
Single-Cell Approaches to Deconvolute the Development of HSCs



Abstract
:1. Efforts on HSC Identification and Generation
1.1. Attempts to Generate HSCs
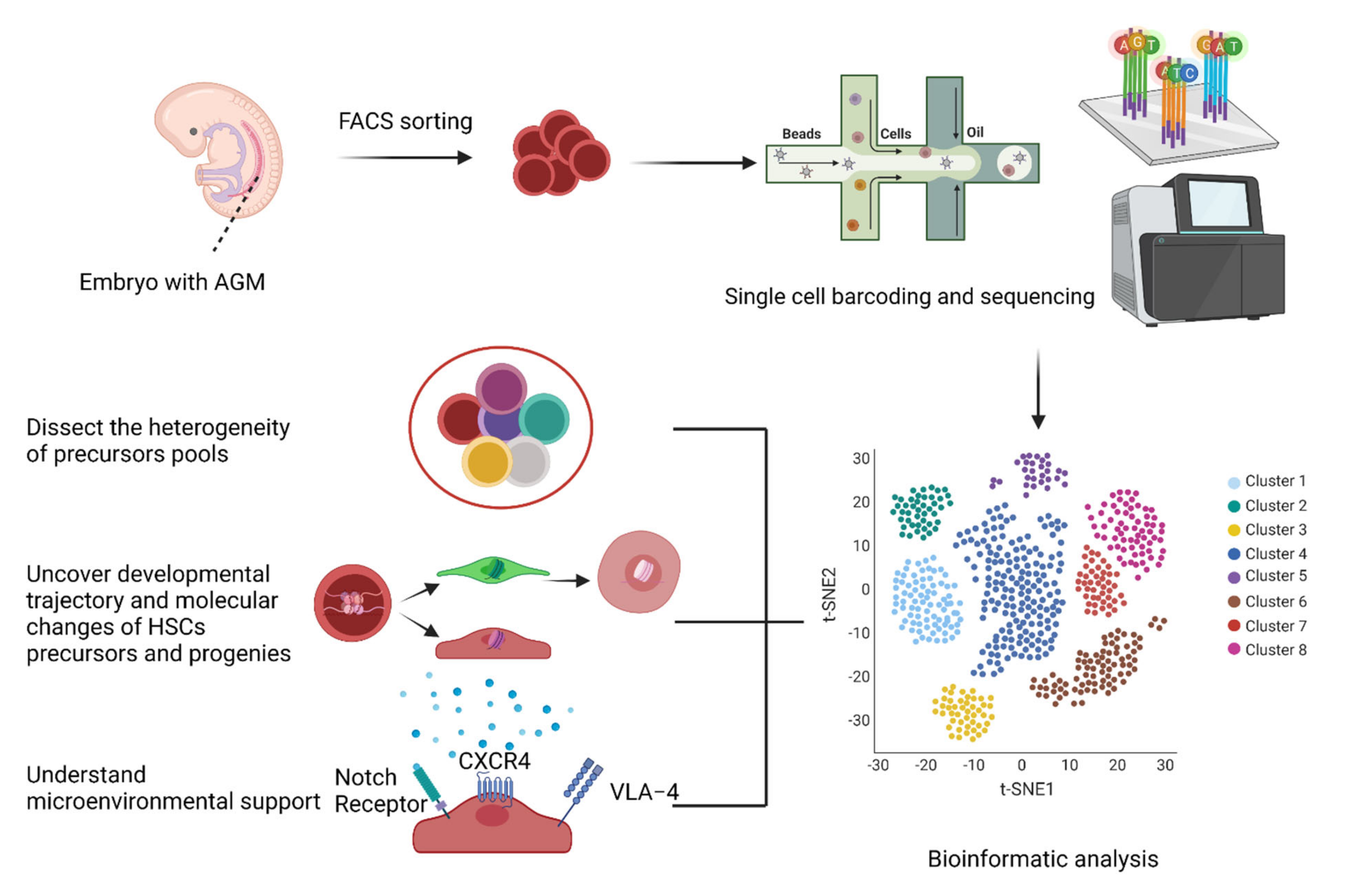
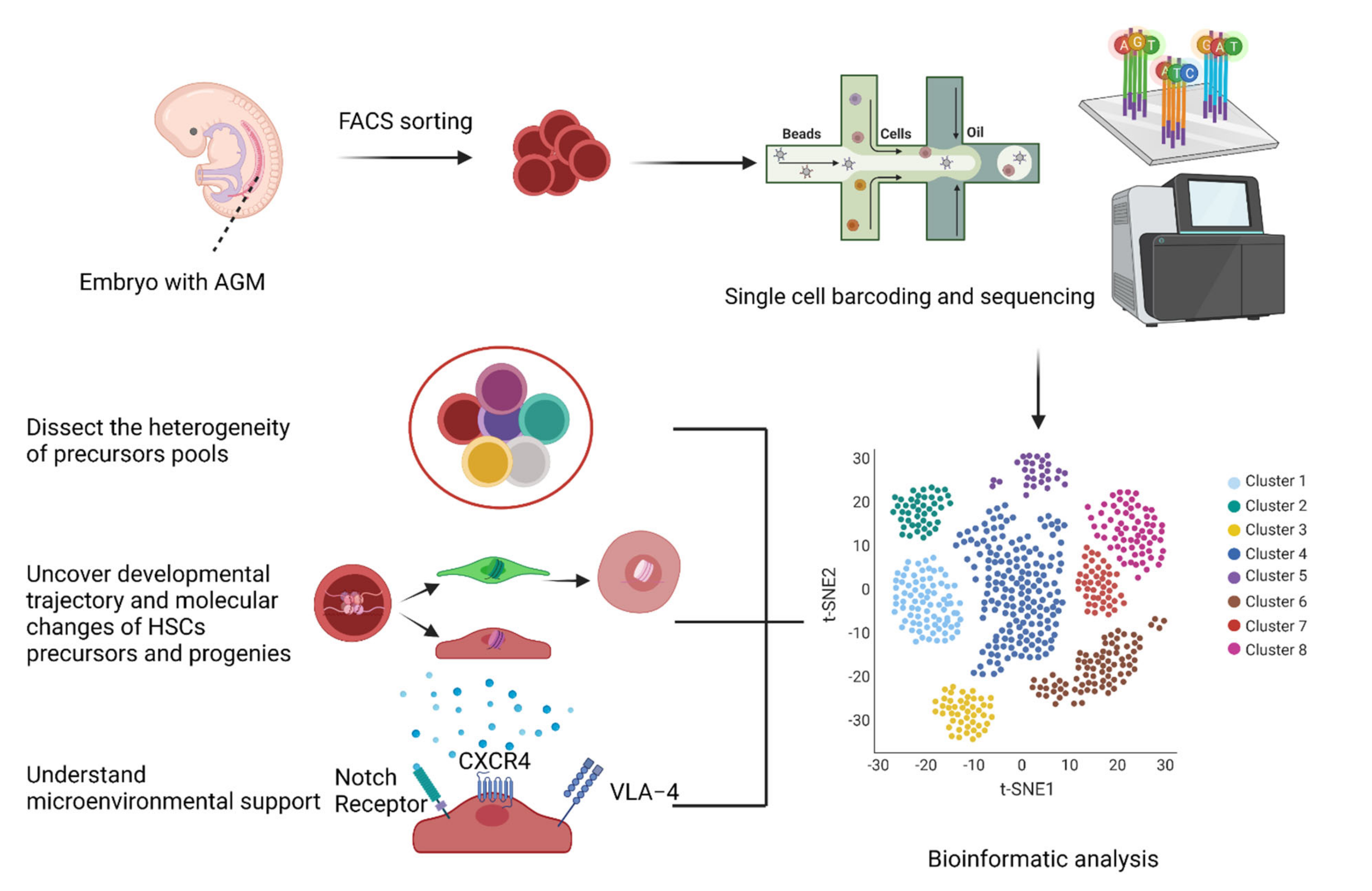
1.2. Single-Cell RNA-Seq Deconvolution for Missing Links in HSC Generation
- The heterogeneity of the precursors pool;
- The developmental trajectory and molecular changes for HSC themselves (such as transcriptome and epigenetic information);
- The microenvironmental support in vitro and in vivo.
2. New Single-Cell Techniques Address HSC Generation at Spatial and Temporal Resolutions
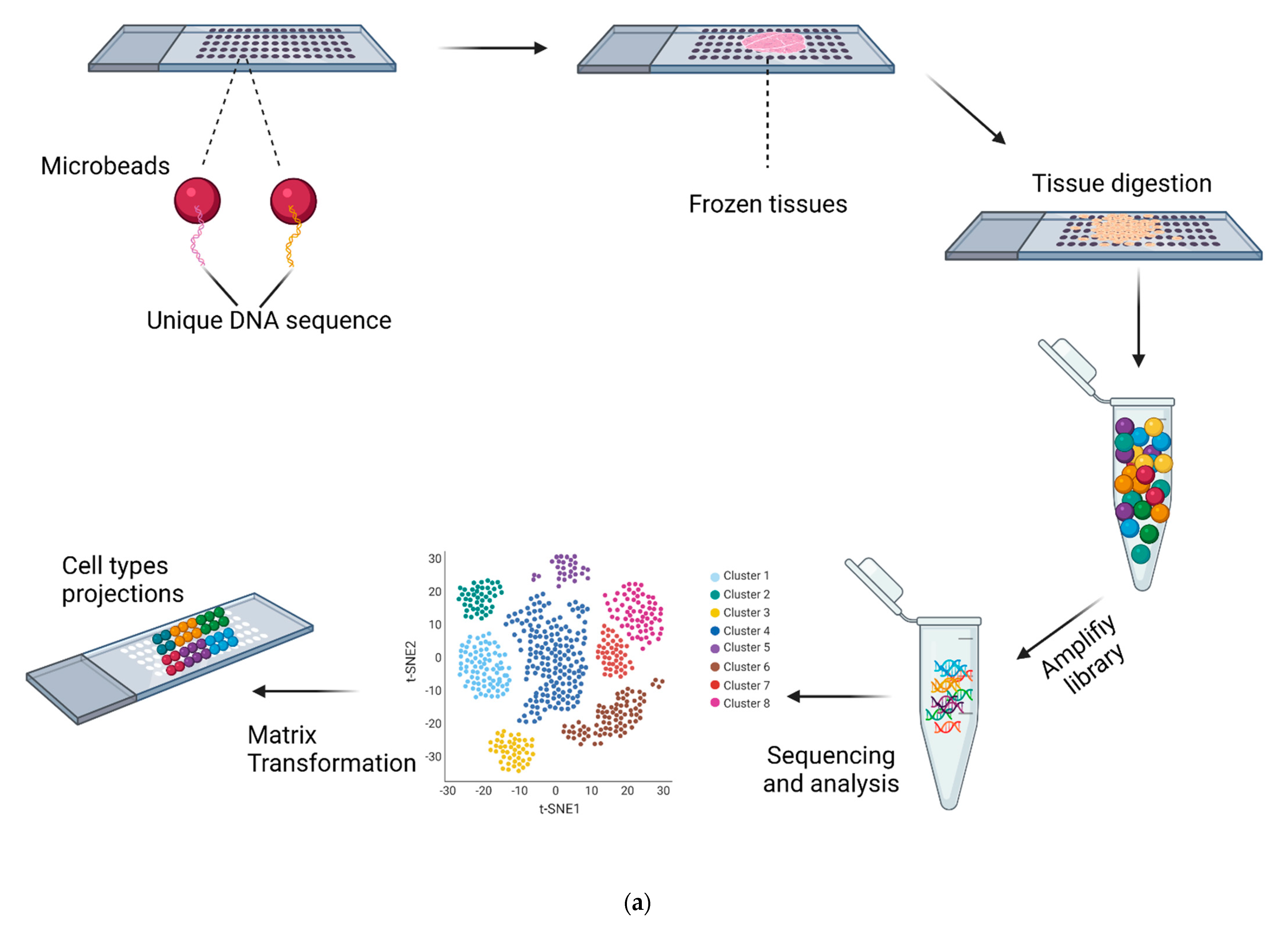
2.1. Spatial Single-Cell RNA-Seq
2.2. Molecular Barcoding
- Transposon (Transposase-mediated random integration barcoding);
- CARLIN (CRISPR array repair lineage tracing);
- LARRY (lineage and RNA recovery);
- PolyloxExpress (Cre-recombinase-dependent barcoding);
- GESTALT (genome editing of synthetic target arrays for lineage tracing).
3. Outlook for HSC Generation
Author Contributions
Funding
Conflicts of Interest
References
- Morrison, S.J.; Scadden, D.T. The bone marrow niche for haematopoietic stem cells. Nature 2014, 505, 327–334. [Google Scholar] [CrossRef] [Green Version]
- Siminovitch, L.; McCulloch, E.A.; Till, J.E. The Distribution of Colony-forming Cells Among Spleen Colonies. J. Cell. Comp. Physiol. 1963, 62, 327–336. [Google Scholar] [CrossRef] [Green Version]
- Gerald, J.; Spangrude, S.H.; Irving, L. Weissman, Purification and characterization of mouse hematopoietic stem cells. Science 1988, 241, 58–62. [Google Scholar]
- Kiel, M.J.; Yilmaz, O.H.; Iwashita, T.; Yilmaz, O.H.; Terhorst, C.; Morrison, S.J. SLAM family receptors distinguish hemato-poietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell 2005, 121, 1109–1121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- North, T.E.; De Bruijn, M.F.; Stacy, T.; Talebian, L.; Lind, E.; Robin, C.; Binder, M.; Dzierzak, E.; Speck, N.A. Runx1 Expression Marks Long-Term Repopulating Hematopoietic Stem Cells in the Midgestation Mouse Embryo. Immunity 2002, 16, 661–672. [Google Scholar] [CrossRef] [Green Version]
- Bertrand, J.; Giroux, S.; Golub, R.; Klaine, M.; Jalil, A.; Boucontet, L.; Godin, I.; Cumano, A. Characterization of purified intraembryonic hematopoietic stem cells as a tool to define their site of origin. Proc. Natl. Acad. Sci. USA 2005, 102, 134–139. [Google Scholar] [CrossRef] [Green Version]
- Boisset, J.-C.; Van Cappellen, W.; Andrieu-Soler, C.; Galjart, N.; Dzierzak, E.; Robin, C. In vivo imaging of haematopoietic cells emerging from the mouse aortic endothelium. Nature 2010, 464, 116–120. [Google Scholar] [CrossRef] [PubMed]
- Butler, J.M.; Nolan, D.J.; Vertes, E.L.; Varnum-Finney, B.; Kobayashi, H.; Hooper, A.T.; Seandel, M.; Shido, K.; White, I.A.; Kobayashi, M.; et al. Endothelial Cells Are Essential for the Self-Renewal and Repopulation of Notch-Dependent Hematopoietic Stem Cells. Cell Stem Cell 2010, 6, 251–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugimura, R.; He, X.C.; Venkatraman, A.; Arai, F.; Box, A.; Semerad, C.; Haug, J.S.; Peng, L.; Zhong, X.-B.; Suda, T.; et al. Noncanonical Wnt Signaling Maintains Hematopoietic Stem Cells in the Niche. Cell 2012, 150, 351–365. [Google Scholar] [CrossRef] [Green Version]
- Decker, M.; Leslie, J.; Liu, Q.; Ding, L. Hepatic thrombopoietin is required for bone marrow hematopoietic stem cell mainte-nance. Science 2018, 360, 106–110. [Google Scholar] [CrossRef] [Green Version]
- Kim, P.G.; Albacker, C.E.; Lu, Y.-F.; Jang, I.-H.; Lim, Y.; Heffner, G.C.; Arora, N.; Bowman, T.V.; Lin, M.I.; Lensch, M.W.; et al. Signaling axis involving Hedgehog, Notch, and Scl promotes the embryonic endothelial-to-hematopoietic transition. Proc. Natl. Acad. Sci. USA 2012, 110, 141–150. [Google Scholar] [CrossRef]
- Rafii, S.; Kloss, C.C.; Butler, J.M.; Ginsberg, M.; Gars, E.; Lis, R.; Zhan, Q.; Josipovic, P.; Ding, B.-S.; Xiang, J.; et al. Human ESC-derived hemogenic endothelial cells undergo distinct waves of endothelial to hematopoietic transition. Blood 2013, 121, 770–780. [Google Scholar] [CrossRef]
- Sturgeon, C.M.; Ditadi, A.; Clarke, R.L.; Keller, G. Defining the path to hematopoietic stem cells. Nat. Biotechnol. 2013, 31, 416–418. [Google Scholar] [CrossRef]
- Doulatov, S.; Vo, L.T.; Chou, S.S.; Kim, P.G.; Arora, N.; Li, H.; Hadland, B.K.; Bernstein, I.D.; Collins, J.J.; Zon, L.I.; et al. Induction of multipotential hematopoietic progenitors from human pluripotent stem cells via respecification of lineage-restricted precursors. Cell Stem Cell 2013, 13, 459–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kennedy, M.; Awong, G.; Sturgeon, C.M.; Ditadi, A.; LaMotte-Mohs, R.; Zúñiga-Pflücker, J.C.; Keller, G. T Lymphocyte Potential Marks the Emergence of Definitive Hematopoietic Progenitors in Human Pluripotent Stem Cell Differentiation Cultures. Cell Rep. 2012, 2, 1722–1735. [Google Scholar] [CrossRef] [Green Version]
- Amabile, G.; Welner, R.S.; Nombela-Arrieta, C.; D’Alise, A.M.; Di Ruscio, A.; Ebralidze, A.K.; Kraytsberg, Y.; Ye, M.; Kocher, O.; Neuberg, D.S.; et al. In vivo generation of transplantable human hematopoietic cells from induced pluripotent stem cells. Blood 2013, 121, 1255–1264. [Google Scholar] [CrossRef]
- Suzuki, N.; Yamazaki, S.; Yamaguchi, T.; Okabe, M.; Masaki, H.; Takaki, S.; Otsu, M.; Nakauchi, H. Generation of Engraftable Hematopoietic Stem Cells from Induced Pluripotent Stem Cells by Way of Teratoma Formation. Mol. Ther. 2013, 21, 1424–1431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elcheva, I.; Brok-Volchanskaya, V.; Kumar, A.; Liu, P.; Lee, J.-H.; Tong, L.; A Vodyanik, M.; Swanson, S.; Stewart, R.; Kyba, M.; et al. Direct induction of haematoendothelial programs in human pluripotent stem cells by transcriptional regulators. Nat. Commun. 2014, 5, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riddell, J.; Gazit, R.; Garrison, B.S.; Guo, G.; Saadatpour, A.; Mandal, P.; Ebina, W.; Volchkov, P.; Yuan, G.-C.; Orkin, S.H.; et al. Reprogramming Committed Murine Blood Cells to Induced Hematopoietic Stem Cells with Defined Factors. Cell 2014, 157, 549–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugimura, R.; Jha, D.; Han, A.; Soria-Valles, C.; da Rocha, E.L.; Lu, Y.-F.; Goettel, J.A.; Serrao, E.; Rowe, R.G.; Malleshaiah, M.; et al. Haematopoietic stem and progenitor cells from human pluripotent stem cells. Nature 2017, 545, 432–438. [Google Scholar] [CrossRef] [Green Version]
- Zhou, F.; Li, X.; Wang, W.; Zhu, P.; Zhou, J.; He, W.; Ding, M.; Xiong, F.; Zheng, X.; Li, Z.; et al. Tracing haematopoietic stem cell formation at single-cell resolution. Nature 2016, 533, 487–492. [Google Scholar] [CrossRef]
- Zeng, Y.; He, J.; Bai, Z.; Li, Z.; Gong, Y.; Liu, C.; Ni, Y.; Du, J.; Ma, C.; Bian, L.; et al. Tracing the first hematopoietic stem cell generation in human embryo by single-cell RNA sequencing. Cell Res. 2019, 29, 881–894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howell, E.D.; Speck, N.A. Forks in the road to the first hematopoietic stem cells. Cell Res. 2020, 30, 457–458. [Google Scholar] [CrossRef] [PubMed]
- Fidanza, A.; Stumpf, P.S.; Ramachandran, P.; Tamagno, S.; Babtie, A.; Lopez-Yrigoyen, M.; Taylor, A.H.; Easterbrook, J.; Henderson, B.E.P.; Axton, R.; et al. Single-cell analyses and machine learning define hematopoietic progenitor and HSC-like cells derived from human PSCs. Blood 2020, 136, 2893–2904. [Google Scholar] [CrossRef]
- Lange, L.; Morgan, M.; Schambach, A. The hemogenic endothelium: A critical source for the generation of PSC-derived hemato-poietic stem and progenitor cells. Cell Mol. Life Sci. 2021, 78, 4143–4160. [Google Scholar] [CrossRef]
- Zhu, Y.; Wang, T.; Gu, J.; Huang, K.; Zhang, T.; Zhang, Z.; Liu, H.; Tang, J.; Mai, Y.; Zhang, Y.; et al. Characterization and generation of human definitive multipotent hematopoietic stem/progenitor cells. Cell Discov. 2020, 6, 1–18. [Google Scholar] [CrossRef]
- Zhou, J.; Liu, B.; Lan, Y. When blood development meets single-cell transcriptomics. Blood Sci. 2019, 1, 65–68. [Google Scholar] [CrossRef]
- Hadland, B.; Varnum-Finney, B.; Dozono, S.; Dignum, T.; Nourigat-McKay, C.; Jackson, D.L.; Itkin, T.; Butler, J.M.; Rafii, S.; Trapnell, C.; et al. Engineering a niche supporting haematopoietic stem cell development using integrated single cell transcriptomics. BioRxiv 2021. [Google Scholar] [CrossRef]
- Satija, R.; Farrell, J.; Gennert, D.; Schier, A.F.; Regev, A. Spatial reconstruction of single-cell gene expression data. Nat. Biotechnol. 2015, 33, 495–502. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.H.; Boettiger, A.N.; Moffitt, J.R.; Wang, S.; Zhuang, X. Spatially resolved, highly multiplexed RNA profiling in single cells. Science 2015, 348, aaa6090. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Liu, M.; Yang, J.; Weissman, S.M.; Pan, X.; Katz, S.G.; Wang, S. Spatial transcriptome profiling by MERFISH reveals fetal liver hematopoietic stem cell niche architecture. Cell Discov. 2021, 7, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Ståhl, P.L.; Salmén, F.; Vickovic, S.; Lundmark, A.; Navarro, J.F.; Magnusson, J.; Giacomello, S.; Asp, M.; Westholm, J.O.; Huss, M.; et al. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science 2016, 353, 78–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriques, S.G.; Stickels, R.R.; Goeva, A.; Martin, C.A.; Murray, E.; Vanderburg, C.R.; Welch, J.; Chen, L.M.; Chen, F.; Macosko, E.Z. Slide-seq: A scalable technology for measuring genome-wide expression at high spatial resolution. Science 2019, 363, 1463–1467. [Google Scholar] [CrossRef] [PubMed]
- Stickels, R.R.; Murray, E.; Kumar, P.; Li, J.; Marshall, J.L.; Di Bella, D.J.; Arlotta, P.; Macosko, E.Z.; Chen, F. Highly sensitive spatial transcriptomics at near-cellular resolution with Slide-seqV2. Nat. Biotechnol. 2021, 39, 313–319. [Google Scholar] [CrossRef]
- VanHorn, S.; Morris, S.A. Next-Generation Lineage Tracing and Fate Mapping to Interrogate Development. Dev. Cell 2021, 56, 7–21. [Google Scholar] [CrossRef] [PubMed]
- Maniatis, S.; Petrescu, J.; Phatnani, H. Spatially resolved transcriptomics and its applications in cancer. Curr. Opin. Genet. Dev. 2021, 66, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Rabbie, R.; Lau, D.; White, R.M.; Adams, D.J. Unraveling the cartography of the cancer ecosystem. Genome Biol. 2021, 22, 1–9. [Google Scholar] [CrossRef]
- Lu, R.; Neff, N.F.; Quake, S.R.; Weissman, I.L. Tracking single hematopoietic stem cells in vivo using high-throughput sequencing in conjunction with viral genetic barcoding. Nat. Biotechnol. 2011, 29, 928–933. [Google Scholar] [CrossRef] [Green Version]
- Cheung, A.M.S.; Nguyen, L.V.; Carles, A.; Beer, P.; Miller, P.H.; Knapp, D.J.H.F.; Dhillon, K.; Hirst, M.; Eaves, C.J. Analysis of the clonal growth and differentiation dynamics of primitive barcoded human cord blood cells in NSG mice. Blood 2013, 122, 3129–3137. [Google Scholar] [CrossRef] [Green Version]
- Stark, R.; Grzelak, M.; Hadfield, J. RNA sequencing: The teenage years. Nat. Rev. Genet. 2019, 20, 631–656. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Ramos, A.; Chapman, B.; Johnnidis, J.B.; Le, L.; Ho, Y.-J.; Klein, A.M.; Hofmann, O.; Camargo, F.D. Clonal dynamics of native haematopoiesis. Nature 2014, 514, 322–327. [Google Scholar] [CrossRef] [PubMed]
- Bowling, S.; Sritharan, D.; Osorio, F.G.; Nguyen, M.; Cheung, P.; Rodriguez-Fraticelli, A.; Patel, S.; Yuan, W.C.; Fujiwara, Y.; Li, B.E.; et al. An Engineered CRISPR-Cas9 Mouse Line for Simultaneous Readout of Lineage Histories and Gene Expression Profiles in Single Cells. Cell 2020, 181, 1410–1422. [Google Scholar] [CrossRef]
- Rodriguez-Fraticelli, A.E.; Weinreb, C.; Wang, S.-W.; Migueles, R.P.; Jankovic, M.; Usart, M.; Klein, A.M.; Lowell, S.; Camargo, F.D. Single-cell lineage tracing unveils a role for TCF15 in haematopoiesis. Nature 2020, 583, 585–589. [Google Scholar] [CrossRef] [PubMed]
- Pei, W.; Shang, F.; Wang, X.; Fanti, A.-K.; Greco, A.; Busch, K.; Klapproth, K.; Zhang, Q.; Quedenau, C.; Sauer, S.; et al. Resolving Fates and Single-Cell Transcriptomes of Hematopoietic Stem Cell Clones by PolyloxExpress Barcoding. Cell Stem Cell 2020, 27, 383–395. [Google Scholar] [CrossRef]
- Simeonov, K.P.; Byrns, C.N.; Clark, M.L.; Norgard, R.J.; Martin, B.; Stanger, B.Z.; McKenna, A.; Shendure, J.; Lengner, C.J. Single-cell lineage and transcriptome reconstruction of metastatic cancer reveals selection of aggressive hybrid EMT states. BioRxiv 2021. [Google Scholar] [CrossRef]
- Fasching, L.; Jang, Y.; Tomasi, S.; Schreiner, J.; Tomasini, L.; Brady, M.V.; Bae, T.; Sarangi, V.; Vasmatzis, N.; Wang, Y.; et al. Early developmental asymmetries in cell lineage trees in living individuals. Science 2021, 371, 1245–1248. [Google Scholar] [CrossRef] [PubMed]
- Bizzotto, S.; Dou, Y.; Ganz, J.; Doan, R.N.; Kwon, M.; Bohrson, C.L.; Kim, S.N.; Bae, T.; Abyzov, A.; Park, P.J.; et al. Landmarks of human embryonic development inscribed in somatic mutations. Science 2021, 371, 1249–1253. [Google Scholar] [CrossRef]
- Dzierzak, E.; Bigas, A. Blood Development: Hematopoietic Stem Cell Dependence and Independence. Cell Stem Cell 2018, 22, 639–651. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Technique | Barcode Type | Static Barcodes or Evolving Barcodes? | Barcode as mRNA? |
|---|---|---|---|
| CARLIN | INDEL | Evolving barcodes | Yes |
| Macs-GESTALT | INDEL | Static barcodes & Evolving barcodes | Yes |
| Poly-Lox Express | Recombination | Evolving barcodes | Yes |
| LARRY | Integration | Static barcodes | Yes |
| Transposon | Random Integration | Evolving barcodes | No |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiang, Y.; Sugimura, R. Single-Cell Approaches to Deconvolute the Development of HSCs. Cells 2021, 10, 2876. https://doi.org/10.3390/cells10112876
Xiang Y, Sugimura R. Single-Cell Approaches to Deconvolute the Development of HSCs. Cells. 2021; 10(11):2876. https://doi.org/10.3390/cells10112876
Chicago/Turabian StyleXiang, Yang, and Ryohichi Sugimura. 2021. "Single-Cell Approaches to Deconvolute the Development of HSCs" Cells 10, no. 11: 2876. https://doi.org/10.3390/cells10112876
APA StyleXiang, Y., & Sugimura, R. (2021). Single-Cell Approaches to Deconvolute the Development of HSCs. Cells, 10(11), 2876. https://doi.org/10.3390/cells10112876