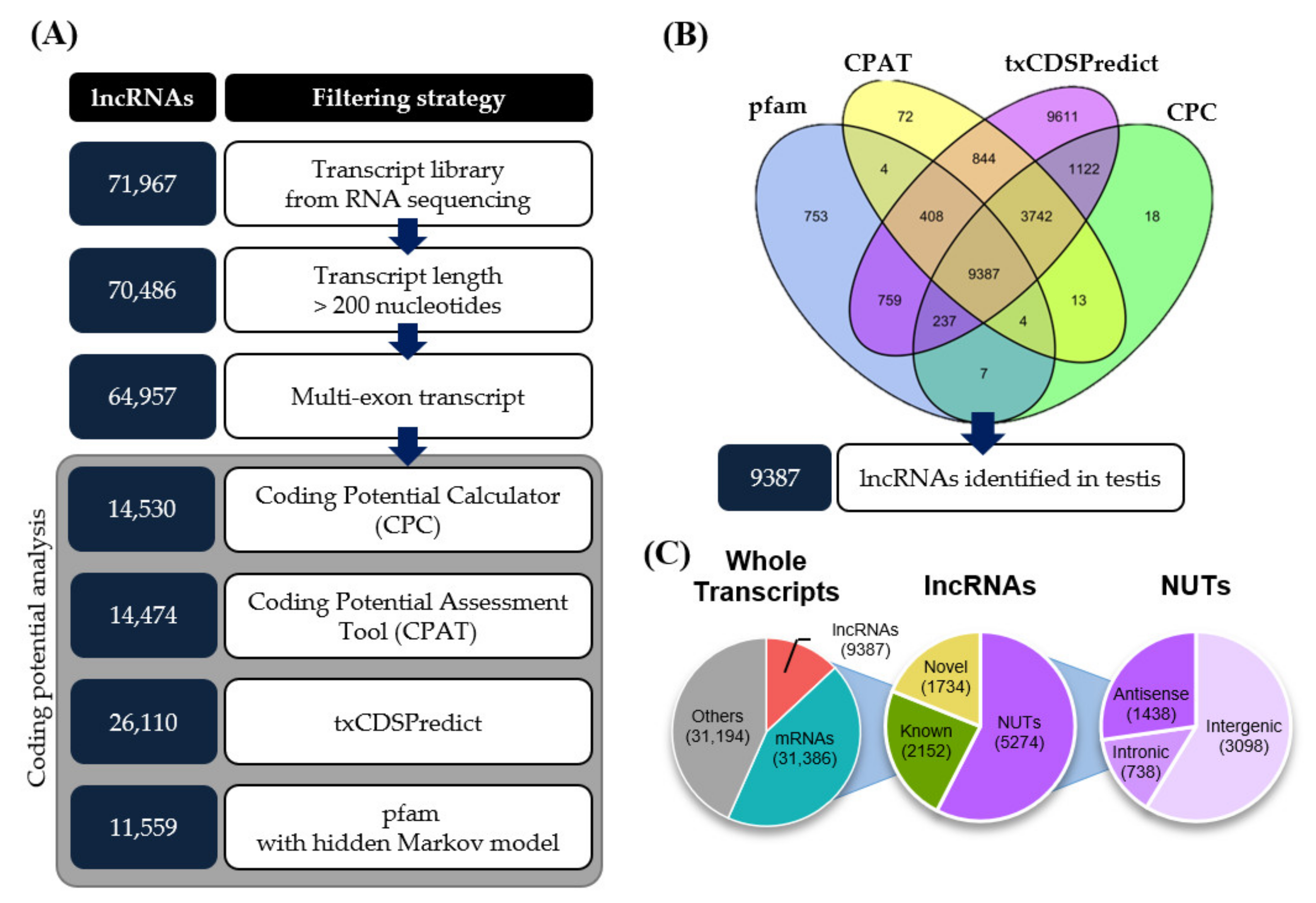
3.1. Identification of mRNAs and lncRNAs in Mouse Testes during Aging
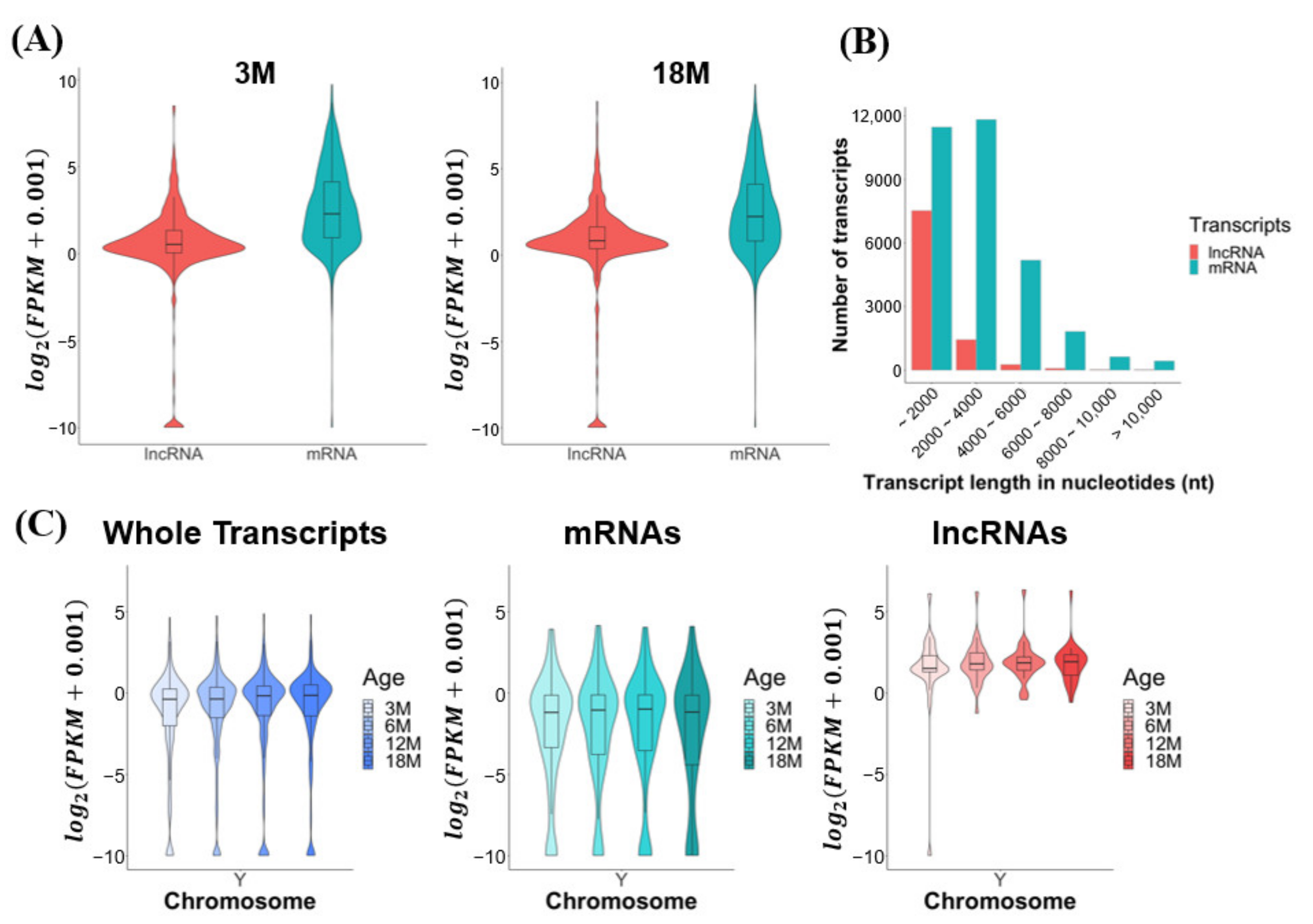
3.2. Global Expression and Transcriptomic Features of mRNAs and lncRNAs Expressed in Mouse Testes during Aging
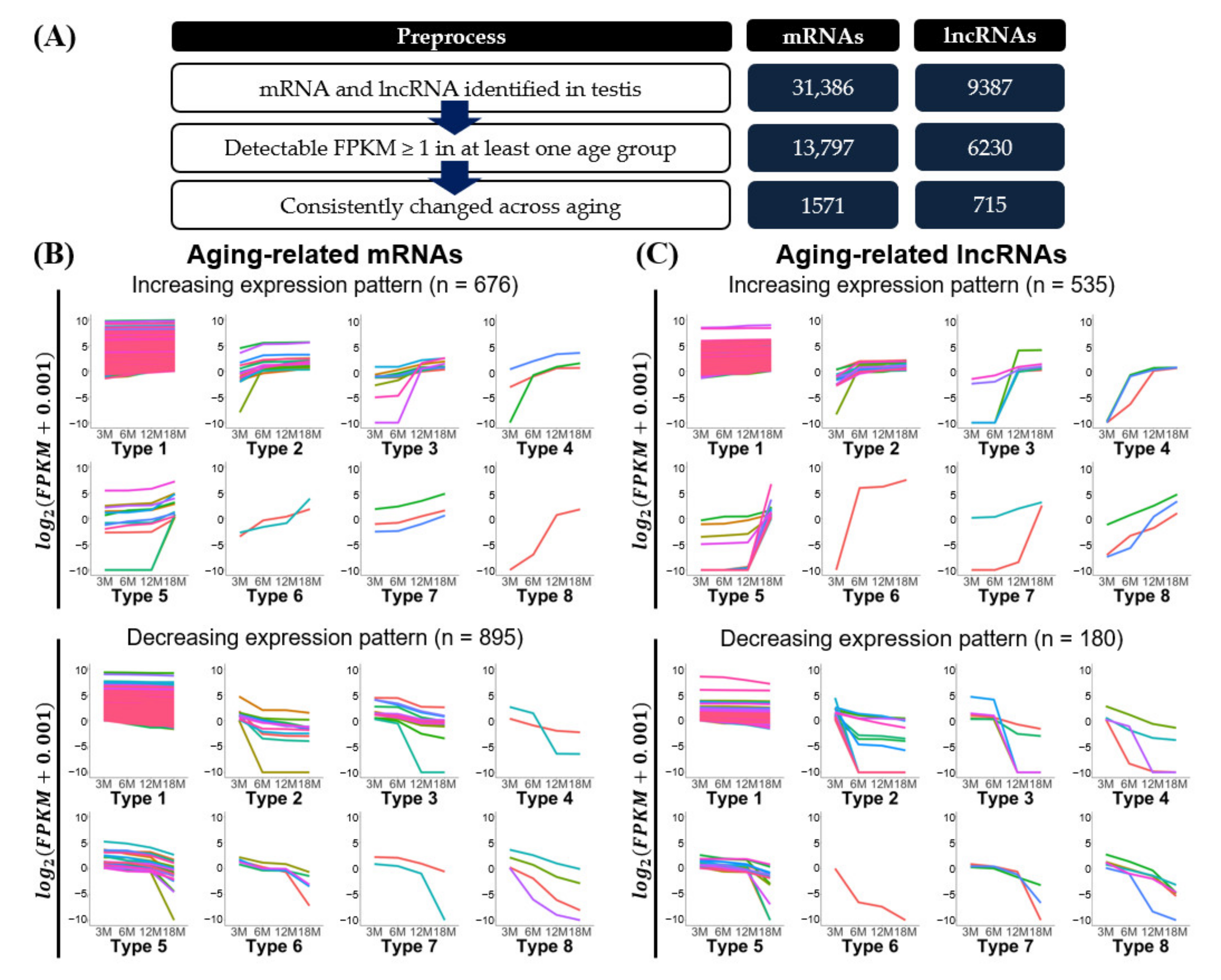
3.3. Aging-Related Expression Patterns of mRNAs and lncRNAs
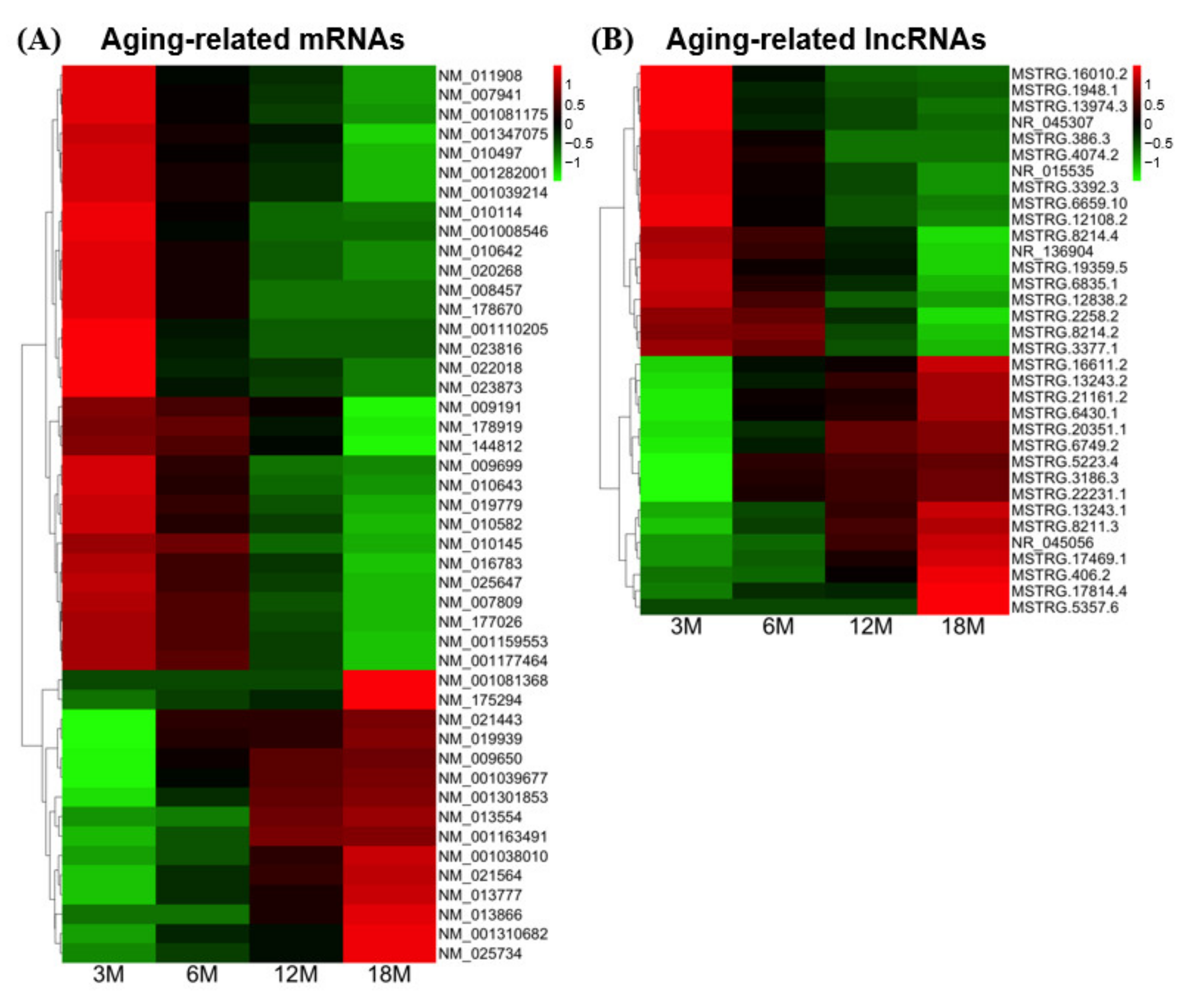
3.4. Testis-Specific Expression of Aging-Related mRNAs and lncRNAs
3.5. Potential Features of Aging-Related Transcripts Showing Changes between 3M and 18M
3.6. Potential Cis-Regulatory Targets of Aging-Related lncRNAs
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Johnson, S.L.; Dunleavy, J.; Gemmell, N.J.; Nakagawa, S. Consistent age-dependent declines in human semen quality: A systematic review and meta-analysis. Ageing Res. Rev. 2015, 19, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Santiago, J.; Silva, J.V.; Alves, M.G.; Oliveira, P.F.; Fardilha, M. Testicular Aging: An Overview of Ultrastructural, Cellular, and Molecular Alterations. J. Gerontol. A Biol. Sci. Med. Sci. 2019, 74, 860–871. [Google Scholar] [CrossRef]
- Sharma, R.; Agarwal, A.; Rohra, V.K.; Assidi, M.; Abu-Elmagd, M.; Turki, R.F. Effects of increased paternal age on sperm quality, reproductive outcome and associated epigenetic risks to offspring. Reprod. Biol. Endocrinol. 2015, 13, 35. [Google Scholar] [CrossRef]
- Harris, I.D.; Fronczak, C.; Roth, L.; Meacham, R.B. Fertility and the aging male. Rev. Urol. 2011, 13, e184–e190. [Google Scholar]
- Eddy, E.M. Male germ cell gene expression. Recent Prog. Horm. Res. 2002, 57, 103–128. [Google Scholar] [CrossRef]
- Sha, J. Identification of testis development and spermatogenesis-related genes in human and mouse testes using cDNA arrays. Mol. Hum. Reprod. 2002, 8, 511–517. [Google Scholar] [CrossRef]
- Hayashi, K.; Chuva De Sousa Lopes, S.M.; Kaneda, M.; Tang, F.; Hajkova, P.; Lao, K.; O’Carroll, D.; Das, P.P.; Tarakhovsky, A.; Miska, E.A.; et al. MicroRNA Biogenesis Is Required for Mouse Primordial Germ Cell Development and Spermatogenesis. PLoS ONE 2008, 3, e1738. [Google Scholar] [CrossRef] [PubMed]
- Gan, H.; Lin, X.; Zhang, Z.; Zhang, W.; Liao, S.; Wang, L.; Han, C. piRNA profiling during specific stages of mouse spermatogenesis. RNA 2011, 17, 1191–1203. [Google Scholar] [CrossRef] [PubMed]
- Anguera, M.C.; Ma, W.; Clift, D.; Namekawa, S.; Kelleher, R.J.; Lee, J.T. Tsx Produces a Long Noncoding RNA and Has General Functions in the Germline, Stem Cells, and Brain. PLoS Genet. 2011, 7, e1002248. [Google Scholar] [CrossRef]
- Li, L.; Wang, M.; Wang, M.; Wu, X.; Geng, L.; Xue, Y.; Wei, X.; Jia, Y.; Wu, X. A long non-coding RNA interacts with Gfra1 and maintains survival of mouse spermatogonial stem cells. Cell Death Dis. 2016, 7, e2140. [Google Scholar] [CrossRef]
- Hosono, Y.; Niknafs, Y.S.; Prensner, J.R.; Iyer, M.K.; Dhanasekaran, S.M.; Mehra, R.; Pitchiaya, S.; Tien, J.; Escara-Wilke, J.; Poliakov, A.; et al. Oncogenic Role of THOR, a Conserved Cancer/Testis Long Non-coding RNA. Cell 2017, 171, 1559–1572. [Google Scholar] [CrossRef]
- Kataruka, S.; Akhade, V.S.; Kayyar, B.; Rao, M.R.S. Mrhl Long Noncoding RNA Mediates Meiotic Commitment of Mouse Spermatogonial Cells by Regulating Sox8 Expression. Mol. Cell. Biol. 2017, 37, e00632-16. [Google Scholar] [CrossRef]
- Hu, K.; Zhang, J.; Liang, M. LncRNA AK015322 promotes proliferation of spermatogonial stem cell C18-4 by acting as a decoy for microRNA-19b-3p. In Vitro Cell. Dev. Biol. Anim. 2017, 53, 277–284. [Google Scholar] [CrossRef]
- Satoh, Y.; Takei, N.; Kawamura, S.; Takahashi, N.; Kotani, T.; Kimura, A.P. A novel testis-specific long noncoding RNA, Tesra, activates the Prss42/Tessp-2 gene during mouse spermatogenesis†. Biol. Reprod. 2019, 100, 833–848. [Google Scholar] [CrossRef] [PubMed]
- Lewandowski, J.P.; Dumbović, G.; Watson, A.R.; Hwang, T.; Jacobs-Palmer, E.; Chang, N.; Much, C.; Turner, K.M.; Kirby, C.; Rubinstein, N.D.; et al. The Tug1 lncRNA locus is essential for male fertility. Genome Biol. 2020, 21, 237. [Google Scholar] [CrossRef]
- Hong, S.H.; Kwon, J.T.; Kim, J.; Jeong, J.; Kim, J.; Lee, S.; Cho, C. Profiling of testis-specific long noncoding RNAs in mice. BMC Genom. 2018, 19, 539. [Google Scholar] [CrossRef]
- Hong, S.H.; Han, G.; Lee, S.J.; Cocquet, J.; Cho, C. Testicular germ cell-specific lncRNA, Teshl, is required for complete expression of Y chromosome genes and a normal offspring sex ratio. Sci. Adv. 2021, 7, eabg5177. [Google Scholar] [CrossRef] [PubMed]
- Marttila, S.; Chatsirisupachai, K.; Palmer, D.; de Magalhaes, J.P. Ageing-associated changes in the expression of lncRNAs in human tissues reflect a transcriptional modulation in ageing pathways. Mech. Ageing Dev. 2020, 185, 111177. [Google Scholar] [CrossRef] [PubMed]
- Mularoni, V.; Esposito, V.; Di Persio, S.; Vicini, E.; Spadetta, G.; Berloco, P.; Fanelli, F.; Mezzullo, M.; Pagotto, U.; Pelusi, C.; et al. Age-related changes in human Leydig cell status. Hum. Reprod. 2020, 35, 2663–2676. [Google Scholar] [CrossRef]
- Tatehana, M.; Kimura, R.; Mochizuki, K.; Inada, H.; Osumi, N. Comprehensive histochemical profiles of histone modification in male germline cells during meiosis and spermiogenesis: Comparison of young and aged testes in mice. PLoS ONE 2020, 15, e0230930. [Google Scholar] [CrossRef]
- Merz, S.E.; Klopfleisch, R.; Breithaupt, A.; Gruber, A.D. Aging and Senescence in Canine Testes. Vet. Pathol. 2019, 56, 715–724. [Google Scholar] [CrossRef]
- Machida, T.; Yonezawa, Y.; Noumura, T. Age-associated changes in plasma testosterone levels in male mice and their relation to social dominance or subordinance. Horm. Behav. 1981, 15, 238–245. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed]
- Mclean, C.Y.; Bristor, D.; Hiller, M.; Clarke, S.L.; Schaar, B.T.; Lowe, C.B.; Wenger, A.M.; Bejerano, G. GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol. 2010, 28, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Kovaka, S.; Zimin, A.V.; Pertea, G.M.; Razaghi, R.; Salzberg, S.L.; Pertea, M. Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome Biol. 2019, 20, 278. [Google Scholar] [CrossRef] [PubMed]
- Pertea, G.; Pertea, M. GFF Utilities: GffRead and GffCompare. F1000Research 2020, 9, 304. [Google Scholar] [CrossRef]
- Kang, Y.J.; Yang, D.C.; Kong, L.; Hou, M.; Meng, Y.Q.; Wei, L.; Gao, G. CPC2: A fast and accurate coding potential calculator based on sequence intrinsic features. Nucleic Acids Res. 2017, 45, W12–W16. [Google Scholar] [CrossRef]
- Wang, L.; Park, H.J.; Dasari, S.; Wang, S.; Kocher, J.P.; Li, W. CPAT: Coding-Potential Assessment Tool using an alignment-free logistic regression model. Nucleic Acids Res. 2013, 41, e74. [Google Scholar] [CrossRef] [PubMed]
- Punta, M.; Coggill, P.C.; Eberhardt, R.Y.; Mistry, J.; Tate, J.; Boursnell, C.; Pang, N.; Forslund, K.; Ceric, G.; Clements, J.; et al. The Pfam protein families database. Nucleic Acids Res. 2012, 40, D290–D301. [Google Scholar] [CrossRef] [PubMed]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The Hallmarks of Aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef]
- Jain, A.; Tuteja, G. TissueEnrich: Tissue-specific gene enrichment analysis. Bioinformatics 2019, 35, 1966–1967. [Google Scholar] [CrossRef]
- Rohe, H.J.; Ahmed, I.S.; Twist, K.E.; Craven, R.J. PGRMC1 (progesterone receptor membrane component 1): A targetable protein with multiple functions in steroid signaling, P450 activation and drug binding. Pharmacol. Ther. 2009, 121, 14–19. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, F.; Ye, L.; Zirkin, B.; Chen, H. Steroidogenesis in Leydig cells: Effects of aging and environmental factors. Reproduction 2017, 154, R111–R122. [Google Scholar] [CrossRef]
- Lawrence, M.G.; Lai, J.; Clements, J.A. Kallikreins on steroids: Structure, function, and hormonal regulation of prostate-specific antigen and the extended kallikrein locus. Endocr. Rev. 2010, 31, 407–446. [Google Scholar] [CrossRef] [PubMed]
- Joshi, M.; Rajender, S. Long non-coding RNAs (lncRNAs) in spermatogenesis and male infertility. Reprod. Biol. Endocrinol. 2020, 18, 103. [Google Scholar] [CrossRef]
- Chen, Y.; Zheng, Y.; Gao, Y.; Lin, Z.; Yang, S.; Wang, T.; Wang, Q.; Xie, N.; Hua, R.; Liu, M.; et al. Single-cell RNA-seq uncovers dynamic processes and critical regulators in mouse spermatogenesis. Cell Res. 2018, 28, 879–896. [Google Scholar] [CrossRef]
- Sun, J.; Lin, Y.; Wu, J. Long Non-Coding RNA Expression Profiling of Mouse Testis during Postnatal Development. PLoS ONE 2013, 8, e75750. [Google Scholar] [CrossRef]
- Bahar, R.; Hartmann, C.H.; Rodriguez, K.A.; Denny, A.D.; Busuttil, R.A.; Dollé, M.E.T.; Calder, R.B.; Chisholm, G.B.; Pollock, B.H.; Klein, C.A.; et al. Increased cell-to-cell variation in gene expression in ageing mouse heart. Nature 2006, 441, 1011–1014. [Google Scholar] [CrossRef]
- Schaum, N.; Lehallier, B.; Hahn, O.; Pálovics, R.; Hosseinzadeh, S.; Lee, S.E.; Sit, R.; Lee, D.P.; Losada, P.M.; Zardeneta, M.E.; et al. Ageing hallmarks exhibit organ-specific temporal signatures. Nature 2020, 583, 596–602. [Google Scholar] [CrossRef]
- Paul, C.; Robaire, B. Impaired Function of the Blood-Testis Barrier during Aging Is Preceded by a Decline in Cell Adhesion Proteins and GTPases. PLoS ONE 2013, 8, e84354. [Google Scholar] [CrossRef]
- Greenbaum, M.P.; Yan, W.; Wu, M.-H.; Lin, Y.-N.; Agno, J.E.; Sharma, M.; Braun, R.E.; Rajkovic, A.; Matzuk, M.M. TEX14 is essential for intercellular bridges and fertility in male mice. Proc. Natl. Acad. Sci. USA 2006, 103, 4982–4987. [Google Scholar] [CrossRef]
- Rodwell, G.E.; Sonu, R.; Zahn, J.M.; Lund, J.; Wilhelmy, J.; Wang, L.; Xiao, W.; Mindrinos, M.; Crane, E.; Segal, E.; et al. A transcriptional profile of aging in the human kidney. PLoS Biol. 2004, 2, e427. [Google Scholar] [CrossRef]
- Berchtold, N.C.; Cribbs, D.H.; Coleman, P.D.; Rogers, J.; Head, E.; Kim, R.; Beach, T.; Miller, C.; Troncoso, J.; Trojanowski, J.Q.; et al. Gene expression changes in the course of normal brain aging are sexually dimorphic. Proc. Natl. Acad. Sci. USA 2008, 105, 15605–15610. [Google Scholar] [CrossRef]
- Lu, T.; Pan, Y.; Kao, S.Y.; Li, C.; Kohane, I.; Chan, J.; Yankner, B.A. Gene regulation and DNA damage in the ageing human brain. Nature 2004, 429, 883–891. [Google Scholar] [CrossRef]
- Kayo, T.; Allison, D.B.; Weindruch, R.; Prolla, T.A. Influences of aging and caloric restriction on the transcriptional profile of skeletal muscle from rhesus monkeys. Proc. Natl. Acad. Sci. USA 2001, 98, 5093–5098. [Google Scholar] [CrossRef]
- Peffers, M.J.; Fang, Y.; Cheung, K.; Wei, T.K.; Clegg, P.D.; Birch, H.L. Transcriptome analysis of ageing in uninjured human Achilles tendon. Arthritis Res. Ther. 2015, 17, 33. [Google Scholar] [CrossRef]
- Cai, H.; Fields, M.A.; Hoshino, R.; Priore, L.V. Effects of aging and anatomic location on gene expression in human retina. Front. Aging Neurosci. 2012, 4, 8. [Google Scholar] [CrossRef]
- Yoshida, S.; Yashar, B.M.; Hiriyanna, S.; Swaroop, A. Microarray analysis of gene expression in the aging human retina. Investig. Ophthalmol. Vis. Sci. 2002, 43, 2554–2560. [Google Scholar]
- Lee, C.K.; Weindruch, R.; Prolla, T.A. Gene-expression profile of the ageing brain in mice. Nat. Genet. 2000, 25, 294–297. [Google Scholar] [CrossRef]
- Bronikowski, A.M.; Carter, P.A.; Morgan, T.J.; Garland, T., Jr.; Ung, N.; Pugh, T.D.; Weindruch, R.; Prolla, T.A. Lifelong voluntary exercise in the mouse prevents age-related alterations in gene expression in the heart. Physiol. Genom. 2003, 12, 129–138. [Google Scholar] [CrossRef]
- Cao, S.X.; Dhahbi, J.M.; Mote, P.L.; Spindler, S.R. Genomic profiling of short- and long-term caloric restriction effects in the liver of aging mice. Proc. Natl. Acad. Sci. USA 2001, 98, 10630–10635. [Google Scholar] [CrossRef] [PubMed]
- Stegeman, R.; Weake, V.M. Transcriptional Signatures of Aging. J. Mol. Biol. 2017, 429, 2427–2437. [Google Scholar] [CrossRef] [PubMed]
- Edwards, M.G.; Sarkar, D.; Klopp, R.; Morrow, J.D.; Weindruch, R.; Prolla, T.A. Age-related impairment of the transcriptional responses to oxidative stress in the mouse heart. Physiol. Genom. 2003, 13, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.; Suen, H.C.; Luk, A.C.S.; Yang, L.; Lee, A.W.T.; Qi, H.; Lee, T.-L. Transcriptomic and epigenomic profiling of young and aged spermatogonial stem cells reveals molecular targets regulating differentiation. PLOS Genet. 2021, 17, e1009369. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | Total Reads | Total Read Bases 1 | Q20(%) 2 | Q30(%) 3 |
|---|---|---|---|---|
| 3M-1 | 67,091,602 | 6,776,251,802 | 98.6 | 96.17 |
| 3M-2 | 73,106,236 | 7,383,729,836 | 98.5 | 95.93 |
| 3M-3 | 73,869,148 | 7,460,783,948 | 98.63 | 96.24 |
| 6M-1 | 66,020,784 | 6,668,099,184 | 98.48 | 95.75 |
| 6M-2 | 65,090,562 | 6,574,146,762 | 98.56 | 96.09 |
| 6M-3 | 71,624,278 | 7,234,052,078 | 98.59 | 96.15 |
| 12M-1 | 75,862,548 | 7,662,117,348 | 98.67 | 96.27 |
| 12M-2 | 69,647,216 | 7,034,368,816 | 98.62 | 96.16 |
| 12M-3 | 79,299,106 | 8,009,209,706 | 98.62 | 96.18 |
| 18M-1 | 72,565,298 | 7,329,095,098 | 98.47 | 95.89 |
| 18M-2 | 67,057,420 | 6,772,799,420 | 98.67 | 96.27 |
| 18M-3 | 90,942,020 | 9,185,144,020 | 98.35 | 95.33 |
| Type | Substantial Expression Change 1 | mRNAs 2 | lncRNAs 2 | Total 2 | |||
|---|---|---|---|---|---|---|---|
| 3M to 6M | 6M to 12M | 12M to 18M | |||||
| Increased | 1 | 629 | 481 | 1110 | |||
| 2 | + | 17 | 17 | 34 | |||
| 3 | + | 9 | 7 | 16 | |||
| 4 | + | + | 3 | 3 | 6 | ||
| 5 | + | 12 | 21 | 33 | |||
| 6 | + | + | 2 | 1 | 3 | ||
| 7 | + | + | 3 | 2 | 5 | ||
| 8 | + | + | + | 1 | 3 | 4 | |
| Decreased | 1 | 834 | 123 | 957 | |||
| 2 | + | 11 | 24 | 35 | |||
| 3 | + | 13 | 5 | 18 | |||
| 4 | + | + | 2 | 4 | 6 | ||
| 5 | + | 24 | 14 | 38 | |||
| 6 | + | + | 5 | 1 | 6 | ||
| 7 | + | + | 2 | 3 | 5 | ||
| 8 | + | + | + | 4 | 6 | 10 | |
| Total | 1571 | 715 | 2286 | ||||
| Transcript ID | Gene Symbol | Average FPKM | 3M–18Mp-Value 1 | Expression Pattern | ||||
|---|---|---|---|---|---|---|---|---|
| 3M | 6M | 12M | 18M | Inc/Dec 2 | Type | |||
| NM_001110205 | Acvr1 | 1.16 | 0.26 | 0.01 | 0.00 | 8.98 × 10−58 | Decreased | 8 |
| NM_010114 | Klk1b22 | 4.36 | 1.63 | 0.33 | 0.14 | 4.52 × 10−4 | Decreased | 8 |
| NM_013866 | Zfp385a | 0.00 | 0.01 | 1.59 | 3.40 | 4.12 × 10−6 | Increased | 8 |
| NM_020268 | Klk1b27 | 12.33 | 5.92 | 1.98 | 0.93 | 3.96 × 10−56 | Decreased | 8 |
| NM_177026 | Tmcc3 | 1.81 | 1.39 | 0.51 | 0.00 | 4.62 × 10−7 | Decreased | 7 |
| NM_001081175 | Itpkb | 2.85 | 1.23 | 0.62 | 0.01 | 1.70 × 10−77 | Decreased | 6 |
| NM_022018 | Fam129a | 3.36 | 0.92 | 0.75 | 0.09 | 7.93 × 10−6 | Decreased | 6 |
| NM_025734 | Kcng4 | 0.09 | 0.77 | 1.28 | 3.50 | 6.89 × 10−5 | Increased | 6 |
| NM_001039214 | Mex3c | 12.72 | 7.02 | 4.52 | 0.46 | 1.13 × 10−7 | Decreased | 5 |
| NM_001081368 | Tbccd1 | 0.15 | 0.15 | 0.16 | 1.01 | 3.88 × 10−7 | Increased | 5 |
| NM_001282001 | Rbl2 | 1.92 | 1.02 | 0.62 | 0.00 | 9.08 × 10−5 | Decreased | 5 |
| NM_001347075 | Xpnpep3 | 2.60 | 1.48 | 1.13 | 0.05 | 8.90 × 10−72 | Decreased | 5 |
| NM_009191 | Clpb | 10.02 | 8.83 | 7.53 | 2.69 | 3.70 × 10−4 | Decreased | 5 |
| NM_010497 | Idh1 | 5.08 | 2.89 | 2.34 | 0.92 | 4.03 × 10−7 | Decreased | 5 |
| NM_016783 | Pgrmc1 | 36.82 | 28.19 | 16.46 | 6.24 | 1.74 × 10−20 | Decreased | 5 |
| NM_025647 | Cmpk1 | 5.82 | 4.32 | 2.46 | 0.99 | 6.39 × 10−117 | Decreased | 5 |
| NM_144812 | Tnrc6b | 1.15 | 1.02 | 0.70 | 0.04 | 2.51 × 10−10 | Decreased | 5 |
| NM_175294 | Nucks1 | 0.24 | 0.40 | 0.50 | 1.37 | 3.43 × 10−5 | Increased | 5 |
| NM_178919 | Lmf2 | 8.59 | 8.09 | 5.64 | 2.06 | 1.52 × 10−3 | Decreased | 5 |
| NM_001163491 | Sema4a | 0.12 | 0.49 | 1.49 | 1.55 | 5.96 × 10−17 | Increased | 4 |
| NM_008457 | Klk1b8 | 6.27 | 2.70 | 0.01 | 0.01 | 9.23 × 10−11 | Decreased | 4 |
| NM_001038010 | Kat2a | 0.62 | 1.18 | 2.49 | 3.72 | 1.12 × 10−3 | Increased | 3 |
| NM_009699 | Aqp2 | 2.79 | 1.82 | 0.76 | 0.62 | 3.70 × 10−10 | Decreased | 3 |
| NM_010642 | Klk1b21 | 16.36 | 8.22 | 3.01 | 1.72 | 4.60 × 10−6 | Decreased | 3 |
| NM_010643 | Klk1b24 | 16.44 | 9.93 | 3.53 | 1.92 | 6.06 × 10−48 | Decreased | 3 |
| NM_013554 | Hoxd10 | 0.50 | 0.57 | 1.45 | 1.59 | 5.51 × 10−5 | Increased | 3 |
| NM_001039677 | Slc30a2 | 1.37 | 4.14 | 5.35 | 5.77 | 2.19 × 10−87 | Increased | 2 |
| NM_001301853 | Stk11 | 0.24 | 1.54 | 2.87 | 3.15 | 1.90 × 10−20 | Increased | 2 |
| NM_019939 | Mpp6 | 11.41 | 37.46 | 39.21 | 46.83 | 4.51 × 10−11 | Increased | 2 |
| NM_021443 | Ccl8 | 0.72 | 2.28 | 2.32 | 2.70 | 4.32 × 10−4 | Increased | 2 |
| NM_021564 | Fetub | 0.40 | 1.59 | 2.61 | 3.71 | 1.63 × 10−12 | Increased | 2 |
| NM_023816 | Ankrd36 | 2.21 | 0.82 | 0.47 | 0.45 | 1.31 × 10−4 | Decreased | 2 |
| NM_023873 | Cep70 | 1.59 | 0.75 | 0.58 | 0.41 | 1.87 × 10−4 | Decreased | 2 |
| NM_001008546 | Tardbp | 1.29 | 0.69 | 0.44 | 0.42 | 2.37 × 10−5 | Decreased | 1 |
| NM_001159553 | H13 | 1.62 | 1.37 | 0.90 | 0.53 | 5.98 × 10−12 | Decreased | 1 |
| NM_001177464 | Zfp516 | 1.43 | 1.26 | 0.86 | 0.57 | 7.70 × 10−7 | Decreased | 1 |
| NM_001310682 | Prkcd | 0.66 | 1.20 | 1.36 | 2.52 | 1.22 × 10−3 | Increased | 1 |
| NM_007809 | Cyp17a1 | 77.11 | 65.52 | 40.81 | 28.29 | 1.12 × 10−15 | Decreased | 1 |
| NM_007941 | Stx2 | 8.06 | 5.07 | 4.08 | 2.82 | 2.63 × 10−4 | Decreased | 1 |
| NM_009650 | Akap3 | 148.84 | 274.92 | 318.80 | 330.13 | 1.21 × 10−8 | Increased | 1 |
| NM_010145 | Ephx1 | 32.38 | 30.42 | 17.68 | 14.66 | 2.59 × 10−7 | Decreased | 1 |
| NM_010582 | Itih2 | 3.83 | 2.76 | 1.97 | 1.27 | 8.63 × 10−10 | Decreased | 1 |
| NM_011908 | Ubl3 | 39.60 | 26.48 | 23.20 | 16.98 | 4.78 × 10−6 | Decreased | 1 |
| NM_013777 | Akr1c12 | 0.74 | 1.29 | 1.67 | 2.36 | 7.77 × 10−21 | Increased | 1 |
| NM_019779 | Cyp11a1 | 34.14 | 25.92 | 16.40 | 11.67 | 2.39 × 10−7 | Decreased | 1 |
| NM_178670 | 8030462N17Rik | 1.49 | 0.94 | 0.52 | 0.51 | 2.31 × 10−4 | Decreased | 1 |
| Transcript ID | Transcript Locus | Nearby Gene | Nearby Gene Expression in Testis 1 |
|---|---|---|---|
| MSTRG.2258.2 | chr10:119413444–119453556 | Grip1 | testis predominant |
| MSTRG.6835.1 | chr15:8350953–8351246 | Nipbl | testis, brain |
| 2410089E03Rik | testis, CNS | ||
| MSTRG.3377.1 | chr11:86811784–86816370 | Dhx40 | testis, bladder, CNS |
| MSTRG.12838.2 | chr3:55055180–55084491 | Ccna1 | testis specific |
| MSTRG.1948.1 | chr10:81383909–81395392 | Smim24 | testis, intestine |
| MSTRG.8214.2 | chr16:59636945–59672993 | Arl6 | testis, CNS |
| MSTRG.8214.4 | chr16:59636956–59672993 | ||
| NR_045307 | chr19:45726555–45730558 | Npm3 | testis predominant |
| MSTRG.12108.2 | chr2:151088381–151472250 | Gm14147 | testis specific |
| 4921509C19Rik | testis specific | ||
| MSTRG.406.2 | chr1:73015899–73025507 | Tnp1 | testis specific |
| MSTRG.13243.2 | chr3:100417896–100420833 | Fam46c | testis specific |
| MSTRG.13243.1 | chr3:100417891–100421080 | ||
| MSTRG.17469.1 | chr6:125803352–125812378 | Ano2 | testis specific |
| MSTRG.21161.2 | chr9:77357336–77363418 | Lrrc1 | testis predominant |
| MSTRG.22231.1 | chrX:123448948–123456209 | Cldn34c2 | testis predominant |
| MSTRG.5223.4 | chr13:49973191–49977078 | Gm906 | testis, kidney |
| MSTRG.3186.3 | chr11:75588273–75594790 | Inpp5k | testis, lung |
| MSTRG.6430.1 | chr14:61648326–61668174 | Kcnrg | testis predominant |
| MSTRG.16010.2 | chr5:116985353–117004737 | Suds3 | testis, colon, ovary |
| MSTRG.3392.3 | chr11:87405065–87555823 | Tex14 | testis specific |
| MSTRG.5357.6 | chr13:59493404–59557347 | Agtpbp1 | testis predominant |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, G.; Hong, S.-H.; Lee, S.-J.; Hong, S.-P.; Cho, C. Transcriptome Analysis of Testicular Aging in Mice. Cells 2021, 10, 2895. https://doi.org/10.3390/cells10112895
Han G, Hong S-H, Lee S-J, Hong S-P, Cho C. Transcriptome Analysis of Testicular Aging in Mice. Cells. 2021; 10(11):2895. https://doi.org/10.3390/cells10112895
Chicago/Turabian StyleHan, Gwidong, Seong-Hyeon Hong, Seung-Jae Lee, Seung-Pyo Hong, and Chunghee Cho. 2021. "Transcriptome Analysis of Testicular Aging in Mice" Cells 10, no. 11: 2895. https://doi.org/10.3390/cells10112895
APA StyleHan, G., Hong, S.-H., Lee, S.-J., Hong, S.-P., & Cho, C. (2021). Transcriptome Analysis of Testicular Aging in Mice. Cells, 10(11), 2895. https://doi.org/10.3390/cells10112895