
Toxicological Analysis of Hepatocytes Using FLIM Technique: In Vitro versus Ex Vivo Models

 , , ,
, , , 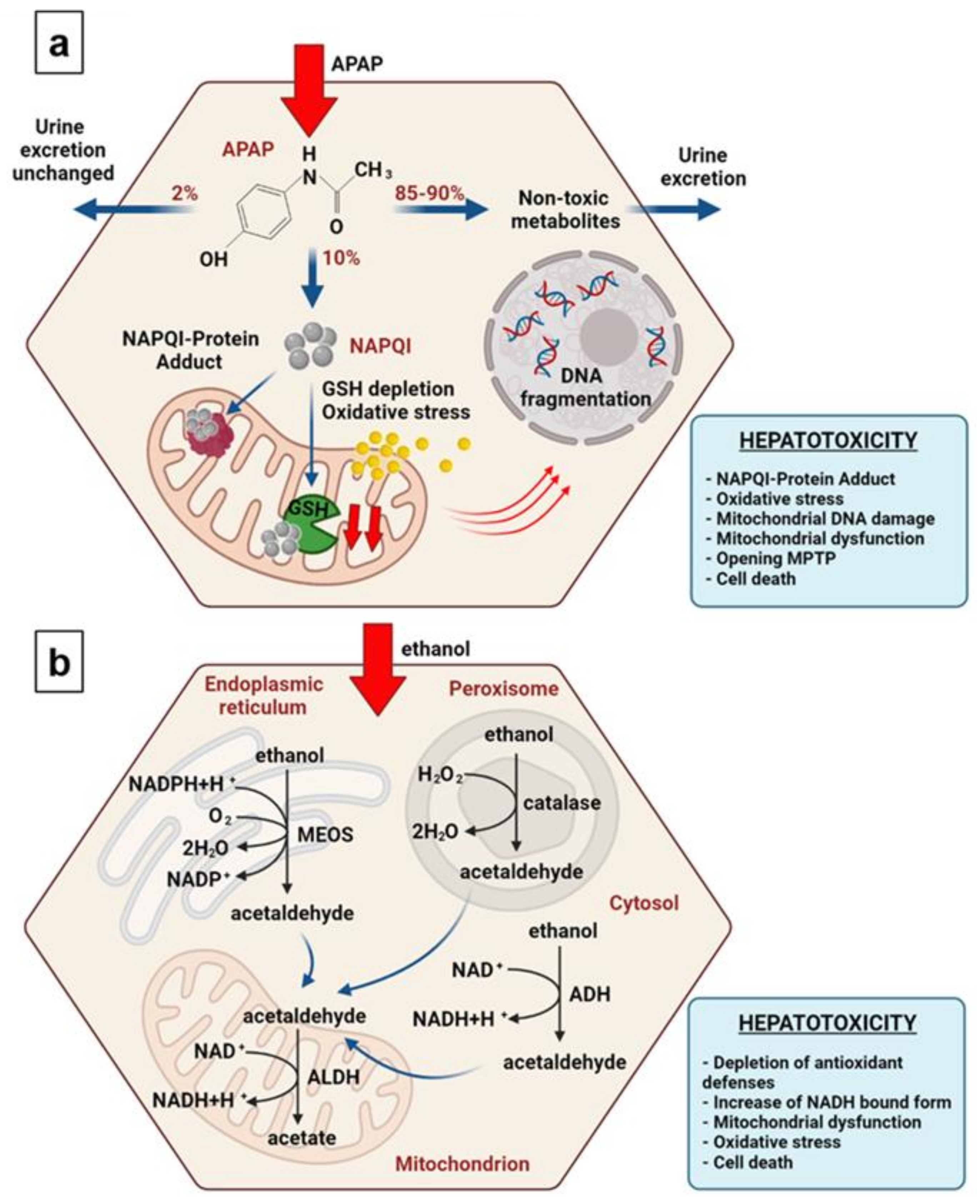{kind=link}
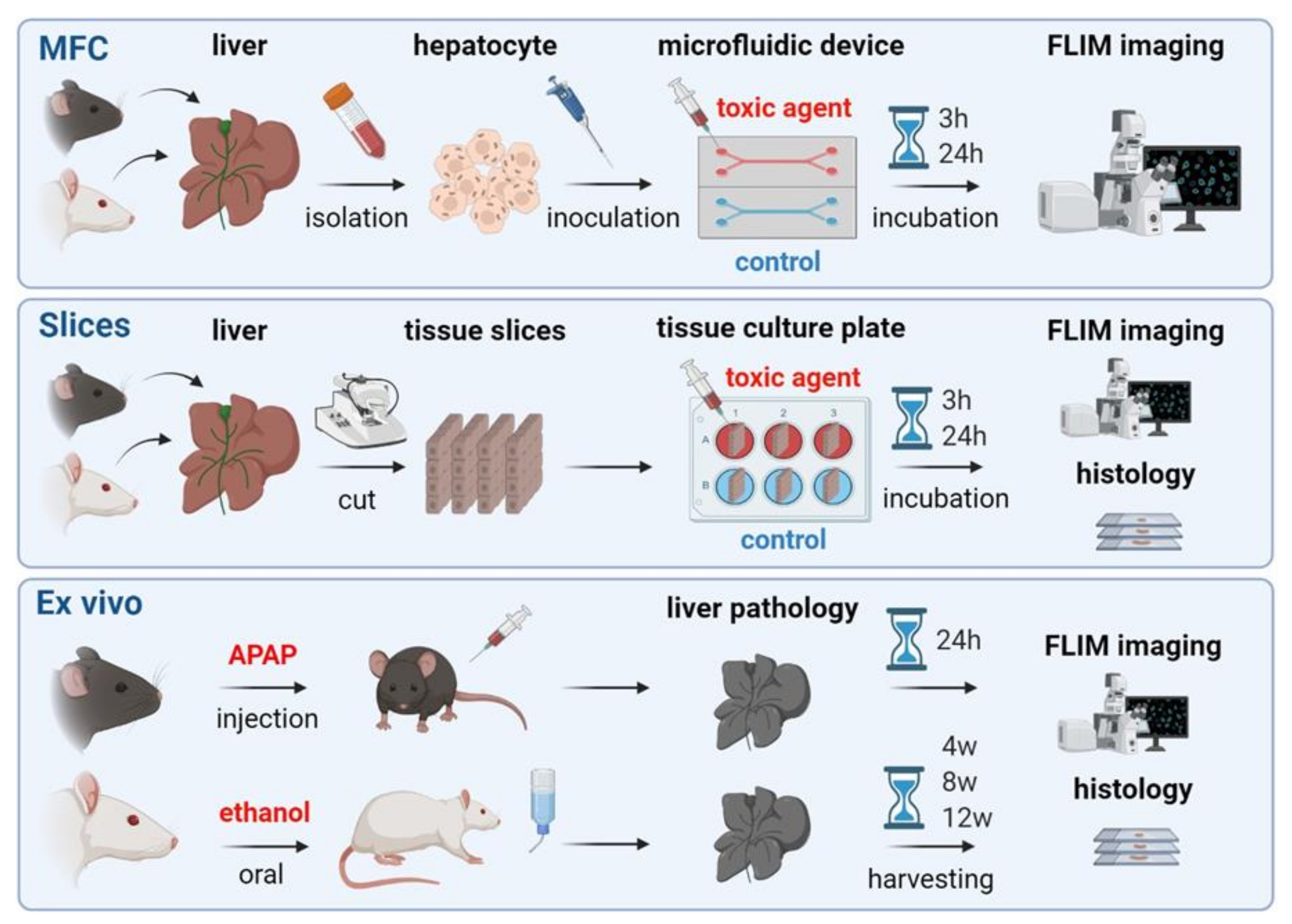{kind=link}
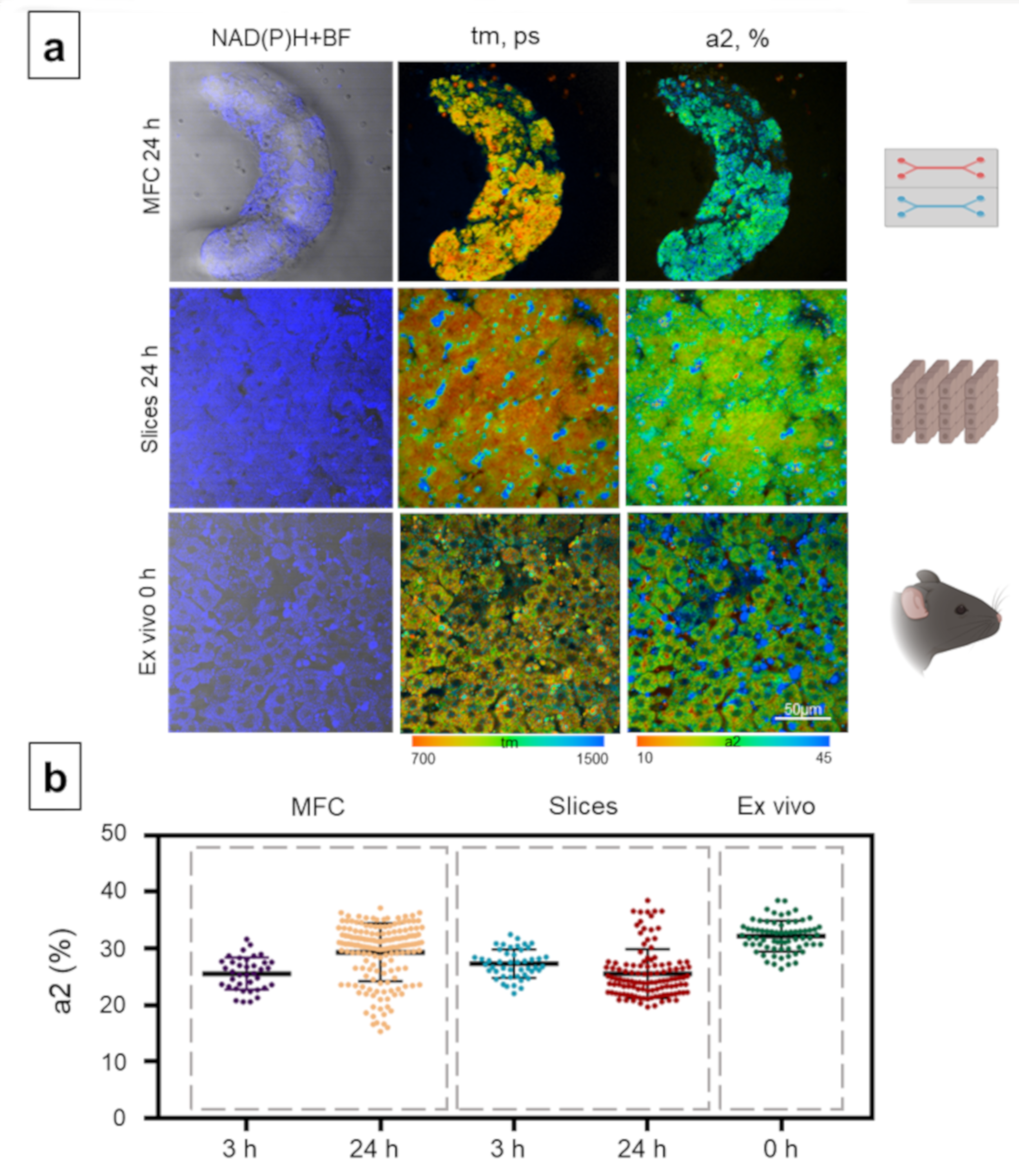{kind=link}
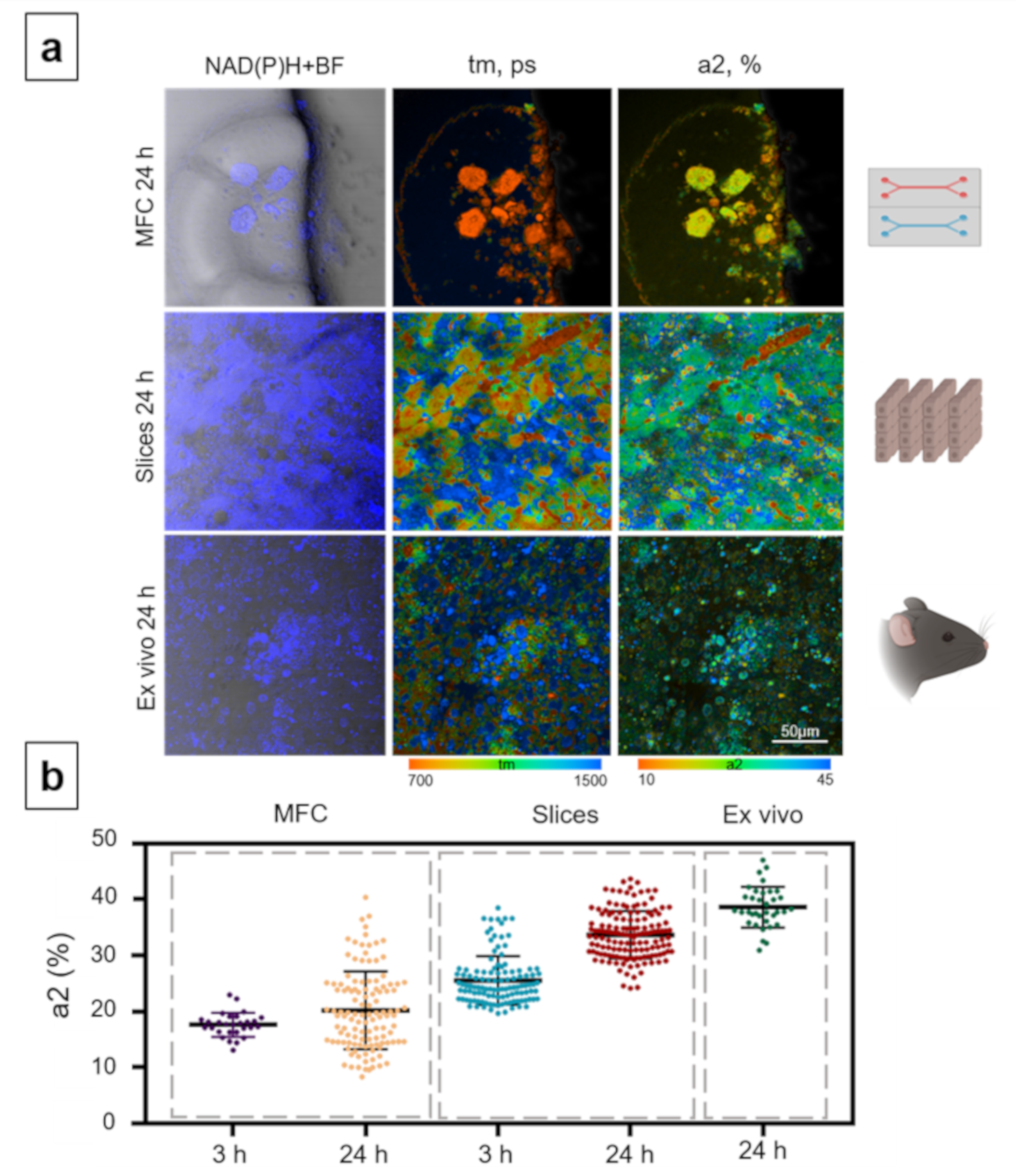{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Fabrication of MFCs and Numerical Simulation of Flow in MFCs
2.2. Experimental Setup with MFCs
2.3. Animal Model
2.4. Precision Cut Liver Slices
2.5. Multiphoton Microscopy
2.6. Histology
3. Results and Discussion
3.1. MFC Use for Hepatocytes Culture
3.2. Analysis of Metabolic State of Normal Hepatocytes from Mice
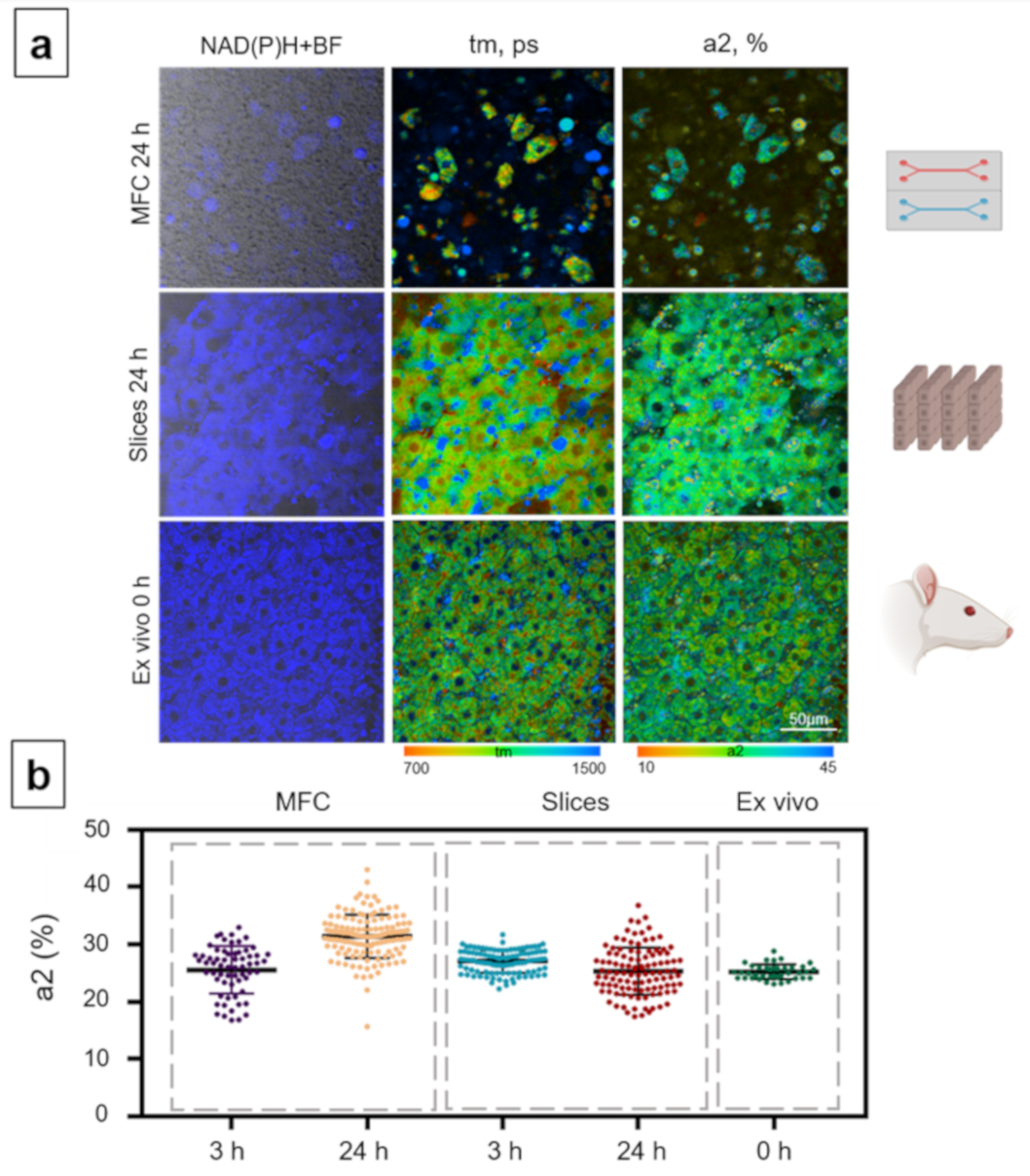
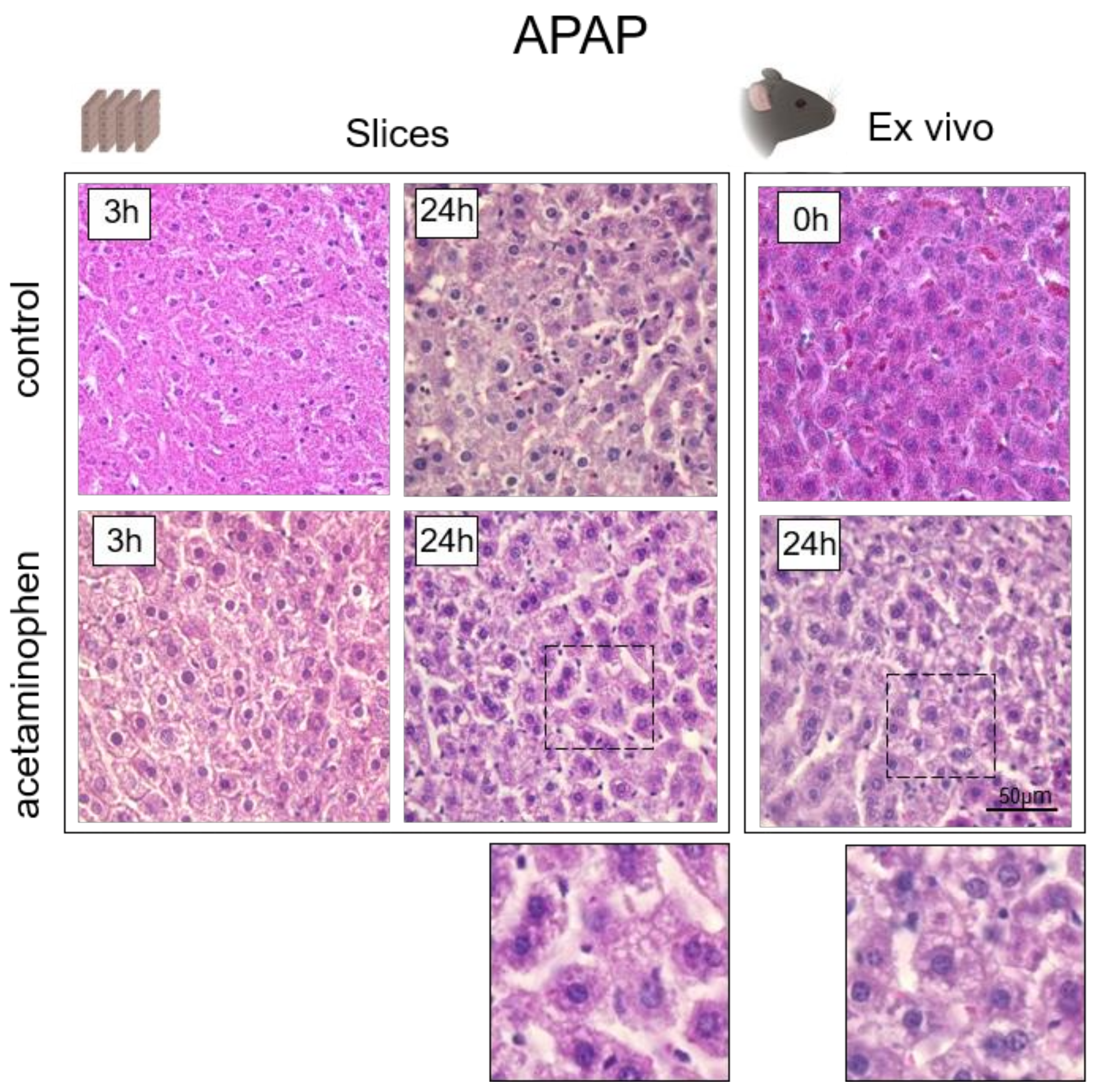
3.3. Analysis of Hepatotoxicity of APAP
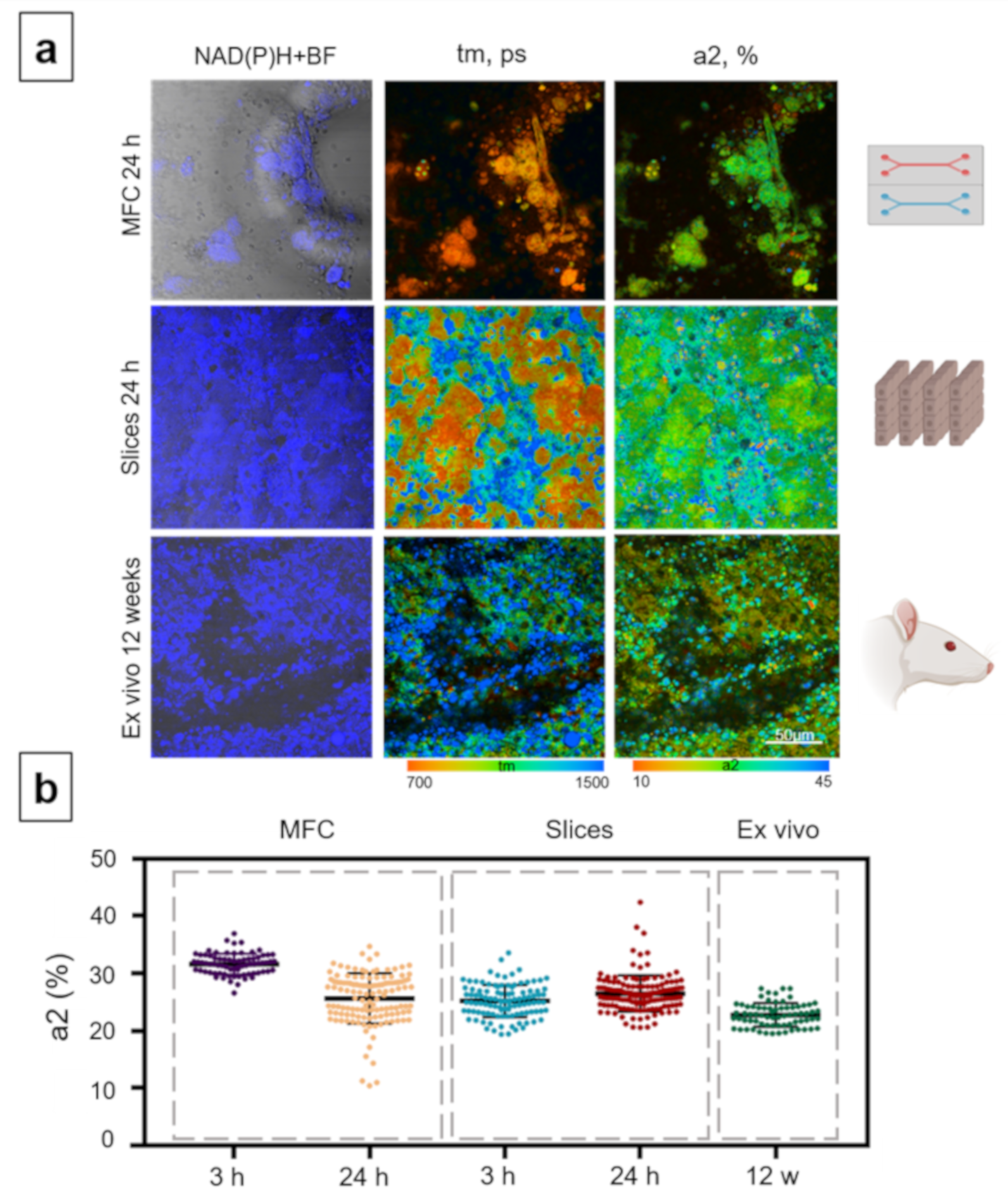
3.4. Analysis of the Metabolic State of Normal Hepatocytes from Rats
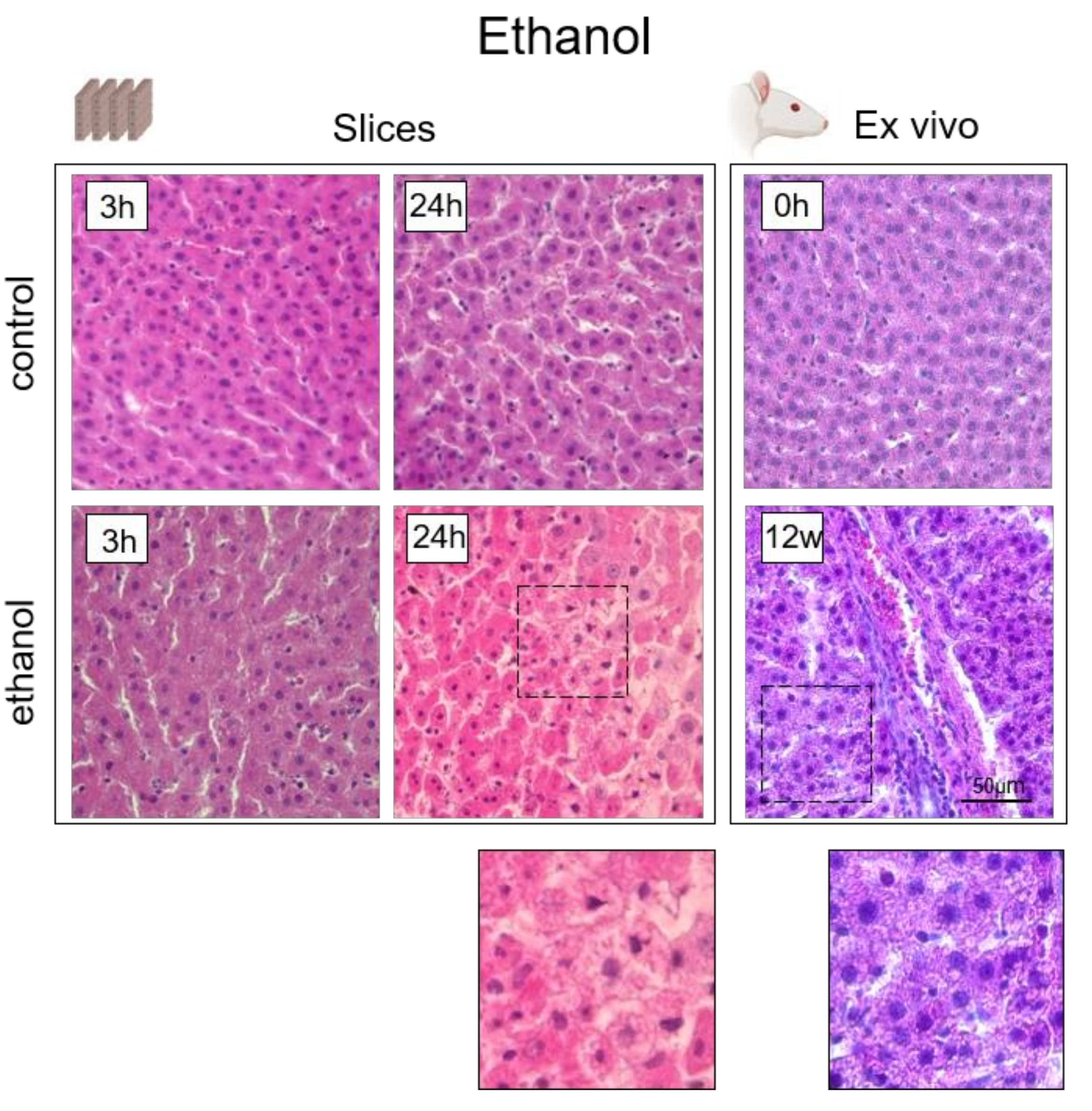
3.5. Analysis of the Hepatotoxicity of Ethanol
4. Histological Analysis of Hepatocytes
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ahmad, F.; Tabassum, N. Experimental models used for the study of antihepatotoxic agents. J. Acute Dis. 2012, 1, 85–89. [Google Scholar] [CrossRef][Green Version]
- Saito, C.; Zwingmann, C.; Jaeschke, H. Novel mechanisms of protection against acetaminophen hepatotoxicity in mice by glutathione and N-acetylcysteine. Hepatology 2010, 51, 246–254. [Google Scholar] [CrossRef]
- O’Shea, R.S.; Dasarathy, S.; McCullough, A.J.; Practice Guideline Committee of the American Association for the Study of Liver Diseases; Practice Parameters Committee of the American College of Gastroenterology. Alcoholic liver disease. Hepatology 2010, 51, 307–328. [Google Scholar] [CrossRef]
- Nguyen, N.T.; Du, K.; Akakpo, J.Y.; Umbaugh, D.S.; Jaeschke, H.; Ramachandran, A. Mitochondrial protein adduct and superoxide generation are prerequisites for early activation of c-jun N-terminal kinase within the cytosol after an acetaminophen overdose in mice. Toxicol. Lett. 2021, 338, 21–31. [Google Scholar] [CrossRef]
- Mossanen, J.C.; Tacke, F. Acetaminophen-induced acute liver injury in mice. Lab. Anim. 2015, 49, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Lancaster, E.M.; Hiatt, J.R.; Zarrinpar, A. Acetaminophen hepatotoxicity: An updated review. Arch. Toxicol. 2015, 89, 193–199. [Google Scholar] [CrossRef]
- Kalinec, G.M.; Thein, P.; Parsa, A.; Yorgason, J.; Luxford, W.; Urrutia, R.; Kalinec, F. Acetaminophen and NAPQI are toxic to auditory cells via oxidative and endoplasmic reticulum stress-dependent pathways. Hear. Res. 2014, 313, 26–37. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Fan, X.; Wang, Y.; Chen, P.; Zeng, H.; Tan, H.; Gonzalez, F.J.; Huang, M.; Bi, H. Schisandrol B protects against acetaminophen-induced hepatotoxicity by inhibition of CYP-mediated bioactivation and regulation of liver regeneration. Toxicol. Sci. 2015, 143, 107–115. [Google Scholar] [CrossRef]
- Jiang, Y.; Fan, X.; Wang, Y.; Tan, H.; Chen, P.; Zeng, H.; Huang, M.; Bi, H. Hepato-protective effects of six schisandra lignans on acetaminophen-induced liver injury are partially associated with the inhibition of CYP-mediated bioactivation. Chem.-Biol. Interact. 2015, 231, 83–89. [Google Scholar] [CrossRef]
- McGill, M.R.; Lebofsky, M.; Norris, H.R.; Slawson, M.H.; Bajt, M.L.; Xie, Y.; Williams, C.D.; Wilkins, D.G.; Rollins, D.E.; Jaeschke, H. Plasma and liver acetaminophen-protein adduct levels in mice after acetaminophen treatment: Dose–response, mechanisms, and clinical implications. Toxicol. Appl. Pharm. 2013, 269, 240–249. [Google Scholar] [CrossRef]
- Moyer, A.M.; Fridley, B.L.; Jenkins, G.D.; Batzler, A.J.; Pelleymounter, L.L.; Kalari, K.R.; Ji, Y.; Chai, Y.; Nordgren, K.K.; Weinshilboum, R.M. Acetaminophen-NAPQI hepatotoxicity: A cell line model system genome-wide association study. Toxicol. Sci. 2011, 120, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Heard, K.J. Acetylcysteine for acetaminophen poisoning. N. Engl. J. Med. 2008, 359, 285–922. [Google Scholar] [CrossRef]
- Waring, W.S.; Stephen, A.F.; Malkowska, A.M.; Robinson, O.D. Acute ethanol coingestion confers a lower risk of hepatotoxicity after deliberate acetaminophen overdose. Acad. Emerg. Med. 2008, 15, 54–58. [Google Scholar] [CrossRef] [PubMed]
- Gyamlani, G.G.; Parikh, C.R. Acetaminophen toxicity: Suicidal vs accidental. Crit Care 2002, 6, 1–5. [Google Scholar] [CrossRef]
- Larson, A.M. Acetaminophen hepatotoxicity. Clin. Liver Dis. 2007, 11, 525–548. [Google Scholar] [CrossRef] [PubMed]
- Cederbaum, A.I. Alcohol metabolism. Clin. Liver Dis. 2012, 16, 667–685. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.S.; Oyama, T.; Isse, T.; Kitagawa, K.; Tanaka, M.; Kawamoto, T. Formation of acetaldehyde-derived DNA adducts due to alcohol exposure. Chem.-Biol. Interact. 2010, 188, 367–375. [Google Scholar] [CrossRef]
- Sun, Q.; Zhong, W.; Zhang, W.; Zhou, Z. Defect of mitochondrial respiratory chain is a mechanism of ROS overproduction in a rat model of alcoholic liver disease: Role of zinc deficiency. Am. J. Physiol. 2016, 310, G205–G214. [Google Scholar] [CrossRef]
- Dou, X.; Shen, C.; Wang, Z.; Li, S.; Zhang, X.; Song, Z. Protection of nicotinic acid against oxidative stress-induced cell death in hepatocytes contributes to its beneficial effect on alcohol-induced liver injury in mice. J. Nutr. Biochem. 2013, 24, 1520–1528. [Google Scholar] [CrossRef]
- Song, Z.; Zhou, Z.; Chen, T.; Hill, D.; Kang, J.; Barve, S.; McClain, C. S-adenosylmethionine (SAMe) protects against acute alcohol induced hepatotoxicity in mice. J. Nutr. Biochem. 2003, 14, 591–597. [Google Scholar] [CrossRef]
- Bao, W.; Li, K.; Rong, S.; Yao, P.; Hao, L.; Ying, C.; Zhang, X.; Nussler, A.; Liu, L. Curcumin alleviates ethanol-induced hepatocytes oxidative damage involving heme oxygenase-1 induction. J. Ethnopharmacol. 2010, 128, 549–553. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, A.; Lebofsky, M.; Baines, C.P.; Lemasters, J.J.; Jaeschke, H. Cyclophilin D deficiency protects against acetaminophen-induced oxidant stress and liver injury. Free Radic. Res. 2011, 45, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Kucera, O.; Cervinkova, Z. Experimental models of non-alcoholic fatty liver disease in rats. World J. Gastroenterol. 2014, 20, 8364. [Google Scholar] [CrossRef] [PubMed]
- Trevisiol, C.H.; Turner, R.T.; Pfaff, J.E.; Hunter, J.C.; Menagh, P.J.; Hardin, K.; Ho, E.; Iwaniec, U.T. Impaired osteoinduction in a rat model for chronic alcohol abuse. Bone 2007, 41, 175–180. [Google Scholar] [CrossRef]
- Lamas-Paz, A.; Hao, F.; Nelson, L.J.; Vázquez, M.T.; Canals, S.; Del Moral, M.G.; Martínez-Naves, E.; Nevzorova, Y.A.; Cubero, F.J. Alcoholic liver disease: Utility of animal models. World J. Gastroenterol. 2018, 24, 5063. [Google Scholar] [CrossRef] [PubMed]
- Antushevich, A.E.; Grebenyuk, A.N.; Khalyutin, D.A.; Yartseva, A.A. Experimental modeling of alcohol-induced liver cirrhosis in rats. Bull. Exp. Biol. Med. 2018, 164, 404–407. [Google Scholar] [CrossRef]
- Prins, G.H.; Luangmonkong, T.; Oosterhuis, D.; Mutsaers, H.A.; Dekker, F.J.; Olinga, P. A pathophysiological model of non-alcoholic fatty liver disease using precision-cut liver slices. Nutrients 2019, 11, 507. [Google Scholar] [CrossRef] [PubMed]
- Palma, E.; Doornebal, E.J.; Chokshi, S. Precision-cut liver slices: A versatile tool to advance liver research. Hepatol. Int. 2019, 13, 51–57. [Google Scholar] [CrossRef]
- Groneberg, D.A.; Grosse-Siestrup, C.; Fischer, A. In vitro models to study hepatotoxicity. Toxicol. Pathol. 2002, 30, 394–399. [Google Scholar] [CrossRef] [PubMed]
- Cong, Y.; Han, X.; Wang, Y.; Chen, Z.; Lu, Y.; Liu, T.; Wu, Z.; Jin, Y.; Luo, Y.; Zhang, X. Drug Toxicity Evaluation Based on Organ-on-a-Chip Technology: A Review. Micromachines 2020, 11, 381. [Google Scholar] [CrossRef]
- Toh, Y.C.; Lim, T.C.; Tai, D.; Xiao, G.; van Noort, D.; Yu, H. A microfluidic 3D hepatocyte chip for drug toxicity testing. Lab Chip 2009, 9, 2026–2035. [Google Scholar] [CrossRef]
- De Ninno, A.; Bertani, F.R.; Gerardino, A.; Schiavoni, G.; Musella, M.; Galassi, C.; Mattei, F.; Sistigu, A.; Businaro, L. Microfluidic Co-Culture Models for Dissecting the Immune Response in in vitro Tumor Microenvironments. J. Vis. Exp. 2021. [Google Scholar] [CrossRef]
- Wang, L.; Li, X.; Chen, C. Inhibition of acetaminophen-induced hepatotoxicity in mice by exogenous thymosinβ4 treatment. Int. Immunopharmacol. 2018, 61, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Huang, X.; Li, Z.; Cao, G.; Zhu, X.; She, S.; Huang, T.; Lu, G. Evaluation of hepatotoxicity induced by 2-ethylhexyldiphenyl phosphate based on transcriptomics and its potential metabolism pathway in human hepatocytes. J. Hazard. Mater. 2021, 413, 125281. [Google Scholar] [CrossRef] [PubMed]
- Muldrew, K.L.; James, L.P.; Coop, L.; McCullough, S.S.; Hendrickson, H.P.; Hinson, J.A.; Mayeux, P.R. Determination of acetaminophen-protein adducts in mouse liver and serum and human serum after hepatotoxic doses of acetaminophen using high-performance liquid chromatography with electrochemical detection. Drug Metab. Dispos. 2002, 30, 446–451. [Google Scholar] [CrossRef] [PubMed]
- Roberts, M.S.; Dancik, Y.; Prow, T.W.; Thorling, C.A.; Lin, L.L.; Grice, J.E.; Robertson, T.A.; König, K.; Becker, W. Non-invasive imaging of skin physiology and percutaneous penetration using fluorescence spectral and lifetime imaging with multiphoton and confocal microscopy. Eur. J. Pharm. Biopharm. 2011, 77, 469–488. [Google Scholar] [CrossRef] [PubMed]
- Bird, D.K.; Yan, L.; Vrotsos, K.M.; Eliceiri, K.W.; Vaughan, E.M.; Keely, P.J.; White, J.G.; Ramanujam, N. Metabolic mapping of MCF10A human breast cells via multiphoton fluorescence lifetime imaging of the coenzyme NADH. Cancer Res. 2005, 65, 8766–8773. [Google Scholar] [CrossRef]
- Lakner, P.H.; Monaghan, M.G.; Möller, Y.; Olayioye, M.A.; Schenke-Layland, K. Applying phasor approach analysis of multiphoton FLIM measurements to probe the metabolic activity of three-dimensional in vitro cell culture models. Sci. Rep. 2017, 7, 42730. [Google Scholar] [CrossRef]
- Wang, H.; Liang, X.; Gravot, G.; Thorling, C.A.; Crawford, D.H.; Xu, Z.P.; Liu, X.; Roberts, M.S. Visualizing liver anatomy, physiology and pharmacology using multiphoton microscopy. J. Biophotonics 2017, 10, 46–60. [Google Scholar] [CrossRef]
- Sun, Y.; Phipps, J.; Elson, D.S.; Stoy, H.; Tinling, S.; Meier, J.; Poirier, B.; Chuang, F.S.; Farwell, D.G.; Marcu, L. Fluorescence lifetime imaging microscopy: In vivo application to diagnosis of oral carcinoma. Opt. Lett. 2009, 34, 2081–2083. [Google Scholar] [CrossRef]
- Sun, Y.; Phipps, J.E.; Meier, J.; Hatami, N.; Poirier, B.; Elson, D.S.; Farwell, D.G.; Marcu, L. Endoscopic fluorescence lifetime imaging for in vivo intraoperative diagnosis of oral carcinoma. Microsc. Microanal. 2013, 19, 791–798. [Google Scholar] [CrossRef]
- Liang, X.; Wang, H.; Liu, X.; Roberts, M. Quantitative optical imaging of paracetamol-induced metabolism changes in the liver. In SPIE BioPhotonics Australasia (10013); International Society for Optics and Photonics: Adelaide, Australia, 2016; p. 100131H. [Google Scholar]
- Roberts, M.S.; Barkauskas, D.S.; Wang, H.; Liu, X.; Studier, H.; Pastore, M.N.; Zhang, R.; Holmes, A.; Grice, J.E.; Xu, Z.; et al. Multiphoton and FLIM imaging in quantifying ex vivo and in vivo body organ kinetics of solutes. In Multiphoton Microscopy in the Biomedical Sciences XX (11244); International Society for Optics and Photonics: Bellingham, WA, USA, 2020; p. 112440S. [Google Scholar]
- Becker, W. Fluorescence lifetime imaging–techniques and applications. J. Microsc. 2012, 247, 119–136. [Google Scholar] [CrossRef]
- Berezin, M.Y.; Achilefu, S. Fluorescence lifetime measurements and biological imaging. Chem. Rev. 2010, 110, 2641–2684. [Google Scholar] [CrossRef] [PubMed]
- Skala, M.C.; Riching, K.M.; Gendron-Fitzpatrick, A.; Eickhoff, J.; Eliceiri, K.W.; White, J.G.; Ramanujam, N. In vivo multiphoton microscopy of NADH and FAD redox states, fluorescence lifetimes, and cellular morphology in precancerous epithelia. Proc. Natl. Acad. Sci. USA 2007, 104, 19494–19499. [Google Scholar] [CrossRef] [PubMed]
- Chorvat, D.; Chorvatova, A. Multi-wavelength fluorescence lifetime spectroscopy: A new approach to the study of endogenous fluorescence in living cells and tissues. Laser Phys. Lett. 2009, 6, 175–193. [Google Scholar] [CrossRef]
- Van Manen, H.J.; Verkuijlen, P.; Wittendorp, P.; Subramaniam, V.; Van den Berg, T.K.; Roos, D.; Otto, C. Refractive index sensing of green fluorescent proteins in living cells using fluorescence lifetime imaging microscopy. Biophys. J. 2008, 94, 67–69. [Google Scholar] [CrossRef] [PubMed]
- Lakowicz, J.R.; Szmacinski, H.; Nowaczyk, K.; Johnson, M.L. Fluorescence lifetime imaging of free and protein-bound NADH. Proc. Natl. Acad. Sci. USA 1992, 89, 1271–1275. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Liang, X.; Mohammed, Y.H.; Thomas, J.A.; Bridle, K.R.; Thorling, C.A.; Grice, J.E.; Xu, Z.P.; Liu, X.; Crawford, D.H.; et al. Real-time histology in liver disease using multiphoton microscopy with fluorescence lifetime imaging. Biomed. Opt. Express 2015, 6, 780–792. [Google Scholar] [CrossRef] [PubMed]
- Thorling, C.A.; Jin, L.; Weiss, M.; Crawford, D.; Liu, X.; Burczynski, F.J.; Liu, D.; Wang, H.; Roberts, M.S. Assessing steatotic liver function after ischemia-reperfusion injury by in vivo multiphoton imaging of fluorescein disposition. Biopharm. Drug Dispos. 2015, 43, 154–162. [Google Scholar] [CrossRef] [PubMed]
- Thorling, C.A.; Crawford, D.; Burczynski, F.J.; Liu, X.; Liau, I.; Roberts, M.S. Multiphoton microscopy in defining liver function. J. Biomed. Opt. 2014, 19, 090901. [Google Scholar] [CrossRef]
- Rodimova, S.A.; Kuznetsova, D.S.; Bobrov, N.V.; Gulin, A.A.; Reunov, D.G.; Karabut, M.M.; Shcheslavskiy, V.I.; Vdovina, N.V.; Zagainov, V.E.; Zagaynova, E.V. Interrogation of the Liver During Regeneration by Fluorescence Lifetime Imaging and Mass Spectrometry. IEEE J. Sel. Top. Quantum Electron. 2021, 27, 1–11. [Google Scholar] [CrossRef]
- Kuznetsova, D.S.; Rodimova, S.A.; Gulin, A.; Reunov, D.; Bobrov, N.; Polozova, A.V.; Vasin, A.; Shcheslavskiy, V.I.; Vdovina, N.; Zagainov, V.E.; et al. Metabolic imaging and secondary ion mass spectrometry to define the structure and function of liver with acute and chronic pathology. J. Biomed. Opt. 2019, 25, 014508. [Google Scholar] [CrossRef] [PubMed]
- Rui, L. Energy metabolism in the liver. Compr. Physiol. 2011, 4, 177–197. [Google Scholar]
- Chaban, Y.; Boekema, E.J.; Dudkina, N.V. Structures of mitochondrial oxidative phosphorylation supercomplexes and mechanisms for their stabilization. Biochim. Biophys. Acta 2014, 1837, 418–426. [Google Scholar] [CrossRef] [PubMed]
- Smeitink, J.; van den Heuvel, L.; DiMauro, S. The genetics and pathology of oxidative phosphorylation. Nat. Rev. Genet 2001, 2, 342–352. [Google Scholar] [CrossRef] [PubMed]
- Cairns, R.A.; Harris, I.S.; Mak, T.W. Regulation of cancer cell metabolism. Nat. Rev. Cancer 2011, 11, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Luengo, A.; Li, Z.; Gui, D.Y.; Sullivan, L.B.; Zagorulya, M.; Do, B.T.; Ferreira, R.; Naamati, A.; Ali, A.; Lewis, C.A.; et al. Increased demand for NAD+ relative to ATP drives aerobic glycolysis. Mol. Cell 2021, 81, 691–707. [Google Scholar] [CrossRef] [PubMed]
- Chan, T.S.; Cassim, S.; Raymond, V.A.; Gottschalk, S.; Merlen, G.; Zwingmann, C.; Lapierre, P.; Darby, P.; Mazer, C.D.; Bilodeau, M. Upregulation of Krebs cycle and anaerobic glycolysis activity early after onset of liver ischemia. PLoS ONE 2018, 13, e0199177. [Google Scholar] [CrossRef]
- Nsiah-Sefaa, A.; McKenzie, M. Combined defects in oxidative phosphorylation and fatty acid β-oxidation in mitochondrial disease. BioSci. Rep. 2016, 36, e00313. [Google Scholar] [CrossRef]
- Li, W.C.; Ralphs, K.L.; Tosh, D. Isolation and culture of adult mouse hepatocytes. In Mouse Cell Culture; Humana Press: Luxembourg, 2010; pp. 185–196. [Google Scholar] [CrossRef]
- Hall, P.D.; Lieber, C.S.; DeCarli, L.M.; French, S.W.; Lindros, K.O.; Järveläinen, H.; Bode, C.; Parlesak, A.; Bode, J.C. Models of alcoholic liver disease in rodents: A critical evaluation. Alcohol. Clin. Exp. Res. 2001, 25, 254S–261S. [Google Scholar] [CrossRef]
- Jaeschke, H.; McGill, M.R.; Williams, C.D.; Ramachandran, A. Current issues with acetaminophen hepatotoxicity—a clinically relevant model to test the efficacy of natural products. Life Sci. 2011, 88, 737–745. [Google Scholar] [CrossRef] [PubMed]
- Van Midwoud, P.M.; Groothuis, G.M.; Merema, M.T.; Verpoorte, E. Microfluidic biochip for the perifusion of precision-cut rat liver slices for metabolism and toxicology studies. Biotechnol. Bioeng. 2010, 105, 184–194. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.C.; Lloyd, W.R.; Kuo, S.; Kim, H.M.; Marcelo, C.L.; Feinberg, S.E.; Mycek, M.A. The potential of label-free nonlinear optical molecular microscopy to non-invasively characterize the viability of engineered human tissue constructs. Biomaterials 2014, 35, 6667–6676. [Google Scholar] [CrossRef]
- Kolenc, O.I.; Quinn, K.P. Evaluating cell metabolism through autofluorescence imaging of NAD(P)H and FAD. Antioxid. Redox Signal. 2019, 30, 875–889. [Google Scholar] [CrossRef]
- Meleshina, A.V.; Dudenkova, V.V.; Bystrova, A.S.; Kuznetsova, D.S.; Shirmanova, M.V.; Zagaynova, E.V. Two-photon FLIM of NAD(P)H and FAD in mesenchymal stem cells undergoing either osteogenic or chondrogenic differentiation. Stem Cell Res. 2017, 8, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Vorrink, S.U.; Zhou, Y.; Ingelman-Sundberg, M.; Lauschke, V.M. Prediction of drug-induced hepatotoxicity using long-term stable primary hepatic 3D spheroid cultures in chemically defined conditions. Toxicol. Sci. 2018, 163, 655–665. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, S.; Nian, F.S.; Tsai, J.W.; Karmenyan, A.; Chiou, A. Quantification of the metabolic state in cell-model of Parkinson’s disease by fluorescence lifetime imaging microscopy. Sci. Rep. 2016, 6, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Lukina, M.M.; Shimolina, L.E.; Kiselev, N.M.; Zagainov, V.E.; Komarov, D.V.; Zagaynova, E.V.; Shirmanova, M.V. Interrogation of tumor metabolism in tissue samples ex vivo using fluorescence lifetime imaging of NAD(P)H. Methods Appl. Fluoresc. 2019, 8, 014002. [Google Scholar] [CrossRef]
- Yang, S.; Tan, T.M.C.; Wee, A.; Leow, C.K. Mitochondrial respiratory function and antioxidant capacity in normal and cirrhotic livers following partial hepatectomy. Cell Mol. Life Sci. 2004, 61, 220–229. [Google Scholar] [CrossRef]
- Liu, C.; Sekine, S.; Ito, K. Assessment of mitochondrial dysfunction-related, drug-induced hepatotoxicity in primary rat hepatocytes. Toxicol. Appl. Pharm. 2016, 302, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Ito, Y.; Bethea, N.W.; Abril, E.R.; McCuskey, R.S. Early hepatic microvascular injury in response to acetaminophen toxicity. Microcirculation 2003, 10, 391–400. [Google Scholar] [CrossRef] [PubMed]
- Ruepp, S.U.; Tonge, R.P.; Shaw, J.; Wallis, N.; Pognan, F. Genomics and proteomics analysis of acetaminophen toxicity in mouse liver. Toxicol. Sci. 2002, 65, 135–150. [Google Scholar] [CrossRef]
- Lee, K.K.; Imaizumi, N.; Chamberland, S.R.; Alder, N.N.; Boelsterli, U.A. Targeting mitochondria with methylene blue protects mice against acetaminophen-induced liver injury. Hepatology 2015, 61, 326–336. [Google Scholar] [CrossRef]
- George, E.; Murdock, J.; Aylott, M.; Westmoreland, C. Comparison of hepatocyte cultures and liver slices in in vitro toxicity testing. Altern. Lab. Anim. 1999, 27, 769–781. [Google Scholar] [CrossRef]
- Granitzny, A.; Knebel, J.; Schaudien, D.; Braun, A.; Steinberg, P.; Dasenbrock, C.; Hansen, T. Maintenance of high quality rat precision cut liver slices during culture to study hepatotoxic responses: Acetaminophen as a model compound. Toxicol. Vitr. 2017, 42, 200–213. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.C.; Carella, M.A.; Papa, S.; Bubici, C. High expression of glycolytic genes in cirrhosis correlates with the risk of developing liver cancer. Front. Cell Dev. Biol. 2018, 6, 138. [Google Scholar] [CrossRef] [PubMed]
- Blacker, T.S.; Mann, Z.F.; Gale, J.E.; Ziegler, M.; Bain, A.J.; Szabadkai, G.; Duchen, M.R. Separating NADH and NADPH fluorescence in live cells and tissues using FLIM. Nat. Commun. 2014, 5, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Venkatraman, A.; Shiva, S.; Wigley, A.; Ulasova, E.; Chhieng, D.; Bailey, S.M.; Darley-Usmar, V.M. The role of iNOS in alcohol-dependent hepatotoxicity and mitochondrial dysfunction in mice. Hepatology 2004, 40, 565–573. [Google Scholar] [CrossRef] [PubMed]
- Vishwasrao, H.D.; Heikal, A.A.; Kasischke, K.A.; Webb, W.W. Conformational dependence of intracellular NADH on metabolic state revealed by associated fluorescence anisotropy. J. Biol. Chem. 2005, 280, 25119–25126. [Google Scholar] [CrossRef]
- Atashi, F.; Modarressi, A.; Pepper, M.S. The role of reactive oxygen species in mesenchymal stem cell adipogenic and osteogenic differentiation: A review. Stem Cells Dev. 2015, 24, 1150–1163. [Google Scholar] [CrossRef] [PubMed]
- Couto, N.; Wood, J.; Barber, J. The role of glutathione reductase and related enzymes on cellular redox homoeostasis network. Free Radic. Biol. Med. 2016, 95, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Bailey, S.M.; Robinson, G.; Pinner, A.; Chamlee, L.; Ulasova, E.; Pompilius, M. S-adenosylmethionine prevents chronic alcohol-induced mitochondrial dysfunction in the rat liver. Am. J. Physiol. 2006, 291, G857–G867. [Google Scholar] [CrossRef] [PubMed]
- Sykora, P.; Kharbanda, K.K.; Crumm, S.E.; Cahill, A. S-adenosyl-L-methionine co-administration prevents the ethanol-elicited dissociation of hepatic mitochondrial ribosomes in male rats. Alcohol. Clin. Exp. Res. 2009, 33, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Yan, M.; Zhu, P.; Liu, H.M.; Zhang, H.T.; Liu, L. Ethanol induced mitochondria injury and permeability transition pore opening: Role of mitochondria in alcoholic liver disease. World J. Gastroenterol. 2007, 13, 2352–2356. [Google Scholar] [CrossRef] [PubMed]
- Kharbanda, K.K.; Todero, S.L.; King, A.L.; Osna, N.A.; McVicker, B.L.; Tuma, D.J.; Wisecarver, J.L.; Bailey, S.M. Betaine treatment attenuates chronic ethanol-induced hepatic steatosis and alterations to the mitochondrial respiratory chain proteome. Int. J. Hepatol. 2012, 2012, 962183. [Google Scholar] [CrossRef]
- Nassir, F.; Ibdah, J.A. Role of mitochondria in alcoholic liver disease. World J. Gastroenterol. 2014, 20, 2136. [Google Scholar] [CrossRef] [PubMed]
- Young, T.A.; Bailey, S.M.; Van Horn, C.G.; Cunningham, C.C. Chronic ethanol consumption decreases mitochondrial and glycolytic production of ATP in liver. Alcohol Alcohol. 2006, 41, 254–260. [Google Scholar] [CrossRef] [PubMed]
- Labonne, B.E.; Gutiérrez, M.; Gómez-Quiroz, L.E.; Fainstein, M.K.; Bucio, L.; Souza, V.; Flores, O.; Ortíz, V.; Hernández, E.; Kershenobich, D.; et al. Acetaldehyde-induced mitochondrial dysfunction sensitizes hepatocytes to oxidative damage. Cell Biol. Toxicol. 2009, 25, 599. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, C.C. Energy availability and alcohol-related liver pathology. Alcohol Res. Health 2003, 27, 291. [Google Scholar] [PubMed]
- Shahid, M.; Subhan, F. Comparative histopathology of acetaminophen induced hepatotoxicity in animal models of mice and rats. Pharm. Online 2014, 3, 32–43. [Google Scholar]
- Turan, A.; Celik, I. Antioxidant and hepatoprotective properties of dried fig against oxidative stress and hepatotoxicity in rats. Int. J. Biol. Macromol. 2016, 91, 554–559. [Google Scholar] [CrossRef] [PubMed]
- Trautwein, C.; Friedman, S.L.; Schuppan, D.; Pinzani, M. Hepatic fibrosis: Concept to treatment. J. Hepatol. 2015, 62, S15–S24. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodimova, S.; Elagin, V.; Karabut, M.; Koryakina, I.; Timin, A.; Zagainov, V.; Zyuzin, M.; Zagaynova, E.; Kuznetsova, D. Toxicological Analysis of Hepatocytes Using FLIM Technique: In Vitro versus Ex Vivo Models. Cells 2021, 10, 2894. https://doi.org/10.3390/cells10112894
Rodimova S, Elagin V, Karabut M, Koryakina I, Timin A, Zagainov V, Zyuzin M, Zagaynova E, Kuznetsova D. Toxicological Analysis of Hepatocytes Using FLIM Technique: In Vitro versus Ex Vivo Models. Cells. 2021; 10(11):2894. https://doi.org/10.3390/cells10112894
Chicago/Turabian StyleRodimova, Svetlana, Vadim Elagin, Maria Karabut, Irina Koryakina, Alexander Timin, Vladimir Zagainov, Mikhail Zyuzin, Elena Zagaynova, and Daria Kuznetsova. 2021. "Toxicological Analysis of Hepatocytes Using FLIM Technique: In Vitro versus Ex Vivo Models" Cells 10, no. 11: 2894. https://doi.org/10.3390/cells10112894
APA StyleRodimova, S., Elagin, V., Karabut, M., Koryakina, I., Timin, A., Zagainov, V., Zyuzin, M., Zagaynova, E., & Kuznetsova, D. (2021). Toxicological Analysis of Hepatocytes Using FLIM Technique: In Vitro versus Ex Vivo Models. Cells, 10(11), 2894. https://doi.org/10.3390/cells10112894