
Electrical Ventricular Remodeling in Dilated Cardiomyopathy

 , ,
, ,
Abstract
1. Introduction: Characteristics of Ventricular Arrhythmias in Patients with Dilated Cardiomyopathy
2. Genetic Basis of Ventricular Arrhythmogenesis in DCM
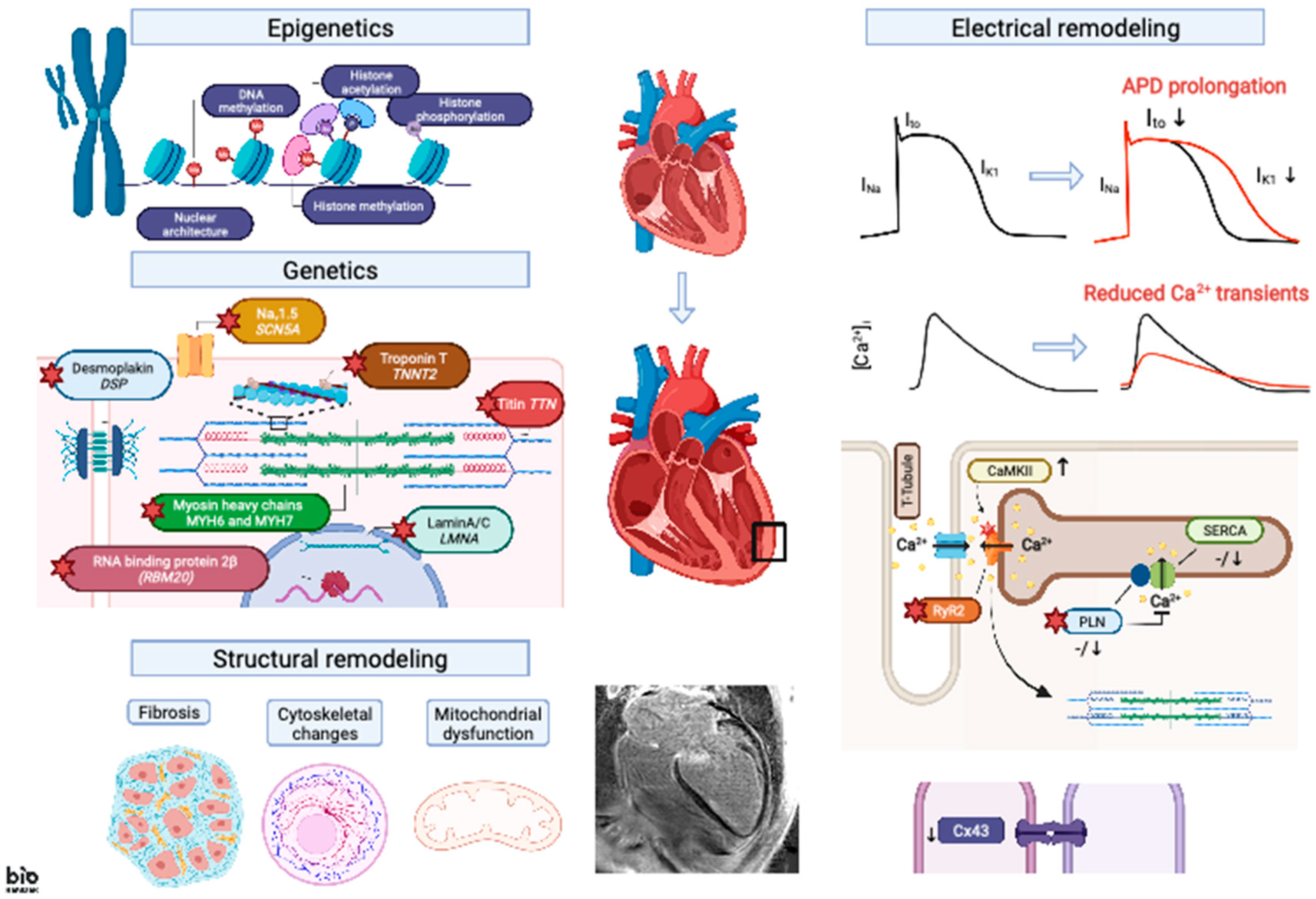
3. Electrical Remodeling
3.1. APD Changes
3.2. EC Coupling
3.3. Cell–Cell Coupling
4. Modulation of Epigenetic Signaling in DCM
5. Structural Remodeling
6. Translational Perspective and Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- McKenna, W.J.; Maron, B.J.; Thiene, G. Classification, Epidemiology, and Global Burden of Cardiomyopathies. Circ. Res. 2017, 121, 722–730. [Google Scholar] [CrossRef] [PubMed]
- Weintraub, R.G.; Semsarian, C.; Macdonald, P. Dilated Cardiomyopathy. Lancet 2017, 390, 400–414. [Google Scholar] [CrossRef]
- Schultheiss, H.-P.; Fairweather, D.; Caforio, A.L.P.; Escher, F.; Hershberger, R.E.; Lipshultz, S.E.; Liu, P.P.; Matsumori, A.; Mazzanti, A.; McMurray, J.; et al. Dilated Cardiomyopathy. Nat. Rev. Dis. Primers 2019, 5, 32. [Google Scholar] [CrossRef] [PubMed]
- Elliott, P.; Andersson, B.; Arbustini, E.; Bilinska, Z.; Cecchi, F.; Charron, P.; Dubourg, O.; Kühl, U.; Maisch, B.; McKenna, W.J.; et al. Classification of the Cardiomyopathies: A Position Statement from the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur. Heart J. 2008, 29, 270–276. [Google Scholar] [CrossRef] [PubMed]
- Pinto, Y.M.; Elliott, P.M.; Arbustini, E.; Adler, Y.; Anastasakis, A.; Böhm, M.; Duboc, D.; Gimeno, J.; de Groote, P.; Imazio, M.; et al. Proposal for a Revised Definition of Dilated Cardiomyopathy, Hypokinetic Non-Dilated Cardiomyopathy, and Its Implications for Clinical Practice: A Position Statement of the ESC Working Group on Myocardial and Pericardial Diseases. Eur. Heart J. 2016, 37, 1850–1858. [Google Scholar] [CrossRef]
- Dec, G.W.; Fuster, V. Idiopathic Dilated Cardiomyopathy. N. Engl. J. Med. 1994, 331, 1564–1575. [Google Scholar] [CrossRef]
- Maron, B.J.; Towbin, J.A.; Thiene, G.; Antzelevitch, C.; Corrado, D.; Arnett, D.; Moss, A.J.; Seidman, C.E.; Young, J.B.; American Heart Association; et al. Contemporary Definitions and Classification of the Cardiomyopathies: An American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation 2006, 113, 1807–1816. [Google Scholar] [CrossRef] [PubMed]
- Finocchiaro, G.; Merlo, M.; Sheikh, N.; De Angelis, G.; Papadakis, M.; Olivotto, I.; Rapezzi, C.; Carr-White, G.; Sharma, S.; Mestroni, L.; et al. The Electrocardiogram in the Diagnosis and Management of Patients with Dilated Cardiomyopathy. Eur. J. Heart Fail. 2020, 22, 1097–1107. [Google Scholar] [CrossRef]
- Priori, S.G.; Blomström-Lundqvist, C.; Mazzanti, A.; Blom, N.; Borggrefe, M.; Camm, J.; Elliott, P.M.; Fitzsimons, D.; Hatala, R.; Hindricks, G.; et al. 2015 ESC Guidelines for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death: The Task Force for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death of the European Society of Cardiology (ESC). Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC). Eur. Heart J. 2015, 36, 2793–2867. [Google Scholar] [CrossRef]
- Towbin, J.A.; McKenna, W.J.; Abrams, D.J.; Ackerman, M.J.; Calkins, H.; Darrieux, F.C.C.; Daubert, J.P.; de Chillou, C.; DePasquale, E.C.; Desai, M.Y.; et al. 2019 HRS Expert Consensus Statement on Evaluation, Risk Stratification, and Management of Arrhythmogenic Cardiomyopathy. Heart Rhythm 2019, 16, e301–e372. [Google Scholar] [CrossRef]
- Brugada, J.; Katritsis, D.G.; Arbelo, E.; Arribas, F.; Bax, J.J.; Blomström-Lundqvist, C.; Calkins, H.; Corrado, D.; Deftereos, S.G.; Diller, G.-P.; et al. 2019 ESC Guidelines for the Management of Patients with Supraventricular TachycardiaThe Task Force for the Management of Patients with Supraventricular Tachycardia of the European Society of Cardiology (ESC). Eur. Heart J. 2020, 41, 655–720. [Google Scholar] [CrossRef]
- Hindricks, G.; Potpara, T.; Dagres, N.; Arbelo, E.; Bax, J.J.; Blomström-Lundqvist, C.; Boriani, G.; Castella, M.; Dan, G.-A.; Dilaveris, P.E.; et al. 2020 ESC Guidelines for the Diagnosis and Management of Atrial Fibrillation Developed in Collaboration with the European Association for Cardio-Thoracic Surgery (EACTS): The Task Force for the Diagnosis and Management of Atrial Fibrillation of the European Society of Cardiology (ESC) Developed with the Special Contribution of the European Heart Rhythm Association (EHRA) of the ESC. Eur. Heart J. 2021, 42, 373–498. [Google Scholar] [CrossRef]
- Nuzzi, V.; Cannatà, A.; Manca, P.; Castrichini, M.; Barbati, G.; Aleksova, A.; Fabris, E.; Zecchin, M.; Merlo, M.; Boriani, G.; et al. Atrial Fibrillation in Dilated Cardiomyopathy: Outcome Prediction from an Observational Registry. Int. J. Cardiol. 2021, 323, 140–147. [Google Scholar] [CrossRef]
- Towbin, J.A.; Lorts, A. Arrhythmias and Dilated Cardiomyopathy. J. Am. Coll. Cardiol. 2011, 57, 2169–2171. [Google Scholar] [CrossRef] [PubMed]
- Liuba, I.; Marchlinski, F.E. The Substrate and Ablation of Ventricular Tachycardia in Patients with Nonischemic Cardiomyopathy. Circ. J. 2013, 77, 1957–1966. [Google Scholar] [CrossRef] [PubMed]
- Bondue, A.; Arbustini, E.; Bianco, A.; Ciccarelli, M.; Dawson, D.; De Rosa, M.; Hamdani, N.; Hilfiker-Kleiner, D.; Meder, B.; Leite-Moreira, A.F.; et al. Complex Roads from Genotype to Phenotype in Dilated Cardiomyopathy: Scientific Update from the Working Group of Myocardial Function of the European Society of Cardiology. Cardiovasc. Res. 2018, 114, 1287–1303. [Google Scholar] [CrossRef] [PubMed]
- Peters, S.; Kumar, S.; Elliott, P.; Kalman, J.M.; Fatkin, D. Arrhythmic Genotypes in Familial Dilated Cardiomyopathy: Implications for Genetic Testing and Clinical Management. Heart Lung Circ. 2019, 28, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Watkins, H.; Ashrafian, H.; Redwood, C. Inherited Cardiomyopathies. N. Engl. J. Med. 2011, 364, 1643–1656. [Google Scholar] [CrossRef] [PubMed]
- Hershberger, R.E.; Hedges, D.J.; Morales, A. Dilated Cardiomyopathy: The Complexity of a Diverse Genetic Architecture. Nat. Rev. Cardiol. 2013, 10, 531–547. [Google Scholar] [CrossRef]
- Zegkos, T.; Panagiotidis, T.; Parcharidou, D.; Efthimiadis, G. Emerging Concepts in Arrhythmogenic Dilated Cardiomyopathy. Heart Fail. Rev. 2020, 4, 1219–1229. [Google Scholar] [CrossRef]
- Spezzacatene, A.; Sinagra, G.; Merlo, M.; Barbati, G.; Graw, S.L.; Brun, F.; Slavov, D.; Di Lenarda, A.; Salcedo, E.E.; Towbin, J.A.; et al. Arrhythmogenic Phenotype in Dilated Cardiomyopathy: Natural History and Predictors of Life-Threatening Arrhythmias. J. Am. Heart Assoc. 2015, 4, e002149. [Google Scholar] [CrossRef]
- Corrado, D.; Perazzolo Marra, M.; Zorzi, A.; Beffagna, G.; Cipriani, A.; Lazzari, M.D.; Migliore, F.; Pilichou, K.; Rampazzo, A.; Rigato, I.; et al. Diagnosis of Arrhythmogenic Cardiomyopathy: The Padua Criteria. Int. J. Cardiol. 2020, 319, 106–114. [Google Scholar] [CrossRef] [PubMed]
- Coronel, R.; Wilders, R.; Verkerk, A.O.; Wiegerinck, R.F.; Benoist, D.; Bernus, O. Electrophysiological Changes in Heart Failure and Their Implications for Arrhythmogenesis. Biochim. Biophys. Acta 2013, 1832, 2432–2441. [Google Scholar] [CrossRef] [PubMed]
- Wilde, A.A.M.; Amin, A.S. Clinical Spectrum of SCN5A Mutations: Long QT Syndrome, Brugada Syndrome, and Cardiomyopathy. JACC Clin. Electrophysiol. 2018, 4, 569–579. [Google Scholar] [CrossRef] [PubMed]
- Beuckelmann, D.J.; Näbauer, M.; Erdmann, E. Intracellular Calcium Handling in Isolated Ventricular Myocytes from Patients with Terminal Heart Failure. Circulation 1992, 85, 1046–1055. [Google Scholar] [CrossRef]
- Koumi, S.; Backer, C.L.; Arentzen, C.E. Characterization of Inwardly Rectifying K+ Channel in Human Cardiac Myocytes. Alterations in Channel Behavior in Myocytes Isolated from Patients with Idiopathic Dilated Cardiomyopathy. Circulation 1995, 92, 164–174. [Google Scholar] [CrossRef] [PubMed]
- Akar, F.G.; Nass, R.D.; Hahn, S.; Cingolani, E.; Shah, M.; Hesketh, G.G.; DiSilvestre, D.; Tunin, R.S.; Kass, D.A.; Tomaselli, G.F. Dynamic Changes in Conduction Velocity and Gap Junction Properties during Development of Pacing-Induced Heart Failure. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H1223–H1230. [Google Scholar] [CrossRef] [PubMed]
- Akar, F.G.; Spragg, D.D.; Tunin, R.S.; Kass, D.A.; Tomaselli, G.F. Mechanisms Underlying Conduction Slowing and Arrhythmogenesis in Nonischemic Dilated Cardiomyopathy. Circ. Res. 2004, 95, 717–725. [Google Scholar] [CrossRef]
- Wiegerinck, R.F.; Verkerk, A.O.; Belterman, C.N.; van Veen, T.A.B.; Baartscheer, A.; Opthof, T.; Wilders, R.; de Bakker, J.M.T.; Coronel, R. Larger Cell Size in Rabbits With Heart Failure Increases Myocardial Conduction Velocity and QRS Duration. Circulation 2006, 113, 806–813. [Google Scholar] [CrossRef] [PubMed]
- Olson, T.M.; Michels, V.V.; Ballew, J.D.; Reyna, S.P.; Karst, M.L.; Herron, K.J.; Horton, S.C.; Rodeheffer, R.J.; Anderson, J.L. Sodium Channel Mutations and Susceptibility to Heart Failure and Atrial Fibrillation. JAMA 2005, 293, 447–454. [Google Scholar] [CrossRef]
- Papadatos, G.A.; Wallerstein, P.M.R.; Head, C.E.G.; Ratcliff, R.; Brady, P.A.; Benndorf, K.; Saumarez, R.C.; Trezise, A.E.O.; Huang, C.L.-H.; Vandenberg, J.I.; et al. Slowed Conduction and Ventricular Tachycardia after Targeted Disruption of the Cardiac Sodium Channel Gene Scn5a. Proc. Natl. Acad. Sci. USA 2002, 99, 6210–6215. [Google Scholar] [CrossRef]
- Suzuki, T.; Shioya, T.; Murayama, T.; Sugihara, M.; Odagiri, F.; Nakazato, Y.; Nishizawa, H.; Chugun, A.; Sakurai, T.; Daida, H.; et al. Multistep Ion Channel Remodeling and Lethal Arrhythmia Precede Heart Failure in a Mouse Model of Inherited Dilated Cardiomyopathy. PLoS ONE 2012, 7, e35353. [Google Scholar] [CrossRef]
- Radicke, S.; Cotella, D.; Graf, E.M.; Banse, U.; Jost, N.; Varró, A.; Tseng, G.-N.; Ravens, U.; Wettwer, E. Functional Modulation of the Transient Outward Current Ito by KCNE Beta-Subunits and Regional Distribution in Human Non-Failing and Failing Hearts. Cardiovasc. Res. 2006, 71, 695–703. [Google Scholar] [CrossRef] [PubMed]
- Rahm, A.-K.; Müller, M.E.; Gramlich, D.; Lugenbiel, P.; Uludag, E.; Rivinius, R.; Ullrich, N.D.; Schmack, B.; Ruhparwar, A.; Heimberger, T.; et al. Inhibition of Cardiac Kv4.3 (Ito) Channel Isoforms by Class I Antiarrhythmic Drugs Lidocaine and Mexiletine. Eur. J. Pharmacol. 2020, 880, 173159. [Google Scholar] [CrossRef]
- Szuts, V.; Ménesi, D.; Varga-Orvos, Z.; Zvara, Á.; Houshmand, N.; Bitay, M.; Bogáts, G.; Virág, L.; Baczkó, I.; Szalontai, B.; et al. Altered Expression of Genes for Kir Ion Channels in Dilated Cardiomyopathy. Can. J. Physiol. Pharmacol. 2013, 91, 648–656. [Google Scholar] [CrossRef]
- Guo, A.; Zhang, C.; Wei, S.; Chen, B.; Song, L.-S. Emerging Mechanisms of T-Tubule Remodelling in Heart Failure. Cardiovasc. Res. 2013, 98, 204–215. [Google Scholar] [CrossRef] [PubMed]
- Crossman, D.J.; Ruygrok, P.N.; Ruygrok, P.R.; Soeller, C.; Cannell, M.B. Changes in the Organization of Excitation-Contraction Coupling Structures in Failing Human Heart. PLoS ONE 2011, 6, e17901. [Google Scholar] [CrossRef]
- Zhang, H.-B.; Li, R.-C.; Xu, M.; Xu, S.-M.; Lai, Y.-S.; Wu, H.-D.; Xie, X.-J.; Gao, W.; Ye, H.; Zhang, Y.-Y.; et al. Ultrastructural Uncoupling between T-Tubules and Sarcoplasmic Reticulum in Human Heart Failure. Cardiovasc. Res. 2013, 98, 269–276. [Google Scholar] [CrossRef]
- Respress, J.L.; van Oort, R.J.; Li, N.; Rolim, N.; Dixit, S.S.; de Almeida, A.; Voigt, N.; Lawrence, W.S.; Skapura, D.G.; Skårdal, K.; et al. Role of RyR2 Phosphorylation at S2814 during Heart Failure Progression. Circ. Res. 2012, 110, 1474–1483. [Google Scholar] [CrossRef]
- Maier, L.S.; Zhang, T.; Chen, L.; DeSantiago, J.; Brown, J.H.; Bers, D.M. Transgenic CaMKIIdeltaC Overexpression Uniquely Alters Cardiac Myocyte Ca2+ Handling: Reduced SR Ca2+ Load and Activated SR Ca2+ Release. Circ. Res. 2003, 92, 904–911. [Google Scholar] [CrossRef] [PubMed]
- Fischer, T.H.; Maier, L.S.; Sossalla, S. The Ryanodine Receptor Leak: How a Tattered Receptor Plunges the Failing Heart into Crisis. Heart Fail. Rev. 2013, 18, 475–483. [Google Scholar] [CrossRef][Green Version]
- Van Oort, R.J.; McCauley, M.D.; Dixit, S.S.; Pereira, L.; Yang, Y.; Respress, J.L.; Wang, Q.; De Almeida, A.C.; Skapura, D.G.; Anderson, M.E.; et al. Ryanodine Receptor Phosphorylation by Calcium/Calmodulin-Dependent Protein Kinase II Promotes Life-Threatening Ventricular Arrhythmias in Mice with Heart Failure. Circulation 2010, 122, 2669–2679. [Google Scholar] [CrossRef] [PubMed]
- Meyer, M.; Schillinger, W.; Pieske, B.; Holubarsch, C.; Heilmann, C.; Posival, H.; Kuwajima, G.; Mikoshiba, K.; Just, H.; Hasenfuss, G. Alterations of Sarcoplasmic Reticulum Proteins in Failing Human Dilated Cardiomyopathy. Circulation 1995, 92, 778–784. [Google Scholar] [CrossRef] [PubMed]
- Schwinger, R.H.; Böhm, M.; Schmidt, U.; Karczewski, P.; Bavendiek, U.; Flesch, M.; Krause, E.G.; Erdmann, E. Unchanged Protein Levels of SERCA II and Phospholamban but Reduced Ca2+ Uptake and Ca(2+)-ATPase Activity of Cardiac Sarcoplasmic Reticulum from Dilated Cardiomyopathy Patients Compared with Patients with Nonfailing Hearts. Circulation 1995, 92, 3220–3228. [Google Scholar] [CrossRef]
- Kubo, H.; Margulies, K.B.; Piacentino, V.; Gaughan, J.P.; Houser, S.R. Patients with End-Stage Congestive Heart Failure Treated with Beta-Adrenergic Receptor Antagonists Have Improved Ventricular Myocyte Calcium Regulatory Protein Abundance. Circulation 2001, 104, 1012–1018. [Google Scholar] [CrossRef]
- Haghighi, K.; Kolokathis, F.; Pater, L.; Lynch, R.A.; Asahi, M.; Gramolini, A.O.; Fan, G.-C.; Tsiapras, D.; Hahn, H.S.; Adamopoulos, S.; et al. Human Phospholamban Null Results in Lethal Dilated Cardiomyopathy Revealing a Critical Difference between Mouse and Human. J. Clin. Investig. 2003, 111, 869–876. [Google Scholar] [CrossRef] [PubMed]
- Posch, M.G.; Perrot, A.; Geier, C.; Boldt, L.-H.; Schmidt, G.; Lehmkuhl, H.B.; Hetzer, R.; Dietz, R.; Gutberlet, M.; Haverkamp, W.; et al. Genetic Deletion of Arginine 14 in Phospholamban Causes Dilated Cardiomyopathy with Attenuated Electrocardiographic R Amplitudes. Heart Rhythm 2009, 6, 480–486. [Google Scholar] [CrossRef]
- Young, H.S.; Ceholski, D.K.; Trieber, C.A. Deception in Simplicity: Hereditary Phospholamban Mutations in Dilated Cardiomyopathy. Biochem. Cell Biol. 2015, 93, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Hof, I.E.; van der Heijden, J.F.; Kranias, E.G.; Sanoudou, D.; de Boer, R.A.; van Tintelen, J.P.; van der Zwaag, P.A.; Doevendans, P.A. Prevalence and Cardiac Phenotype of Patients with a Phospholamban Mutation. Neth. Heart J. 2019, 27, 64–69. [Google Scholar] [CrossRef]
- Van der Zwaag, P.A.; van Rijsingen, I.A.W.; Asimaki, A.; Jongbloed, J.D.H.; van Veldhuisen, D.J.; Wiesfeld, A.C.P.; Cox, M.G.P.J.; van Lochem, L.T.; de Boer, R.A.; Hofstra, R.M.W.; et al. Phospholamban R14del Mutation in Patients Diagnosed with Dilated Cardiomyopathy or Arrhythmogenic Right Ventricular Cardiomyopathy: Evidence Supporting the Concept of Arrhythmogenic Cardiomyopathy. Eur. J. Heart Fail. 2012, 14, 1199–1207. [Google Scholar] [CrossRef]
- Schmitt, J.P.; Kamisago, M.; Asahi, M.; Li, G.H.; Ahmad, F.; Mende, U.; Kranias, E.G.; MacLennan, D.H.; Seidman, J.G.; Seidman, C.E. Dilated Cardiomyopathy and Heart Failure Caused by a Mutation in Phospholamban. Science 2003, 299, 1410–1413. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.-S.; Morales, A.; Vafiadaki, E.; Lam, C.K.; Cai, W.-F.; Haghighi, K.; Adly, G.; Hershberger, R.E.; Kranias, E.G. A Novel Human R25C-Phospholamban Mutation Is Associated with Super-Inhibition of Calcium Cycling and Ventricular Arrhythmia. Cardiovasc. Res. 2015, 107, 164–174. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, H.; Ohnishi, Y.; Yoshida, A.; Okajima, K.; Azumi, H.; Ishida, A.; Galeano, E.J.; Kubo, S.; Hayashi, Y.; Itoh, H.; et al. Heterogeneous Loss of Connexin43 Protein in Nonischemic Dilated Cardiomyopathy with Ventricular Tachycardia. J. Cardiovasc. Electrophysiol. 2002, 13, 865–870. [Google Scholar] [CrossRef]
- Salameh, A.; Krautblatter, S.; Karl, S.; Blanke, K.; Gomez, D.R.; Dhein, S.; Pfeiffer, D.; Janousek, J. The Signal Transduction Cascade Regulating the Expression of the Gap Junction Protein Connexin43 by β-Adrenoceptors. Br. J. Pharmacol. 2009, 158, 198–208. [Google Scholar] [CrossRef]
- Guerrero, P.A.; Schuessler, R.B.; Davis, L.M.; Beyer, E.C.; Johnson, C.M.; Yamada, K.A.; Saffitz, J.E. Slow Ventricular Conduction in Mice Heterozygous for a Connexin43 Null Mutation. J. Clin. Investig. 1997, 99, 1991–1998. [Google Scholar] [CrossRef]
- Van Rijen, H.V.M.; Eckardt, D.; Degen, J.; Theis, M.; Ott, T.; Willecke, K.; Jongsma, H.J.; Opthof, T.; de Bakker, J.M.T. Slow Conduction and Enhanced Anisotropy Increase the Propensity for Ventricular Tachyarrhythmias in Adult Mice with Induced Deletion of Connexin43. Circulation 2004, 109, 1048–1055. [Google Scholar] [CrossRef]
- Jansen, J.A.; van Veen, T.A.B.; de Jong, S.; van der Nagel, R.; van Stuijvenberg, L.; Driessen, H.; Labzowski, R.; Oefner, C.M.; Bosch, A.A.; Nguyen, T.Q.; et al. Reduced Cx43 Expression Triggers Increased Fibrosis Due to Enhanced Fibroblast Activity. Circ. Arrhythm. Electrophysiol. 2012, 5, 380–390. [Google Scholar] [CrossRef]
- Poelzing, S.; Rosenbaum, D.S. Altered Connexin43 Expression Produces Arrhythmia Substrate in Heart Failure. Am. J. Physiol. Heart Circ. Physiol. 2004, 287, H1762–H1770. [Google Scholar] [CrossRef]
- Waddington, C.H. The Epigenotype. Endeavour 1942, 1942, 18–20. [Google Scholar] [CrossRef]
- Berger, S.L.; Kouzarides, T.; Shiekhattar, R.; Shilatifard, A. An Operational Definition of Epigenetics. Genes Dev. 2009, 23, 781–783. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Zeng, C.; Wang, Y. Epigenetics in Dilated Cardiomyopathy. Curr. Opin. Cardiol. 2019, 34, 260–269. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, A.T.; Xiao, B.; Neppl, R.L.; Kallin, E.M.; Li, J.; Chen, T.; Wang, D.-Z.; Xiao, X.; Zhang, Y. DOT1L Regulates Dystrophin Expression and Is Critical for Cardiac Function. Genes Dev. 2011, 25, 263–274. [Google Scholar] [CrossRef]
- Hohl, M.; Wagner, M.; Reil, J.-C.; Müller, S.-A.; Tauchnitz, M.; Zimmer, A.M.; Lehmann, L.H.; Thiel, G.; Böhm, M.; Backs, J.; et al. HDAC4 Controls Histone Methylation in Response to Elevated Cardiac Load. J. Clin. Investig. 2013, 123, 1359–1370. [Google Scholar] [CrossRef]
- Ito, E.; Miyagawa, S.; Fukushima, S.; Yoshikawa, Y.; Saito, S.; Saito, T.; Harada, A.; Takeda, M.; Kashiyama, N.; Nakamura, Y.; et al. Histone Modification Is Correlated With Reverse Left Ventricular Remodeling in Nonischemic Dilated Cardiomyopathy. Ann. Thorac. Surg. 2017, 104, 1531–1539. [Google Scholar] [CrossRef]
- Jiang, D.-S.; Yi, X.; Li, R.; Su, Y.-S.; Wang, J.; Chen, M.-L.; Liu, L.-G.; Hu, M.; Cheng, C.; Zheng, P.; et al. The Histone Methyltransferase Mixed Lineage Leukemia (MLL) 3 May Play a Potential Role on Clinical Dilated Cardiomyopathy. Mol. Med. 2017, 23, 196–203. [Google Scholar] [CrossRef]
- Montgomery, R.L.; Davis, C.A.; Potthoff, M.J.; Haberland, M.; Fielitz, J.; Qi, X.; Hill, J.A.; Richardson, J.A.; Olson, E.N. Histone Deacetylases 1 and 2 Redundantly Regulate Cardiac Morphogenesis, Growth, and Contractility. Genes Dev. 2007, 21, 1790–1802. [Google Scholar] [CrossRef]
- Syren, P.; Rahm, A.-K.; Schweizer, P.A.; Bruehl, C.; Katus, H.A.; Frey, N.; Thomas, D.; Lugenbiel, P. Histone Deacetylase 2-Dependent Ventricular Electrical Remodeling in a Porcine Model of Early Heart Failure. Life Sci. 2021, 281, 119769. [Google Scholar] [CrossRef] [PubMed]
- Awad, S.; Kunhi, M.; Little, G.H.; Bai, Y.; An, W.; Bers, D.; Kedes, L.; Poizat, C. Nuclear CaMKII Enhances Histone H3 Phosphorylation and Remodels Chromatin during Cardiac Hypertrophy. Nucleic Acids Res. 2013, 41, 7656–7672. [Google Scholar] [CrossRef]
- Awad, S.; Al-Haffar, K.M.A.; Marashly, Q.; Quijada, P.; Kunhi, M.; Al-Yacoub, N.; Wade, F.S.; Mohammed, S.F.; Al-Dayel, F.; Sutherland, G.; et al. Control of Histone H3 Phosphorylation by CaMKIIδ in Response to Haemodynamic Cardiac Stress. J. Pathol. 2015, 235, 606–618. [Google Scholar] [CrossRef] [PubMed]
- Perrin, M.J.; Gollob, M.H. The Role of Atrial Natriuretic Peptide in Modulating Cardiac Electrophysiology. Heart Rhythm 2012, 9, 610–615. [Google Scholar] [CrossRef] [PubMed]
- Moghtadaei, M.; Polina, I.; Rose, R.A. Electrophysiological Effects of Natriuretic Peptides in the Heart Are Mediated by Multiple Receptor Subtypes. Prog. Biophys. Mol. Biol. 2016, 120, 37–49. [Google Scholar] [CrossRef] [PubMed]
- Goetze, J.P.; Bruneau, B.G.; Ramos, H.R.; Ogawa, T.; Bold, M.K.; Bold, A.J. Cardiac Natriuretic Peptides. Nat. Rev. Cardiol. 2020, 17, 698–717. [Google Scholar] [CrossRef]
- Miao, L.; Wang, M.; Yin, W.-X.; Yuan, Q.; Chen, Y.-X.; Fleischmann, B.; Hescheler, J.; Ji, G. Atrial Natriuretic Peptide Regulates Ca Channel in Early Developmental Cardiomyocytes. PLoS ONE 2010, 5, e8847. [Google Scholar] [CrossRef]
- Burley, D.S.; Cox, C.D.; Zhang, J.; Wann, K.T.; Baxter, G.F. Natriuretic Peptides Modulate ATP-Sensitive K(+) Channels in Rat Ventricular Cardiomyocytes. Basic Res. Cardiol. 2014, 109, 402. [Google Scholar] [CrossRef] [PubMed]
- Stull, J.T.; Manning, D.R.; High, C.W.; Blumenthal, D.K. Phosphorylation of Contractile Proteins in Heart and Skeletal Muscle. Fed. Proc. 1980, 39, 1552–1557. [Google Scholar] [PubMed]
- McKinsey, T.A.; Zhang, C.L.; Lu, J.; Olson, E.N. Signal-Dependent Nuclear Export of a Histone Deacetylase Regulates Muscle Differentiation. Nature 2000, 408, 106–111. [Google Scholar] [CrossRef]
- Backs, J.; Song, K.; Bezprozvannaya, S.; Chang, S.; Olson, E.N. CaM Kinase II Selectively Signals to Histone Deacetylase 4 during Cardiomyocyte Hypertrophy. J. Clin. Investig. 2006, 116, 1853–1864. [Google Scholar] [CrossRef]
- Wu, X.; Zhang, T.; Bossuyt, J.; Li, X.; McKinsey, T.A.; Dedman, J.R.; Olson, E.N.; Chen, J.; Brown, J.H.; Bers, D.M. Local InsP3-Dependent Perinuclear Ca2+ Signaling in Cardiac Myocyte Excitation-Transcription Coupling. J. Clin. Investig. 2006, 116, 675–682. [Google Scholar] [CrossRef]
- Ago, T.; Liu, T.; Zhai, P.; Chen, W.; Li, H.; Molkentin, J.D.; Vatner, S.F.; Sadoshima, J. A Redox-Dependent Pathway for Regulating Class II HDACs and Cardiac Hypertrophy. Cell 2008, 133, 978–993. [Google Scholar] [CrossRef]
- Kim, S.Y.; Zhang, X.; Schiattarella, G.G.; Altamirano, F.; Ramos, T.A.R.; French, K.M.; Jiang, N.; Szweda, P.A.; Evers, B.M.; May, H.I.; et al. Epigenetic Reader BRD4 (Bromodomain-Containing Protein 4) Governs Nucleus-Encoded Mitochondrial Transcriptome to Regulate Cardiac Function. Circulation 2020, 142, 2356–2370. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Wang, X.; Zhang, Y.; Ru, X.; Zhou, L.; Tian, Y. H3K9 Histone Methyltransferase G9a Ameliorates Dilated Cardiomyopathy via the Downregulation of Cell Adhesion Molecules. Mol. Med. Rep. 2015, 11, 3872–3879. [Google Scholar] [CrossRef]
- Koenig, X.; Rubi, L.; Obermair, G.J.; Cervenka, R.; Dang, X.B.; Lukacs, P.; Kummer, S.; Bittner, R.E.; Kubista, H.; Todt, H.; et al. Enhanced Currents through L-Type Calcium Channels in Cardiomyocytes Disturb the Electrophysiology of the Dystrophic Heart. Am. J. Physiol. Heart Circ. Physiol. 2014, 306, H564–H573. [Google Scholar] [CrossRef]
- Haas, J.; Frese, K.S.; Park, Y.J.; Keller, A.; Vogel, B.; Lindroth, A.M.; Weichenhan, D.; Franke, J.; Fischer, S.; Bauer, A.; et al. Alterations in Cardiac DNA Methylation in Human Dilated Cardiomyopathy. EMBO Mol. Med. 2013, 5, 413–429. [Google Scholar] [CrossRef]
- Jo, B.-S.; Koh, I.-U.; Bae, J.-B.; Yu, H.-Y.; Jeon, E.-S.; Lee, H.-Y.; Kim, J.-J.; Choi, M.; Choi, S.S. Methylome Analysis Reveals Alterations in DNA Methylation in the Regulatory Regions of Left Ventricle Development Genes in Human Dilated Cardiomyopathy. Genomics 2016, 108, 84–92. [Google Scholar] [CrossRef]
- Meder, B.; Haas, J.; Sedaghat-Hamedani, F.; Kayvanpour, E.; Frese, K.; Lai, A.; Nietsch, R.; Scheiner, C.; Mester, S.; Bordalo, D.M.; et al. Epigenome-Wide Association Study Identifies Cardiac Gene Patterning and a Novel Class of Biomarkers for Heart Failure. Circulation 2017, 136, 1528–1544. [Google Scholar] [CrossRef]
- Gi, W.-T.; Haas, J.; Sedaghat-Hamedani, F.; Kayvanpour, E.; Tappu, R.; Lehmann, D.H.; Shirvani Samani, O.; Wisdom, M.; Keller, A.; Katus, H.A.; et al. Epigenetic Regulation of Alternative MRNA Splicing in Dilated Cardiomyopathy. J. Clin. Med. 2020, 9, 1499. [Google Scholar] [CrossRef] [PubMed]
- Rosa-Garrido, M.; Chapski, D.J.; Schmitt, A.D.; Kimball, T.H.; Karbassi, E.; Monte, E.; Balderas, E.; Pellegrini, M.; Shih, T.-T.; Soehalim, E.; et al. High-Resolution Mapping of Chromatin Conformation in Cardiac Myocytes Reveals Structural Remodeling of the Epigenome in Heart Failure. Circulation 2017, 136, 1613–1625. [Google Scholar] [CrossRef] [PubMed]
- Salvarani, N.; Crasto, S.; Miragoli, M.; Bertero, A.; Paulis, M.; Kunderfranco, P.; Serio, S.; Forni, A.; Lucarelli, C.; Dal Ferro, M.; et al. The K219T-Lamin Mutation Induces Conduction Defects through Epigenetic Inhibition of SCN5A in Human Cardiac Laminopathy. Nat. Commun. 2019, 10, 2267. [Google Scholar] [CrossRef] [PubMed]
- Skinner, M.K.; Manikkam, M.; Guerrero-Bosagna, C. Epigenetic Transgenerational Actions of Environmental Factors in Disease Etiology. Trends Endocrinol. Metab. 2010, 21, 214–222. [Google Scholar] [CrossRef]
- Shah, M.H.; Binkley, P.; Chan, K.; Xiao, J.; Arbogast, D.; Collamore, M.; Farra, Y.; Young, D.; Grever, M. Cardiotoxicity of Histone Deacetylase Inhibitor Depsipeptide in Patients with Metastatic Neuroendocrine Tumors. Clin. Cancer Res. 2006, 12, 3997–4003. [Google Scholar] [CrossRef]
- Olsen, E.A.; Kim, Y.H.; Kuzel, T.M.; Pacheco, T.R.; Foss, F.M.; Parker, S.; Frankel, S.R.; Chen, C.; Ricker, J.L.; Arduino, J.M.; et al. Phase IIb Multicenter Trial of Vorinostat in Patients with Persistent, Progressive, or Treatment Refractory Cutaneous T-Cell Lymphoma. J. Clin. Oncol. 2007, 25, 3109–3115. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Dong, M.; Hong, X.; Zhang, W.; Feng, J.; Zhu, J.; Yu, L.; Ke, X.; Huang, H.; Shen, Z.; et al. Results from a Multicenter, Open-Label, Pivotal Phase II Study of Chidamide in Relapsed or Refractory Peripheral T-Cell Lymphoma. Ann. Oncol. 2015, 26, 1766–1771. [Google Scholar] [CrossRef] [PubMed]
- Shah, R.R. Safety and Tolerability of Histone Deacetylase (HDAC) Inhibitors in Oncology. Drug Saf. 2019, 42, 235–245. [Google Scholar] [CrossRef] [PubMed]
- Spence, S.; Deurinck, M.; Ju, H.; Traebert, M.; McLean, L.; Marlowe, J.; Emotte, C.; Tritto, E.; Tseng, M.; Shultz, M.; et al. Histone Deacetylase Inhibitors Prolong Cardiac Repolarization through Transcriptional Mechanisms. Toxicol. Sci. 2016, 153, 39–54. [Google Scholar] [CrossRef]
- Morita, H.; Seidman, J.; Seidman, C.E. Genetic Causes of Human Heart Failure. J. Clin. Investig. 2005, 115, 518–526. [Google Scholar] [CrossRef]
- Verdonschot, J.A.J.; Hazebroek, M.R.; Derks, K.W.J.; Barandiarán Aizpurua, A.; Merken, J.J.; Wang, P.; Bierau, J.; van den Wijngaard, A.; Schalla, S.M.; Abdul Hamid, M.A.; et al. Titin Cardiomyopathy Leads to Altered Mitochondrial Energetics, Increased Fibrosis and Long-Term Life-Threatening Arrhythmias. Eur. Heart J. 2018, 39, 864–873. [Google Scholar] [CrossRef]
- Chatzifrangkeskou, M.; Le Dour, C.; Wu, W.; Morrow, J.P.; Joseph, L.C.; Beuvin, M.; Sera, F.; Homma, S.; Vignier, N.; Mougenot, N.; et al. ERK1/2 Directly Acts on CTGF/CCN2 Expression to Mediate Myocardial Fibrosis in Cardiomyopathy Caused by Mutations in the Lamin A/C Gene. Hum. Mol. Genet. 2016, 25, 2220–2233. [Google Scholar] [CrossRef]
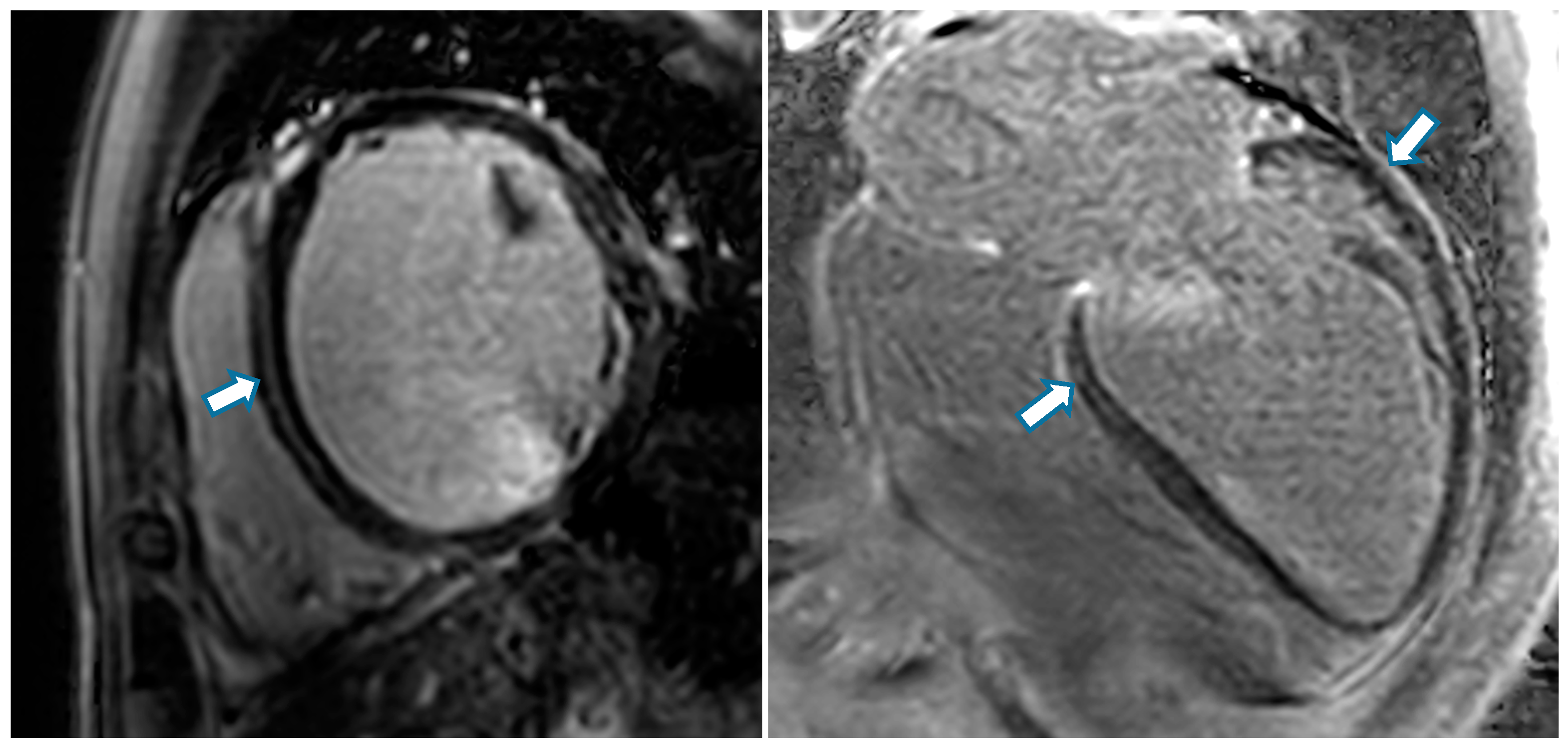
- Lehrke, S.; Lossnitzer, D.; Schöb, M.; Steen, H.; Merten, C.; Kemmling, H.; Pribe, R.; Ehlermann, P.; Zugck, C.; Korosoglou, G.; et al. Use of Cardiovascular Magnetic Resonance for Risk Stratification in Chronic Heart Failure: Prognostic Value of Late Gadolinium Enhancement in Patients with Non-Ischaemic Dilated Cardiomyopathy. Heart 2011, 97, 727–732. [Google Scholar] [CrossRef]
- Gulati, A.; Jabbour, A.; Ismail, T.F.; Guha, K.; Khwaja, J.; Raza, S.; Morarji, K.; Brown, T.D.H.; Ismail, N.A.; Dweck, M.R.; et al. Association of Fibrosis with Mortality and Sudden Cardiac Death in Patients with Nonischemic Dilated Cardiomyopathy. JAMA 2013, 309, 896–908. [Google Scholar] [CrossRef] [PubMed]
- Barison, A.; Del Torto, A.; Chiappino, S.; Aquaro, G.D.; Todiere, G.; Vergaro, G.; Passino, C.; Lombardi, M.; Emdin, M.; Masci, P.G. Prognostic Significance of Myocardial Extracellular Volume Fraction in Nonischaemic Dilated Cardiomyopathy. J. Cardiovasc. Med. 2015, 16, 681–687. [Google Scholar] [CrossRef] [PubMed]
- Puntmann, V.O.; Carr-White, G.; Jabbour, A.; Yu, C.-Y.; Gebker, R.; Kelle, S.; Hinojar, R.; Doltra, A.; Varma, N.; Child, N.; et al. T1-Mapping and Outcome in Nonischemic Cardiomyopathy: All-Cause Mortality and Heart Failure. JACC Cardiovasc. Imaging 2016, 9, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Aus dem Siepen, F.; Buss, S.J.; Messroghli, D.; Andre, F.; Lossnitzer, D.; Seitz, S.; Keller, M.; Schnabel, P.A.; Giannitsis, E.; Korosoglou, G.; et al. T1 Mapping in Dilated Cardiomyopathy with Cardiac Magnetic Resonance: Quantification of Diffuse Myocardial Fibrosis and Comparison with Endomyocardial Biopsy. Eur. Heart J. Cardiovasc. Imaging 2015, 16, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Bello, D.; Kipper, S.; Valderrábano, M.; Shivkumar, K. Catheter Ablation of Ventricular Tachycardia Guided by Contrast-Enhanced Cardiac Computed Tomography. Heart Rhythm 2004, 1, 490–492. [Google Scholar] [CrossRef] [PubMed]
- Crean, A.M.; Spears, D.A.; Suszko, A.M.; Chauhan, V.S. High-Resolution 3D Scar Imaging Using a Novel Late Iodine Enhancement Multidetector CT Protocol to Guide Ventricular Tachycardia Catheter Ablation. J. Cardiovasc. Electrophysiol. 2013, 24, 708–710. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Song, D.; Dong, J.; Zhu, P.; Liu, J.; Liu, W.; Ma, X.; Zhao, L.; Ling, S. Current Understanding of the Pathophysiology of Myocardial Fibrosis and Its Quantitative Assessment in Heart Failure. Front. Physiol. 2017, 8, 238. [Google Scholar] [CrossRef]
- Mitropoulou, P.; Georgiopoulos, G.; Figliozzi, S.; Klettas, D.; Nicoli, F.; Masci, P.G. Multi-Modality Imaging in Dilated Cardiomyopathy: With a Focus on the Role of Cardiac Magnetic Resonance. Front. Cardiovasc. Med. 2020, 7, 97. [Google Scholar] [CrossRef] [PubMed]
- Di Marco, A.; Anguera, I.; Schmitt, M.; Klem, I.; Neilan, T.G.; White, J.A.; Sramko, M.; Masci, P.G.; Barison, A.; Mckenna, P.; et al. Late Gadolinium Enhancement and the Risk for Ventricular Arrhythmias or Sudden Death in Dilated Cardiomyopathy: Systematic Review and Meta-Analysis. JACC Heart Fail. 2017, 5, 28–38. [Google Scholar] [CrossRef]
- Elming, M.B.; Hammer-Hansen, S.; Voges, I.; Nyktari, E.; Raja, A.A.; Svendsen, J.H.; Pehrson, S.; Signorovitch, J.; Køber, L.; Prasad, S.K.; et al. Myocardial Fibrosis and the Effect of Primary Prophylactic Defibrillator Implantation in Patients with Non-Ischemic Systolic Heart Failure-DANISH-MRI. Am. Heart J. 2020, 221, 165–176. [Google Scholar] [CrossRef]
- Pedretti, S.; Vargiu, S.; Baroni, M.; Dellegrottaglie, S.; Lanzarin, B.; Roghi, A.; Milazzo, A.; Quattrocchi, G.; Lunati, M.; Pedrotti, P. Complexity of Scar and Ventricular Arrhythmias in Dilated Cardiomyopathy of Any Etiology: Long-Term Data from the SCARFEAR (Cardiovascular Magnetic Resonance Predictors of Appropriate Implantable Cardioverter-Defibrillator Therapy Delivery) Registry. Clin. Cardiol. 2018, 41, 494–501. [Google Scholar] [CrossRef]
- Klem, I.; Klein, M.; Khan, M.; Yang, E.Y.; Nabi, F.; Ivanov, A.; Bhatti, L.; Hayes, B.; Graviss, E.A.; Nguyen, D.T.; et al. Relationship of LVEF and Myocardial Scar to Long-Term Mortality Risk and Mode of Death in Patients With Nonischemic Cardiomyopathy. Circulation 2021, 143, 1343–1358. [Google Scholar] [CrossRef]
- Balaban, G.; Halliday, B.P.; Porter, B.; Bai, W.; Nygåard, S.; Owen, R.; Hatipoglu, S.; Ferreira, N.D.; Izgi, C.; Tayal, U.; et al. Late-Gadolinium Enhancement Interface Area and Electrophysiological Simulations Predict Arrhythmic Events in Patients With Nonischemic Dilated Cardiomyopathy. JACC Clin. Electrophysiol. 2021, 7, 238–249. [Google Scholar] [CrossRef] [PubMed]
- McDonagh, T.A.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Böhm, M.; Burri, H.; Butler, J.; Čelutkienė, J.; Chioncel, O.; et al. 2021 ESC Guidelines for the Diagnosis and Treatment of Acute and Chronic Heart Failure. Eur. Heart J. 2021, 42, 3599–3726. [Google Scholar] [CrossRef] [PubMed]
- De Diego, C.; González-Torres, L.; Núñez, J.M.; Centurión Inda, R.; Martin-Langerwerf, D.A.; Sangio, A.D.; Chochowski, P.; Casasnovas, P.; Blazquéz, J.C.; Almendral, J. Effects of Angiotensin-Neprilysin Inhibition Compared to Angiotensin Inhibition on Ventricular Arrhythmias in Reduced Ejection Fraction Patients under Continuous Remote Monitoring of Implantable Defibrillator Devices. Heart Rhythm 2018, 15, 395–402. [Google Scholar] [CrossRef]
- Curtain, J.P.; Docherty, K.F.; Jhund, P.S.; Petrie, M.C.; Inzucchi, S.E.; Køber, L.; Kosiborod, M.N.; Martinez, F.A.; Ponikowski, P.; Sabatine, M.S.; et al. Effect of Dapagliflozin on Ventricular Arrhythmias, Resuscitated Cardiac Arrest, or Sudden Death in DAPA-HF. Eur. Heart J. 2021, 42, 3727–3738. [Google Scholar] [CrossRef]
- Køber, L.; Thune, J.J.; Nielsen, J.C.; Haarbo, J.; Videbæk, L.; Korup, E.; Jensen, G.; Hildebrandt, P.; Steffensen, F.H.; Bruun, N.E.; et al. Defibrillator Implantation in Patients with Nonischemic Systolic Heart Failure. N. Engl. J. Med. 2016, 375, 1221–1230. [Google Scholar] [CrossRef]
- Gatzoulis, K.A.; Dilaveris, P.; Arsenos, P.; Tsiachris, D.; Antoniou, C.-K.; Sideris, S.; Kolettis, T.; Kanoupakis, E.; Sideris, A.; Flevari, P.; et al. Arrhythmic Risk Stratification in Nonischemic Dilated Cardiomyopathy: The ReCONSIDER Study Design-A Two-Step, Multifactorial, Electrophysiology-Inclusive Approach. Hell. J. Cardiol. 2021, 62, 169–172. [Google Scholar] [CrossRef]
- Akhtar, M.; Elliott, P.M. Risk Stratification for Sudden Cardiac Death in Non-Ischaemic Dilated Cardiomyopathy. Curr. Cardiol. Rep. 2019, 21, 155. [Google Scholar] [CrossRef] [PubMed]
- Halliday, B.P.; Gulati, A.; Ali, A.; Newsome, S.; Lota, A.; Tayal, U.; Vassiliou, V.S.; Arzanauskaite, M.; Izgi, C.; Krishnathasan, K.; et al. Sex- and Age-Based Differences in the Natural History and Outcome of Dilated Cardiomyopathy. Eur. J. Heart Fail. 2018, 20, 1392–1400. [Google Scholar] [CrossRef]
- Seferović, P.M.; Polovina, M.; Bauersachs, J.; Arad, M.; Gal, T.B.; Lund, L.H.; Felix, S.B.; Arbustini, E.; Caforio, A.L.P.; Farmakis, D.; et al. Heart Failure in Cardiomyopathies: A Position Paper from the Heart Failure Association of the European Society of Cardiology. Eur. J. Heart Fail. 2019, 21, 553–576. [Google Scholar] [CrossRef] [PubMed]
- Siontis, K.C.; Kim, H.M.; Sharaf Dabbagh, G.; Latchamsetty, R.; Stojanovska, J.; Jongnarangsin, K.; Morady, F.; Bogun, F.M. Association of Preprocedural Cardiac Magnetic Resonance Imaging with Outcomes of Ventricular Tachycardia Ablation in Patients with Idiopathic Dilated Cardiomyopathy. Heart Rhythm 2017, 14, 1487–1493. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Protein | Gene | Protein Function |
|---|---|---|
| Titin | TTN | Sarcomere structural protein |
| Lamin A/C | LMNA | Inner nuclear membrane |
| Cardiac sodium channel Nav1.5 | SCN5A | Cardiac sodium channel α-subunit |
| Filamin C | FLNC | Actin cytoskeleton |
| Desmoplakin | DSP | Desmosomal protein |
| RNA-binding motif protein 20 | RBM20 | RNA binding and splicing regulation |
| Phospholamban | PLN | Sarcoplasmic reticulum protein involved in calcium homeostasis |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mages, C.; Gampp, H.; Syren, P.; Rahm, A.-K.; André, F.; Frey, N.; Lugenbiel, P.; Thomas, D. Electrical Ventricular Remodeling in Dilated Cardiomyopathy. Cells 2021, 10, 2767. https://doi.org/10.3390/cells10102767
Mages C, Gampp H, Syren P, Rahm A-K, André F, Frey N, Lugenbiel P, Thomas D. Electrical Ventricular Remodeling in Dilated Cardiomyopathy. Cells. 2021; 10(10):2767. https://doi.org/10.3390/cells10102767
Chicago/Turabian StyleMages, Christine, Heike Gampp, Pascal Syren, Ann-Kathrin Rahm, Florian André, Norbert Frey, Patrick Lugenbiel, and Dierk Thomas. 2021. "Electrical Ventricular Remodeling in Dilated Cardiomyopathy" Cells 10, no. 10: 2767. https://doi.org/10.3390/cells10102767
APA StyleMages, C., Gampp, H., Syren, P., Rahm, A.-K., André, F., Frey, N., Lugenbiel, P., & Thomas, D. (2021). Electrical Ventricular Remodeling in Dilated Cardiomyopathy. Cells, 10(10), 2767. https://doi.org/10.3390/cells10102767