
Towards Splicing Therapy for Lysosomal Storage Disorders: Methylxanthines and Luteolin Ameliorate Splicing Defects in Aspartylglucosaminuria and Classic Late Infantile Neuronal Ceroid Lipofuscinosis



Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Minigene Constructs and Transfection
2.3. Treatment of Patient Fibroblasts with Recombinant AGA or Splicing Modulating Compounds
2.4. Antibodies
2.5. Enzyme Activity Measurements
2.6. Quantitative Real-Time PCR, Standard PCR and Sequencing of PCR Products
2.7. Immunofluorescence
2.8. SRSF1 and SRSF2 Overexpression
2.9. Statistical Analysis
3. Results
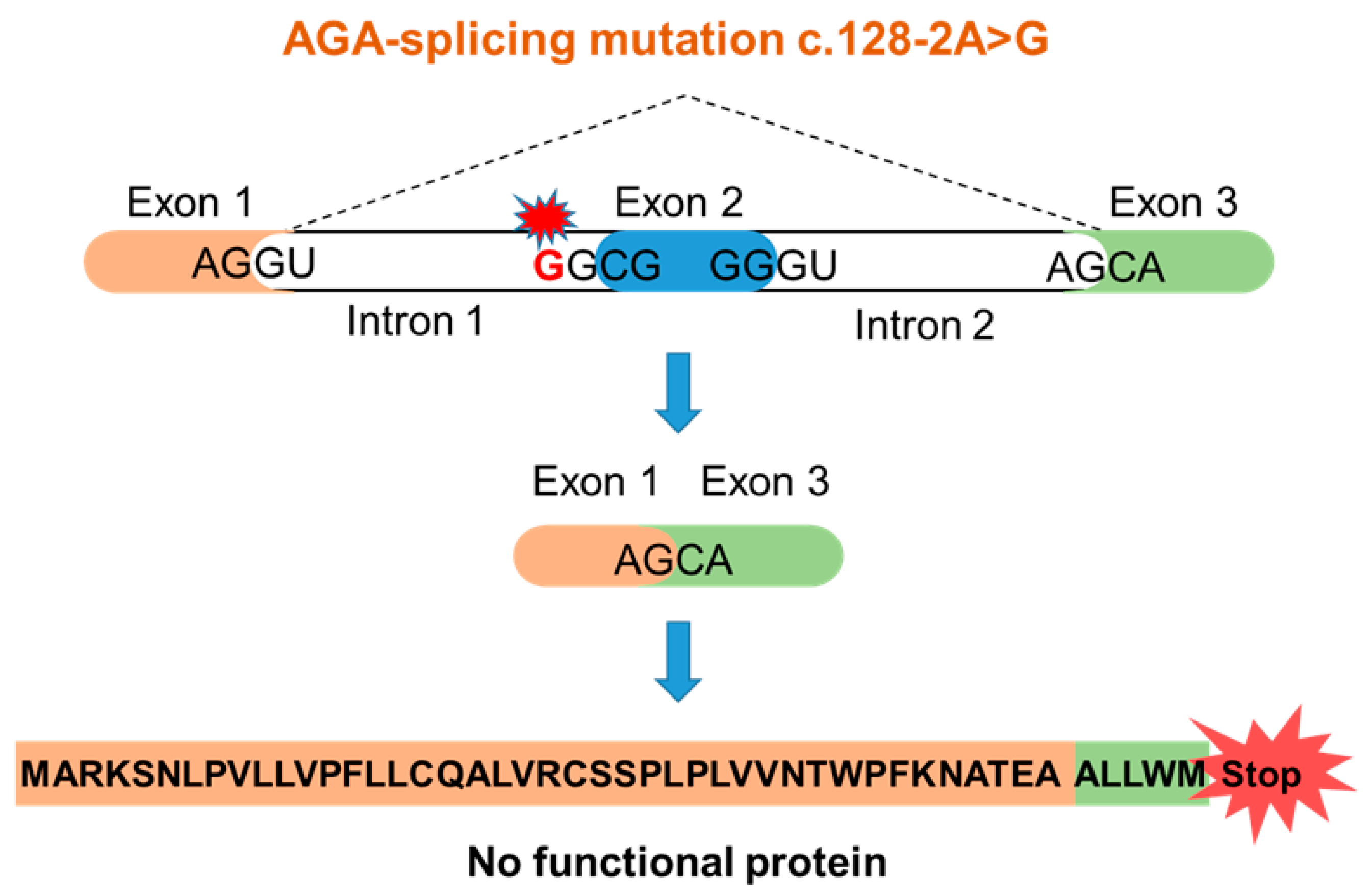
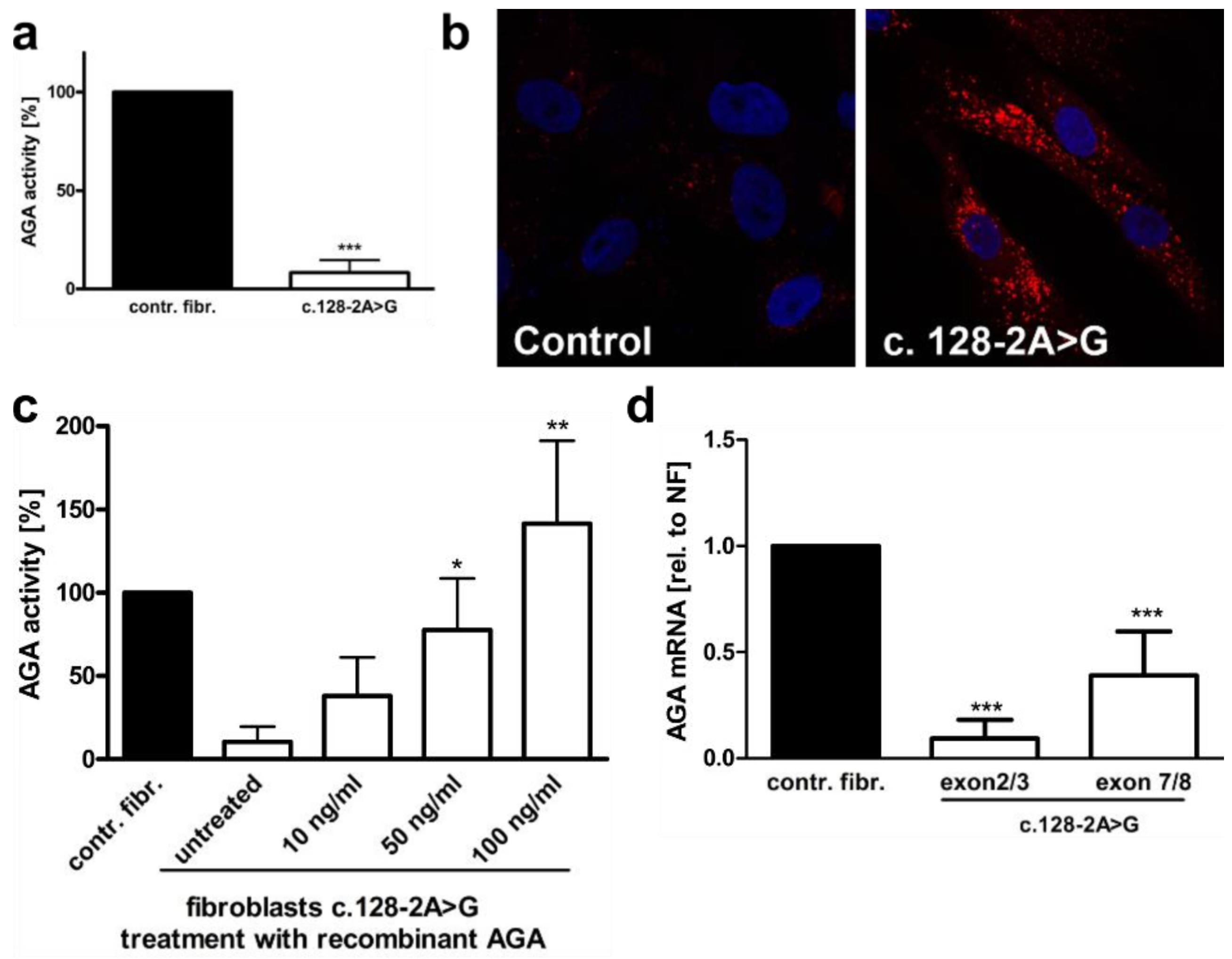
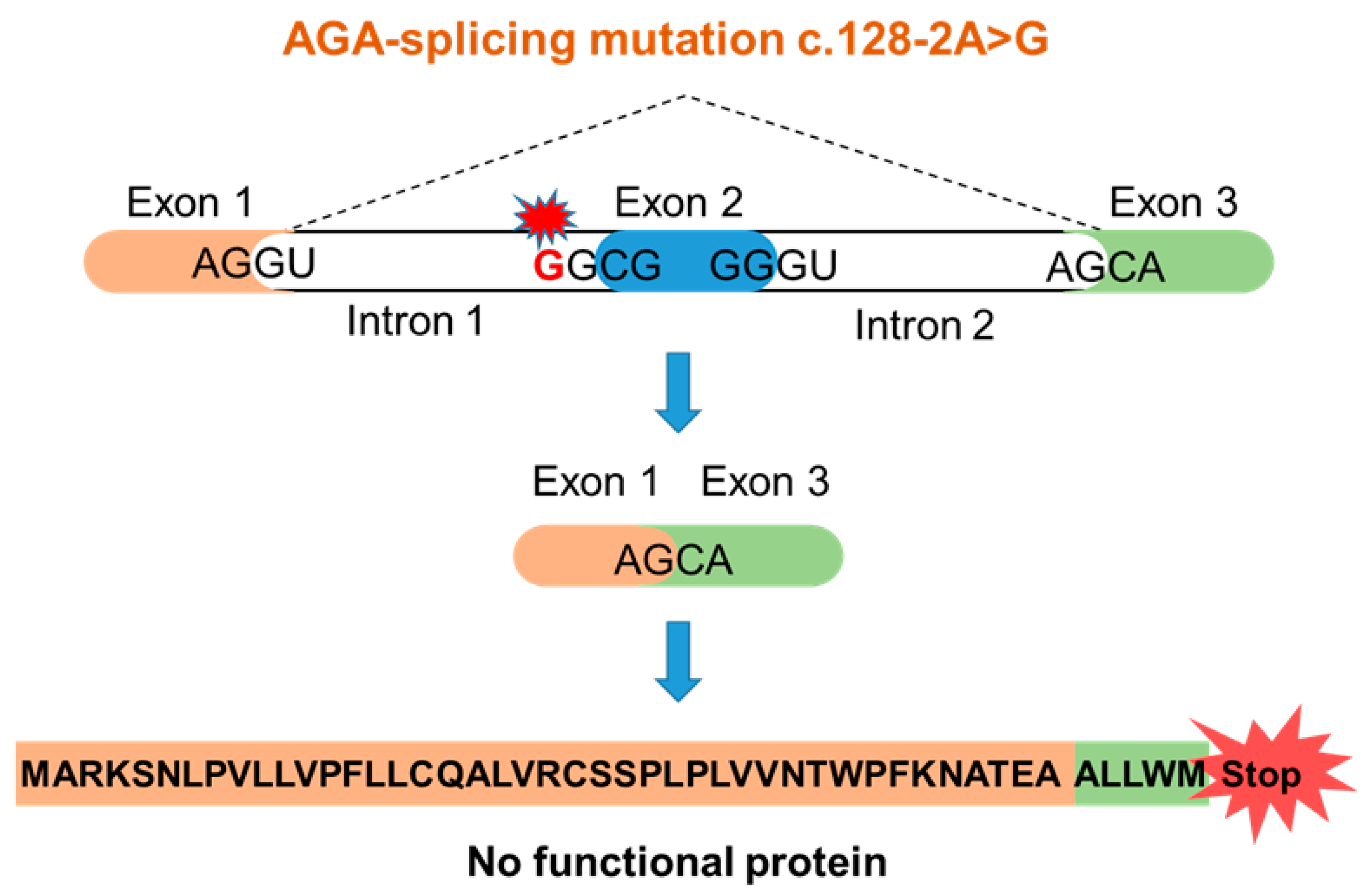
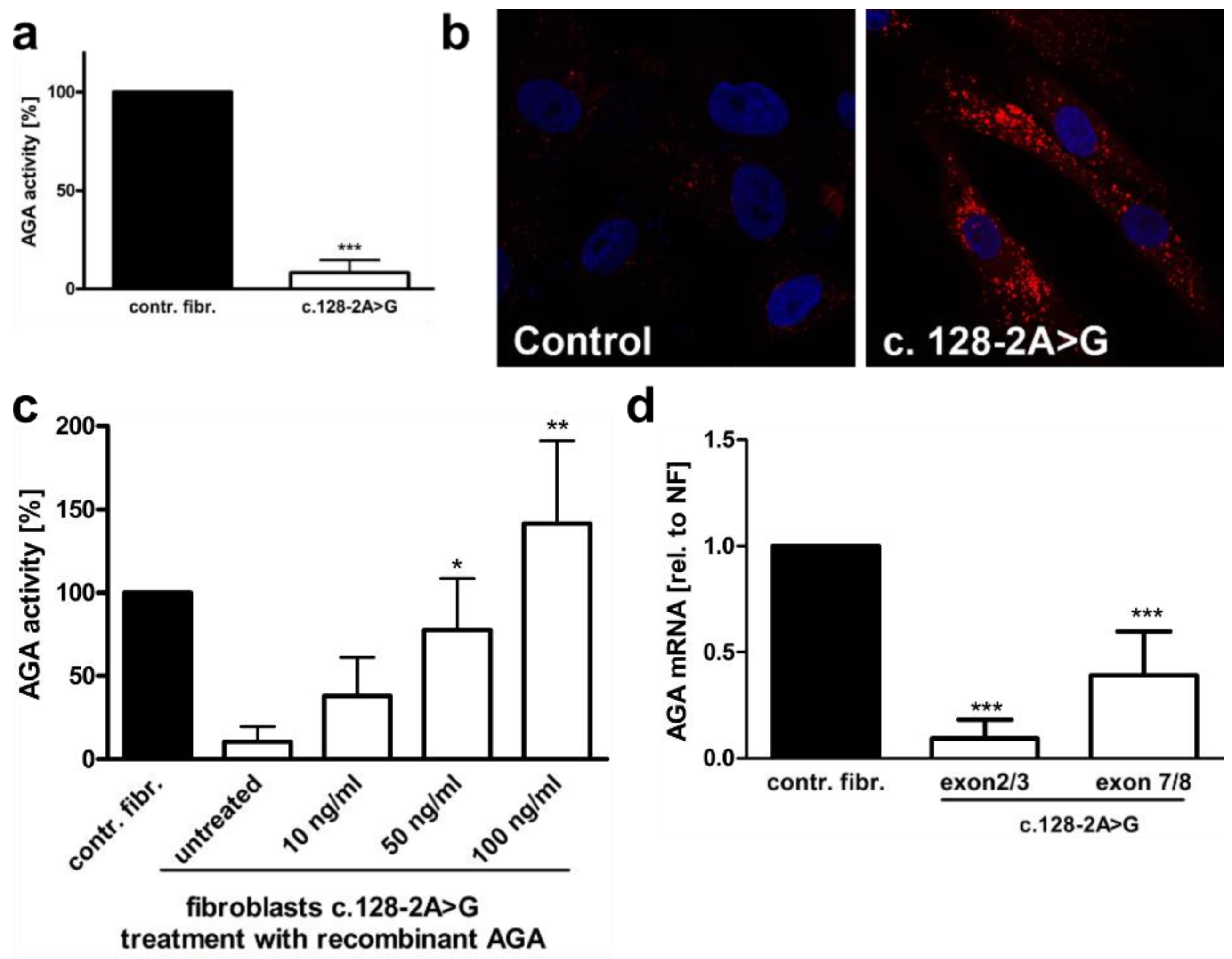
3.1. An AGU Patient with a Homozygous Splice Acceptor Site Mutation
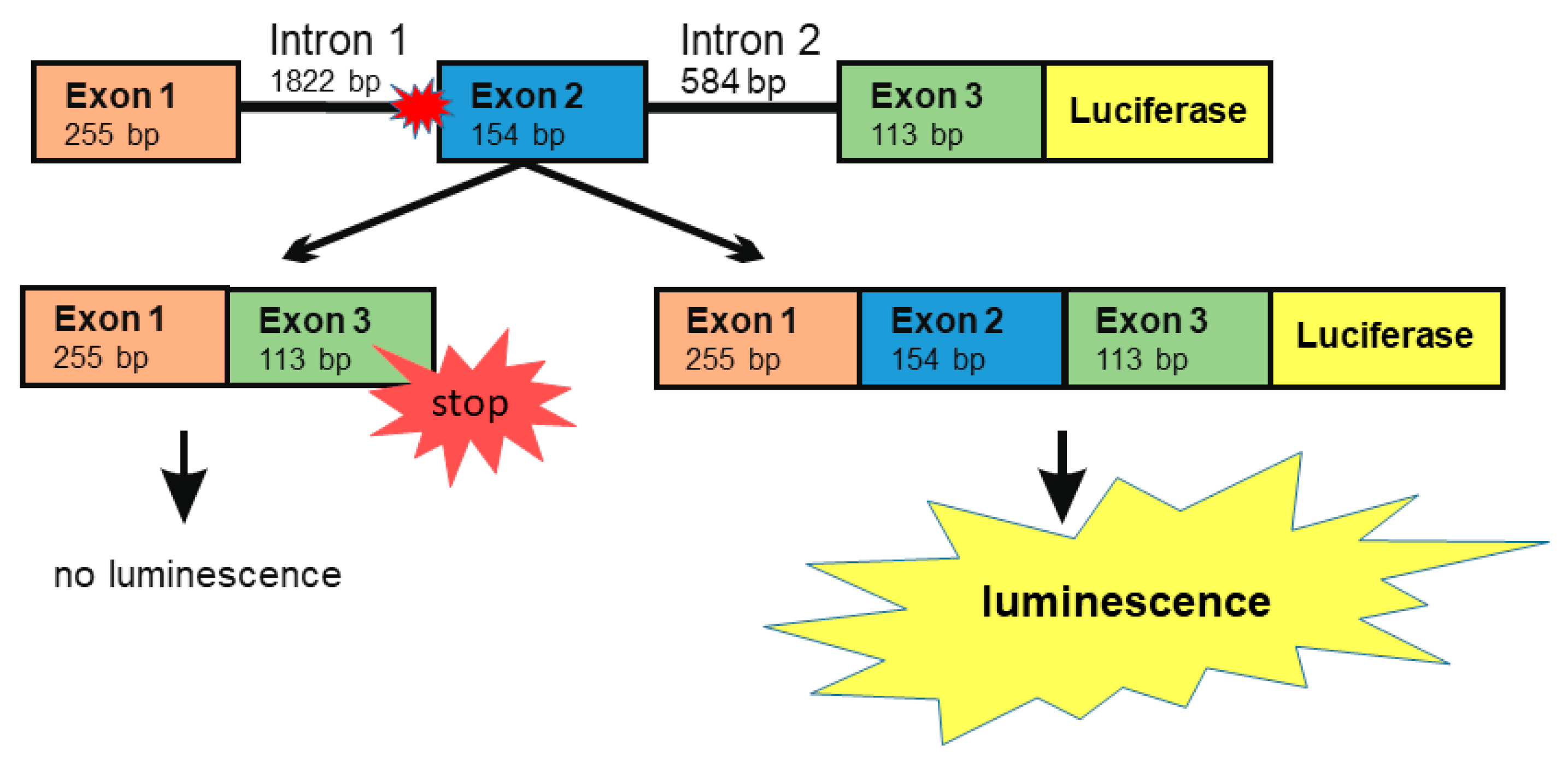
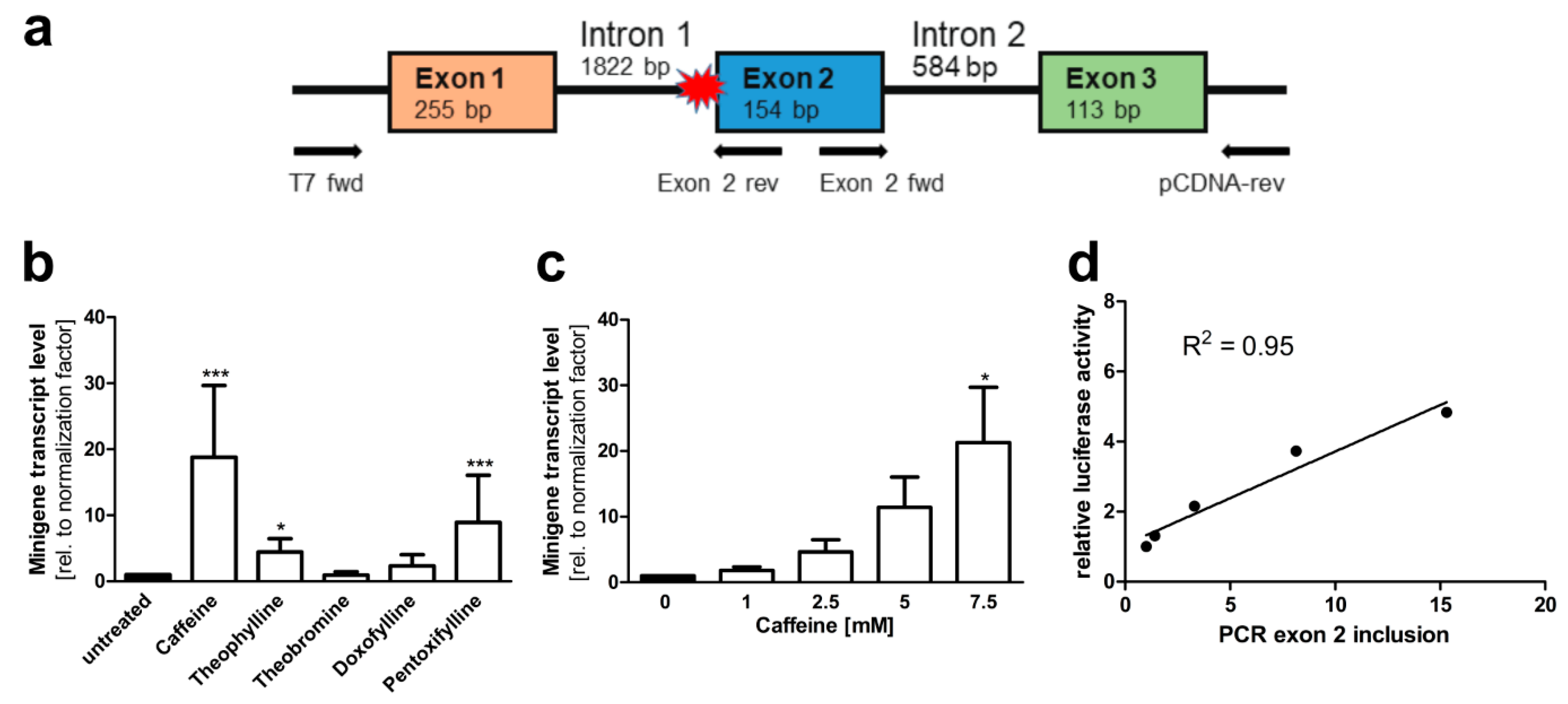
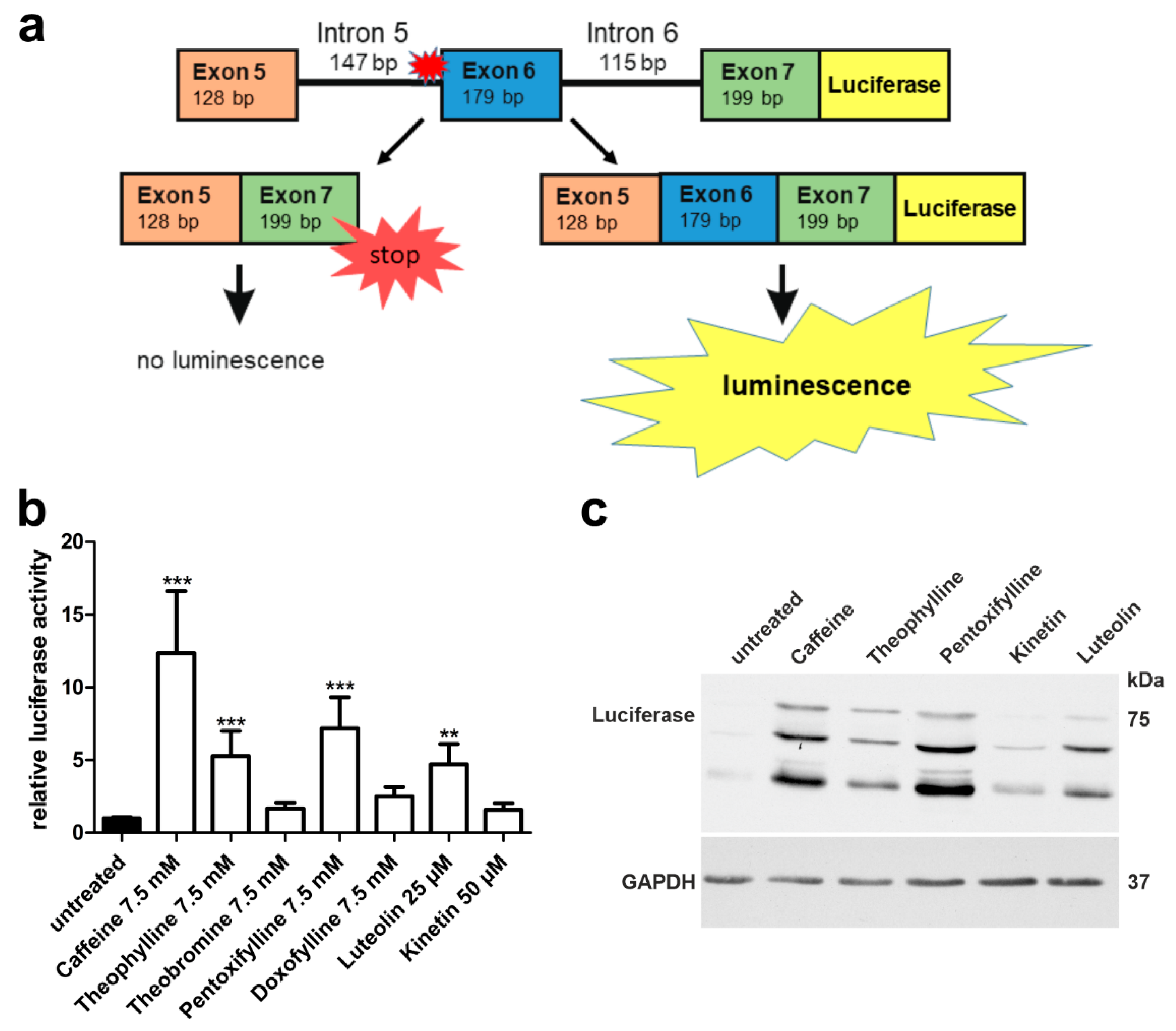
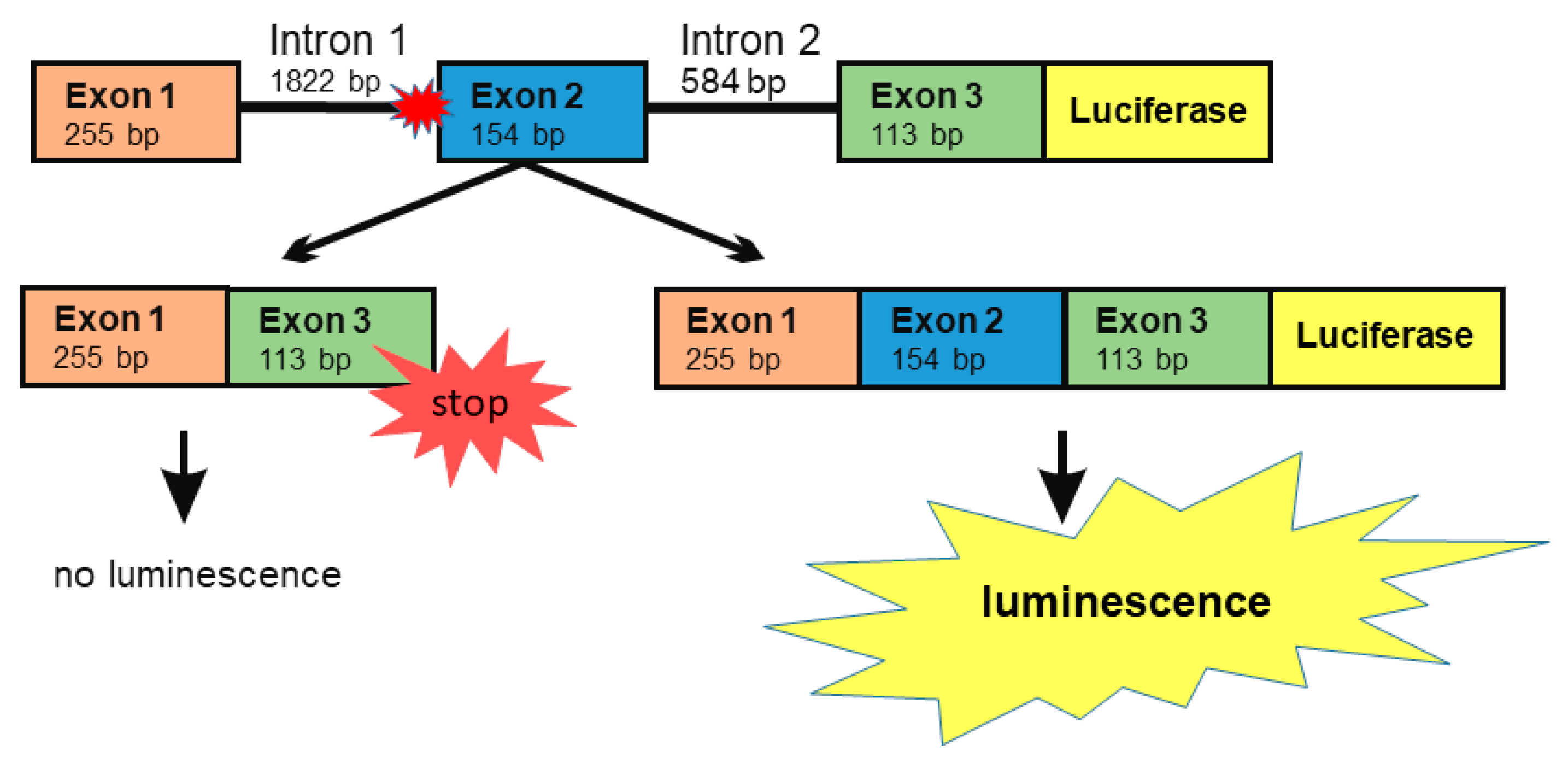
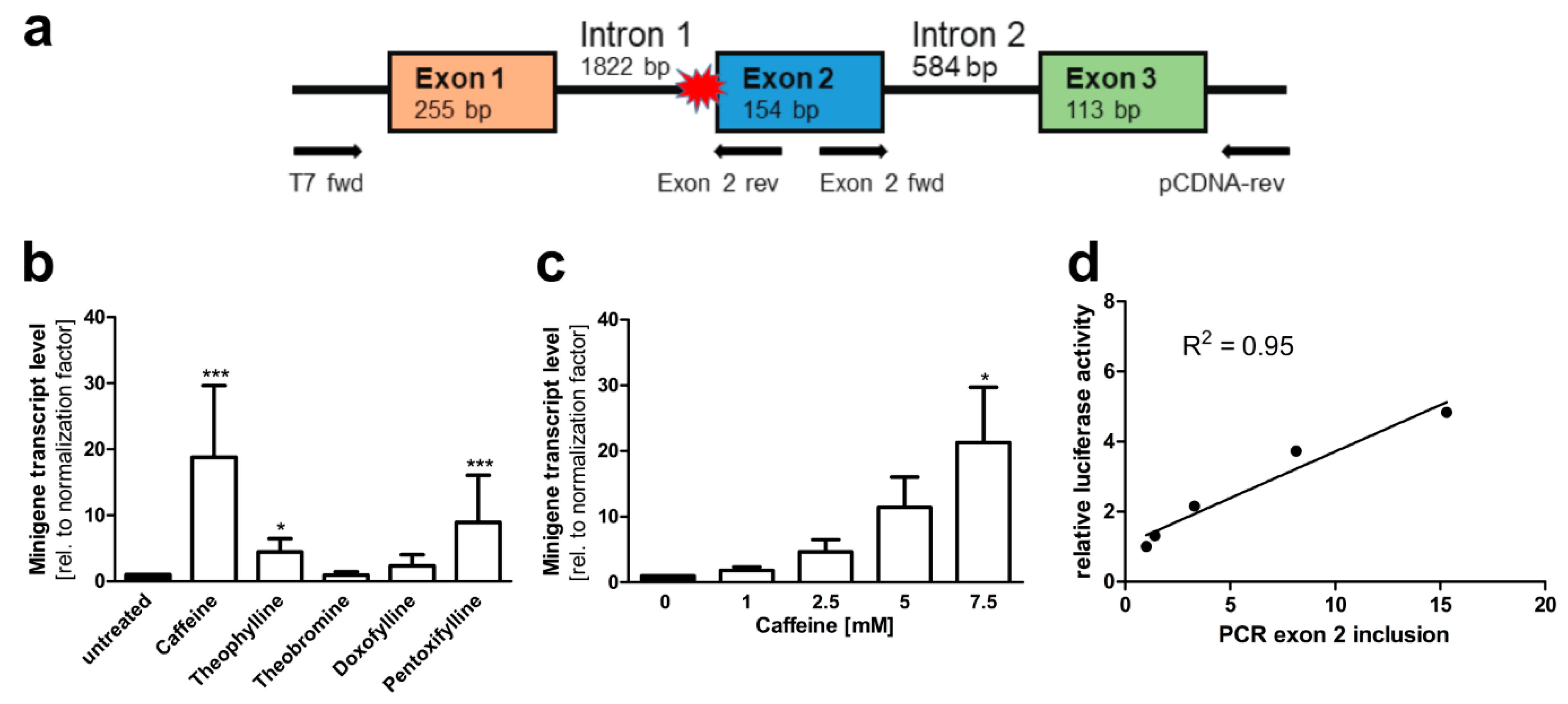
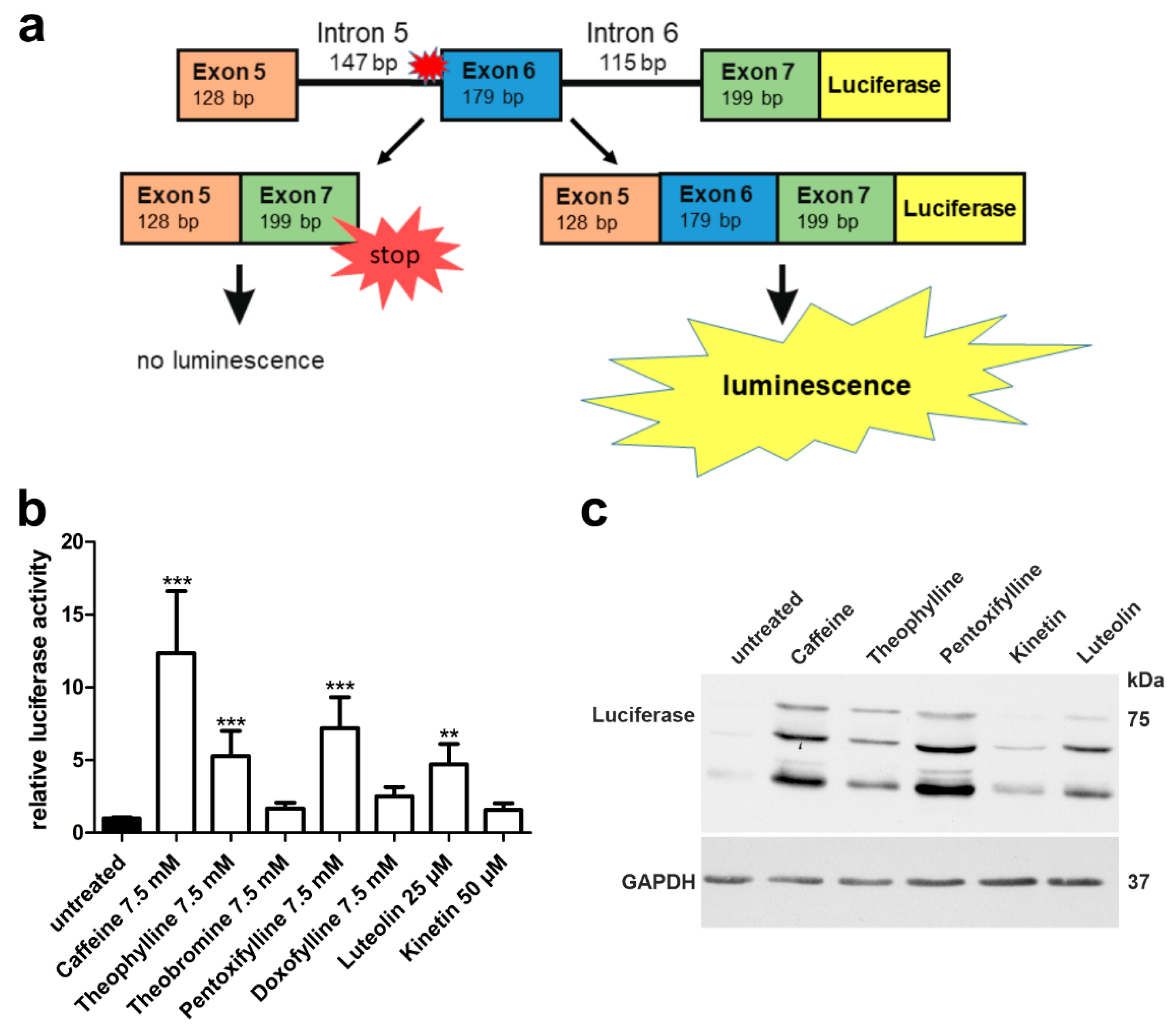
3.2. A Luciferase Minigene Assay for Testing Substances That Affect AGA Splicing
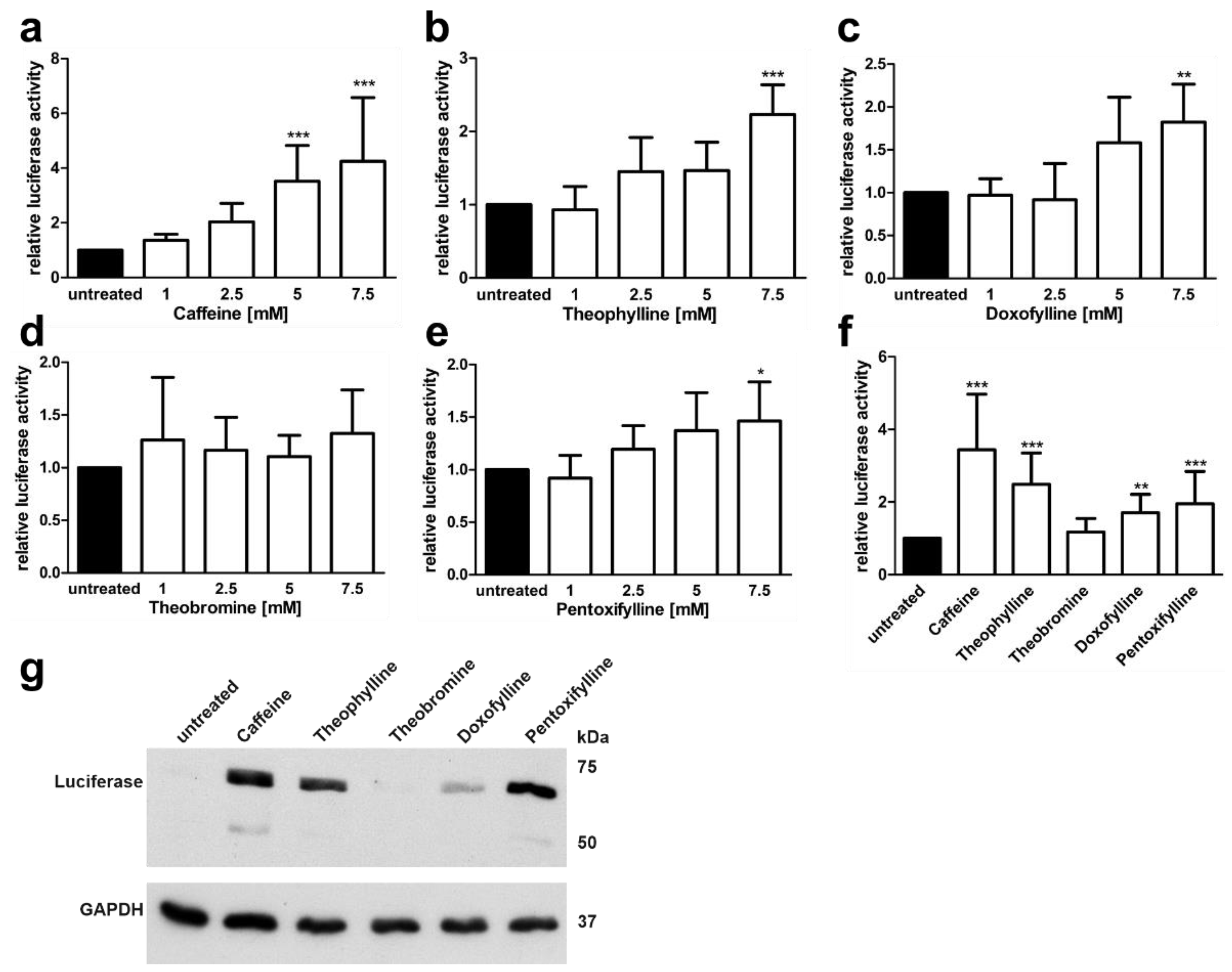
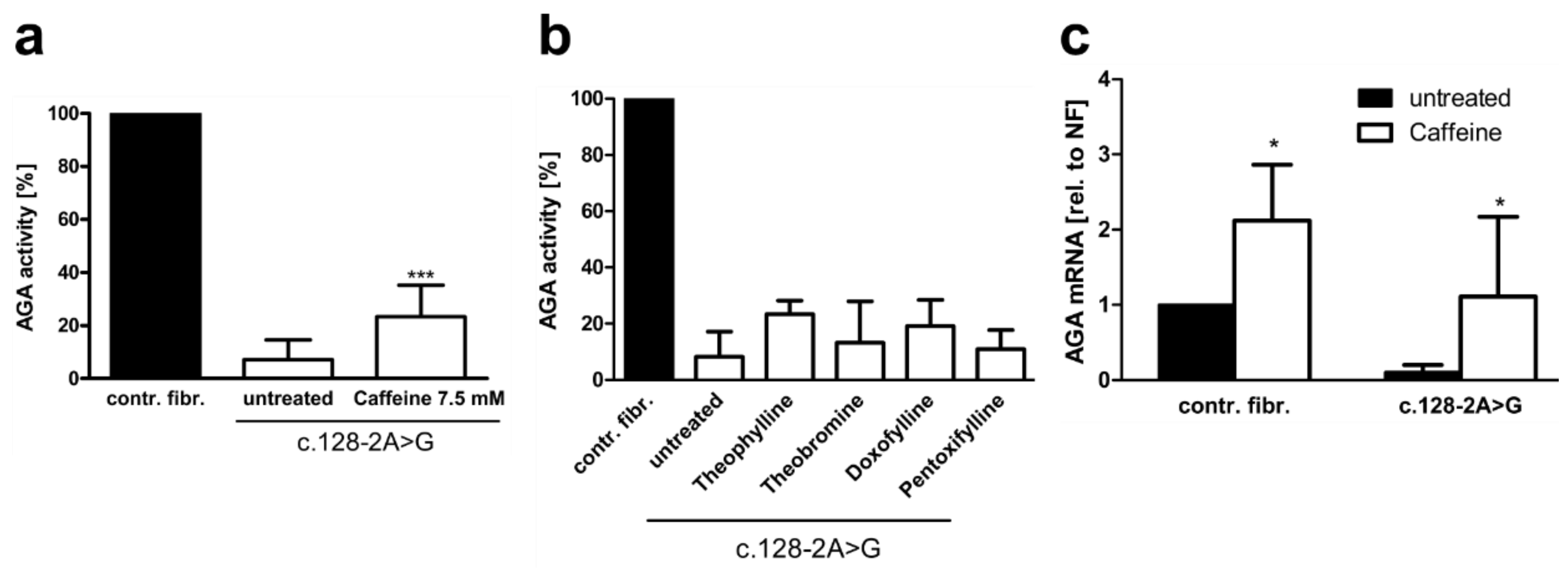
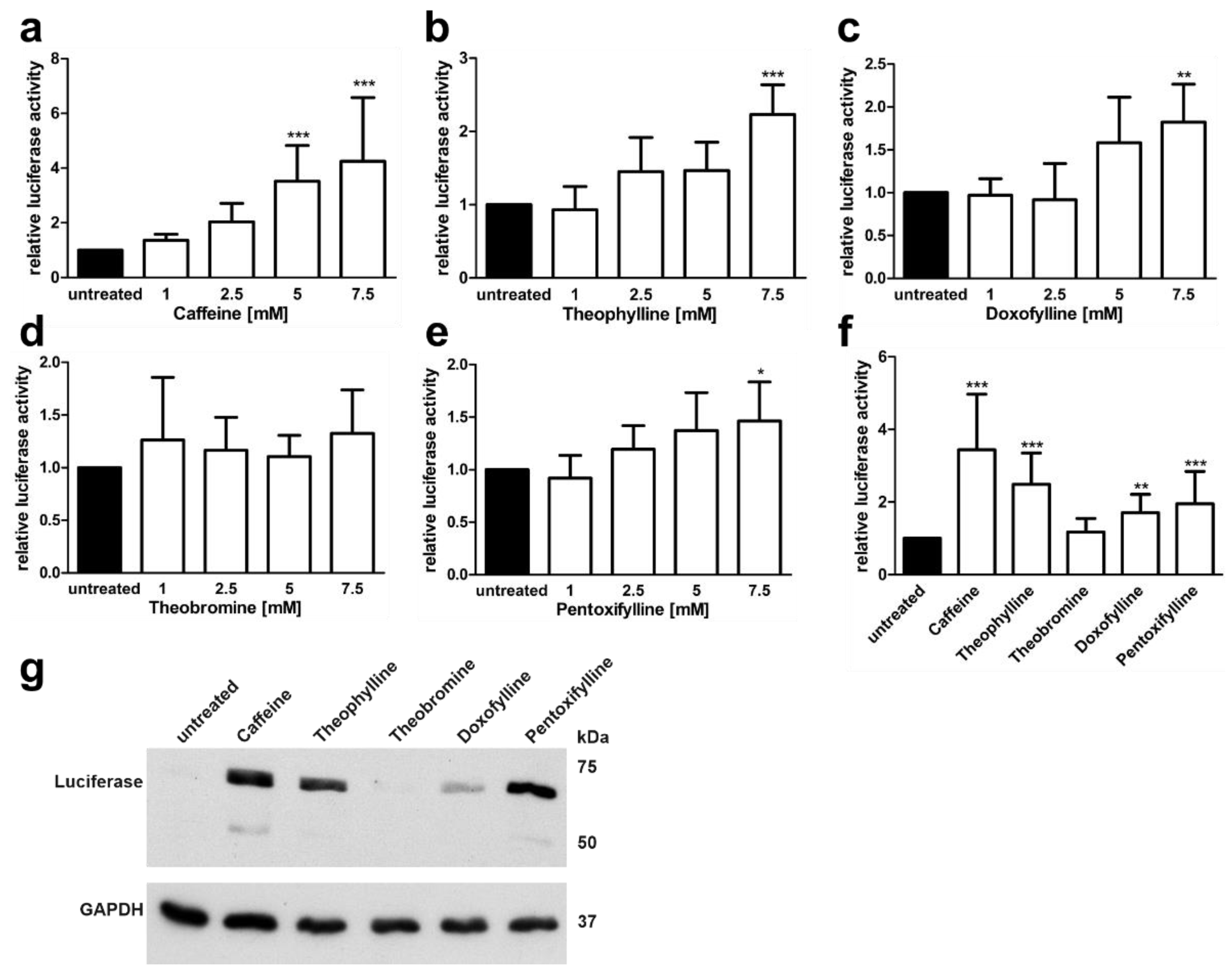
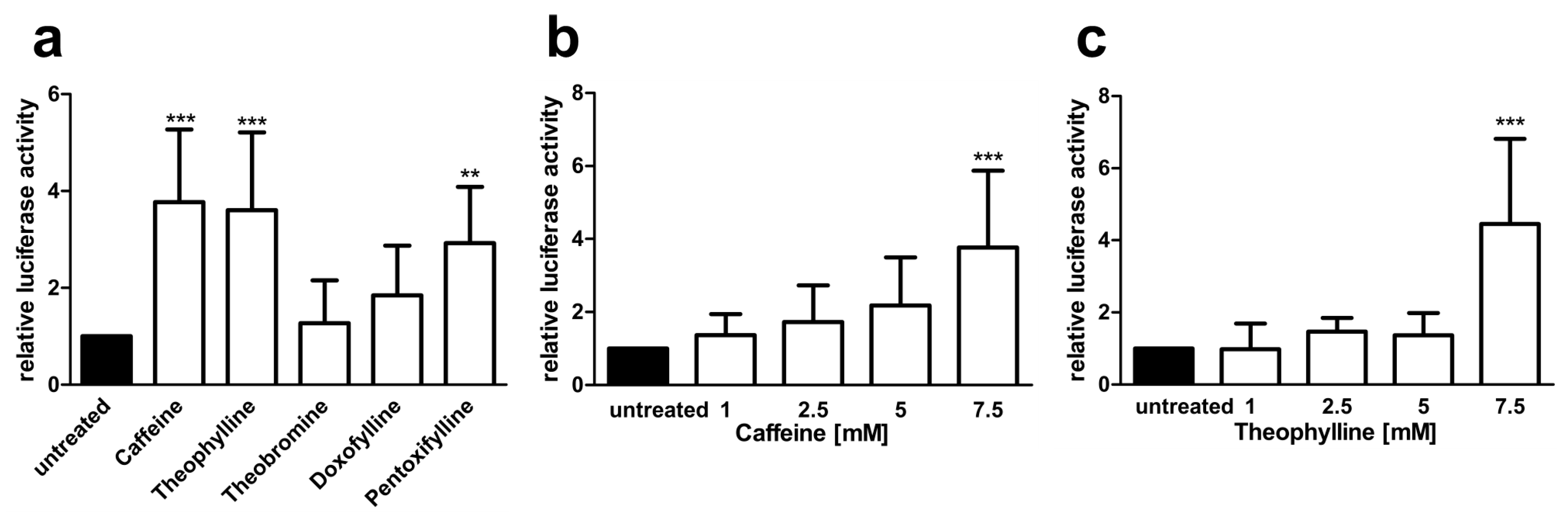
3.3. Xanthine Derivatives Enhance the Correct Splicing of the Mutated AGA mRNA
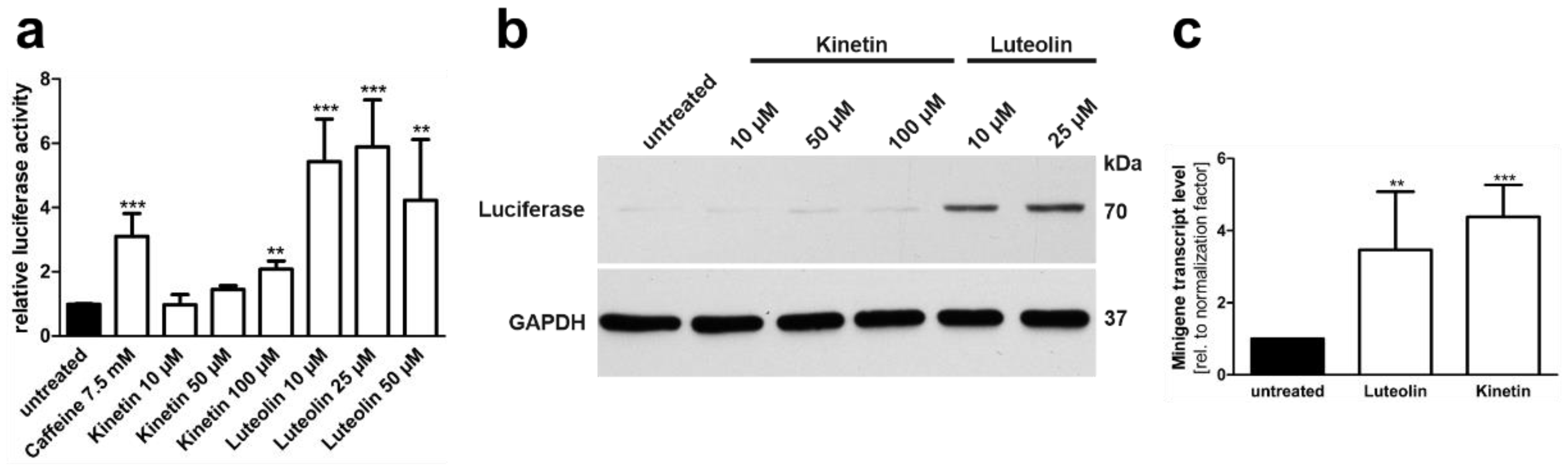
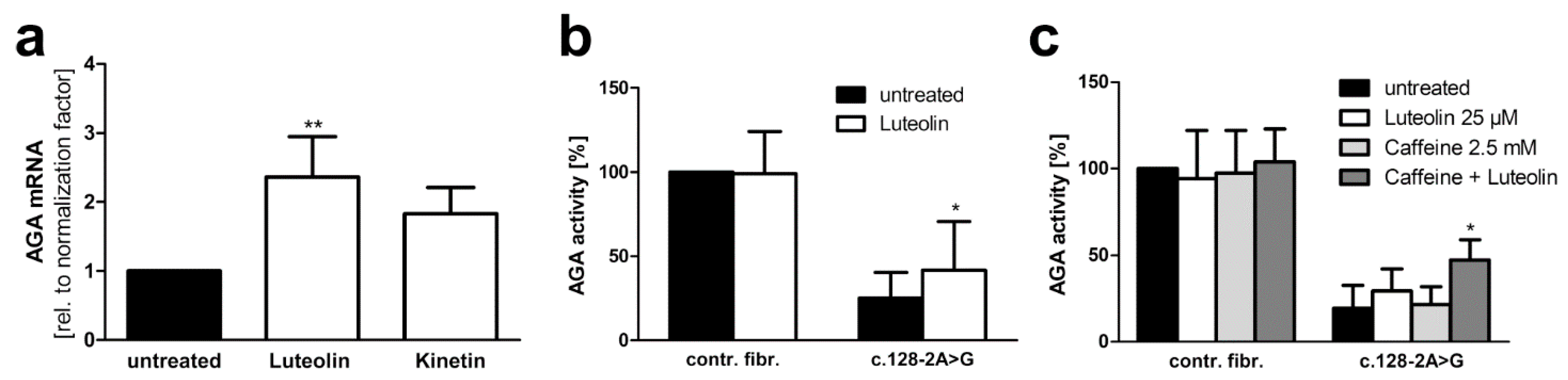
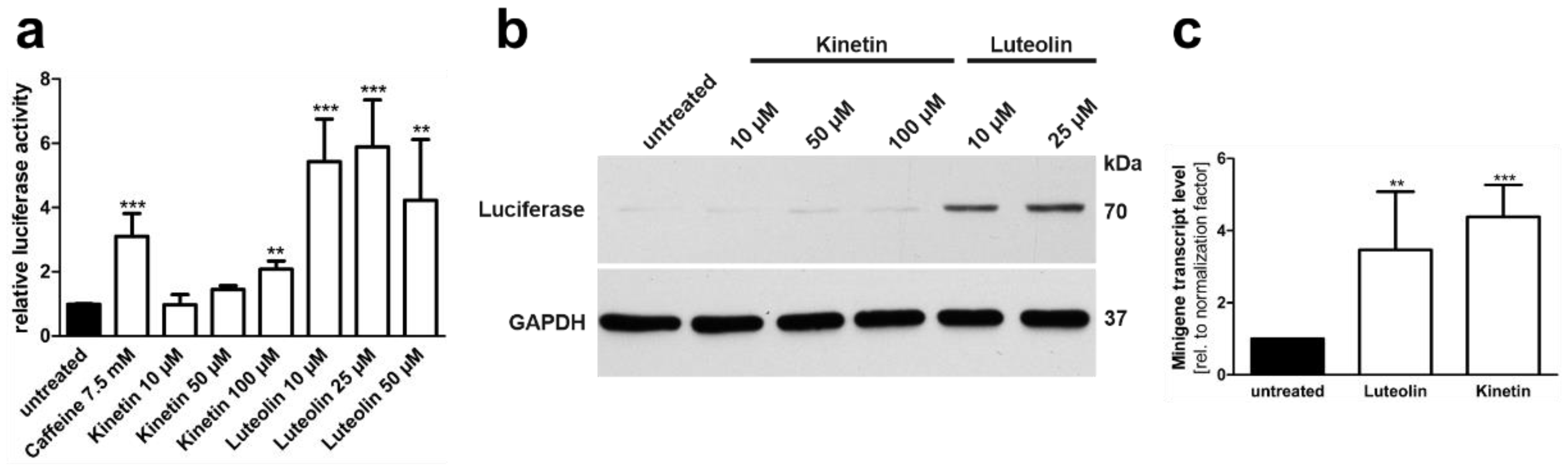
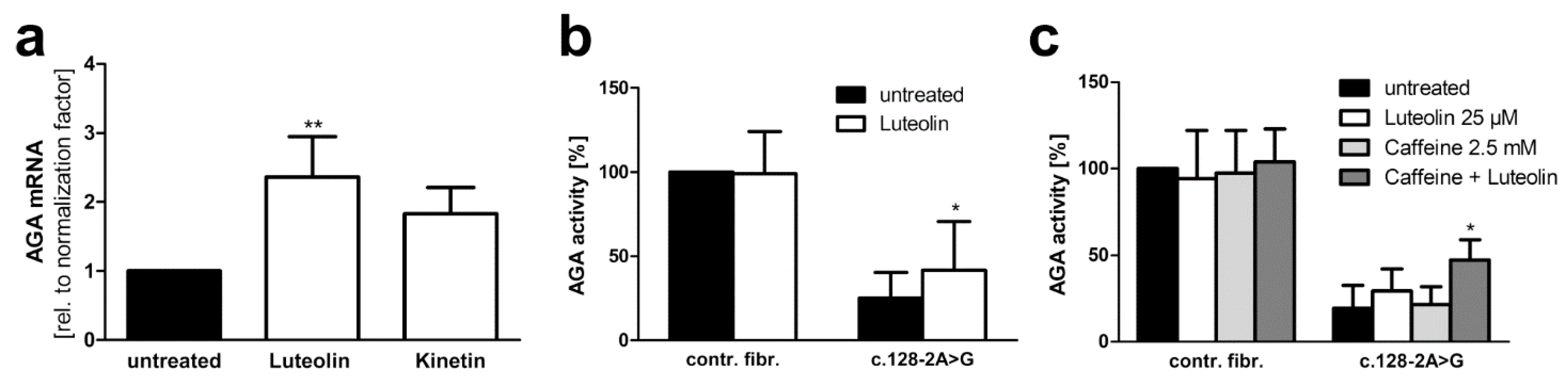
3.4. Plant-Derived Bioactive Compounds Enhance the Splicing of the AGA mRNA
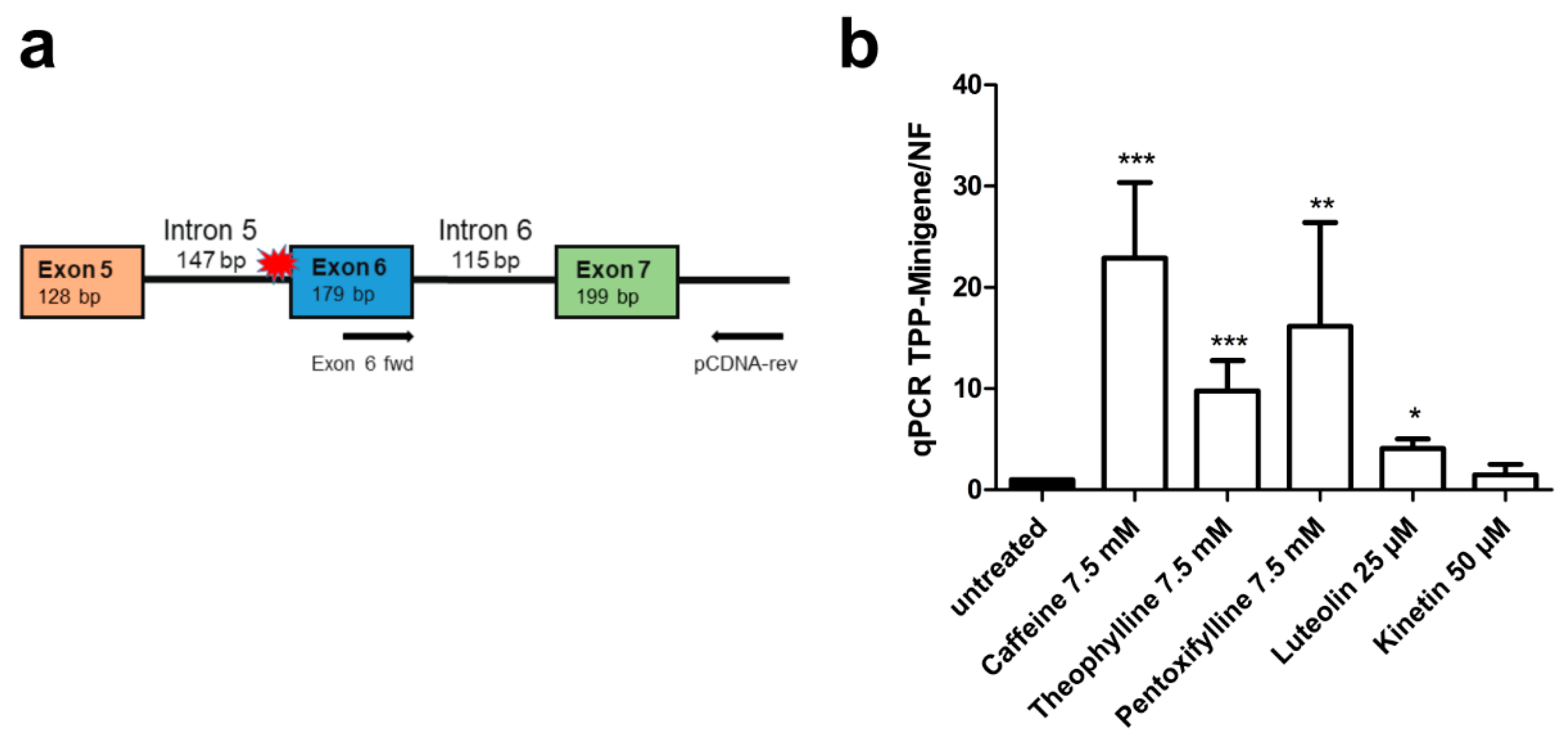
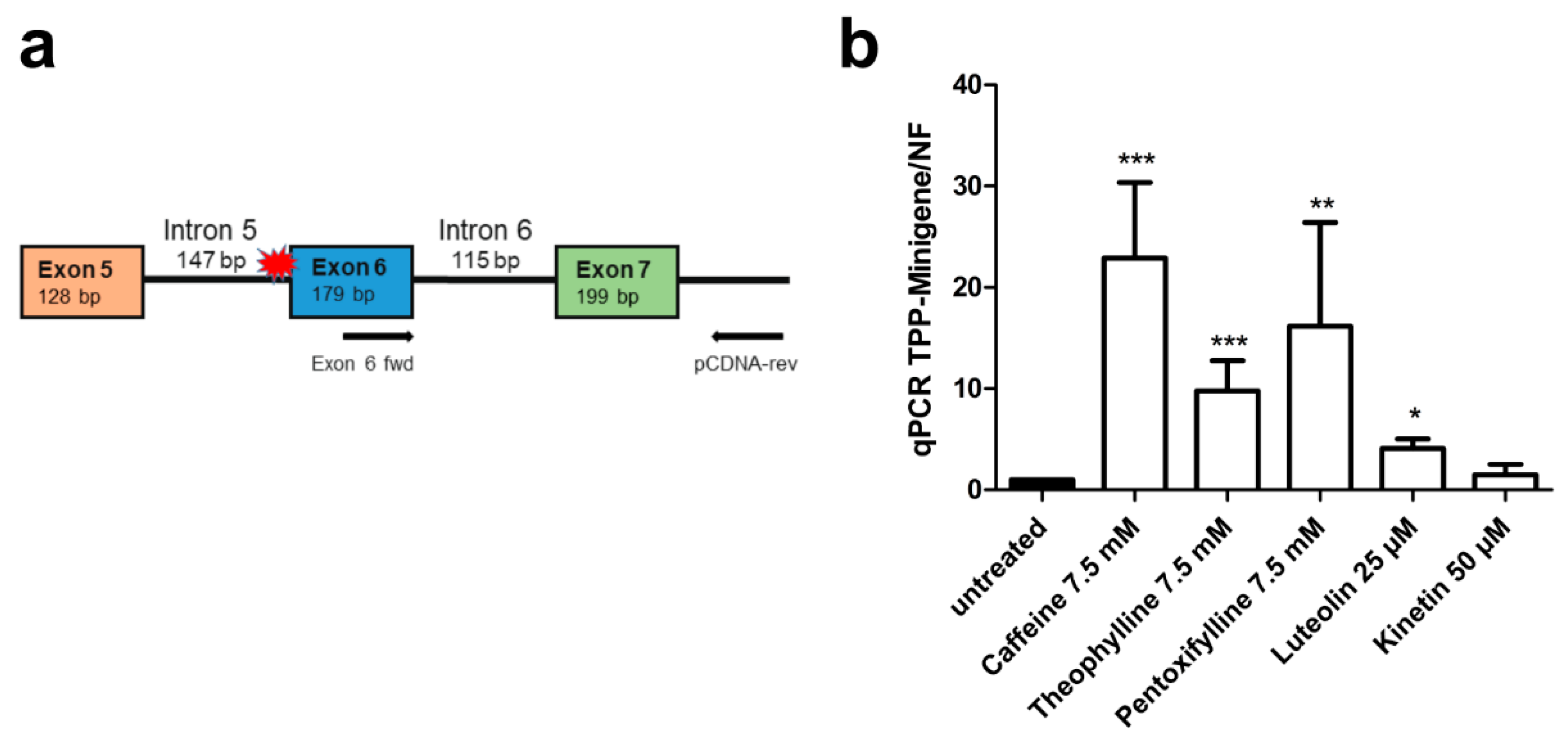
3.5. Xanthine Derivatives and Luteolin Correct the Splicing of a Common cLINCL/TPP1 Variant
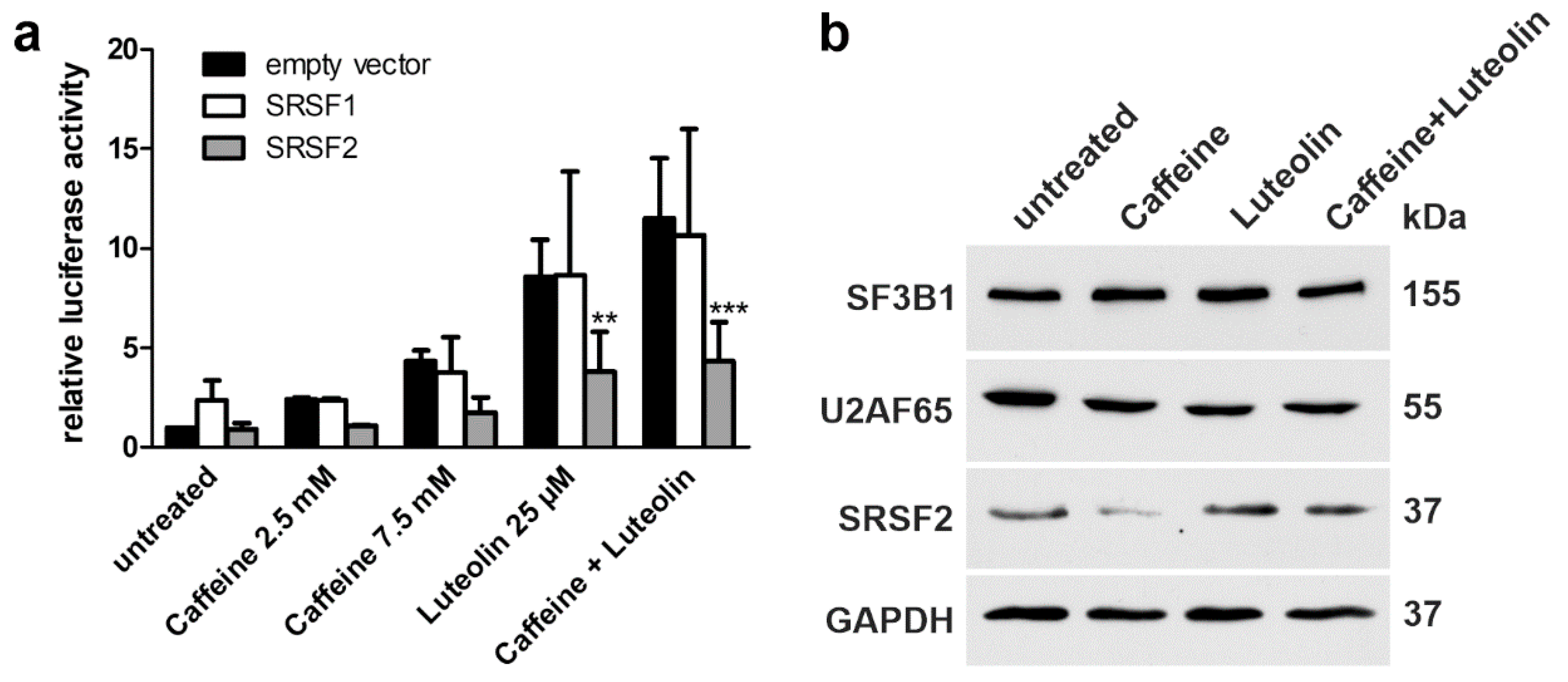
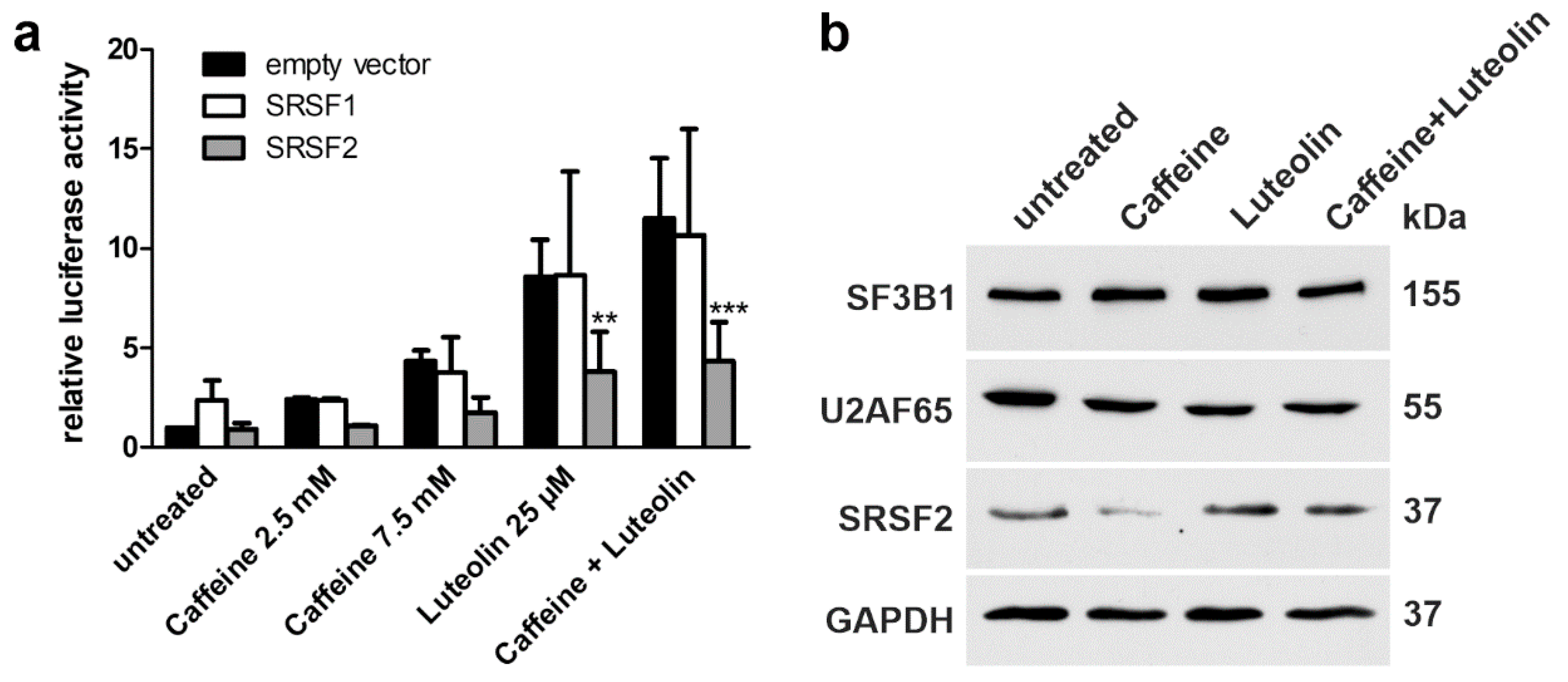
3.6. Role of Splicing Factors in the Splicing of the Mutant AGA Gene
4. Discussion
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Arvio, M.; Mononen, I. Aspartylglycosaminuria: A review. Orphanet. J. Rare Dis. 2016, 11, 162. [Google Scholar] [CrossRef] [Green Version]
- Goodspeed, K.; Feng, C.; Laine, M.; Lund, T.C. Aspartylglucosaminuria: Clinical Presentation and Potential Therapies. J. Child Neurol. 2021, 36, 403–414. [Google Scholar] [CrossRef]
- Saarela, J.; Laine, M.; Oinonen, C.; von Schantz, C.; Jalanko, A.; Rouvinen, J.; Peltonen, L. Molecular pathogenesis of a disease: Structural consequences of aspartylglucosaminuria mutations. Hum. Mol. Genet. 2001, 10, 983–995. [Google Scholar] [CrossRef] [Green Version]
- Banning, A.; Gulec, C.; Rouvinen, J.; Gray, S.J.; Tikkanen, R. Identification of Small Molecule Compounds for Pharmacological Chaperone Therapy of Aspartylglucosaminuria. Sci. Rep. 2016, 6, 37583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- EU Clinical Trials Register. Available online: https://www.clinicaltrialsregister.eu/ctr-search/trial/2017-000645-48/FI (accessed on 9 October 2020).
- Banning, A.; Schiff, M.; Tikkanen, R. Amlexanox provides a potential therapy for nonsense mutations in the lysosomal storage disorder Aspartylglucosaminuria. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 668–675. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Snanoudj-Verber, S.; Pollard, L.; Hu, Y.; Cathey, S.S.; Tikkanen, R.; Gray, S.J. Pre-clinical Gene Therapy with AAV9/AGA in Aspartylglucosaminuria Mice Provides Evidence for Clinical Translation. Mol. Ther. 2021, 29, 989–1000. [Google Scholar] [CrossRef] [PubMed]
- Human Gene Mutation Database. Available online: http://www.hgmd.cf.ac.uk/ac/index.php (accessed on 29 September 2020).
- Baralle, D.; Lucassen, A.; Buratti, E. Missed threads. The impact of pre-mRNA splicing defects on clinical practice. EMBO Rep. 2009, 10, 810–816. [Google Scholar] [CrossRef] [Green Version]
- Aartsma-Rus, A.; Fokkema, I.; Verschuuren, J.; Ginjaar, I.; van Deutekom, J.; van Ommen, G.J.; den Dunnen, J.T. Theoretic applicability of antisense-mediated exon skipping for Duchenne muscular dystrophy mutations. Hum. Mutat. 2009, 30, 293–299. [Google Scholar] [CrossRef]
- Cirak, S.; Arechavala-Gomeza, V.; Guglieri, M.; Feng, L.; Torelli, S.; Anthony, K.; Abbs, S.; Garralda, M.E.; Bourke, J.; Wells, D.J.; et al. Exon skipping and dystrophin restoration in patients with Duchenne muscular dystrophy after systemic phosphorodiamidate morpholino oligomer treatment: An open-label, phase 2, dose-escalation study. Lancet 2011, 378, 595–605. [Google Scholar] [CrossRef] [Green Version]
- Goemans, N.M.; Tulinius, M.; van den Akker, J.T.; Burm, B.E.; Ekhart, P.F.; Heuvelmans, N.; Holling, T.; Janson, A.A.; Platenburg, G.J.; Sipkens, J.A.; et al. Systemic administration of PRO051 in Duchenne’s muscular dystrophy. N. Engl. J. Med. 2011, 364, 1513–1522. [Google Scholar] [CrossRef]
- van Deutekom, J.C.; Janson, A.A.; Ginjaar, I.B.; Frankhuizen, W.S.; Aartsma-Rus, A.; Bremmer-Bout, M.; den Dunnen, J.T.; Koop, K.; van der Kooi, A.J.; Goemans, N.M.; et al. Local dystrophin restoration with antisense oligonucleotide PRO051. N. Engl. J. Med. 2007, 357, 2677–2686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, L. Motor neuron disease: Nusinersen potentially effective in SMA. Nat. Rev. Neurol. 2017, 13, 66. [Google Scholar] [CrossRef] [PubMed]
- Passini, M.A.; Bu, J.; Richards, A.M.; Kinnecom, C.; Sardi, S.P.; Stanek, L.M.; Hua, Y.; Rigo, F.; Matson, J.; Hung, G.; et al. Antisense oligonucleotides delivered to the mouse CNS ameliorate symptoms of severe spinal muscular atrophy. Sci. Transl. Med. 2011, 3, 72ra18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Centa, J.L.; Jodelka, F.M.; Hinrich, A.J.; Johnson, T.B.; Ochaba, J.; Jackson, M.; Duelli, D.M.; Weimer, J.M.; Rigo, F.; Hastings, M.L. Therapeutic efficacy of antisense oligonucleotides in mouse models of CLN3 Batten disease. Nat. Med. 2020, 26, 1444–1451. [Google Scholar] [CrossRef]
- Kim, J.; Hu, C.; Moufawad El Achkar, C.; Black, L.E.; Douville, J.; Larson, A.; Pendergast, M.K.; Goldkind, S.F.; Lee, E.A.; Kuniholm, A.; et al. Patient-Customized Oligonucleotide Therapy for a Rare Genetic Disease. N. Engl. J. Med. 2019, 381, 1644–1652. [Google Scholar] [CrossRef]
- Baranello, G.; Darras, B.T.; Day, J.W.; Deconinck, N.; Klein, A.; Masson, R.; Mercuri, E.; Rose, K.; El-Khairi, M.; Gerber, M.; et al. Risdiplam in Type 1 Spinal Muscular Atrophy. N. Engl. J. Med. 2021, 384, 915–923. [Google Scholar] [CrossRef]
- Ratni, H.; Ebeling, M.; Baird, J.; Bendels, S.; Bylund, J.; Chen, K.S.; Denk, N.; Feng, Z.; Green, L.; Guerard, M.; et al. Discovery of Risdiplam, a Selective Survival of Motor Neuron-2 (SMN2) Gene Splicing Modifier for the Treatment of Spinal Muscular Atrophy (SMA). J. Med. Chem. 2018, 61, 6501–6517. [Google Scholar] [CrossRef] [Green Version]
- Ando, S.; Suzuki, S.; Okubo, S.; Ohuchi, K.; Takahashi, K.; Nakamura, S.; Shimazawa, M.; Fuji, K.; Hara, H. Discovery of a CNS penetrant small molecule SMN2 splicing modulator with improved tolerability for spinal muscular atrophy. Sci. Rep. 2020, 10, 17472. [Google Scholar] [CrossRef]
- Palacino, J.; Swalley, S.E.; Song, C.; Cheung, A.K.; Shu, L.; Zhang, X.; Van Hoosear, M.; Shin, Y.; Chin, D.N.; Keller, C.G.; et al. SMN2 splice modulators enhance U1-pre-mRNA association and rescue SMA mice. Nat. Chem. Biol. 2015, 11, 511–517. [Google Scholar] [CrossRef]
- Sergott, R.C.; Amorelli, G.M.; Baranello, G.; Barreau, E.; Beres, S.; Kane, S.; Mercuri, E.; Orazi, L.; SantaMaria, M.; Tremolada, G.; et al. Risdiplam treatment has not led to retinal toxicity in patients with spinal muscular atrophy. Ann. Clin. Transl. Neurol. 2021, 8, 54–65. [Google Scholar] [CrossRef]
- Kapahnke, M.; Banning, A.; Tikkanen, R. Random Splicing of Several Exons Caused by a Single Base Change in the Target Exon of CRISPR/Cas9 Mediated Gene Knockout. Cells 2016, 5, 45. [Google Scholar] [CrossRef]
- Fujita, K.I.; Ishizuka, T.; Mitsukawa, M.; Kurata, M.; Masuda, S. Regulating Divergent Transcriptomes through mRNA Splicing and Its Modulation Using Various Small Compounds. Int. J. Mol. Sci. 2020, 21, 2026. [Google Scholar] [CrossRef] [Green Version]
- Keeling, K.M.; Wang, D.; Dai, Y.; Murugesan, S.; Chenna, B.; Clark, J.; Belakhov, V.; Kandasamy, J.; Velu, S.E.; Baasov, T.; et al. Attenuation of nonsense-mediated mRNA decay enhances in vivo nonsense suppression. PLoS ONE 2013, 8, e60478. [Google Scholar] [CrossRef] [Green Version]
- Chiba, M.; Ariga, H.; Maita, H. A Splicing Reporter Tuned to Non-AG Acceptor Sites Reveals that Luteolin Enhances the Recognition of Non-canonical Acceptor Sites. Chem. Biol. Drug Des. 2016, 87, 275–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hims, M.M.; Ibrahim, E.C.; Leyne, M.; Mull, J.; Liu, L.; Lazaro, C.; Shetty, R.S.; Gill, S.; Gusella, J.F.; Reed, R.; et al. Therapeutic potential and mechanism of kinetin as a treatment for the human splicing disease familial dysautonomia. J. Mol. Med. 2007, 85, 149–161. [Google Scholar] [CrossRef] [PubMed]
- Slaugenhaupt, S.A.; Mull, J.; Leyne, M.; Cuajungco, M.P.; Gill, S.P.; Hims, M.M.; Quintero, F.; Axelrod, F.B.; Gusella, J.F. Rescue of a human mRNA splicing defect by the plant cytokinin kinetin. Hum. Mol. Genet. 2004, 13, 429–436. [Google Scholar] [CrossRef] [PubMed]
- Schoefer, L.; Braune, A.; Blaut, M. A fluorescence quenching test for the detection of flavonoid transformation. FEMS Microbiol. Lett. 2001, 204, 277–280. [Google Scholar] [CrossRef]
- Tikkanen, R.; Enomaa, N.; Riikonen, A.; Ikonen, E.; Peltonen, L. Intracellular sorting of aspartylglucosaminidase: The role of N-linked oligosaccharides and evidence of Man-6-P-independent lysosomal targeting. DNA Cell Biol. 1995, 14, 305–312. [Google Scholar] [CrossRef]
- Gardner, E.; Bailey, M.; Schulz, A.; Aristorena, M.; Miller, N.; Mole, S.E. Mutation update: Review of TPP1 gene variants associated with neuronal ceroid lipofuscinosis CLN2 disease. Hum. Mutat. 2019, 40, 1924–1938. [Google Scholar] [CrossRef] [Green Version]
- Howard, J.M.; Sanford, J.R. The RNAissance family: SR proteins as multifaceted regulators of gene expression. Wiley Interdiscip. Rev. RNA 2015, 6, 93–110. [Google Scholar] [CrossRef]
- Jeong, S. SR Proteins: Binders, Regulators, and Connectors of RNA. Mol. Cells 2017, 40, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Lu, G.Y.; Huang, S.M.; Liu, S.T.; Liu, P.Y.; Chou, W.Y.; Lin, W.S. Caffeine induces tumor cytotoxicity via the regulation of alternative splicing in subsets of cancer-associated genes. Int. J. Biochem. Cell Biol. 2014, 47, 83–92. [Google Scholar] [CrossRef]
- Shi, J.; Hu, Z.; Pabon, K.; Scotto, K.W. Caffeine regulates alternative splicing in a subset of cancer-associated genes: A role for SC35. Mol. Cell Biol. 2008, 28, 883–895. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.; Pabon, K.; Scotto, K.W. Methylxanthines Increase Expression of the Splicing Factor SRSF2 by Regulating Multiple Post-transcriptional Mechanisms. J. Biol. Chem. 2015, 290, 14986–15003. [Google Scholar] [CrossRef] [Green Version]
- Kurata, M.; Fujiwara, N.; Fujita, K.I.; Yamanaka, Y.; Seno, S.; Kobayashi, H.; Miyamae, Y.; Takahashi, N.; Shibuya, Y.; Masuda, S. Food-Derived Compounds Apigenin and Luteolin Modulate mRNA Splicing of Introns with Weak Splice Sites. iScience 2019, 22, 336–352. [Google Scholar] [CrossRef]
- Kitamura, K.; Nimura, K. Regulation of RNA Splicing: Aberrant Splicing Regulation and Therapeutic Targets in Cancer. Cells 2021, 10, 923. [Google Scholar] [CrossRef]
- Lee, Y.; Rio, D.C. Mechanisms and Regulation of Alternative Pre-mRNA Splicing. Annu. Rev. Biochem. 2015, 84, 291–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bode, A.M.; Dong, Z. The enigmatic effects of caffeine in cell cycle and cancer. Cancer Lett. 2007, 247, 26–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamashita, A.; Ohnishi, T.; Kashima, I.; Taya, Y.; Ohno, S. Human SMG-1, a novel phosphatidylinositol 3-kinase-related protein kinase, associates with components of the mRNA surveillance complex and is involved in the regulation of nonsense-mediated mRNA decay. Genes Dev. 2001, 15, 2215–2228. [Google Scholar] [CrossRef] [Green Version]
- Harada, N.; Hatakeyama, A.; Okuyama, M.; Miyatake, Y.; Nakagawa, T.; Kuroda, M.; Masumoto, S.; Tsutsumi, R.; Nakaya, Y.; Sakaue, H. Readthrough of ACTN3 577X nonsense mutation produces full-length alpha-actinin-3 protein. Biochem. Biophys. Res. Commun. 2018, 502, 422–428. [Google Scholar] [CrossRef] [PubMed]
- Lentini, L.; Melfi, R.; Cancemi, P.; Pibiri, I.; Di Leonardo, A. Caffeine boosts Ataluren’s readthrough activity. Heliyon 2019, 5, e01963. [Google Scholar] [CrossRef] [Green Version]
- Kolahdouzan, M.; Hamadeh, M.J. The neuroprotective effects of caffeine in neurodegenerative diseases. CNS Neurosci. Ther. 2017, 23, 272–290. [Google Scholar] [CrossRef]
- Matera, M.G.; Page, C.; Cazzola, M. Doxofylline is not just another theophylline! Int. J. Chron. Obstr. Pulmon. Dis. 2017, 12, 3487–3493. [Google Scholar] [CrossRef] [Green Version]
- Barnes, P.J. Theophylline. Am. J. Respir. Crit. Care Med. 2013, 188, 901–906. [Google Scholar] [CrossRef]
- Chang, Y.L.; Hsu, Y.J.; Chen, Y.; Wang, Y.W.; Huang, S.M. Theophylline exhibits anti-cancer activity via suppressing SRSF3 in cervical and breast cancer cell lines. Oncotarget 2017, 8, 101461–101474. [Google Scholar] [CrossRef] [Green Version]
- Ito, K.; Lim, S.; Caramori, G.; Cosio, B.; Chung, K.F.; Adcock, I.M.; Barnes, P.J. A molecular mechanism of action of theophylline: Induction of histone deacetylase activity to decrease inflammatory gene expression. Proc. Natl. Acad. Sci. USA 2002, 99, 8921–8926. [Google Scholar] [CrossRef] [Green Version]
- Barnes, P.J. Theophylline: New perspectives for an old drug. Am. J. Respir. Crit. Care Med. 2003, 167, 813–818. [Google Scholar] [CrossRef]
- Annamaraju, P.; Baradhi, K.M. Pentoxifylline; StatPearls: Treasure Island, FL, USA, 2020. [Google Scholar]
- Brie, D.; Sahebkar, A.; Penson, P.E.; Dinca, M.; Ursoniu, S.; Serban, M.C.; Zanchetti, A.; Howard, G.; Ahmed, A.; Aronow, W.S.; et al. Effects of pentoxifylline on inflammatory markers and blood pressure: A systematic review and meta-analysis of randomized controlled trials. J. Hypertens. 2016, 34, 2318–2329. [Google Scholar] [CrossRef]
- Salani, M.; Urbina, F.; Brenner, A.; Morini, E.; Shetty, R.; Gallagher, C.S.; Law, E.A.; Sunshine, S.; Finneran, D.J.; Johnson, G.; et al. Development of a Screening Platform to Identify Small Molecules That Modify ELP1 Pre-mRNA Splicing in Familial Dysautonomia. SLAS Discov. 2019, 24, 57–67. [Google Scholar] [CrossRef]
- Axelrod, F.B.; Liebes, L.; Gold-Von Simson, G.; Mendoza, S.; Mull, J.; Leyne, M.; Norcliffe-Kaufmann, L.; Kaufmann, H.; Slaugenhaupt, S.A. Kinetin improves IKBKAP mRNA splicing in patients with familial dysautonomia. Pediatr. Res. 2011, 70, 480–483. [Google Scholar] [CrossRef]
- Arango, D.; Morohashi, K.; Yilmaz, A.; Kuramochi, K.; Parihar, A.; Brahimaj, B.; Grotewold, E.; Doseff, A.I. Molecular basis for the action of a dietary flavonoid revealed by the comprehensive identification of apigenin human targets. Proc. Natl. Acad. Sci. USA 2013, 110, E2153–E2162. [Google Scholar] [CrossRef] [Green Version]
- Nabavi, S.F.; Braidy, N.; Gortzi, O.; Sobarzo-Sanchez, E.; Daglia, M.; Skalicka-Wozniak, K.; Nabavi, S.M. Luteolin as an anti-inflammatory and neuroprotective agent: A brief review. Brain Res. Bull. 2015, 119, 1–11. [Google Scholar] [CrossRef]
- Kwon, K.; Kwon, Y.S.; Kim, S.W.; Yu, K.; Lee, K.H.; Kwon, O.Y. Luteolin-induced apoptosis through activation of endoplasmic reticulum stress sensors in pheochromocytoma cells. Mol. Med. Rep. 2017, 16, 380–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imran, M.; Rauf, A.; Abu-Izneid, T.; Nadeem, M.; Shariati, M.A.; Khan, I.A.; Imran, A.; Erdogan Orhan, I.; Rizwan, M.; Atif, M.; et al. Corrigendum to “Luteolin, a flavonoid, as an anticancer agent: A review” [Biomed. Pharmacother. 112 (2019) 108612]. Biomed. Pharmacother. 2019, 116, 109084. [Google Scholar] [CrossRef] [PubMed]
- Kang, K.A.; Piao, M.J.; Hyun, Y.J.; Zhen, A.X.; Cho, S.J.; Ahn, M.J.; Yi, J.M.; Hyun, J.W. Luteolin promotes apoptotic cell death via upregulation of Nrf2 expression by DNA demethylase and the interaction of Nrf2 with p53 in human colon cancer cells. Exp. Mol. Med. 2019, 51, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez-Lazaro, M. Distribution and biological activities of the flavonoid luteolin. Mini Rev. Med. Chem. 2009, 9, 31–59. [Google Scholar] [CrossRef] [PubMed]
- Gates, M.A.; Tworoger, S.S.; Hecht, J.L.; De Vivo, I.; Rosner, B.; Hankinson, S.E. A prospective study of dietary flavonoid intake and incidence of epithelial ovarian cancer. Int. J. Cancer 2007, 121, 2225–2232. [Google Scholar] [CrossRef] [PubMed]
- Marniemi, J.; Alanen, E.; Impivaara, O.; Seppanen, R.; Hakala, P.; Rajala, T.; Ronnemaa, T. Dietary and serum vitamins and minerals as predictors of myocardial infarction and stroke in elderly subjects. Nutr. Metab. Cardiovasc. Dis. 2005, 15, 188–197. [Google Scholar] [CrossRef] [PubMed]
- Taliou, A.; Zintzaras, E.; Lykouras, L.; Francis, K. An open-label pilot study of a formulation containing the anti-inflammatory flavonoid luteolin and its effects on behavior in children with autism spectrum disorders. Clin. Ther. 2013, 35, 592–602. [Google Scholar] [CrossRef] [PubMed]
- Theoharides, T.C.; Asadi, S.; Panagiotidou, S. A case series of a luteolin formulation (NeuroProtek(R)) in children with autism spectrum disorders. Int. J. Immunopathol. Pharmacol. 2012, 25, 317–323. [Google Scholar] [CrossRef] [Green Version]
- Coleta, M.; Campos, M.G.; Cotrim, M.D.; Lima, T.C.; Cunha, A.P. Assessment of luteolin (3′,4′,5,7-tetrahydroxyflavone) neuropharmacological activity. Behav. Brain Res. 2008, 189, 75–82. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Name | Primer Sequence 5′ to 3’ |
|---|---|
| AGA-Minigene fwd HindIII | CTATAAAGCTTAGGGACGCCTGAGCGAACCC |
| AGA-Minigene rev XhoI | CTATACTCGAGCTGACTCTCCTACTAAAAGTGTGT |
| TPP-IVS-Minigene fwd BamHI | CTAGGATCCATGCAAGCAGAGCTGCTGCTCCCTG |
| TPP-IVS-Minigene rev XhoI | CTATACTCGAGCAGGGCTACTGTAGACCCAGGTG |
| Luciferase fwd XhoI | CTATACTCGAGAAGACGCCAAAAACATAAAGAAAGG |
| Luciferase rev XbaI | CTATATCTAGATTACACGGCGATCTTTCCGCC |
| Primer Name | Primer Sequence 5′ to 3′ |
|---|---|
| B2M-fwd | AGATGAGTATGCCTGCCGTGTG |
| B2M-rev | TGCGGCATCTTCAAACCTCCA |
| Rpl13a-fwd | CCTGGAGGAGAAGAGGAAAGAGA |
| Rpl13a-rev | TTGAGGACCTCTGTGTATTTGTCAA |
| Ywhaz-fwd | AGGTTGCCGCTGGTGATGAC |
| Ywhaz-rev | GGCCAGACCCAGTCTGATAGGA |
| TBP-fwd | GACTATTGGTGTTCTGAATAGGC |
| TBP-rev | GGAATCCCTATCTTTAGTCCAAT |
| AGA exon2 fwd | CGGCTCTGTAGGCTTTGGAGGA |
| AGA exon2 rev | GCCTCCAGATGCTAATGCCCTC |
| AGA exon3 rev | CCAGTACTTTCCGTGCCACACC |
| AGA exon 7/8 fwd | ATGGCCGTGTAGGAGACTCACC |
| AGA exon 8/9 rev | ACAGCTTGGTAGCTTGGCAGGA |
| TPP1 exon 6/7 fwd | CTGTGCCCAGTTCCTGGAGC |
| pCDNA3-rev | GGCAACTAGAAGGCACAGTC |
| T7 fwd | TAATACGACTCACTATAGGG |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Banning, A.; Tikkanen, R. Towards Splicing Therapy for Lysosomal Storage Disorders: Methylxanthines and Luteolin Ameliorate Splicing Defects in Aspartylglucosaminuria and Classic Late Infantile Neuronal Ceroid Lipofuscinosis. Cells 2021, 10, 2813. https://doi.org/10.3390/cells10112813
Banning A, Tikkanen R. Towards Splicing Therapy for Lysosomal Storage Disorders: Methylxanthines and Luteolin Ameliorate Splicing Defects in Aspartylglucosaminuria and Classic Late Infantile Neuronal Ceroid Lipofuscinosis. Cells. 2021; 10(11):2813. https://doi.org/10.3390/cells10112813
Chicago/Turabian StyleBanning, Antje, and Ritva Tikkanen. 2021. "Towards Splicing Therapy for Lysosomal Storage Disorders: Methylxanthines and Luteolin Ameliorate Splicing Defects in Aspartylglucosaminuria and Classic Late Infantile Neuronal Ceroid Lipofuscinosis" Cells 10, no. 11: 2813. https://doi.org/10.3390/cells10112813
APA StyleBanning, A., & Tikkanen, R. (2021). Towards Splicing Therapy for Lysosomal Storage Disorders: Methylxanthines and Luteolin Ameliorate Splicing Defects in Aspartylglucosaminuria and Classic Late Infantile Neuronal Ceroid Lipofuscinosis. Cells, 10(11), 2813. https://doi.org/10.3390/cells10112813