
NLRP3 Activation and Its Relationship to Endothelial Dysfunction and Oxidative Stress: Implications for Preeclampsia and Pharmacological Interventions



Abstract
1. Introduction
2. Preeclampsia and Endothelial Dysfunction
3. NLRP3 Inflammasome Activation and Regulation in Preeclampsia
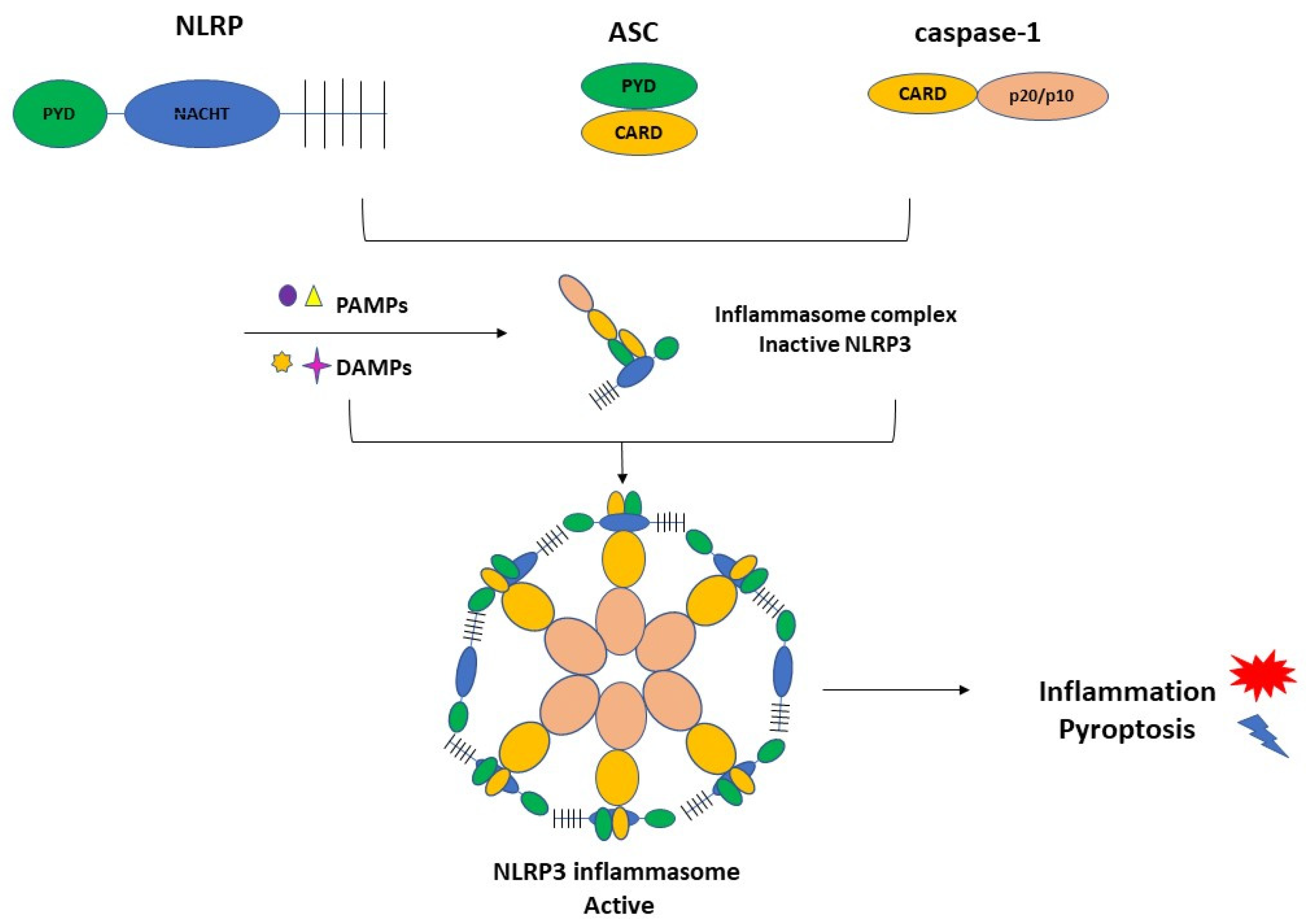
3.1. Inflammasome Formation and the Role of NLRP3 in the Pathogenesis of PE
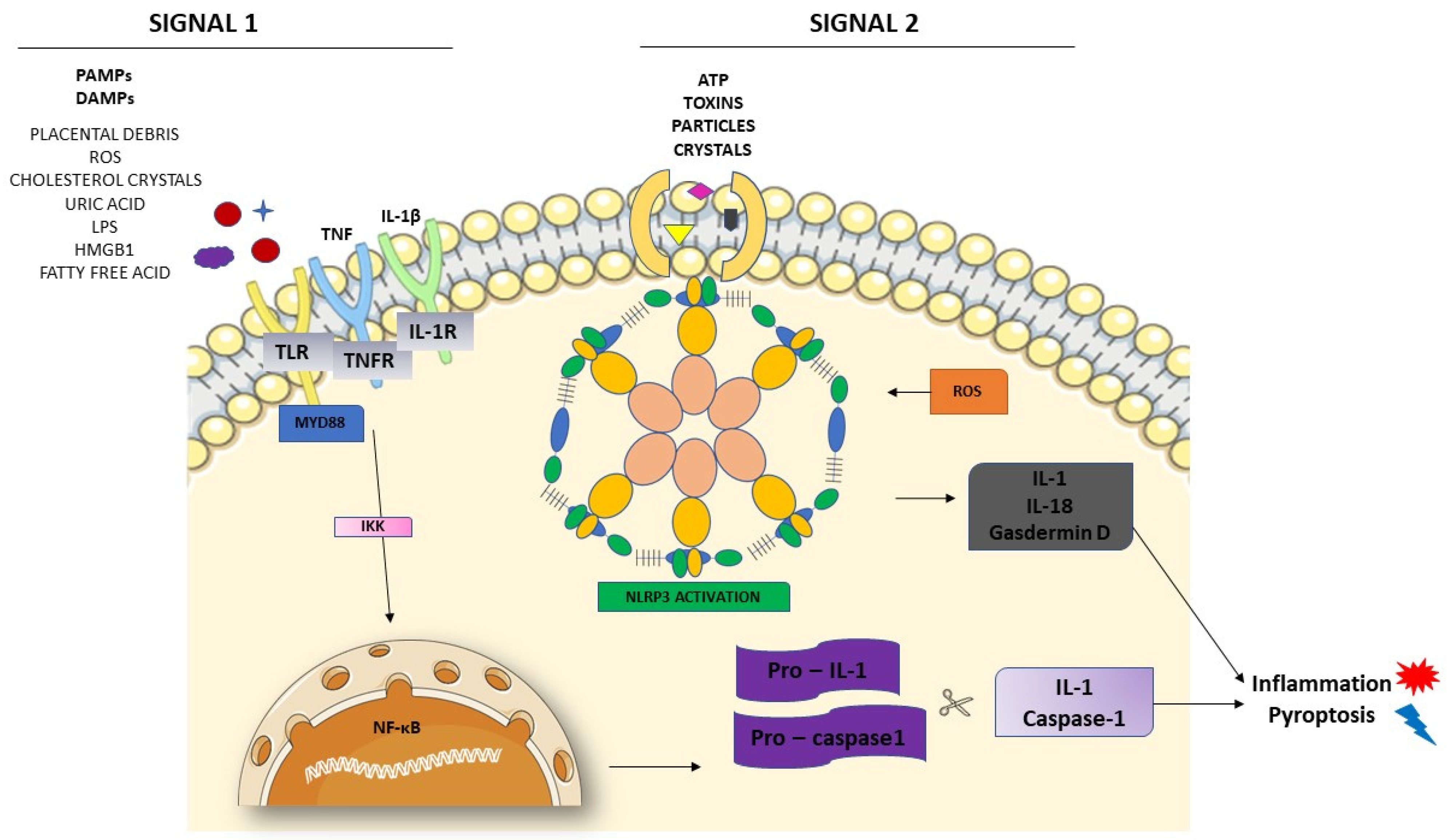
3.2. Activation of NLRP3 and Pyroptosis: The Cell Death Related to Inflammatory Processes
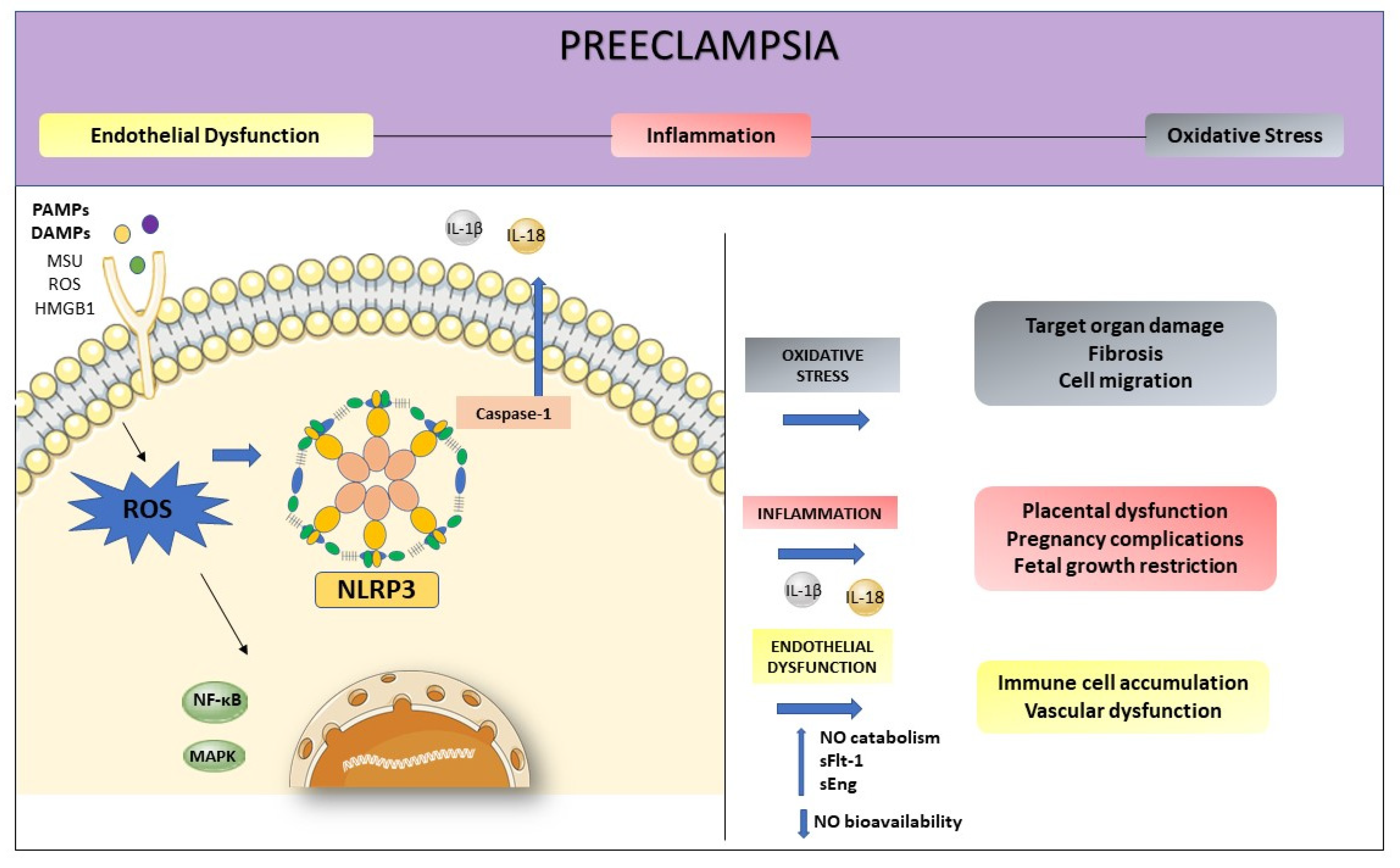
4. NLRP3 and its Relation with Endothelial Dysfunction and Oxidative Stress
5. Pharmacological Interventions: Selective and Non-Selective Drugs
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- American College of Obstetricians and Gynecologists. ACOG practice bulletin number 222. Gestational Hypertension and Preeclampsia. Obstet. Gynecol. 2020, 135, 237–260. [Google Scholar] [CrossRef]
- Tranquilli, A.L.; Dekker, G.; Magee, L.; Roberts, J.; Sibai, B.M.; Steyn, W.; Zeeman, G.G.; Brown, M.A. The classification, diagnosis, and management of the hypertensive disorders of pregnancy: A revised statement from the ISSHP. Pregnancy Hypertens. 2014, 4, 97–104. [Google Scholar] [CrossRef]
- Mol, B.W.; Roberts, C.T.; Thangaratinam, S.; Magee, L.A.; de Groot, C.J.; Hofmeyr, G.J. Pre-eclampsia. Lancet 2016, 387, 999–1011. [Google Scholar] [CrossRef]
- Kintiraki, E.; Papakatsika, S.; Kotronis, G.; Goulis, D.G.; Kotsis, V. Pregnancy-Induced hypertension. Horm 2015, 14, 211–223. [Google Scholar] [CrossRef]
- Collins, S.L.; Birks, J.S.; Stevenson, G.N.; Papageorghiou, A.T.; Noble, J.A.; Impey, L. Measurement of spiral artery jets: General principles and differences observed in small-for-gestational-age pregnancies. Ultrasound Obstet. Gynecol. 2012, 40, 171–178. [Google Scholar] [CrossRef]
- Von Dadelszen, P.; Magee, L.A.; Roberts, J.M. Subclassification of Preeclampsia. Hypertens. Pregnancy 2003, 22, 143–148. [Google Scholar] [CrossRef]
- George, E.M.; Granger, J.P. Mechanisms and potential therapies for preeclampsia. Curr. Hypertens. Rep. 2011, 13, 269–275. [Google Scholar] [CrossRef]
- Turco, M.Y.; Moffett, A. Development of the human placenta. Development 2019, 146, dev163428. [Google Scholar] [CrossRef]
- Burton, G.J.; Redman, C.W.; Roberts, J.M.; Moffett, A. Pre-eclampsia: Pathophysiology and clinical implications. BMJ 2019, 366, l2381. [Google Scholar] [CrossRef] [PubMed]
- Pober, J.S.; Sessa, W.C. Evolving functions of endothelial cells in inflammation. Nat. Rev. Immunol. 2007, 7, 803–815. [Google Scholar] [CrossRef] [PubMed]
- Opitz, B.; Hippenstiel, S.; Eitel, J.; Suttorp, N. Extra- and intracellular innate immune recognition in endothelial cells. Thromb. Haemost. 2007, 98, 319–326. [Google Scholar]
- Li, Y.X.; Wang, P.; Yang, X.; Wang, W.; Zhang, J.; He, Y.; Zhang, W.; Jing, T.; Wang, B.; Lin, R. SIRT1 inhibits inflammatory response partly through regulation of NLRP3 inflammasome in vascular endothelial cells. Mol. Immunol. 2016, 77, 148–156. [Google Scholar] [CrossRef]
- Chen, Y.; Pitzer, A.L.; Li, X.; Li, P.L.; Wang, L.; Zhang, Y. Instigation of endothelial Nlrp3 inflammasome by adipokine visfatin promotes inter-endothelial junction disruption: Role of HMGB1. J. Cell Mol. Med. 2015, 19, 2715–2727. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wang, L.; Pitzer, A.L.; Li, X.; Li, P.L.; Zhang, Y. Contribution of redox-dependent activation of endothelial Nlrp3 inflammasomes to hyperglycemia-induced endothelial dysfunction. J. Mol. Med. 2016, 94, 1335–1347. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, X.; Pitzer, A.L.; Chen, Y.; Wang, L.; Li, P.L. Coronary endothelial dysfunction induced by nucleotide oligomerization domain-like receptor protein with pyrin domain containing 3 inflammasome activation during hypercholesterolemia: Beyond inflammation. Antioxid. Redox Signal 2015, 22, 1084–1096. [Google Scholar] [CrossRef]
- Weel, I.C.; Romão-Veiga, M.; Matias, M.L.; Fioratti, E.G.; Peraçoli, J.C.; Borges, V.T.; Araujo Jr, J.P.; Peraçoli, M.T. Increased expression of NLRP3 inflammasome in placentas from pregnant women with severe preeclampsia. J. Reprod. Immunol. 2017, 123, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Li, S.; Liu, Z.; Jiang, S.; Wang, J.; Guo, M.; Zhao, X.; Song, W.; Liu, S. The NLRP3 rs10754558 polymorphism is a risk factor for preeclampsia in a Chinese Han population. J. Matern. Fetal Neonatal Med. 2019, 32, 1792–1799. [Google Scholar] [CrossRef] [PubMed]
- Pontillo, A.; Reis, E.C.; Bricher, P.N.; Vianna, P.; Diniz, S.; Fernandes, K.S.; Chies, J.A.; Sandrim, V. NLRP1 L155H Polymorphism is a Risk Factor for Preeclampsia Development. Am. J. Reprod. Immunol. 2015, 73, 577–581. [Google Scholar] [CrossRef]
- Gimbrone, M.A.; García-cardeña, G. Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Circ. Res. 2016, 118, 620–636. [Google Scholar] [CrossRef]
- Xu, S.; Ilyas, I.; Little, P.J.; Li, H.; Kamato, D.; Zheng, X.; Luo, S.; Li, Z.; Liu, P.; Han, J.; et al. Endothelial dysfunction in atherosclerotic cardiovascular diseases and beyond: From mechanism to pharmacotherapies. Pharmacol. Rev. 2021, 73, 924–967. [Google Scholar] [CrossRef] [PubMed]
- Chesley, L.C.; Talledo, E.; Bohler, C.S.; Zuspan, F.P. Vascular reactivity to angiotensin II and norepinephrine in pregnant and nonpregnant women. Am. J. Obstet. Gynecol. 1965, 91, 837–842. [Google Scholar] [CrossRef]
- Duvekot, J.J.; Cheriex, E.C.; Pieters, F.A.; Menheere, P.P.; Peeters, L.H. Early pregnancy changes in hemodynamics and volume homeostasis are consecutive adjustments triggered by a primary fall in systemic vascular tone. Am. J. Obstet. Gynecol. 1993, 169, 382–392. [Google Scholar] [CrossRef]
- Amaral, L.M.; Wallace, K.; Owens, M.; LaMarca, B. Pathophysiology and Current Clinical Management of Preeclampsia. Curr. Hypertens. Rep. 2017, 19, 19–21. [Google Scholar] [CrossRef] [PubMed]
- Rana, S.; Lemoine, E.; Granger, J.P.; Karumanchi, A. Preeclampsia: Pathophysiology, Challenges, and Perspectives. Circ. Res. 2019, 124, 1094–1112. [Google Scholar] [CrossRef]
- Redman, C.W.G. Pre-eclampsia and the placenta. Placenta 1991, 12, 301–308. [Google Scholar] [CrossRef]
- Yoshida, A.; Nakao, S.; Kobayashi, M.; Kobayashi, H. Flow-mediated vasodilation and plasma fibronectin levels in preeclampsia. Hypertension 2000, 36, 400–404. [Google Scholar] [CrossRef]
- Maynard, S.E.; Min, J.; Merchan, J.; Lim, K.; Li, J.; Mondal, S. Excess placental soluble fms-like tyrosine kinase 1 (sFlt1) may contribute to endothelial dysfunction hypertension and proteinuria in preeclampsia. J. Clin. Investig. 2003, 111, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Rinehart, B.K.; Terrone, D.A.; Lagoo-Deenadayalan, S.; Barber, W.H.; Hale, E.A.; Martin, J.N., Jr.; Bennett, W.A. Expression of the placental cytokines tumor necrosis factor-alpha, interleukin 1beta, and interleukin 10 is increased in preeclampsia. Am. J. Obstet. Gynecol. 1999, 181, 915–920. [Google Scholar] [CrossRef]
- Myers, J.; Mires, G.; Macleod, M.; Baker, P. In preeclampsia, the circulating factors capable of altering in vitro endothelial function precede clinical disease. Hypertension 2005, 45, 258–263. [Google Scholar] [CrossRef]
- Roberts, J. Endothelial Dysfunction in Preeclampsia. Semin. Reprod. Endocrinol. 1998, 16, 5–15. [Google Scholar] [CrossRef]
- Akhilesh, M.; Mahalingam, V.; Nalliah, S.; Ali, R.M.; Ganesalingam, M.; Haleagrahara, N. Hypoxia-inducible factor-1α as a predictive marker in pre-eclampsia. Biomed. Rep. 2013, 1, 257–258. [Google Scholar] [CrossRef] [PubMed]
- Rana, S.; Karumanchi, S.A.; Levine, R.J.; Venkatesha, S.; Rauh-Hain, J.A.; Tamez, H.; Thadhani, R. Sequential changes in antiangiogenic factors in early pregnancy and risk of developing preeclampsia. Hypertension 2007, 50, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Romero, R.; Nien, J.K.; Espinoza, J.; Todem, D.; Fu, W.; Chung, W.; Kusanovic, J.P.; Gotsch, F.; Erez, O.; Mazaki-Tovi, S.; et al. A longitudinal study of angiogenic (placental growth factor) and anti-angiogenic (soluble endoglin and soluble vascular endothelial growth factor receptor-1) factors in normal pregnancy and patients destined to develop preeclampsia and deliver a small for gestational age neonate. J. Matern. Fetal Neonatal Med. 2008, 21, 9–23. [Google Scholar] [PubMed]
- Cakmak, M.; Yilmaz, H.; Bağlar, E.; Darcin, T.; Inan, O.; Aktas, A.; Celik, H.T.; Ozdemir, O.; Atalay, C.R.; Akcay, A. Serum levels of endocan correlate with the presence and severity of pre-eclampsia. Clin. Exp. Hypertens. 2016, 38, 137–142. [Google Scholar] [CrossRef]
- Pinheiro, M.B.; Gomes, K.B.; Ronda, C.R.S.C.; Guimarães, G.G.; Freitas, L.G.; Teixeira-Carvalho, A.; Martins-Filho, O.A.; Dusse, L.M. Severe preeclampsia: Association of genes polymorphisms and maternal cytokines production in Brazilian population. Cytokine 2014, 71, 232–237. [Google Scholar] [CrossRef]
- Ramma, W.; Buhimschi, I.A.; Zhao, G.; Dulay, A.T.; Nayeri, U.A.; Buhimschi, C.S.; Ahmed, A. The elevation in circulating anti-angiogenic factors is independent of markers of neutrophil activation in preeclampsia. Angiogenesis 2012, 15, 333–340. [Google Scholar] [CrossRef]
- Siljee, J.E.; Wortelboer, E.J.; Koster, M.P.H.; Imholz, S.; Rodenburg, W.; Visser, G.H.A.; de Vries, A.; Schielen, P.C.J.I.; Pennings, J.L.A. Identification of interleukin-1 beta, but no other inflammatory proteins, as an early onset pre-eclampsia biomarker in first-trimester serum by bead-based multiplexed immunoassays. Prenat. Diagn. 2013, 33, 1183–1188. [Google Scholar] [CrossRef]
- Brennan, L.J.; Morton, J.S.; Davidge, S.T. Vascular Dysfunction in Preeclampsia. Microcirculation 2014, 21, 4–14. [Google Scholar] [CrossRef]
- Ives, C.W.; Sinkey, R.; Rajapreyar, I.; Tita, A.T.N.; Oparil, S. Preeclampsia-Pathophysiology, and Clinical Presentations: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2020, 76, 1690–1702. [Google Scholar] [CrossRef]
- Shah, D.A.; Khalil, R.A. Bioactive factors in uteroplacental and systemic circulation link placental ischemia to generalized vascular dysfunction in hypertensive pregnancy and preeclampsia. Biochem. Pharmacol. 2015, 95, 211–226. [Google Scholar] [CrossRef] [PubMed]
- Socha, M.W.; Malinowski, B.; Puk, O.; Dubiel, M.; Wiciński, M. The NLRP3 Inflammasome Role in the Pathogenesis of Pregnancy Induced Hypertension and Preeclampsia. Cells 2020, 9, 1642. [Google Scholar] [CrossRef]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef]
- Xie, F.; Hu, Y.; Turvey, S.E.; Magee, L.A.; Brunham, R.M.; Choi, K.C.; Krajden, M.; Leung, P.C.K.; Money, D.M.; Patrick, D.M.; et al. Toll-like receptors 2 and 4 and the cryopyrin inflammasome in normal pregnancy and pre-eclampsia. BJOG 2010, 117, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Matias, M.L.; Romão, M.; Weel, I.C.; Ribeiro, V.R.; Nunes, P.R.; Borges, V.T.M.; Araújo, J.P., Jr.; Peraçoli., J.C.; de Oliveira, L.; Peraçoli, M.T. Endogenous and uric acid-induced activation of NLRP3 inflammasome in pregnant women with preeclampsia. PLoS ONE 2015, 10, e0129095. [Google Scholar] [CrossRef] [PubMed]
- Mulla, M.J.; Myrtolli, K.; Potter, J.; Boeras, C.; Kavathas, P.B.; Sfakianaki, A.K.; Tadesse, S.; Norwitz, E.R.; Guller, S.; Abrahams, V.M. Uric acid induces trophoblast IL-1beta production via the inflammasome: Implications for the pathogenesis of preeclampsia. Am. J. Reprod. Immunol. 2011, 65, 542–548. [Google Scholar] [CrossRef] [PubMed]
- Shirasuna, K.; Usui, F.; Karasawa, T.; Kimura, H.; Kawashima, A.; Mizukami, H.; Ohkuchi, A.; Nishimura, S.; Sagara, J.; Noda, T.; et al. Nanosilica-induced placental inflammation and pregnancy complications: Different roles of the inflammasome components NLRP3 and ASC. Nanotoxicology 2015, 9, 554–567. [Google Scholar] [CrossRef]
- Tamura, K.; Ishikawa, G.; Yoshie, M.; Ohneda, W.; Nakai, A.; Takeshita, T.; Tachikawa, E. Glibenclamide inhibits NLRP3 inflammasome-mediated IL-1beta secretion in human trophoblasts. J. Pharmacol. Sci. 2017, 135, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Brien, M.E.; Duval, J.C.; Palacios, J.; Boufaied, I.; Hudon-Thibeault, A.-A.; Nadeau-Valle’e, M.; Vaillancourt, C.; Sibley, C.P.; Abrahams, V.M.; Jones, R.L.; et al. Uric acid crystals induce placental inflammation and alter trophoblast function via an IL-1-dependent pathway: Implications for fetal growth restriction. J. Immunol. 2017, 198, 443–451. [Google Scholar] [CrossRef]
- Shirasuna, K.; Karasawa, F.T.; Usui, M.; Kobayashi, T.; Komada, H.; Kimura, A.; Kawashima, A.; Ohkuchi, A.; Taniguchi, S.; Takahashi, M. NLRP3 deficiency improves angiotensin II-induced hypertension but not fetal growth restriction during pregnancy. Endocrinology 2015, 156, 4281–4292. [Google Scholar] [CrossRef]
- Seno, K.; Sase, S.; Ozeki, A.; Takahashi, H.; Ohkuchi, A.; Suzuki, H.; Matsubara, S.; Iwata, H.; Kuwayama, T.; Shirasuna, K. Advanced glycation end products regulate interleukin-1b production in human placenta. J. Reprod. Dev. 2017, 63, 401–408. [Google Scholar] [CrossRef]
- Gomez-Lopez, N.; Motomura, K.; Miller, D.; Garcia-Flores, V.; Galaz, J.; Romero, R. Inflammasomes: Their Role in Normal and Complicated Pregnancies. J. Immunol. 2019, 203, 2757–2769. [Google Scholar] [CrossRef]
- Sutterwala, F.S.; Haasken, S.; Cassel, S.L. Mechanism of NLRP3 inflammasome activation. Ann. N. Y. Acad. Sci. 2014, 1319, 82–95. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Zeng, X.; Li, X.; Mehta, J.L.; Wang, X. Role of NLRP3 inflammasome in the pathogenesis of cardiovascular diseases. Basic Res. Cardiol. 2017, 113, 5. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.; Nakashima, A.; Huber, W.J.; Davis, S.; Banerjee, S.; Huang, Z.; Saito, S.; Sadovsky, Y.; Sharma, S. Pyroptosis is a critical inflammatory pathway in the placenta from early-onset preeclampsia and in human trophoblasts exposed to hypoxia and endoplasmic reticulum stressors. Cell Death Dis. 2019, 10, 927. [Google Scholar] [CrossRef] [PubMed]
- Keyel, P.A. How is inflammation initiated? Individual influences of IL-1, IL-18, and HMGB1. Cytokine 2014, 69, 136–145. [Google Scholar] [CrossRef]
- Shi, J.; Gao, W.; Shao, F. Pyroptosis: Gasdermin-mediated programmed necrotic cell death. Trends Biochem. Sci. 2017, 42, 245–254. [Google Scholar] [CrossRef]
- Goulopoulou, S.; Davidge, S.T. Molecular mechanisms of maternal vascular dysfunction in preeclampsia. Trends Mol. Med. 2015, 21, 88–97. [Google Scholar] [CrossRef]
- Komarova, Y.A.; Kruse, K.; Mehta, D.; Malik, A.B. Protein interactions at endothelial junctions and signaling mechanisms regulating endothelial permeability. Circ. Res. 2017, 120, 179–206. [Google Scholar] [CrossRef]
- Radeva, M.Y.; Waschke, J. Mind the gap: Mechanisms regulating the endothelial barrier. Acta Physiol. 2018, 222, e12860. [Google Scholar] [CrossRef]
- Gong, T.; Liu, L.; Jiang, W.; Zhou, R. DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat. Rev. Immunol. 2020, 20, 95–112. [Google Scholar] [CrossRef]
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef]
- Chen, Z.; Martin, M.; Li, Z.; Shyy, J.Y. Endothelial dysfunction: The role of sterol regulatory element-binding protein-induced NOD-like receptor Family pyrin domain-containing protein 3 inflammasome in atherosclerosis. Curr. Opin. Lipido. 2014, 25, 339–349. [Google Scholar] [CrossRef]
- Wang, J.; Williams, J.C.; Davis, B.K.; Jacobson, K.; Doerschuk, C.M.; Ting, J.P.; Mackman, N. Monocytic microparticles activate endothelial cells in an IL- 1beta-dependent manner. Blood 2011, 118, 2366–2374. [Google Scholar] [CrossRef]
- Grebe, A.; Hoss, F.; Latz, E. NLRP3 inflammasome and the IL-1 pathway in atherosclerosis. Circ. Res. 2018, 122, 1722–1740. [Google Scholar] [CrossRef]
- He, Y.; Hara, H.; Nunez, G. Mechanism and regulation of NLRP3 inflammasome activation. Trends Biochem. Sci. 2016, 41, 1012–1021. [Google Scholar] [CrossRef]
- Abais, J.M.; Xia, M.; Zhang, Y.; Boini, K.M.; Li, P.L. Redox regulation of NLRP3 inflammasomes: ROS as trigger or effector? Antioxid. Redox Signal 2015, 22, 1111–1129. [Google Scholar] [CrossRef]
- Dunn, D.; Alvarez, L.A.; Zhang, X.; Soldati, T. Reactive oxygen species, and mitochondria: A nexus of cellular homeostasis. Redox Biol. 2015, 6, 472–485. [Google Scholar] [CrossRef] [PubMed]
- Shihata, W.A.; Michell, D.L.; Andrews, K.L.; Chin-Dusting, J.P. Caveolae: A role in endothelial inflammation and mechanotransduction? Front. Physiol. 2016, 7, 628. [Google Scholar] [CrossRef] [PubMed]
- Lamkanfi, M.; Mueller, J.L.; Vitari, A.C.; Misaghi, S.; Fedorova, A.; Deshayes, K.; Lee, W.P.; Hoffman, H.M.; Dixit, V.M. Glyburide inhibits the Cryopyrin/Nalp3 inflammasome. J. Cell Biol. 2009, 187, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Perregaux, D.G.; McNiff, P.; Laliberte, R. Hawryluk, N.; Peurano, H.; Stam Eggler, J.; Griffiths, R.; Dombroski, M.A.; Gabel, C.A. Identification and characterization of a novel class of interleukin-1 post-translational processing inhibitors. J. Pharmacol. Exp. Ther. 2001, 299, 187–197. [Google Scholar]
- Marchetti, C.; Toldo, S.; Chojnacki, J.; Mezzaroma, E.; Liu, K.; Salloum, F.N.; Nordio, A.; Carbone, S.; Mauro, A.G.; Das, A.; et al. Pharmacologic inhibition of the NLRP3 inflammasome preserves cardiac function after ischemic and non-ischemic injury in the mouse. J. Cardiovasc. Pharmacol. 2015, 66, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, C.; Chojnacki, J.; Toldo, S.; Mezzaroma, E.; Tranchida, N.; Rose, S.W.; Federici, M.; Van Tassell, B.W.; Zhang, S.; Abbate, A. A novel pharmacologic inhibitor of the NLRP3 inflammasome limits myocardial injury following ischemia-reperfusion in the mouse. J. Cardiovasc. Pharmacol. 2014, 63, 316–322. [Google Scholar] [CrossRef] [PubMed]
- Hamon, Y.; Luciani, M.F.; Becq, F.; Verrier, B.; Rubartelli, A.; Chimini, G. Interleukin-1beta secretion is impaired by inhibitors of the Atp binding cassette transporter, ABC1. Blood 1997, 90, 2911–2915. [Google Scholar] [CrossRef] [PubMed]
- Lottaz, D.; Beleznay, Z.; Bickel, M. Inhibition of ATP-binding cassette transporter downregulates interleukin-1beta-mediated autocrine activation of human dermal fibroblasts. J. Investig. Dermatol. 2001, 117, 871–876. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mangan, M.S.; Olhava, E.J.; Roush, W.R.; Seidel, H.M.; Glick, G.D.; Latz, E. Targeting the NLRP3 inflammasome in inflammatory diseases. Nat. Rev. Drug Discov. 2018, 17, 588–606. [Google Scholar] [CrossRef] [PubMed]
- Kuwar, R.; Rolfe, A.; Di, L.; Xu, H.; He, L.; Jiang, Y.; Zhang, S.; Sun, D. A novel small molecular NLRP3 inflammasome inhibitor alleviates neuroinflammatory response following traumatic brain injury. J. Neuroinflammation 2019, 16, 81. [Google Scholar] [CrossRef] [PubMed]
- Fulp, J.; He, L.; Toldo, S.; Jiang, Y.; Boice, A.; Guo, C.; Li, X.; Rolfe, A.; Sun, D.; Abbate, A.; et al. Structural insights of benzenesulfonamide analogues as NLRP3 inflammasome inhibitors: Design, synthesis, and biological characterization. J. Med. Chem. 2018, 61, 5412–5423. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Guo, W.; Wu, J.; Luo, Q.; Tao, F.; Gu, Y.; Shen, Y.; Li, J.; Tan, R.; Xu, Q.; et al. A novel benzo [d] imidazole derivate prevents the development of dextran sulfate sodium-induced murine experimental colitis via inhibition of NLRP3 inflammasome. Biochem. Pharmacol. 2013, 85, 1504–1512. [Google Scholar] [CrossRef]
- Coll, R.C.; Robertson, A.A.; Chae, J.J.; Higgins, S.C.; Muñoz-Planillo, R.; Inserra, M.C.; Vetter, I.; Dungan, L.S.; Monks, B.G.; Stutz, A.; et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat. Med. 2015, 21, 248–255. [Google Scholar] [CrossRef]
- Coll, R.C.; Hill, J.R.; Day, C.J.; Zamoshnikova, A.; Boucher, D.; Massey, N.L.; Chitty, J.L.; Fraser, J.A.; Jennings, M.P.; Robertson, A.A.B.; et al. MCC950 directly targets the NLRP3 ATP-hydrolysis motif for inflammasome inhibition. Nat. Chem. Biol. 2019, 15, 556–559. [Google Scholar] [CrossRef]
- Ferreira, N.S.; Bruder-Nascimento, T.; Pereira, C.A.; Zanotto, C.Z.; Prado, D.S.; Silva, J.F.; Rassi, D.M.; Foss-Freitas, M.C.; Alves-Filho, J.C.; Carlos, D.; et al. NLRP3 Inflammasome and Mineralocorticoid Receptors Are Associated with Vascular Dysfunction in Type 2 Diabetes Mellitus. Cells 2019, 8, 1595. [Google Scholar] [CrossRef]
- Cornelius, D.C.; Travis, O.K.; Tramel, R.W.; Borges-Rodriguez, M.; Baik, C.H.; Greer, M.; Giachelli, C.A.; Tardo, G.A.; Williams, J.M. NLRP3 inflammasome inhibition attenuates sepsis induced platelet activation and prevents multi-organ injury in cecal-ligation puncture. PLoS ONE 2020, 15, e0234039. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Meng, J.; Yan, J.; Shang, F.; Zhang, T.; Lv, D.; Li, C.; Yang, X.; Luo, S. Role of the nucleotide-binding domain-like receptor protein 3 inflammasome in the endothelial dysfunction of early sepsis. Inflammation 2020, 43, 1561–1571. [Google Scholar] [CrossRef]
- Primiano, M.J.; Lefker, B.A.; Bowman, M.R.; Bree, A.G.; Hubeau, C.; Bonin, P.D.; Mangan, M.; Dower, K.; Monks, B.G.; Cushing, L.; et al. Efficacy and pharmacology of the NLRP3 inflammasome inhibitor CP-456,773 (CRID3) in murine models of dermal and pulmonary inflammation. J. Immunol. 2016, 197, 2421–2433. [Google Scholar] [CrossRef]
- Ma, T.; Thiagarajah, J.R.; Yang, H.; Sonawane, N.D.; Folli, C.; Galietta, L.J.; Verkman, A.S. Thiazolidinone CFTR inhibitor identified by high-throughput screening blocks cholera toxin–induced intestinal fluid secretion. J. Clin. Investig. 2002, 110, 1651–1658. [Google Scholar] [CrossRef]
- Jiang, H.; He, H.; Chen, Y.; Huang, W.; Cheng, J.; Ye, J.; Wang, A.; Tao, J.; Wang, C.; Liu, Q.; et al. Identification of a selective and direct NLRP3 inhibitor to treat inflammatory disorders. J. Exp. Med. 2017, 214, 3219–3238. [Google Scholar] [CrossRef]
- Zahid, A.; Li, B.; Kombe, A.J.; Jin, T.; Tao, J. Pharmacological Inhibitors of the NLRP3 Inflammasome. Front. Immunol. 2019, 10, 2538. [Google Scholar] [CrossRef]
- Darakhshan, S.; Pour, A.B. Tranilast: A review of its therapeutic applications. Pharmacol. Res. 2015, 91, 15–28. [Google Scholar] [CrossRef]
- Konneh, M. Tranilast Kissei pharmaceutical. Idrugs 1998, 1, 141–146. [Google Scholar] [PubMed]
- Platten, M.; Ho, P.P.; Youssef, S.; Fontoura, P.; Garren, H.; Hur, E.M.; Gupta, R.; Lee, L.Y.; Kidd, B.A.; Robinson, W.H.; et al. Treatment of autoimmune neuroinflammation with a synthetic tryptophan metabolite. Science 2005, 310, 850–855. [Google Scholar] [CrossRef] [PubMed]
- Toldo, S.; Abbate, A. The NLRP3 inflammasome in acute myocardial infarction. Nat. Rev. Cardiol. 2018, 15, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, C.; Swartzwelter, B.; Gamboni, F.; Neff, C.P.; Richter, K.; Azam, T.; Carta, S.; Tengesdal, I.; Nemkov, T.; D′Alessandro, A.; et al. OLT1177, a β-sulfonyl nitrile compound, safe in humans, inhibits the NLRP3 inflammasome and reverses the metabolic cost of inflammation. Proc. Natl. Acad. Sci. USA 2018, 115, E1530–E1539. [Google Scholar] [CrossRef] [PubMed]
- Kadota, S.; Basnet, P.; Ishii, E.; Tamura, T.; Namba, T. Antibacterial activity of trichorabdal A from Rabdosia trichocarpa against Helicobacter pylori. Zent. Für Bakteriol. 1997, 286, 63–67. [Google Scholar] [CrossRef]
- Kuo, L.-M.; Kuo, C.-Y.; Lin, C.-Y.; Hung, M.-F.; Shen, J.-J.; Hwang, T.-L. Intracellular glutathione depletion by oridonin leads to apoptosis in hepatic stellate cells. Molecules 2014, 19, 3327–3344. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Wu, L.; Tashiro, S.; Onodera, S.; Ikejima, T. A comparison of the signal pathways between the TNFα-and oridonin-induced Murine L929 fibrosarcoma cell death. Acta Med. Okayama 2005, 59, 261–270. [Google Scholar]
- Zhao, G.; Zhang, T.; Ma, X.; Jiang, K.; Wu, H.; Qiu, C.; Guo, M.; Deng, G. Oridonin attenuates the release of pro-inflammatory cytokines in lipopolysaccharide-induced RAW264. 7 cells and acute lung injury. Oncotarget 2017, 8, 68153. [Google Scholar] [CrossRef]
- He, H.; Jiang, H.; Chen, Y.; Ye, J.; Wang, A.; Wang, C.; Liu, Q.; Liang, G.; Deng, X.; Jiang, W.; et al. Oridonin is a covalent NLRP3 inhibitor with strong anti-inflammasome activity. Nat. Commun. 2018, 9, 2550. [Google Scholar] [CrossRef]
- Heinrich, M.; Robles, M.; West, J.E.; Ortiz de Montellano, B.R.; Rodriguez, E. Ethnopharmacology of Mexican asteraceae (compositae). Ann. Rev. Pharmacol. Toxicol. 1998, 38, 539–565. [Google Scholar] [CrossRef]
- Juliana, C.; Fernandes-Alnemri, T.; Wu, J.; Datta, P.; Solorzano, L.; Yu, J.-W.; Meng, R.; Quong, A.A.; Latz, E.; Scott, C.P.; et al. Anti-inflammatory compounds parthenolide and Bay 11–7082 are direct inhibitors of the inflammasome. J. Biol. Chem. 2010, 285, 9792–9802. [Google Scholar] [CrossRef]
- Guzman, M.L.; Rossi, R.M.; Neelakantan, S.; Li, X.; Corbett, C.A.; Hassane, D.C.; Becker, M.W.; Bennett, J.M.; Sullivan, E.; Lachowicz, J.L.; et al. An orally bioavailable parthenolide analog selectively eradicates acute myelogenous leukemia stem and progenitor cells. Blood 2007, 110, 4427–4435. [Google Scholar] [CrossRef]
- D’anneo, A.; Carlisi, D.; Lauricella, M.; Puleio, R.; Martinez, R.; Di Bella, S.; Di Marco, P.; Emanuele, S.; Di Fiore, R.; Guercio, A.; et al. Parthenolide generates reactive oxygen species and autophagy in MDA-MB231 cells. A soluble parthenolide analogue inhibits tumour growth and metastasis in a xenograft model of breast cancer. Cell Death Dis. 2013, 4, e891. [Google Scholar] [CrossRef] [PubMed]
- Wannamaker, W.; Davies, R.; Namchuk, M.; Pollard, J.; Ford, P.; Ku, G.; Decker, C.; Charifson, P.; Weber, P.; Germann, U.A.; et al. (S)-1-((S)-2-{[1-(4-Amino-3-chloro-phenyl)-methanoyl]-amino}-3,3-dimethyl-butanoyl)-pyrrolidine-2-carboxylic acid ((2R,3S)-2-ethoxy-5-oxo-tetrahydro-furan-3-yl)-amide (VX-765), an Orally Available Selective Interleukin (IL)-Converting Enzyme/Caspase-1 Inhi. J. Pharmacol. Exp. Ther. 2007, 321, 509–516. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, S.H.; Schipper, J.L.; Clark, A.C. The potential for caspases in drug discovery. Curr. Opin. Drug Discov. Devel. 2010, 13, 568–576. [Google Scholar]
- Boxer, M.B.; Shen, M.; Auld, D.S.; Wells, J.A.; Thomas, C.J. A Small Molecule Inhibitor of Caspase 1. Probe Reports from the NIH Molecular Libraries Program [Internet]; National Center for Biotechnology Information: Bethesda, MD, USA, 2010.
- Siegmund, B.; Zeitz, M. Pralnacasan (vertex pharmaceuticals). Idrugs 2003, 6, 154–158. [Google Scholar]
- Strand, V.; Sokolove, J. Randomized controlled trial design in rheumatoid arthritis: The past decade. Arthritis Res. Ther. 2009, 11, 1–11. [Google Scholar] [CrossRef]
- Fischer, U.; Schulzeosthoff, K. Apoptosis-based therapies and drug targets. Cell Death Differ. 2005, 12, 942–961. [Google Scholar] [CrossRef]
- Maroso, M.; Balosso, S.; Ravizza, T.; Liu, J.; Bianchi, M.E.; Vezzani, A. Interleukin-1 type 1 receptor/Toll-like receptor signalling in epilepsy: The importance of IL-1beta and high-mobility group box 1. J. Int. Med. 2011, 270, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Youm, Y.-H.; Nguyen, K.Y.; Grant, R.W.; Goldberg, E.L.; Bodogai, M.; Kim, D.; D′Agostino, D.; Planavsky, N.; Lupfer, C.; Kanneganti, T.D.; et al. The ketone metabolite β-hydroxybutyrate blocks NLRP3 inflammasome–mediated inflammatory disease. Nat. Med. 2015, 21, 263–269. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Drug | NLRP3-Specific | Direct Inhibition | Action |
|---|---|---|---|
| Glyburide [17,69,70,71,72,73,74,75] | Yes | No | Induces the closure of ATP-sensitive K+ channels; Raises the intracellular K+ concentration |
| 16673-34-0 [71,72] | Yes | No | Interferes with downstream events involved in NLRP3 conformational changes secondary to activation or binding to ASC |
| JC124 [76,77] | Yes | No | Blocks ASC aggregation, caspase-1 activation, and IL-1β secretion |
| FC11A-2 [78] | Yes | No | Repress IL-1β/18 release; induces autocleavage of procaspase-1, resulting in a reduced amount of activated caspase-1 |
| MCC950 [79,80,81,82,83,84] | Yes | Yes | Blocks the release of IL-1β induced by NLRP3 activators |
| CY-09 [85,86,87] | Yes | Yes | Blocks the ATP, monosodium urate (MSU), and nigericin-induced activation of caspase-1 and resultant release of IL-1β |
| Tranilast [80,88,89,90] | Yes | Yes | Impairs the endogenous NLRP3-ASC interaction |
| OLT1177 [91,92] | Yes | Yes | Binds with NLRP3 to block its ATPase activity |
| Oridonin [93,94,95,96,97] | Yes | Yes | Inhibits the NF-κB or MAPK activation and repress the release of inflammasome-independent proinflammatory cytokines release |
| Parthenolide [98,99,100,101] | No | No | Inhibits caspase-1 activation; Targets ATPase activity of NLRP3 |
| VX-740/VX-765 [102,103,104,105,106,107,108] | No | No | Block caspase-1 and resultant cleavage of pro-IL-1β/18 |
| Bay 11-7082 [87,99] | No | No | Prevents the organization of ASC pyroptosome |
| BHB [109] | No | No | Lowered the production of IL-1ß and IL-1; reduces the oligomerization and speck formation of ASC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nunes, P.R.; Mattioli, S.V.; Sandrim, V.C. NLRP3 Activation and Its Relationship to Endothelial Dysfunction and Oxidative Stress: Implications for Preeclampsia and Pharmacological Interventions. Cells 2021, 10, 2828. https://doi.org/10.3390/cells10112828
Nunes PR, Mattioli SV, Sandrim VC. NLRP3 Activation and Its Relationship to Endothelial Dysfunction and Oxidative Stress: Implications for Preeclampsia and Pharmacological Interventions. Cells. 2021; 10(11):2828. https://doi.org/10.3390/cells10112828
Chicago/Turabian StyleNunes, Priscila Rezeck, Sarah Viana Mattioli, and Valeria Cristina Sandrim. 2021. "NLRP3 Activation and Its Relationship to Endothelial Dysfunction and Oxidative Stress: Implications for Preeclampsia and Pharmacological Interventions" Cells 10, no. 11: 2828. https://doi.org/10.3390/cells10112828
APA StyleNunes, P. R., Mattioli, S. V., & Sandrim, V. C. (2021). NLRP3 Activation and Its Relationship to Endothelial Dysfunction and Oxidative Stress: Implications for Preeclampsia and Pharmacological Interventions. Cells, 10(11), 2828. https://doi.org/10.3390/cells10112828