
Involvement of Autophagy in Ageing and Chronic Cholestatic Diseases


Abstract
:1. Introduction
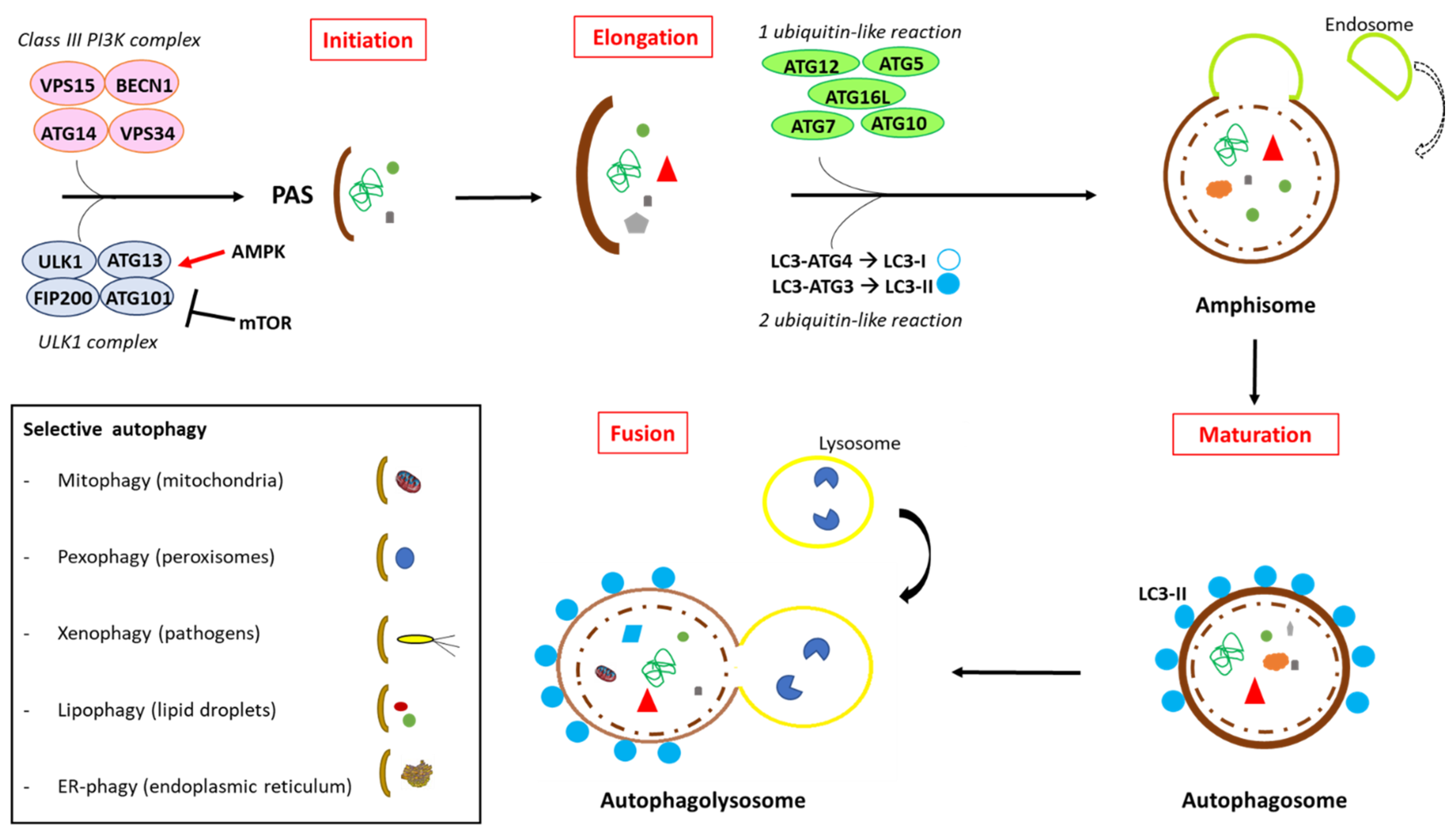
2. Molecular Aspects of the Autophagy Process
2.1. Autophagy Evaluation in Experimental Settings
2.2. Autophagy in Liver Physiology
2.3. Autophagy in Liver Disease
3. Autophagy in Cholestatic Liver Diseases
3.1. Autophagy as a Therapeutic Strategy in Cholestasis Treatment
3.2. The Emerging Role of Nuclear Receptors in Autophagy and Cholestatic Disease
4. Ageing in Cholestatic Diseases
4.1. Ageing and Autophagy
4.2. Molecular Mechanisms and Pathways Involved in Autophagy
5. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Takeshige, K.; Baba, M.; Tsuboi, S.; Noda, T.; Ohsumi, Y. Autophagy in Yeast Demonstrated with Proteinase-Deficient Mutants and Conditions for Its Induction. J. Cell Biol. 1992, 119, 301–311. [Google Scholar] [CrossRef] [Green Version]
- Tsukada, M.; Ohsumi, Y. Isolation and Characterization of Autophagy-Defective Mutants of Saccharomyces Cerevisiae. FEBS Lett. 1993, 333, 169–174. [Google Scholar] [CrossRef] [Green Version]
- Gatica, D.; Lahiri, V.; Klionsky, D.J. Cargo Recognition and Degradation by Selective Autophagy. Nat. Cell Biol. 2018, 20, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Zaffagnini, G.; Martens, S. Mechanisms of Selective Autophagy. J. Mol. Biol. 2016, 428, 1714–1724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and Molecular Mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.; He, D.; Yao, Z.; Klionsky, D.J. The Machinery of Macroautophagy. Cell Res. 2014, 24, 24–41. [Google Scholar] [CrossRef] [Green Version]
- Kounakis, K.; Chaniotakis, M.; Markaki, M.; Tavernarakis, N. Emerging Roles of Lipophagy in Health and Disease. Front. Cell Dev. Biol. 2019, 7, 185. [Google Scholar] [CrossRef]
- Wilkinson, S. ER-Phagy: Shaping up and Destressing the Endoplasmic Reticulum. FEBS J. 2019, 286, 2645–2663. [Google Scholar] [CrossRef] [Green Version]
- Loi, M.; Fregno, I.; Guerra, C.; Molinari, M. Eat It Right: ER-Phagy and RecovER-Phagy. Biochem. Soc. Trans. 2018, 46, 699–706. [Google Scholar] [CrossRef] [Green Version]
- Fregno, I.; Molinari, M. Endoplasmic Reticulum Turnover: ER-Phagy and Other Flavors in Selective and Non-Selective ER Clearance. F1000Research 2018, 7, 454. [Google Scholar] [CrossRef]
- Fregno, I.; Fasana, E.; Soldà, T.; Galli, C.; Molinari, M. N-Glycan Processing Selects ERAD-Resistant Misfolded Proteins for ER-to-Lysosome-Associated Degradation. EMBO J. 2021, 40, e107240. [Google Scholar] [CrossRef]
- Molinari, M. ER-Phagy Responses in Yeast, Plants, and Mammalian Cells and Their Crosstalk with UPR and ERAD. Dev. Cell 2021, 56, 949–966. [Google Scholar] [CrossRef]
- Fregno, I.; Molinari, M. Proteasomal and Lysosomal Clearance of Faulty Secretory Proteins: ER-Associated Degradation (ERAD) and ER-to-Lysosome-Associated Degradation (ERLAD) Pathways. Crit. Rev. Biochem. Mol. Biol. 2019, 54, 153–163. [Google Scholar] [CrossRef]
- Kroemer, G.; Mariño, G.; Levine, B. Autophagy and the Integrated Stress Response. Mol. Cell 2010, 40, 280–293. [Google Scholar] [CrossRef] [Green Version]
- Zhan, L.; Li, J.; Wei, B. Autophagy Therapeutics: Preclinical Basis and Initial Clinical Studies. Cancer Chemother. Pharm. 2018, 82, 923–934. [Google Scholar] [CrossRef]
- Parzych, K.R.; Klionsky, D.J. An Overview of Autophagy: Morphology, Mechanism, and Regulation. Antioxid. Redox Signal. 2014, 20, 460–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuck, S. Microautophagy—Distinct Molecular Mechanisms Handle Cargoes of Many Sizes. J. Cell Sci. 2020, 133, jcs246322. [Google Scholar] [CrossRef]
- Yang, Q.; Wang, R.; Zhu, L. Chaperone-Mediated Autophagy. Adv. Exp. Med. Biol. 2019, 1206, 435–452. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, S.; Bandyopadhyay, U.; Sridhar, S.; Kiffin, R.; Martinez-Vicente, M.; Kon, M.; Orenstein, S.J.; Wong, E.; Cuervo, A.M. Chaperone-Mediated Autophagy at a Glance. J. Cell Sci. 2011, 124, 495–499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Axe, E.L.; Walker, S.A.; Manifava, M.; Chandra, P.; Roderick, H.L.; Habermann, A.; Griffiths, G.; Ktistakis, N.T. Autophagosome Formation from Membrane Compartments Enriched in Phosphatidylinositol 3-Phosphate and Dynamically Connected to the Endoplasmic Reticulum. J. Cell Biol. 2008, 182, 685–701. [Google Scholar] [CrossRef] [Green Version]
- Ravikumar, B.; Moreau, K.; Rubinsztein, D.C. Plasma Membrane Helps Autophagosomes Grow. Autophagy 2010, 6, 1184–1186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hailey, D.W.; Rambold, A.S.; Satpute-Krishnan, P.; Mitra, K.; Sougrat, R.; Kim, P.K.; Lippincott-Schwartz, J. Mitochondria Supply Membranes for Autophagosome Biogenesis during Starvation. Cell 2010, 141, 656–667. [Google Scholar] [CrossRef] [Green Version]
- Van der Vaart, A.; Griffith, J.; Reggiori, F. Exit from the Golgi Is Required for the Expansion of the Autophagosomal Phagophore in Yeast Saccharomyces Cerevisiae. Mol. Biol. Cell 2010, 21, 2270–2284. [Google Scholar] [CrossRef] [Green Version]
- Ohashi, Y.; Tremel, S.; Williams, R.L. VPS34 Complexes from a Structural Perspective. J. Lipid Res. 2019, 60, 229–241. [Google Scholar] [CrossRef] [Green Version]
- Russell, R.C.; Tian, Y.; Yuan, H.; Park, H.W.; Chang, Y.-Y.; Kim, J.; Kim, H.; Neufeld, T.P.; Dillin, A.; Guan, K.-L. ULK1 Induces Autophagy by Phosphorylating Beclin-1 and Activating VPS34 Lipid Kinase. Nat. Cell Biol. 2013, 15, 741–750. [Google Scholar] [CrossRef] [Green Version]
- Hanada, T.; Noda, N.N.; Satomi, Y.; Ichimura, Y.; Fujioka, Y.; Takao, T.; Inagaki, F.; Ohsumi, Y. The Atg12-Atg5 Conjugate Has a Novel E3-like Activity for Protein Lipidation in Autophagy. J. Biol. Chem. 2007, 282, 37298–37302. [Google Scholar] [CrossRef] [Green Version]
- Fujita, N.; Itoh, T.; Omori, H.; Fukuda, M.; Noda, T.; Yoshimori, T. The Atg16L Complex Specifies the Site of LC3 Lipidation for Membrane Biogenesis in Autophagy. Mol. Biol. Cell 2008, 19, 2092–2100. [Google Scholar] [CrossRef] [Green Version]
- Søreng, K.; Neufeld, T.P.; Simonsen, A. Membrane Trafficking in Autophagy. Int. Rev. Cell Mol. Biol. 2018, 336, 1–92. [Google Scholar] [CrossRef]
- Ravikumar, B.; Futter, M.; Jahreiss, L.; Korolchuk, V.I.; Lichtenberg, M.; Luo, S.; Massey, D.C.O.; Menzies, F.M.; Narayanan, U.; Renna, M.; et al. Mammalian Macroautophagy at a Glance. J. Cell Sci. 2009, 122, 1707–1711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravikumar, B.; Sarkar, S.; Davies, J.E.; Futter, M.; Garcia-Arencibia, M.; Green-Thompson, Z.W.; Jimenez-Sanchez, M.; Korolchuk, V.I.; Lichtenberg, M.; Luo, S.; et al. Regulation of Mammalian Autophagy in Physiology and Pathophysiology. Physiol. Rev. 2010, 90, 1383–1435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klionsky, D.J.; Abdel-Aziz, A.K.; Abdelfatah, S.; Abdellatif, M.; Abdoli, A.; Abel, S.; Abeliovich, H.; Abildgaard, M.H.; Abudu, Y.P.; Acevedo-Arozena, A.; et al. Guidelines for the Use and Interpretation of Assays for Monitoring Autophagy (4th Edition) 1. Autophagy 2021, 17, 1–382. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Yoshimori, T.; Levine, B. Methods in Mammalian Autophagy Research. Cell 2010, 140, 313–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moulis, M.; Vindis, C. Methods for Measuring Autophagy in Mice. Cells 2017, 6, 14. [Google Scholar] [CrossRef] [Green Version]
- Qian, H.; Chao, X.; Williams, J.; Fulte, S.; Li, T.; Yang, L.; Ding, W.-X. Autophagy in Liver Diseases: A Review. Mol. Asp. Med. 2021, 100973. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.-M.; Ding, W.-X.; Gao, W. Autophagy in the Liver. Hepatology 2008, 47, 1773–1785. [Google Scholar] [CrossRef]
- Ueno, T.; Komatsu, M. Autophagy in the Liver: Functions in Health and Disease. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 170–184. [Google Scholar] [CrossRef]
- Ezaki, J.; Matsumoto, N.; Takeda-Ezaki, M.; Komatsu, M.; Takahashi, K.; Hiraoka, Y.; Taka, H.; Fujimura, T.; Takehana, K.; Yoshida, M.; et al. Liver Autophagy Contributes to the Maintenance of Blood Glucose and Amino Acid Levels. Autophagy 2011, 7, 727–736. [Google Scholar] [CrossRef] [Green Version]
- Madrigal-Matute, J.; Cuervo, A.M. Regulation of Liver Metabolism by Autophagy. Gastroenterology 2016, 150, 328–339. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Guan, K.-L. MTOR as a Central Hub of Nutrient Signalling and Cell Growth. Nat. Cell Biol. 2019, 21, 63–71. [Google Scholar] [CrossRef]
- Rabanal-Ruiz, Y.; Korolchuk, V.I. MTORC1 and Nutrient Homeostasis: The Central Role of the Lysosome. Int. J. Mol. Sci. 2018, 19, 818. [Google Scholar] [CrossRef] [Green Version]
- Cheng, X.; Ma, X.; Zhu, Q.; Song, D.; Ding, X.; Li, L.; Jiang, X.; Wang, X.; Tian, R.; Su, H.; et al. Pacer Is a Mediator of MTORC1 and GSK3-TIP60 Signaling in Regulation of Autophagosome Maturation and Lipid Metabolism. Mol. Cell 2019, 73, 788–802.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Ni, H.-M.; Ding, W.-X. The Double-Edged Sword of MTOR in Autophagy Deficiency Induced-Liver Injury and Tumorigenesis. Autophagy 2019, 15, 1671–1673. [Google Scholar] [CrossRef] [PubMed]
- Ni, H.-M.; Chao, X.; Yang, H.; Deng, F.; Wang, S.; Bai, Q.; Qian, H.; Cui, Y.; Cui, W.; Shi, Y.; et al. Dual Roles of Mammalian Target of Rapamycin in Regulating Liver Injury and Tumorigenesis in Autophagy-Defective Mouse Liver. Hepatology 2019, 70, 2142–2155. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.C.; Guan, K.-L. MTOR: A Pharmacologic Target for Autophagy Regulation. J. Clin. Investig. 2015, 125, 25–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, X.; Paliogiannis, P.; Calvisi, D.F.; Chen, X. Role of the Mammalian Target of Rapamycin Pathway in Liver Cancer: From Molecular Genetics to Targeted Therapies. Hepatology 2021, 73, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Tsai, T.-H.; Chen, E.; Li, L.; Saha, P.; Lee, H.-J.; Huang, L.-S.; Shelness, G.S.; Chan, L.; Chang, B.H.-J. The Constitutive Lipid Droplet Protein PLIN2 Regulates Autophagy in Liver. Autophagy 2017, 13, 1130–1144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bataller, R.; Brenner, D.A. Liver Fibrosis. J. Clin. Investig. 2005, 115, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Roehlen, N.; Crouchet, E.; Baumert, T.F. Liver Fibrosis: Mechanistic Concepts and Therapeutic Perspectives. Cells 2020, 9, 875. [Google Scholar] [CrossRef] [Green Version]
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global Burden of NAFLD and NASH: Trends, Predictions, Risk Factors and Prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20. [Google Scholar] [CrossRef]
- Sumida, Y.; Yoneda, M. Current and Future Pharmacological Therapies for NAFLD/NASH. J. Gastroenterol. 2018, 53, 362–376. [Google Scholar] [CrossRef] [Green Version]
- Hammoutene, A.; Biquard, L.; Lasselin, J.; Kheloufi, M.; Tanguy, M.; Vion, A.-C.; Mérian, J.; Colnot, N.; Loyer, X.; Tedgui, A.; et al. A Defect in Endothelial Autophagy Occurs in Patients with Non-Alcoholic Steatohepatitis and Promotes Inflammation and Fibrosis. J. Hepatol. 2020, 72, 528–538. [Google Scholar] [CrossRef] [PubMed]
- Ruart, M.; Chavarria, L.; Campreciós, G.; Suárez-Herrera, N.; Montironi, C.; Guixé-Muntet, S.; Bosch, J.; Friedman, S.L.; Garcia-Pagán, J.C.; Hernández-Gea, V. Impaired Endothelial Autophagy Promotes Liver Fibrosis by Aggravating the Oxidative Stress Response during Acute Liver Injury. J. Hepatol. 2019, 70, 458–469. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Xin, T.; Li, D.; Wang, C.; Zhu, H.; Zhou, H. Therapeutic Effect of Sirtuin 3 on Ameliorating Nonalcoholic Fatty Liver Disease: The Role of the ERK-CREB Pathway and Bnip3-Mediated Mitophagy. Redox Biol. 2018, 18, 229–243. [Google Scholar] [CrossRef] [PubMed]
- Challa, T.D.; Wueest, S.; Lucchini, F.C.; Dedual, M.; Modica, S.; Borsigova, M.; Wolfrum, C.; Blüher, M.; Konrad, D. Liver ASK1 Protects from Non-Alcoholic Fatty Liver Disease and Fibrosis. EMBO Mol. Med. 2019, 11, e10124. [Google Scholar] [CrossRef]
- Zhao, J.; Peng, L.; Cui, R.; Guo, X.; Yan, M. Dimethyl α-Ketoglutarate Reduces CCl4-Induced Liver Fibrosis through Inhibition of Autophagy in Hepatic Stellate Cells. Biochem. Biophys. Res. Commun. 2016, 481, 90–96. [Google Scholar] [CrossRef]
- Gao, J.; Wei, B.; de Assuncao, T.M.; Liu, Z.; Hu, X.; Ibrahim, S.; Cooper, S.A.; Cao, S.; Shah, V.H.; Kostallari, E. Hepatic Stellate Cell Autophagy Inhibits Extracellular Vesicle Release to Attenuate Liver Fibrosis. J. Hepatol. 2020, 73, 1144–1154. [Google Scholar] [CrossRef]
- Liu, P.; de la Vega, M.R.; Dodson, M.; Yue, F.; Shi, B.; Fang, D.; Chapman, E.; Liu, L.; Zhang, D.D. Spermidine Confers Liver Protection by Enhancing NRF2 Signaling Through a MAP1S-Mediated Noncanonical Mechanism. Hepatology 2019, 70, 372–388. [Google Scholar] [CrossRef]
- Yue, F.; Li, W.; Zou, J.; Jiang, X.; Xu, G.; Huang, H.; Liu, L. Spermidine Prolongs Lifespan and Prevents Liver Fibrosis and Hepatocellular Carcinoma by Activating MAP1S-Mediated Autophagy. Cancer Res. 2017, 77, 2938–2951. [Google Scholar] [CrossRef] [Green Version]
- Liu, N.; Feng, J.; Lu, X.; Yao, Z.; Liu, Q.; Lv, Y.; Han, Y.; Deng, J.; Zhou, Y. Isorhamnetin Inhibits Liver Fibrosis by Reducing Autophagy and Inhibiting Extracellular Matrix Formation via the TGF-Β1/Smad3 and TGF-Β1/P38 MAPK Pathways. Mediat. Inflamm. 2019, 2019, 6175091. [Google Scholar] [CrossRef] [Green Version]
- Wan, J.; Weiss, E.; Ben Mkaddem, S.; Mabire, M.; Choinier, P.-M.; Picq, O.; Thibault-Sogorb, T.; Hegde, P.; Pishvaie, D.; Bens, M.; et al. LC3-Associated Phagocytosis Protects against Inflammation and Liver Fibrosis via Immunoreceptor Inhibitory Signaling. Sci. Transl. Med. 2020, 12, eaaw8523. [Google Scholar] [CrossRef]
- Wang, H.Y.; Li, C.; Liu, W.H.; Deng, F.M.; Ma, Y.; Guo, L.N.; Kong, D.H.; Hu, K.A.; Liu, Q.; Wu, J.; et al. Autophagy Inhibition via Becn1 Downregulation Improves the Mesenchymal Stem Cells Antifibrotic Potential in Experimental Liver Fibrosis. J. Cell Physiol. 2020, 235, 2722–2737. [Google Scholar] [CrossRef] [Green Version]
- Strazzabosco, M. Transport Systems in Cholangiocytes: Their Role in Bile Formation and Cholestasis. Yale J. Biol. Med. 1997, 70, 427–434. [Google Scholar]
- Pastore, N.; Vainshtein, A.; Herz, N.J.; Huynh, T.; Brunetti, L.; Klisch, T.J.; Mutarelli, M.; Annunziata, P.; Kinouchi, K.; Brunetti-Pierri, N.; et al. Nutrient-Sensitive Transcription Factors TFEB and TFE3 Couple Autophagy and Metabolism to the Peripheral Clock. EMBO J. 2019, 38, e101347. [Google Scholar] [CrossRef]
- Arduini, A.; Serviddio, G.; Tormos, A.M.; Monsalve, M.; Sastre, J. Mitochondrial Dysfunction in Cholestatic Liver Diseases. Front. Biosci. (Elite Ed.) 2012, 4, 2233–2252. [Google Scholar] [CrossRef] [PubMed]
- Woolbright, B.L.; Jaeschke, H. Measuring Apoptosis and Necrosis in Cholestatic Liver Injury. Methods Mol. Biol. 2019, 1981, 133–147. [Google Scholar] [CrossRef] [PubMed]
- Pinzani, M.; Luong, T.V. Pathogenesis of Biliary Fibrosis. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 1279–1283. [Google Scholar] [CrossRef] [PubMed]
- Malhi, H.; Kaufman, R.J. Endoplasmic Reticulum Stress in Liver Disease. J. Hepatol. 2011, 54, 795–809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.; Han, S.Y.; Yu, K.S.; Han, D.; Ahn, H.-J.; Jo, J.-E.; Kim, J.-H.; Shin, J.; Park, H.-W. Impaired Autophagy Promotes Bile Acid-Induced Hepatic Injury and Accumulation of Ubiquitinated Proteins. Biochem. Biophys. Res. Commun. 2018, 495, 1541–1547. [Google Scholar] [CrossRef] [PubMed]
- Fickert, P.; Trauner, M.; Fuchsbichler, A.; Stumptner, C.; Zatloukal, K.; Denk, H. Bile Acid-Induced Mallory Body Formation in Drug-Primed Mouse Liver. Am. J. Pathol. 2002, 161, 2019–2026. [Google Scholar] [CrossRef] [Green Version]
- Harada, M.; Hanada, S.; Toivola, D.M.; Ghori, N.; Omary, M.B. Autophagy Activation by Rapamycin Eliminates Mouse Mallory-Denk Bodies and Blocks Their Proteasome Inhibitor-Mediated Formation. Hepatology 2008, 47, 2026–2035. [Google Scholar] [CrossRef]
- Carlson, J.A.; Rogers, B.B.; Sifers, R.N.; Finegold, M.J.; Clift, S.M.; DeMayo, F.J.; Bullock, D.W.; Woo, S.L. Accumulation of PiZ Alpha 1-Antitrypsin Causes Liver Damage in Transgenic Mice. J. Clin. Investig. 1989, 83, 1183–1190. [Google Scholar] [CrossRef] [Green Version]
- Teckman, J.H.; Perlmutter, D.H. Retention of Mutant Alpha(1)-Antitrypsin Z in Endoplasmic Reticulum Is Associated with an Autophagic Response. Am. J. Physiol. Gastrointest. Liver Physiol. 2000, 279, G821–G974. [Google Scholar] [CrossRef]
- Greene, C.M.; Marciniak, S.J.; Teckman, J.; Ferrarotti, I.; Brantly, M.L.; Lomas, D.A.; Stoller, J.K.; McElvaney, N.G. A1-Antitrypsin Deficiency. Nat. Rev. Dis. Primers 2016, 2, 16051. [Google Scholar] [CrossRef]
- Tang, Y.; Fickert, P.; Trauner, M.; Marcus, N.; Blomenkamp, K.; Teckman, J. Autophagy Induced by Exogenous Bile Acids Is Therapeutic in a Model of α-1-AT Deficiency Liver Disease. Am. J. Physiol. Gastrointest. Liver Physiol. 2016, 311, G156–G165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khambu, B.; Li, T.; Yan, S.; Yu, C.; Chen, X.; Goheen, M.; Li, Y.; Lin, J.; Cummings, O.W.; Lee, Y.A.; et al. Hepatic Autophagy Deficiency Compromises Farnesoid X Receptor Functionality and Causes Cholestatic Injury. Hepatology 2019, 69, 2182–2213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, L.; Lv, G.; Guo, X.; Jing, Y.; Han, Z.; Zhang, S.; Sun, K.; Li, R.; Yang, Y.; Wei, L. Activation of Autophagy Protects against Cholestasis-Induced Hepatic Injury. Cell Biosci. 2014, 4, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strnad, P.; Zatloukal, K.; Stumptner, C.; Kulaksiz, H.; Denk, H. Mallory-Denk-Bodies: Lessons from Keratin-Containing Hepatic Inclusion Bodies. Biochim. Biophys. Acta 2008, 1782, 764–774. [Google Scholar] [CrossRef] [Green Version]
- Fickert, P.; Trauner, M.; Fuchsbichler, A.; Stumptner, C.; Zatloukal, K.; Denk, H. Mallory Body Formation in Primary Biliary Cirrhosis Is Associated with Increased Amounts and Abnormal Phosphorylation and Ubiquitination of Cytokeratins. J. Hepatol. 2003, 38, 387–394. [Google Scholar] [CrossRef]
- Luciani, A.; Villella, V.R.; Esposito, S.; Brunetti-Pierri, N.; Medina, D.; Settembre, C.; Gavina, M.; Pulze, L.; Giardino, I.; Pettoello-Mantovani, M.; et al. Defective CFTR Induces Aggresome Formation and Lung Inflammation in Cystic Fibrosis through ROS-Mediated Autophagy Inhibition. Nat. Cell Biol. 2010, 12, 863–875. [Google Scholar] [CrossRef]
- Panzitt, K.; Jungwirth, E.; Krones, E.; Lee, J.M.; Pollheimer, M.; Thallinger, G.G.; Kolb-Lenz, D.; Xiao, R.; Thorell, A.; Trauner, M.; et al. FXR-Dependent Rubicon Induction Impairs Autophagy in Models of Human Cholestasis. J. Hepatol. 2020, 72, 1122–1131. [Google Scholar] [CrossRef] [Green Version]
- Tsai, M.-S.; Lee, H.-M.; Huang, S.-C.; Sun, C.-K.; Chiu, T.-C.; Chen, P.-H.; Lin, Y.-C.; Hung, T.-M.; Lee, P.-H.; Kao, Y.-H. Nerve Growth Factor Induced Farnesoid X Receptor Upregulation Modulates Autophagy Flux and Protects Hepatocytes in Cholestatic Livers. Arch. Biochem. Biophys. 2020, 682, 108281. [Google Scholar] [CrossRef] [PubMed]
- Salas-Silva, S.; Simoni-Nieves, A.; Lopez-Ramirez, J.; Bucio, L.; Gómez-Quiroz, L.E.; Gutiérrez-Ruiz, M.C.; Roma, M.G. Cholangiocyte Death in Ductopenic Cholestatic Cholangiopathies: Mechanistic Basis and Emerging Therapeutic Strategies. Life Sci. 2019, 218, 324–339. [Google Scholar] [CrossRef] [PubMed]
- Pinto, C.; Ninfole, E.; Benedetti, A.; Maroni, L.; Marzioni, M. Aging-Related Molecular Pathways in Chronic Cholestatic Conditions. Front. Med. 2019, 6, 332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindor, K.D.; Gershwin, M.E.; Poupon, R.; Kaplan, M.; Bergasa, N.V.; Heathcote, E.J. American Association for Study of Liver Diseases Primary Biliary Cirrhosis. Hepatology 2009, 50, 291–308. [Google Scholar] [CrossRef] [PubMed]
- Roma, M.G.; Toledo, F.D.; Boaglio, A.C.; Basiglio, C.L.; Crocenzi, F.A.; Sánchez Pozzi, E.J. Ursodeoxycholic Acid in Cholestasis: Linking Action Mechanisms to Therapeutic Applications. Clin. Sci. 2011, 121, 523–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dilger, K.; Hohenester, S.; Winkler-Budenhofer, U.; Bastiaansen, B.A.J.; Schaap, F.G.; Rust, C.; Beuers, U. Effect of Ursodeoxycholic Acid on Bile Acid Profiles and Intestinal Detoxification Machinery in Primary Biliary Cirrhosis and Health. J. Hepatol. 2012, 57, 133–140. [Google Scholar] [CrossRef]
- Wagner, M.; Trauner, M. Recent Advances in Understanding and Managing Cholestasis. F1000Research 2016, 5. [Google Scholar] [CrossRef] [Green Version]
- Banales, J.M.; Huebert, R.C.; Karlsen, T.; Strazzabosco, M.; LaRusso, N.F.; Gores, G.J. Cholangiocyte Pathobiology. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 269–281. [Google Scholar] [CrossRef]
- Amaral, J.D.; Viana, R.J.S.; Ramalho, R.M.; Steer, C.J.; Rodrigues, C.M.P. Bile Acids: Regulation of Apoptosis by Ursodeoxycholic Acid. J. Lipid Res. 2009, 50, 1721–1734. [Google Scholar] [CrossRef] [Green Version]
- Basiglio, C.L.; Mottino, A.D.; Roma, M.G. Tauroursodeoxycholate Counteracts Hepatocellular Lysis Induced by Tensioactive Bile Salts by Preventing Plasma Membrane-Micelle Transition. Chem. Biol. Interact. 2010, 188, 386–392. [Google Scholar] [CrossRef]
- Ozcan, U.; Yilmaz, E.; Ozcan, L.; Furuhashi, M.; Vaillancourt, E.; Smith, R.O.; Görgün, C.Z.; Hotamisligil, G.S. Chemical Chaperones Reduce ER Stress and Restore Glucose Homeostasis in a Mouse Model of Type 2 Diabetes. Science 2006, 313, 1137–1140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Q.; Khaoustov, V.I.; Chung, C.C.; Sohn, J.; Krishnan, B.; Lewis, D.E.; Yoffe, B. Effect of Tauroursodeoxycholic Acid on Endoplasmic Reticulum Stress-Induced Caspase-12 Activation. Hepatology 2002, 36, 592–601. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Dutta, A.; Kresge, C.; Bugde, A.; Feranchak, A.P. Bile Acids Stimulate Cholangiocyte Fluid Secretion by Activation of Transmembrane Member 16A Cl- Channels. Hepatology 2018, 68, 187–199. [Google Scholar] [CrossRef] [Green Version]
- Mueller, M.; Thorell, A.; Claudel, T.; Jha, P.; Koefeler, H.; Lackner, C.; Hoesel, B.; Fauler, G.; Stojakovic, T.; Einarsson, C.; et al. Ursodeoxycholic Acid Exerts Farnesoid X Receptor-Antagonistic Effects on Bile Acid and Lipid Metabolism in Morbid Obesity. J. Hepatol. 2015, 62, 1398–1404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, P.; Zhao, J.; Guo, Y.; Yu, Y.; Wu, X.; Xiao, H. Ursodeoxycholic Acid Alleviates Nonalcoholic Fatty Liver Disease by Inhibiting Apoptosis and Improving Autophagy via Activating AMPK. Biochem. Biophys. Res. Commun. 2020, 529, 834–838. [Google Scholar] [CrossRef]
- Fickert, P.; Fuchsbichler, A.; Wagner, M.; Zollner, G.; Kaser, A.; Tilg, H.; Krause, R.; Lammert, F.; Langner, C.; Zatloukal, K.; et al. Regurgitation of Bile Acids from Leaky Bile Ducts Causes Sclerosing Cholangitis in Mdr2 (Abcb4) Knockout Mice. Gastroenterology 2004, 127, 261–274. [Google Scholar] [CrossRef] [PubMed]
- Fickert, P.; Wagner, M.; Marschall, H.-U.; Fuchsbichler, A.; Zollner, G.; Tsybrovskyy, O.; Zatloukal, K.; Liu, J.; Waalkes, M.P.; Cover, C.; et al. 24-NorUrsodeoxycholic Acid Is Superior to Ursodeoxycholic Acid in the Treatment of Sclerosing Cholangitis in Mdr2 (Abcb4) Knockout Mice. Gastroenterology 2006, 130, 465–481. [Google Scholar] [CrossRef]
- Halilbasic, E.; Fiorotto, R.; Fickert, P.; Marschall, H.-U.; Moustafa, T.; Spirli, C.; Fuchsbichler, A.; Gumhold, J.; Silbert, D.; Zatloukal, K.; et al. Side Chain Structure Determines Unique Physiologic and Therapeutic Properties of Norursodeoxycholic Acid in Mdr2-/- Mice. Hepatology 2009, 49, 1972–1981. [Google Scholar] [CrossRef] [Green Version]
- Moustafa, T.; Fickert, P.; Magnes, C.; Guelly, C.; Thueringer, A.; Frank, S.; Kratky, D.; Sattler, W.; Reicher, H.; Sinner, F.; et al. Alterations in Lipid Metabolism Mediate Inflammation, Fibrosis, and Proliferation in a Mouse Model of Chronic Cholestatic Liver Injury. Gastroenterology 2012, 142, 140–151.e12. [Google Scholar] [CrossRef]
- Sombetzki, M.; Fuchs, C.D.; Fickert, P.; Österreicher, C.H.; Mueller, M.; Claudel, T.; Loebermann, M.; Engelmann, R.; Langner, C.; Sahin, E.; et al. 24-nor-Ursodeoxycholic Acid Ameliorates Inflammatory Response and Liver Fibrosis in a Murine Model of Hepatic Schistosomiasis. J. Hepatol. 2015, 62, 871–878. [Google Scholar] [CrossRef] [Green Version]
- Hofmann, A.F.; Zakko, S.F.; Lira, M.; Clerici, C.; Hagey, L.R.; Lambert, K.K.; Steinbach, J.H.; Schteingart, C.D.; Olinga, P.; Groothuis, G.M.M. Novel Biotransformation and Physiological Properties of Norursodeoxycholic Acid in Humans. Hepatology 2005, 42, 1391–1398. [Google Scholar] [CrossRef]
- Hofmann, A.F. Bile Acids: Trying to Understand Their Chemistry and Biology with the Hope of Helping Patients. Hepatology 2009, 49, 1403–1418. [Google Scholar] [CrossRef]
- Fickert, P.; Hirschfield, G.M.; Denk, G.; Marschall, H.-U.; Altorjay, I.; Färkkilä, M.; Schramm, C.; Spengler, U.; Chapman, R.; Bergquist, A.; et al. NorUrsodeoxycholic Acid Improves Cholestasis in Primary Sclerosing Cholangitis. J. Hepatol. 2017, 67, 549–558. [Google Scholar] [CrossRef] [Green Version]
- Tang, Y.; Blomenkamp, K.S.; Fickert, P.; Trauner, M.; Teckman, J.H. NorUDCA Promotes Degradation of A1-Antitrypsin Mutant Z Protein by Inducing Autophagy through AMPK/ULK1 Pathway. PLoS ONE 2018, 13, e0200897. [Google Scholar] [CrossRef] [PubMed]
- Liang, C. Negative Regulation of Autophagy. Cell Death Differ. 2010, 17, 1807–1815. [Google Scholar] [CrossRef] [Green Version]
- Wagner, M.; Fickert, P. Drug Therapies for Chronic Cholestatic Liver Diseases. Annu. Rev. Pharmacol. Toxicol. 2020, 60, 503–527. [Google Scholar] [CrossRef] [PubMed]
- Wagner, M.; Zollner, G.; Trauner, M. Nuclear Receptors in Liver Disease. Hepatology 2011, 53, 1023–1034. [Google Scholar] [CrossRef] [PubMed]
- Nevens, F.; Andreone, P.; Mazzella, G.; Strasser, S.I.; Bowlus, C.; Invernizzi, P.; Drenth, J.P.H.; Pockros, P.J.; Regula, J.; Beuers, U.; et al. A Placebo-Controlled Trial of Obeticholic Acid in Primary Biliary Cholangitis. N. Engl. J. Med. 2016, 375, 631–643. [Google Scholar] [CrossRef]
- Corpechot, C.; Chazouillères, O.; Rousseau, A.; Le Gruyer, A.; Habersetzer, F.; Mathurin, P.; Goria, O.; Potier, P.; Minello, A.; Silvain, C.; et al. A Placebo-Controlled Trial of Bezafibrate in Primary Biliary Cholangitis. N. Engl. J. Med. 2018, 378, 2171–2181. [Google Scholar] [CrossRef]
- Massafra, V.; van Mil, S.W.C. Farnesoid X Receptor: A “Homeostat” for Hepatic Nutrient Metabolism. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 45–59. [Google Scholar] [CrossRef]
- Manley, S.; Ni, H.-M.; Kong, B.; Apte, U.; Guo, G.; Ding, W.-X. Suppression of Autophagic Flux by Bile Acids in Hepatocytes. Toxicol. Sci. Off. J. Soc. Toxicol. 2014, 137, 478–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Gunewardena, S.; Li, F.; Matye, D.J.; Chen, C.; Chao, X.; Jung, T.; Zhang, Y.; Czerwiński, M.; Ni, H.-M.; et al. An FGF15/19-TFEB Regulatory Loop Controls Hepatic Cholesterol and Bile Acid Homeostasis. Nat. Commun. 2020, 11, 3612. [Google Scholar] [CrossRef]
- Deutschmann, K.; Reich, M.; Klindt, C.; Dröge, C.; Spomer, L.; Häussinger, D.; Keitel, V. Bile Acid Receptors in the Biliary Tree: TGR5 in Physiology and Disease. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 1319–1325. [Google Scholar] [CrossRef]
- Chiang, J.Y.L.; Ferrell, J.M. Bile Acids as Metabolic Regulators and Nutrient Sensors. Annu. Rev. Nutr. 2019, 39, 175–200. [Google Scholar] [CrossRef]
- Perino, A.; Demagny, H.; Velazquez-Villegas, L.; Schoonjans, K. Molecular Physiology of Bile Acid Signaling in Health, Disease, and Aging. Physiol. Rev. 2021, 101, 683–731. [Google Scholar] [CrossRef]
- Lee, J.M.; Wagner, M.; Xiao, R.; Kim, K.H.; Feng, D.; Lazar, M.A.; Moore, D.D. Nutrient-Sensing Nuclear Receptors Coordinate Autophagy. Nature 2014, 516, 112–115. [Google Scholar] [CrossRef]
- Seok, S.; Fu, T.; Choi, S.-E.; Li, Y.; Zhu, R.; Kumar, S.; Sun, X.; Yoon, G.; Kang, Y.; Zhong, W.; et al. Transcriptional Regulation of Autophagy by an FXR-CREB Axis. Nature 2014, 516, 108–111. [Google Scholar] [CrossRef] [Green Version]
- Chawla, A.; Repa, J.J.; Evans, R.M.; Mangelsdorf, D.J. Nuclear Receptors and Lipid Physiology: Opening the X-Files. Science 2001, 294, 1866–1870. [Google Scholar] [CrossRef] [Green Version]
- Lien, F.; Berthier, A.; Bouchaert, E.; Gheeraert, C.; Alexandre, J.; Porez, G.; Prawitt, J.; Dehondt, H.; Ploton, M.; Colin, S.; et al. Metformin Interferes with Bile Acid Homeostasis through AMPK-FXR Crosstalk. J. Clin. Investig. 2014, 124, 1037–1051. [Google Scholar] [CrossRef] [Green Version]
- Noh, K.; Kim, Y.M.; Kim, Y.W.; Kim, S.G. Farnesoid X Receptor Activation by Chenodeoxycholic Acid Induces Detoxifying Enzymes through AMP-Activated Protein Kinase and Extracellular Signal-Regulated Kinase 1/2-Mediated Phosphorylation of CCAAT/Enhancer Binding Protein β. Drug Metab. Dispos. 2011, 39, 1451–1459. [Google Scholar] [CrossRef] [Green Version]
- Ferdinandusse, S.; Houten, S.M. Peroxisomes and Bile Acid Biosynthesis. Biochim. Biophys. Acta 2006, 1763, 1427–1440. [Google Scholar] [CrossRef] [Green Version]
- Sheedfar, F.; Di Biase, S.; Koonen, D.; Vinciguerra, M. Liver Diseases and Aging: Friends or Foes? Aging Cell 2013, 12, 950–954. [Google Scholar] [CrossRef] [PubMed]
- Pibiri, M. Liver Regeneration in Aged Mice: New Insights. Aging 2018, 10, 1801–1824. [Google Scholar] [CrossRef] [PubMed]
- Pinto, C.; Ninfole, E.; Gaggiano, L.; Benedetti, A.; Marzioni, M.; Maroni, L. Aging and the Biological Response to Liver Injury. Semin. Liver Dis. 2020, 40, 225–232. [Google Scholar] [CrossRef]
- Hunt, N.J.; Kang, S.W.S.; Lockwood, G.P.; Le Couteur, D.G.; Cogger, V.C. Hallmarks of Aging in the Liver. Comput. Struct. Biotechnol. J. 2019, 17, 1151–1161. [Google Scholar] [CrossRef]
- Kim, I.H.; Kisseleva, T.; Brenner, D.A. Aging and Liver Disease. Curr. Opin. Gastroenterol. 2015, 31, 184–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, D.; Li, J.; Chen, K.; Yin, Y.; Fang, Z.; Pang, H.; Rong, X.; Guo, J. Study on Metabolic Trajectory of Liver Aging and the Effect of Fufang Zhenzhu Tiaozhi on Aging Mice. Front. Pharmacol. 2019, 10, 926. [Google Scholar] [CrossRef]
- Nguyen, P.; Valanejad, L.; Cast, A.; Wright, M.; Garcia, J.M.; El-Serag, H.B.; Karns, R.; Timchenko, N.A. Elimination of Age-Associated Hepatic Steatosis and Correction of Aging Phenotype by Inhibition of Cdk4-C/EBPα-P300 Axis. Cell Rep. 2018, 24, 1597–1609. [Google Scholar] [CrossRef] [Green Version]
- Niccoli, T.; Partridge, L. Ageing as a Risk Factor for Disease. Curr. Biol. 2012, 22, R741–R752. [Google Scholar] [CrossRef] [Green Version]
- Cheung, A.C.; Lammers, W.J.; Murillo Perez, C.F.; van Buuren, H.R.; Gulamhusein, A.; Trivedi, P.J.; Lazaridis, K.N.; Ponsioen, C.Y.; Floreani, A.; Hirschfield, G.M.; et al. Effects of Age and Sex of Response to Ursodeoxycholic Acid and Transplant-Free Survival in Patients With Primary Biliary Cholangitis. Clin. Gastroenterol. Hepatol. 2019, 17, 2076–2084.e2. [Google Scholar] [CrossRef]
- Weismüller, T.J.; Trivedi, P.J.; Bergquist, A.; Imam, M.; Lenzen, H.; Ponsioen, C.Y.; Holm, K.; Gotthardt, D.; Färkkilä, M.A.; Marschall, H.-U.; et al. Patient Age, Sex, and Inflammatory Bowel Disease Phenotype Associate with Course of Primary Sclerosing Cholangitis. Gastroenterology 2017, 152, 1975–1984.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Randall, H.B.; Cao, S.; deVera, M.E. Transplantation in Elderly Patients. Arch. Surg. 2003, 138, 1089–1092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tabibian, J.H.; O’Hara, S.P.; Splinter, P.L.; Trussoni, C.E.; LaRusso, N.F. Cholangiocyte Senescence by Way of N-Ras Activation Is a Characteristic of Primary Sclerosing Cholangitis. Hepatology 2014, 59, 2263–2275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, A.; Guo, E.; Yang, J.; Yang, Y.; Liu, S.; Jiang, X.; Hu, Q.; Dirsch, O.; Dahmen, U.; Zhang, C.; et al. Young Plasma Reverses Age-Dependent Alterations in Hepatic Function through the Restoration of Autophagy. Aging Cell 2018, 17. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Cai, M.; Wang, J.; Gao, Q.; Guo, X.; Jia, X.; Xu, S.; Zhu, H. Decreased Ovarian Function and Autophagy Gene Methylation in Aging Rats. J. Ovarian. Res. 2020, 13, 12. [Google Scholar] [CrossRef]
- Lipinski, M.M.; Zheng, B.; Lu, T.; Yan, Z.; Py, B.F.; Ng, A.; Xavier, R.J.; Li, C.; Yankner, B.A.; Scherzer, C.R.; et al. Genome-Wide Analysis Reveals Mechanisms Modulating Autophagy in Normal Brain Aging and in Alzheimer’s Disease. Proc. Natl. Acad. Sci. USA 2010, 107, 14164–14169. [Google Scholar] [CrossRef] [Green Version]
- Uddin, M.N.; Nishio, N.; Ito, S.; Suzuki, H.; Isobe, K. Autophagic Activity in Thymus and Liver during Aging. Age 2012, 34, 75–85. [Google Scholar] [CrossRef] [Green Version]
- Chang, J.T.; Kumsta, C.; Hellman, A.B.; Adams, L.M.; Hansen, M. Spatiotemporal Regulation of Autophagy during Caenorhabditis Elegans Aging. Elife 2017, 6, e18459. [Google Scholar] [CrossRef]
- Donati, A.; Cavallini, G.; Paradiso, C.; Vittorini, S.; Pollera, M.; Gori, Z.; Bergamini, E. Age-Related Changes in the Autophagic Proteolysis of Rat Isolated Liver Cells: Effects of Antiaging Dietary Restrictions. J. Gerontol. A Biol. Sci. Med. Sci. 2001, 56, B375–B383. [Google Scholar] [CrossRef] [Green Version]
- Simonsen, A.; Cumming, R.C.; Brech, A.; Isakson, P.; Schubert, D.R.; Finley, K.D. Promoting Basal Levels of Autophagy in the Nervous System Enhances Longevity and Oxidant Resistance in Adult Drosophila. Autophagy 2008, 4, 176–184. [Google Scholar] [CrossRef] [Green Version]
- Meléndez, A.; Tallóczy, Z.; Seaman, M.; Eskelinen, E.-L.; Hall, D.H.; Levine, B. Autophagy Genes Are Essential for Dauer Development and Life-Span Extension in C. elegans. Science 2003, 301, 1387–1391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lapierre, L.R.; Gelino, S.; Meléndez, A.; Hansen, M. Autophagy and Lipid Metabolism Coordinately Modulate Life Span in Germline-Less C. Elegans. Curr. Biol. 2011, 21, 1507–1514. [Google Scholar] [CrossRef] [Green Version]
- Hansen, M.; Chandra, A.; Mitic, L.L.; Onken, B.; Driscoll, M.; Kenyon, C. A Role for Autophagy in the Extension of Lifespan by Dietary Restriction in C. Elegans. PLoS Genet. 2008, 4, e24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tóth, M.L.; Sigmond, T.; Borsos, E.; Barna, J.; Erdélyi, P.; Takács-Vellai, K.; Orosz, L.; Kovács, A.L.; Csikós, G.; Sass, M.; et al. Longevity Pathways Converge on Autophagy Genes to Regulate Life Span in Caenorhabditis Elegans. Autophagy 2008, 4, 330–338. [Google Scholar] [CrossRef] [Green Version]
- Jia, K.; Levine, B. Autophagy Is Required for Dietary Restriction-Mediated Life Span Extension in C. Elegans. Autophagy 2007, 3, 597–599. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, T.; Takabatake, Y.; Kimura, T.; Takahashi, A.; Namba, T.; Matsuda, J.; Minami, S.; Kaimori, J.-Y.; Matsui, I.; Kitamura, H.; et al. Time-Dependent Dysregulation of Autophagy: Implications in Aging and Mitochondrial Homeostasis in the Kidney Proximal Tubule. Autophagy 2016, 12, 801–813. [Google Scholar] [CrossRef] [Green Version]
- Eisenberg, T.; Abdellatif, M.; Schroeder, S.; Primessnig, U.; Stekovic, S.; Pendl, T.; Harger, A.; Schipke, J.; Zimmermann, A.; Schmidt, A.; et al. Cardioprotection and Lifespan Extension by the Natural Polyamine Spermidine. Nat. Med. 2016, 22, 1428–1438. [Google Scholar] [CrossRef]
- Pyo, J.-O.; Yoo, S.-M.; Ahn, H.-H.; Nah, J.; Hong, S.-H.; Kam, T.-I.; Jung, S.; Jung, Y.-K. Overexpression of Atg5 in Mice Activates Autophagy and Extends Lifespan. Nat. Commun. 2013, 4, 2300. [Google Scholar] [CrossRef] [Green Version]
- Eisenberg, T.; Knauer, H.; Schauer, A.; Büttner, S.; Ruckenstuhl, C.; Carmona-Gutierrez, D.; Ring, J.; Schroeder, S.; Magnes, C.; Antonacci, L.; et al. Induction of Autophagy by Spermidine Promotes Longevity. Nat. Cell. Biol. 2009, 11, 1305–1314. [Google Scholar] [CrossRef]
- Mercken, E.M.; Hu, J.; Krzysik-Walker, S.; Wei, M.; Li, Y.; McBurney, M.W.; de Cabo, R.; Longo, V.D. SIRT1 but Not Its Increased Expression Is Essential for Lifespan Extension in Caloric-Restricted Mice. Aging Cell 2014, 13, 193–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madeo, F.; Zimmermann, A.; Maiuri, M.C.; Kroemer, G. Essential Role for Autophagy in Life Span Extension. J. Clin. Investig. 2015, 125, 85–93. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Budanov, A.V.; Park, E.J.; Birse, R.; Kim, T.E.; Perkins, G.A.; Ocorr, K.; Ellisman, M.H.; Bodmer, R.; Bier, E.; et al. Sestrin as a Feedback Inhibitor of TOR That Prevents Age-Related Pathologies. Science 2010, 327, 1223–1228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Kreutzenberg, S.V.; Ceolotto, G.; Papparella, I.; Bortoluzzi, A.; Semplicini, A.; Dalla Man, C.; Cobelli, C.; Fadini, G.P.; Avogaro, A. Downregulation of the Longevity-Associated Protein Sirtuin 1 in Insulin Resistance and Metabolic Syndrome: Potential Biochemical Mechanisms. Diabetes 2010, 59, 1006–1015. [Google Scholar] [CrossRef] [Green Version]
- Caramés, B.; Taniguchi, N.; Otsuki, S.; Blanco, F.J.; Lotz, M. Autophagy Is a Protective Mechanism in Normal Cartilage, and Its Aging-Related Loss Is Linked with Cell Death and Osteoarthritis. Arthritis Rheum. 2010, 62, 791–801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubinsztein, D.C.; Mariño, G.; Kroemer, G. Autophagy and Aging. Cell 2011, 146, 682–695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inami, Y.; Yamashina, S.; Izumi, K.; Ueno, T.; Tanida, I.; Ikejima, K.; Watanabe, S. Hepatic Steatosis Inhibits Autophagic Proteolysis via Impairment of Autophagosomal Acidification and Cathepsin Expression. Biochem. Biophys. Res. Commun. 2011, 412, 618–625. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Kaushik, S.; Wang, Y.; Xiang, Y.; Novak, I.; Komatsu, M.; Tanaka, K.; Cuervo, A.M.; Czaja, M.J. Autophagy Regulates Lipid Metabolism. Nature 2009, 458, 1131–1135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Han, S.-L.; Lu, D.-L.; Li, L.-Y.; Limbu, S.M.; Li, D.-L.; Zhang, M.-L.; Du, Z.-Y. Inhibited Lipophagy Suppresses Lipid Metabolism in Zebrafish Liver Cells. Front. Physiol. 2019, 10, 1077. [Google Scholar] [CrossRef] [PubMed]
- Nassir, F.; Ibdah, J.A. Role of Mitochondria in Nonalcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2014, 15, 8713–8742. [Google Scholar] [CrossRef] [Green Version]
- Ogrodnik, M.; Miwa, S.; Tchkonia, T.; Tiniakos, D.; Wilson, C.L.; Lahat, A.; Day, C.P.; Burt, A.; Palmer, A.; Anstee, Q.M.; et al. Cellular Senescence Drives Age-Dependent Hepatic Steatosis. Nat. Commun. 2017, 8, 15691. [Google Scholar] [CrossRef]
- Cao, L.; Quan, X.-B.; Zeng, W.-J.; Yang, X.-O.; Wang, M.-J. Mechanism of Hepatocyte Apoptosis. J. Cell Death 2016, 9, 19–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koek, G.H.; Liedorp, P.R.; Bast, A. The Role of Oxidative Stress in Non-Alcoholic Steatohepatitis. Clin. Chim. Acta 2011, 412, 1297–1305. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Tian, R.; She, Z.; Cai, J.; Li, H. Role of Oxidative Stress in the Pathogenesis of Nonalcoholic Fatty Liver Disease. Free Radic. Biol. Med. 2020, 152, 116–141. [Google Scholar] [CrossRef] [PubMed]
- Sun, N.; Yun, J.; Liu, J.; Malide, D.; Liu, C.; Rovira, I.I.; Holmström, K.M.; Fergusson, M.M.; Yoo, Y.H.; Combs, C.A.; et al. Measuring In Vivo Mitophagy. Mol. Cell 2015, 60, 685–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stahl, E.C.; Haschak, M.J.; Popovic, B.; Brown, B.N. Macrophages in the Aging Liver and Age-Related Liver Disease. Front. Immunol. 2018, 9, 2795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thoen, L.F.R.; Guimarães, E.L.M.; Dollé, L.; Mannaerts, I.; Najimi, M.; Sokal, E.; van Grunsven, L.A. A Role for Autophagy during Hepatic Stellate Cell Activation. J. Hepatol. 2011, 55, 1353–1360. [Google Scholar] [CrossRef]
- Testerink, N.; Ajat, M.; Houweling, M.; Brouwers, J.F.; Pully, V.V.; van Manen, H.-J.; Otto, C.; Helms, J.B.; Vaandrager, A.B. Replacement of Retinyl Esters by Polyunsaturated Triacylglycerol Species in Lipid Droplets of Hepatic Stellate Cells during Activation. PLoS ONE 2012, 7, e34945. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.; Zhao, Y.; Wang, F.; Tao, L.; Xiao, J.; Yang, C. Autophagy in Hepatic Fibrosis. BioMed Res. Int. 2014, 2014, 436242. [Google Scholar] [CrossRef] [Green Version]
- Ding, W.-X.; Manley, S.; Ni, H.-M. The Emerging Role of Autophagy in Alcoholic Liver Disease. Exp. Biol. Med. 2011, 236, 546–556. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Wu, J.; Frizell, E.; Liu, S.L.; Bashey, R.; Rubin, R.; Norton, P.; Zern, M.A. Rapamycin Inhibits Hepatic Stellate Cell Proliferation in Vitro and Limits Fibrogenesis in an in Vivo Model of Liver Fibrosis. Gastroenterology 1999, 117, 1198–1204. [Google Scholar] [CrossRef]
- Martinez-Lopez, N.; Singh, R. Autophagy and Lipid Droplets in the Liver. Annu. Rev. Nutr. 2015, 35, 215–237. [Google Scholar] [CrossRef]
- Czaja, M.J.; Ding, W.-X.; Donohue, T.M.; Friedman, S.L.; Kim, J.-S.; Komatsu, M.; Lemasters, J.J.; Lemoine, A.; Lin, J.D.; Ou, J.J.; et al. Functions of Autophagy in Normal and Diseased Liver. Autophagy 2013, 9, 1131–1158. [Google Scholar] [CrossRef] [Green Version]
- Schneider, J.L.; Cuervo, A.M. Liver Autophagy: Much More than Just Taking out the Trash. Nat. Rev. Gastroenterol. Hepatol. 2014, 11, 187–200. [Google Scholar] [CrossRef] [Green Version]
- Komatsu, M.; Waguri, S.; Ueno, T.; Iwata, J.; Murata, S.; Tanida, I.; Ezaki, J.; Mizushima, N.; Ohsumi, Y.; Uchiyama, Y.; et al. Impairment of Starvation-Induced and Constitutive Autophagy in Atg7-Deficient Mice. J. Cell Biol. 2005, 169, 425–434. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Cuervo, A.M. Autophagy in the Cellular Energetic Balance. Cell Metab. 2011, 13, 495–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salminen, A.; Kaarniranta, K. AMP-Activated Protein Kinase (AMPK) Controls the Aging Process via an Integrated Signaling Network. Ageing Res. Rev. 2012, 11, 230–241. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Kaarniranta, K.; Kauppinen, A. Age-Related Changes in AMPK Activation: Role for AMPK Phosphatases and Inhibitory Phosphorylation by Upstream Signaling Pathways. Ageing Res. Rev. 2016, 28, 15–26. [Google Scholar] [CrossRef]
- Blagosklonny, M.V. TOR-Driven Aging: Speeding Car without Brakes. Cell Cycle 2009, 8, 4055–4059. [Google Scholar] [CrossRef] [Green Version]
- Khalil, H.; Tazi, M.; Caution, K.; Ahmed, A.; Kanneganti, A.; Assani, K.; Kopp, B.; Marsh, C.; Dakhlallah, D.; Amer, A.O. Aging Is Associated with Hypermethylation of Autophagy Genes in Macrophages. Epigenetics 2016, 11, 381–388. [Google Scholar] [CrossRef] [Green Version]
- Double, K.L.; Dedov, V.N.; Fedorow, H.; Kettle, E.; Halliday, G.M.; Garner, B.; Brunk, U.T. The Comparative Biology of Neuromelanin and Lipofuscin in the Human Brain. Cell Mol. Life Sci. 2008, 65, 1669–1682. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Hua, C.; Tautenhahn, H.-M.; Dirsch, O.; Dahmen, U. The Role of Autophagy for the Regeneration of the Aging Liver. Int. J. Mol. Sci. 2020, 21, 3606. [Google Scholar] [CrossRef]
- Terman, A.; Brunk, U.T. Autophagy in Cardiac Myocyte Homeostasis, Aging, and Pathology. Cardiovasc. Res. 2005, 68, 355–365. [Google Scholar] [CrossRef] [Green Version]
- Brunk, U.T.; Terman, A. The Mitochondrial-Lysosomal Axis Theory of Aging: Accumulation of Damaged Mitochondria as a Result of Imperfect Autophagocytosis. Eur. J. Biochem. 2002, 269, 1982–2002. [Google Scholar] [CrossRef]
- Cuervo, A.M.; Bergamini, E.; Brunk, U.T.; Dröge, W.; Ffrench, M.; Terman, A. Autophagy and Aging: The Importance of Maintaining “Clean” Cells. Autophagy 2005, 1, 131–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreno-García, A.; Kun, A.; Calero, O.; Medina, M.; Calero, M. An Overview of the Role of Lipofuscin in Age-Related Neurodegeneration. Front. Neurosci. 2018, 12, 464. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Chen, Y.; Peng, T.; Li, L.; Zhu, W.; Liu, F.; Liu, S.; An, X.; Luo, R.; Cheng, J.; et al. Mitochondrial ROS-Induced Lysosomal Dysfunction Impairs Autophagic Flux and Contributes to M1 Macrophage Polarization in a Diabetic Condition. Clin. Sci. 2019, 133, 1759–1777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernández, Á.F.; Sebti, S.; Wei, Y.; Zou, Z.; Shi, M.; McMillan, K.L.; He, C.; Ting, T.; Liu, Y.; Chiang, W.-C.; et al. Disruption of the Beclin 1-BCL2 Autophagy Regulatory Complex Promotes Longevity in Mice. Nature 2018, 558, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Kuro-o, M. Klotho and Aging. Biochim. Biophys. Acta 2009, 1790, 1049–1058. [Google Scholar] [CrossRef] [PubMed]
- Kuro-o, M.; Matsumura, Y.; Aizawa, H.; Kawaguchi, H.; Suga, T.; Utsugi, T.; Ohyama, Y.; Kurabayashi, M.; Kaname, T.; Kume, E.; et al. Mutation of the Mouse Klotho Gene Leads to a Syndrome Resembling Ageing. Nature 1997, 390, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Tsujikawa, H.; Kurotaki, Y.; Fujimori, T.; Fukuda, K.; Nabeshima, Y.-I. Klotho, a Gene Related to a Syndrome Resembling Human Premature Aging, Functions in a Negative Regulatory Circuit of Vitamin D Endocrine System. Mol. Endocrinol. 2003, 17, 2393–2403. [Google Scholar] [CrossRef] [PubMed]
- Kurosu, H.; Yamamoto, M.; Clark, J.D.; Pastor, J.V.; Nandi, A.; Gurnani, P.; McGuinness, O.P.; Chikuda, H.; Yamaguchi, M.; Kawaguchi, H.; et al. Suppression of Aging in Mice by the Hormone Klotho. Science 2005, 309, 1829–1833. [Google Scholar] [CrossRef] [Green Version]
- Chen, T.-H.; Kuro-o, M.; Chen, C.-H.; Sue, Y.-M.; Chen, Y.-C.; Wu, H.-H.; Cheng, C.-Y. The Secreted Klotho Protein Restores Phosphate Retention and Suppresses Accelerated Aging in Klotho Mutant Mice. Eur. J. Pharmacol. 2013, 698, 67–73. [Google Scholar] [CrossRef]
- Shi, M.; Flores, B.; Gillings, N.; Bian, A.; Cho, H.J.; Yan, S.; Liu, Y.; Levine, B.; Moe, O.W.; Hu, M.C. AKlotho Mitigates Progression of AKI to CKD through Activation of Autophagy. J. Am. Soc. Nephrol. 2016, 27, 2331–2345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsunaga, K.; Saitoh, T.; Tabata, K.; Omori, H.; Satoh, T.; Kurotori, N.; Maejima, I.; Shirahama-Noda, K.; Ichimura, T.; Isobe, T.; et al. Two Beclin 1-Binding Proteins, Atg14L and Rubicon, Reciprocally Regulate Autophagy at Different Stages. Nat. Cell Biol. 2009, 11, 385–396. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, S.; Oba, M.; Suzuki, M.; Takahashi, A.; Yamamuro, T.; Fujiwara, M.; Ikenaka, K.; Minami, S.; Tabata, N.; Yamamoto, K.; et al. Suppression of Autophagic Activity by Rubicon Is a Signature of Aging. Nat. Commun. 2019, 10, 847. [Google Scholar] [CrossRef]
- Tanaka, S.; Hikita, H.; Tatsumi, T.; Sakamori, R.; Nozaki, Y.; Sakane, S.; Shiode, Y.; Nakabori, T.; Saito, Y.; Hiramatsu, N.; et al. Rubicon Inhibits Autophagy and Accelerates Hepatocyte Apoptosis and Lipid Accumulation in Nonalcoholic Fatty Liver Disease in Mice. Hepatology 2016, 64, 1994–2014. [Google Scholar] [CrossRef]
- Singh, R. Autophagy and Regulation of Lipid Metabolism. Results Probl. Cell Differ. 2010, 52, 35–46. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Fang, F.; Goldstein, J.L.; Brown, M.S.; Zhao, T.-J. Reduced Autophagy in Livers of Fasted, Fat-Depleted, Ghrelin-Deficient Mice: Reversal by Growth Hormone. Proc. Natl. Acad. Sci. USA 2015, 112, 1226–1231. [Google Scholar] [CrossRef] [Green Version]
- He, Q.; Sha, S.; Sun, L.; Zhang, J.; Dong, M. GLP-1 Analogue Improves Hepatic Lipid Accumulation by Inducing Autophagy via AMPK/MTOR Pathway. Biochem. Biophys. Res. Commun. 2016, 476, 182–203. [Google Scholar] [CrossRef]

{kind=link}
| Overview of the Autophagy Process | References |
|---|---|
| Two types of autophagy | |
| Selective | [3,5] |
| Non-selective | [5] |
| Investigation of autophagy | |
| Physical methods | |
| Transmission electron microscopy | [31] |
| Biochemical methods | |
| Fluorescence microscopy | [32] |
| Immunoblotting, SDS-PAGE | [31] |
| Drug | Effects | Ref |
|---|---|---|
| UDCA | ↓ BA pool hydrophobicity | [86,87] |
| ↑ Bile flow and bicarbonate secretion | [88] | |
| Rebalances autophagic response | [94] | |
| ↑ Autophagy ↓ Apoptosis | [95] | |
| UDCA & TUDCA | ↓ Apoptosis | [89] |
| ↓ Necrotic properties | [90] | |
| ↓ ER stress | [91] | |
 Casp-12 activation Casp-12 activation | [92] | |
| norUDCA | ↓ Inflammation, fibrosis and proliferation | [98] |
| ↑ Autophagy via AMPK | [104] | |
| ↓ ATZ globules in PIZZ mice ↑ Ameliorates parameters | [74] | |
| OCA | ↓ BA synthesis | [107] |
| Impairs autophagic flux | [80] | |
| Overcomes adverse effects of reduced autophagy | [108] | |
| Fibrates | ↑ Autophagy | [106,109] |
| Relevance of Autophagy | Key References |
|---|---|
| Cholestatic diseases | |
| Macroautophay | [68,69,70,71,72,73,74,80,81,96,105] |
| Mitophagy | [53,101,121] |
| Lipophagy | [155,156,157,158,159,166,167,168,169] |
| Pexophagy | [121] |
| ERphagy | [72] |
| Age related cholestatic diseases | |
| Macroautophay | [122,123,124,125,126,133] |
| Mitophagy | [164,166,167,168,169] |
| Lipophagy | [127,128,156,157,158,166,167,168,169] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pinto, C.; Ninfole, E.; Benedetti, A.; Marzioni, M.; Maroni, L. Involvement of Autophagy in Ageing and Chronic Cholestatic Diseases. Cells 2021, 10, 2772. https://doi.org/10.3390/cells10102772
Pinto C, Ninfole E, Benedetti A, Marzioni M, Maroni L. Involvement of Autophagy in Ageing and Chronic Cholestatic Diseases. Cells. 2021; 10(10):2772. https://doi.org/10.3390/cells10102772
Chicago/Turabian StylePinto, Claudio, Elisabetta Ninfole, Antonio Benedetti, Marco Marzioni, and Luca Maroni. 2021. "Involvement of Autophagy in Ageing and Chronic Cholestatic Diseases" Cells 10, no. 10: 2772. https://doi.org/10.3390/cells10102772
APA StylePinto, C., Ninfole, E., Benedetti, A., Marzioni, M., & Maroni, L. (2021). Involvement of Autophagy in Ageing and Chronic Cholestatic Diseases. Cells, 10(10), 2772. https://doi.org/10.3390/cells10102772