
Ventricular Arrhythmias in Ischemic Cardiomyopathy—New Avenues for Mechanism-Guided Treatment

 , , , and
, , , and
Abstract
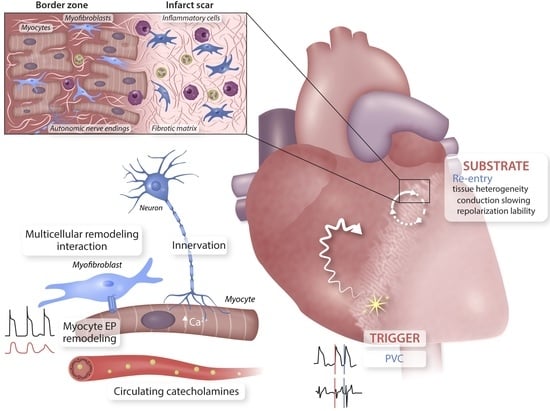
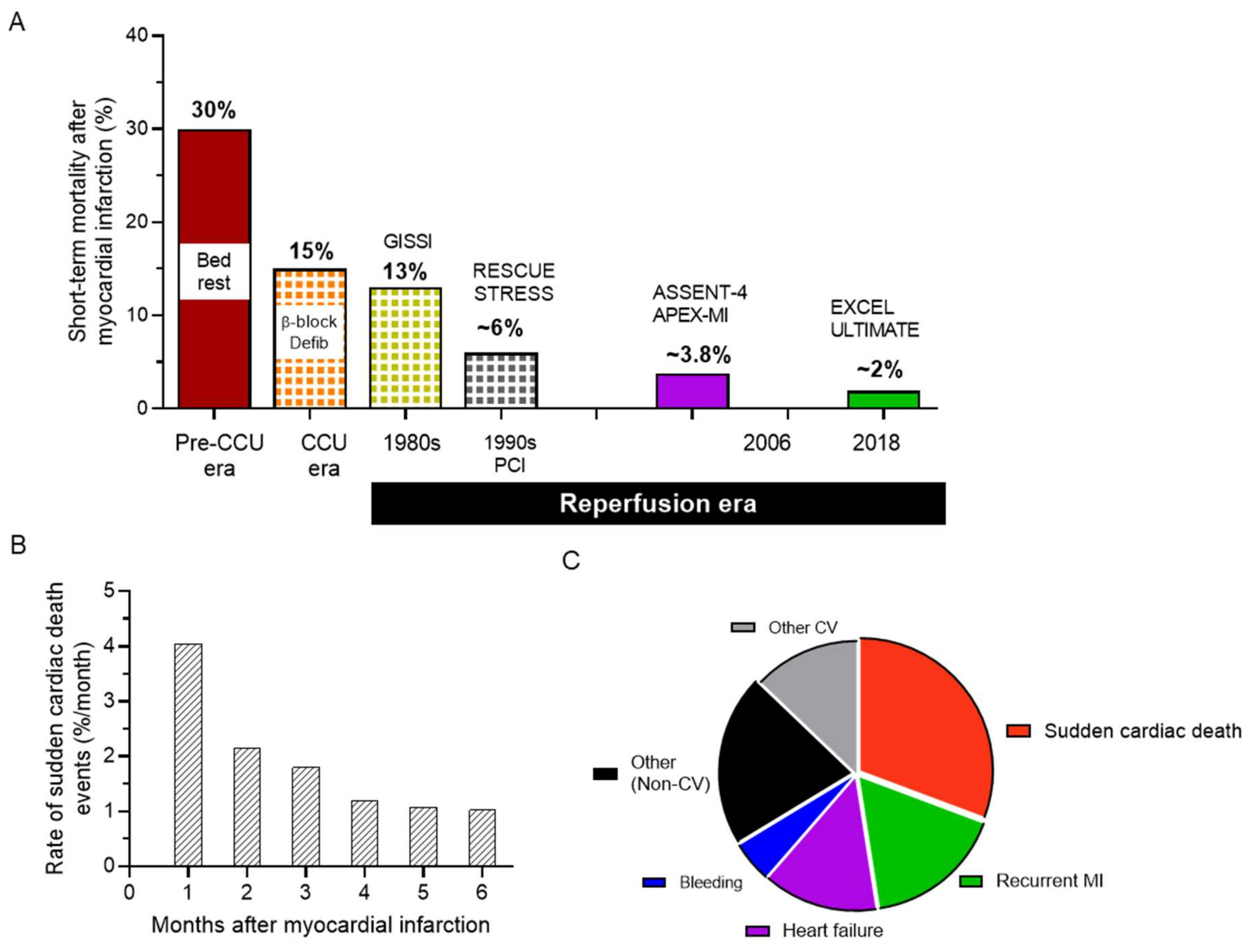
1. The Health Challenge of Arrhythmias in Ischemic Heart Disease
2. Current Management: Much to Be Desired from Pharmacotherapy
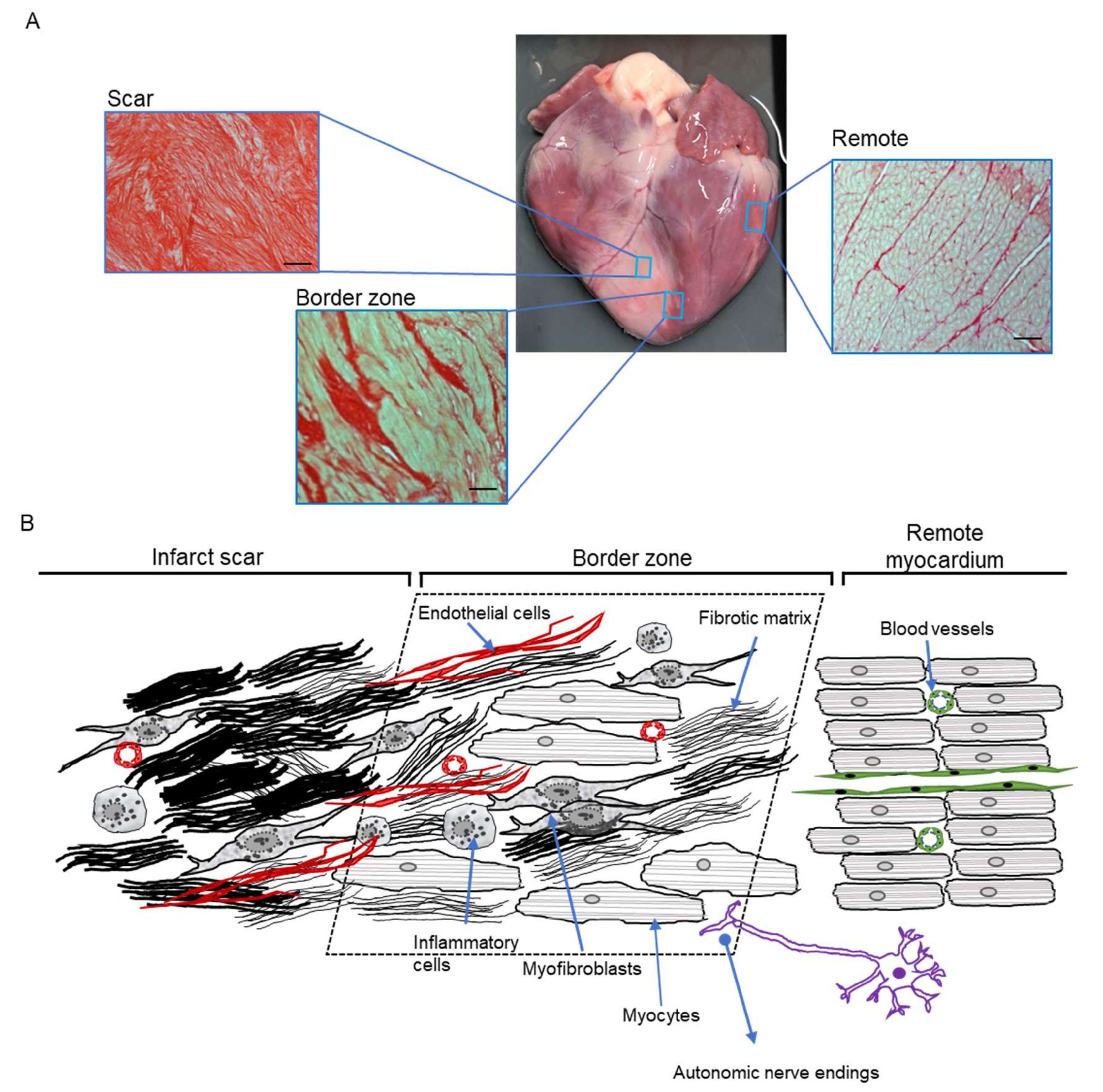
3. The Unique Nature and Central Role of the Border Zone
4. Origin and Maintenance of VT–Mechanistic Insights Obtained In Vivo
4.1. The Conceptual Framework
4.2. Triggers and Arrhythmia Initiation
4.3. Arrhythmia Substrates for Progression and Sustenance/Maintenance
5. Cellular Remodelling in the BZ Underlying In Vivo Arrhythmogenesis
5.1. Animal Models for Post-MI Remodelling
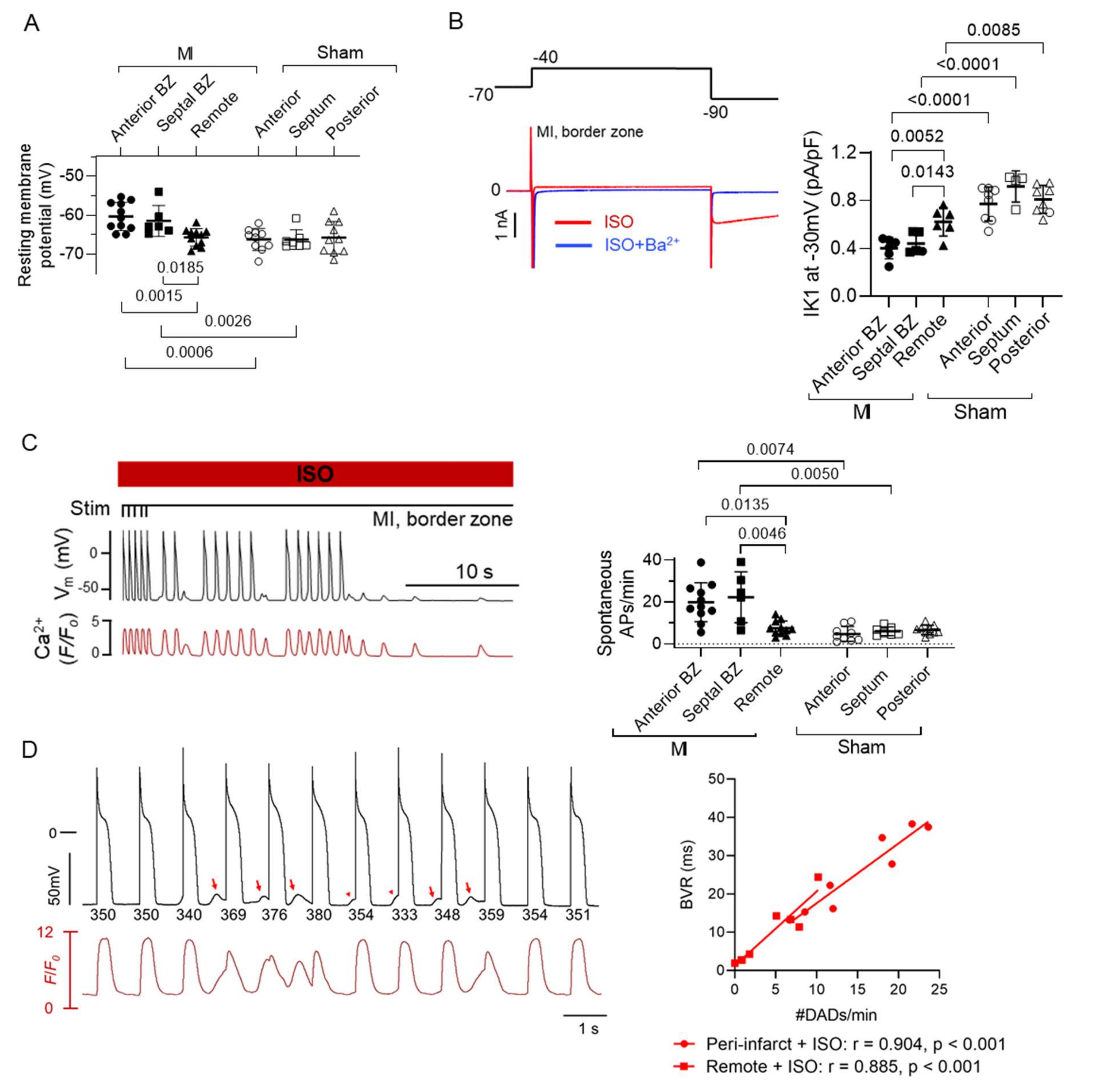
5.2. Myocyte Electrical Remodelling in the BZ–Role in Re-Entry and Triggered Activity
5.3. Role of Autonomic Inputs in the BZ for Arrhythmogenesis
6. Emerging Concepts and Future Research Directions for Post-MI Arrhythmia Management
6.1. Systemic Small Molecule Therapy or Local BZ Targeted Therapy
6.2. Understanding the Complexity of the Arrhythmia Sites
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Timmis, A.; Townsend, N.; Gale, C.P.; Torbica, A.; Lettino, M.; Petersen, S.E.; Mossialos, E.A.; Maggioni, A.P.; Kazakiewicz, D.; May, H.T.; et al. European Society of Cardiology: Cardiovascular Disease Statistics 2019. Eur. Heart J. 2020, 41, 12–85. [Google Scholar] [CrossRef]
- Virani, S.S.; Alonso, A.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Delling, F.N.; et al. Heart Disease and Stroke Statistics—2020 Update: A Report from the American Heart Association. Circulation 2020, 141, e139–e596. [Google Scholar] [CrossRef]
- Berg, D.D.; Wiviott, S.D.; Braunwald, E.; Guo, J.; Im, K.; Kashani, A.; Gibson, C.M.; Cannon, C.P.; Morrow, D.A.; Bhatt, D.L.; et al. Modes and timing of death in 66 252 patients with non-ST-segment elevation acute coronary syndromes enrolled in 14 TIMI trials. Eur. Heart J. 2018, 39, 3810–3820. [Google Scholar] [CrossRef]
- Grey, C.; Jackson, R.; Schmidt, M.; Ezzati, M.; Asaria, P.; Exeter, D.J.; Kerr, A.J. One in four major ischaemic heart disease events are fatal and 60% are pre-hospital deaths: A national data-linkage study (ANZACS-QI 8). Eur. Heart J. 2015, 38, 172–180. [Google Scholar] [CrossRef]
- Solomon, S.D.; Zelenkofske, S.; Mcmurray, J.; Finn, P.V.; Velazquez, E.; Ertl, G.; Harsanyi, A.; Rouleau, J.L.; Maggioni, A.P.; Kober, L.; et al. Sudden Death in Patients with Myocardial Infarction and Left Ventricular Dysfunction, Heart Failure, or Both. N. Engl. J. Med. 2005, 352, 2581–2588. [Google Scholar] [CrossRef]
- Naghavi, M.; Libby, P.; Falk, E.; Casscells, S.W.; Litovsky, S.; Rumberger, J.; Badimon, J.J.; Stefanadis, C.; Moreno, P.; Pasterkamp, G.; et al. From Vulnerable Plaque to Vulnerable Patient: A call for new definitions and risk assessment strategies: Part I. Circulation 2003, 108, 1664–1672. [Google Scholar] [CrossRef] [PubMed]
- Braunwald, E. Cardiovascular Medicine at the Turn of the Millennium: Triumphs, Concerns, and Opportunities. N. Engl. J. Med. 1997, 337, 1360–1369. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, P.; Gershlick, A.H.; Goldstein, P.; Wilcox, R.; Danays, T.; Lambert, Y.; Sulimov, V.; Rosell-Ortiz, F.; Ostojic, M.; Welsh, R.; et al. Fibrinolysis or Primary PCI in ST-Segment Elevation Myocardial Infarction. N. Engl. J. Med. 2013, 368, 1379–1387. [Google Scholar] [CrossRef] [PubMed]
- Dan, G.-A.; Martinez-Rubio, A.; Agewall, S.; Boriani, G.; Borggrefe, M.; Gaita, F.; Van Gelder, I.; Gorenek, B.; Kaski, J.C.; Kjeldsen, K.; et al. Antiarrhythmic drugs–clinical use and clinical decision making: A consensus document from the European Heart Rhythm Association (EHRA) and European Society of Cardiology (ESC) Working Group on Cardiovascular Pharmacology, endorsed by the Heart Rhythm Society (HRS), Asia-Pacific Heart Rhythm Society (APHRS) and International Society of Cardiovascular Pharmacotherapy (ISCP). EP Eur. 2018, 20, 731–732. [Google Scholar] [CrossRef]
- Amsterdam, E.A.; Wenger, N.K.; Brindis, R.G.; Casey, D.; Ganiats, T.G.; Holmes, D.R.; Jaffe, A.S.; Jneid, H.; Kelly, R.F.; Kontos, M.C.; et al. 2014 AHA/ACC Guideline for the Management of Patients with Non–ST-Elevation Acute Coronary Syndromes: Executive Summary. Circulation 2014, 130, 2354–2394. [Google Scholar] [CrossRef] [PubMed]
- Levine, G.N.; Bates, E.R.; Blankenship, J.C.; Bailey, S.R.; Bittl, J.A.; Cercek, B.; Chambers, C.E.; Ellis, S.G.; Guyton, R.A.; Hollenberg, S.M.; et al. 2015 ACC/AHA/SCAI Focused Update on Primary Percutaneous Coronary Intervention for Patients with ST-Elevation Myocardial Infarction: An Update of the 2011 ACCF/AHA/SCAI Guideline for Percutaneous Coronary Intervention and the 2013 ACCF/AHA Guideline for the Management of ST-Elevation Myocardial Infarction. J. Am. Coll. Cardiol. 2016, 67, 1235–1250. [Google Scholar] [CrossRef] [PubMed]
- Mehta, D.; Curwin, J.; Gomes, J.A.; Fuster, V. Sudden death in coronary artery disease: Acute ischemia versus myocardial substrate. Circulation 1997, 96, 3215–3223. [Google Scholar] [CrossRef] [PubMed]
- Davidson, S.M.; Ferdinandy, P.; Andreadou, I.; Botker, H.E.; Heusch, G.; Ibanez, B.; Ovize, M.; Schulz, R.; Yellon, D.M.; Hausenloy, D.J.; et al. Multitarget Strategies to Reduce Myocardial Ischemia/Reperfusion Injury: JACC Review Topic of the Week. J. Am. Coll. Cardiol. 2019, 73, 89–99. [Google Scholar] [CrossRef]
- Echt, D.S.; Liebson, P.R.; Mitchell, L.B.; Peters, R.W.; Obias-Manno, D.; Barker, A.H.; Arensberg, D.; Baker, A.; Friedman, L.; Greene, H.L.; et al. Mortality and morbidity in patients receiving encainide, flecainide, or placebo. The Cardiac Arrhythmia Suppression Trial. N. Engl. J. Med. 1991, 324, 781–788. [Google Scholar] [CrossRef] [PubMed]
- Waldo, A.L.; Camm, A.J.; Deruyter, H.; Friedman, P.L.; MacNeil, D.J.; Pauls, J.F.; Pitt, B.; Pratt, C.M.; Schwartz, P.J.; Veltri, E.P. Effect of d-sotalol on mortality in patients with left ventricular dysfunction after recent and remote myocardial infarction. Lancet 1996, 348, 7–12. [Google Scholar] [CrossRef]
- Al-Khatib, S.M.; Stevenson, W.G.; Ackerman, M.J.; Bryant, W.J.; Callans, D.J.; Curtis, A.B.; Deal, B.J.; Dickfeld, T.; Field, M.E.; Fonarow, G.C.; et al. 2017 AHA/ACC/HRS guideline for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: Executive summary. Heart Rhythm. 2018, 15, e190–e252. [Google Scholar] [CrossRef]
- Al-Gobari, M.; El Khatib, C.; Pillon, F.; Gueyffier, F. Beta-blockers for the prevention of sudden cardiac death in heart failure patients: A meta-analysis of randomized controlled trials. BMC Cardiovasc. Disord. 2013, 13, 1–52. [Google Scholar] [CrossRef] [PubMed]
- Viskin, S.; Chorin, E.; Viskin, D.; Hochstadt, A.; Halkin, A.; Tovia-Brodie, O.; Lee, J.K.; Asher, E.; Laish-Farkash, A.; Amit, G.; et al. Quinidine-Responsive Polymorphic Ventricular Tachycardia in Patients with Coronary Heart Disease. Circulation 2019, 139, 2304–2314. [Google Scholar] [CrossRef]
- Zabel, M.; Willems, R.; Lubinski, A.; Bauer, A.; Brugada, J.; Conen, D.; Flevari, P.; Hasenfuß, G.; Svetlosak, M.; Huikuri, H.V.; et al. Clinical effectiveness of primary prevention implantable cardioverter-defibrillators: Results of the EU-CERT-ICD controlled multicentre cohort study. Eur. Heart J. 2020, 41, 3437–3447. [Google Scholar] [CrossRef]
- Myerburg, R.J.; Junttila, M.J. Sudden Cardiac Death Caused by Coronary Heart Disease. Circulation 2012, 125, 1043–1052. [Google Scholar] [CrossRef]
- Chatterjee, N.A.; Moorthy, M.V.; Pester, J.; Schaecter, A.; Panicker, G.K.; Narula, D.; Lee, D.; Goldberger, J.J.; Kadish, A.; Cook, N.R.; et al. Sudden Death in Patients with Coronary Heart Disease without Severe Systolic Dysfunction. JAMA Cardiol. 2018, 3, 591–600. [Google Scholar] [CrossRef]
- Priori, S.; Blomström-Lundqvist, C.; Mazzanti, A.; Blom, N.A.; Borggrefe, M.; Camm, J.; Elliott, P.; Fitzsimons, D.; Hatala, R.; Hindricks, G.; et al. 2015 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death. Eur. Heart J. 2015, 36, 2793–2867. [Google Scholar] [CrossRef]
- Pedersen, C.T.; Kay, G.N.; Kalman, J.; Borggrefe, M.; Della-Bella, P.; Dickfeld, T.; Dorian, P.; Huikuri, H.; Kim, Y.-H.; Knight, B.; et al. EHRA/HRS/APHRS Expert Consensus on Ventricular Arrhythmias. Heart Rhythm. 2014, 11, e166–e196. [Google Scholar] [CrossRef]
- Buxton, A.E.; Lee, K.L.; Dicarlo, L.; Gold, M.R.; Greer, G.S.; Prystowsky, E.N.; O’Toole, M.F.; Tang, A.; Fisher, J.; Coromilas, J.; et al. Electrophysiologic Testing to Identify Patients with Coronary Artery Disease Who Are at Risk for Sudden Death. N. Engl. J. Med. 2000, 342, 1937–1945. [Google Scholar] [CrossRef]
- Daubert, J.P.; Zareba, W.; Hall, W.J.; Schuger, C.; Corsello, A.; Leon, A.R.; Andrews, M.L.; McNitt, S.; Huang, D.T.; Moss, A.J. Predictive Value of Ventricular Arrhythmia Inducibility for Subsequent Ventricular Tachycardia or Ventricular Fibrillation in Multicenter Automatic Defibrillator Implantation Trial (MADIT) II Patients. J. Am. Coll. Cardiol. 2006, 47, 98–107. [Google Scholar] [CrossRef]
- Wellens, H.J.J.; Schwartz, P.J.; Lindemans, F.W.; Buxton, A.E.; Goldberger, J.J.; Hohnloser, S.H.; Huikuri, H.V.; Kaab, S.; La Rovere, M.T.; Malik, M.; et al. Risk stratification for sudden cardiac death: Current status and challenges for the future. Eur. Heart J. 2014, 35, 1642–1651. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, J.C.; Lin, Y.; Figueiredo, M.J.D.O.; Shamloo, A.S.; Alfie, A.; Boveda, S.; Dagres, N.; Di Toro, D.; Eckhardt, L.L.; Ellenbogen, K.; et al. European Heart Rhythm Association (EHRA)/Heart Rhythm Society (HRS)/Asia Pacific Heart Rhythm Society (APHRS)/Latin American Heart Rhythm Society (LAHRS) expert consensus on risk assessment in cardiac arrhythmias: Use the right tool for the right outcome, in the right population. J. Arrhythmia 2020, 36, 553–607. [Google Scholar] [CrossRef]
- Schmidt, A.; Azevedo, C.F.; Cheng, A.; Gupta, S.N.; Bluemke, D.; Foo, T.K.; Gerstenblith, G.; Weiss, R.G.; Marbán, E.; Tomaselli, G.F.; et al. Infarct Tissue Heterogeneity by Magnetic Resonance Imaging Identifies Enhanced Cardiac Arrhythmia Susceptibility in Patients with Left Ventricular Dysfunction. Circulation 2007, 115, 2006–2014. [Google Scholar] [CrossRef]
- Reichlin, T.; Asatryan, B.; Vos, M.A.; Willems, R.; Huikuri, H.V.; Junttila, M.J.; Schlögl, S.C.; Hnatkova, K.; Schaer, B.A.; Malik, M.; et al. Automated electrocardiographic quantification of myocardial scar in patients undergoing primary prevention implantable cardioverter-defibrillator implantation: Association with mortality and subsequent appropriate and inappropriate therapies. Heart Rhythm. 2020, 17, 1664–1671. [Google Scholar] [CrossRef] [PubMed]
- Goldberger, J.J.; Cain, M.E.; Hohnloser, S.H.; Kadish, A.H.; Knight, B.P.; Lauer, M.S.; Maron, B.J.; Page, R.L.; Passman, R.S.; Siscovick, D.; et al. American Heart Association/American College of Cardiology Foundation/Heart Rhythm Society Scientific Statement on Noninvasive Risk Stratification Techniques for Identifying Patients at Risk for Sudden Cardiac Death: A Scientific Statement From the American Heart Association Council on Clinical Cardiology Committee on Electrocardiography and Arrhythmias and Council on Epidemiology and Prevention. J. Am. Coll. Cardiol. 2008, 52, 1179–1199. [Google Scholar] [CrossRef] [PubMed]
- De Sousa, M.R.; Morillo, C.A.; Rabelo, F.T.; Filho, A.M.N.; Ribeiro, A. Non-sustained ventricular tachycardia as a predictor of sudden cardiac death in patients with left ventricular dysfunction: A meta-analysis. Eur. J. Heart Fail. 2008, 10, 1007–1014. [Google Scholar] [CrossRef]
- Bauer, A.; Klemm, M.; Rizas, K.; Hamm, W.; von Stülpnagel, L.; Dommasch, M.; Steger, A.; Lubinski, A.; Flevari, P.; Harden, M.; et al. Prediction of mortality benefit based on periodic repolarisation dynamics in patients undergoing prophylactic implantation of a defibrillator: A prospective, controlled, multicentre cohort study. Lancet 2019, 394, 1344–1351. [Google Scholar] [CrossRef]
- Seegers, J.; Bergau, L.; Expósito, P.M.; Bauer, A.; Fischer, T.H.; Lüthje, L.; Hasenfuß, G.; Friede, T.; Zabel, M. Prediction of Appropriate Shocks Using 24-Hour Holter Variables and T-Wave Alternans After First Implantable Cardioverter-Defibrillator Implantation in Patients with Ischemic or Nonischemic Cardiomyopathy. Am. J. Cardiol. 2016, 118, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Amoni, M.; Vandenberk, B.; Moeyersons, J.; Van Huffel, S.; Sipido, K.R.; Willems, R. Temporal beat-to-beat variability of repolarizationTemporal beat-to-beat variability of repolarization (BVR) changes predict imminent non-sustained ventricular tachycardia in ischaemic heart disease patients. Europace 2018, 20, i41. [Google Scholar] [CrossRef]
- Smoczyńska, A.; Loen, V.; Sprenkeler, D.J.; Tuinenburg, A.E.; Van Eck, H.J.R.; Malik, M.; Schmidt, G.; Meine, M.; Vos, M.A. Short-Term Variability of the QT Interval Can be Used for the Prediction of Imminent Ventricular Arrhythmias in Patients with Primary Prophylactic Implantable Cardioverter Defibrillators. J. Am. Heart Assoc. 2020, 9, e018133. [Google Scholar] [CrossRef] [PubMed]
- Vandenberk, B.; Junttila, M.J.; Robyns, T.; Garweg, C.; Ector, J.; Huikuri, H.V.; Willems, R. Combining noninvasive risk stratification parameters improves the prediction of mortality and appropriate ICD shocks. Ann. Noninvasive Electrocardiol. 2018, 24, e12604. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, N.A.; Tikkanen, J.T.; Panicker, G.K.; Narula, D.; Lee, D.C.; Kenttä, T.; Junttila, J.M.; Cook, N.R.; Kadish, A.; Goldberger, J.J.; et al. Simple electrocardiographic measures improve sudden arrhythmic death prediction in coronary disease. Eur. Heart J. 2020, 41, 1988–1999. [Google Scholar] [CrossRef]
- Verstraelen, T.E.; van Barreveld, M.; van Dessel, P.H.F.M.; Boersma, L.V.A.; Delnoy, P.-P.P.H.M.; Tuinenburg, A.E.; Theuns, D.A.M.J.; van der Voort, P.H.; Kimman, G.P.; Buskens, E.; et al. Development and external validation of prediction models to predict implantable cardioverter-defibrillator efficacy in primary prevention of sudden cardiac death. EP Eur. 2021, 23, 887–897. [Google Scholar] [CrossRef]
- Li, A.; Kaura, A.; Sunderland, N.; Dhillon, P.S.; Scott, P.A. The Significance of Shocks in Implantable Cardioverter Defibrillator Recipients. Arrhythmia Electrophysiol. Rev. 2016, 5, 110–116. [Google Scholar] [CrossRef]
- Mitchell, L.; Pineda, E.A.; Titus, J.L.; Bartosch, P.M.; Benditt, D.G. Sudden death in patients with implantable cardioverter defibrillators: The importance of post-shock electromechanical dissociation. J. Am. Coll. Cardiol. 2002, 39, 1323–1328. [Google Scholar] [CrossRef]
- Thylén, I.; Moser, D.K.; Strömberg, A.; Dekker, R.A.; Chung, M.L. Concerns about implantable cardioverter-defibrillator shocks mediate the relationship between actual shocks and psychological distress. Europace 2016, 18, 828–835. [Google Scholar] [CrossRef]
- Cronin, E.M.; Bogun, F.M.; Maury, P.; Peichl, P.; Chen, M.; Namboodiri, N.; Aguinaga, L.; Leite, L.R.; Al-Khatib, S.M.; Anter, E.; et al. 2019 HRS/EHRA/APHRS/LAHRS expert consensus statement on catheter ablation of ventricular arrhythmias: Executive summary. Europace 2020, 22, 450–495. [Google Scholar] [CrossRef]
- Sapp, J.L.; Wells, G.A.; Parkash, R.; Stevenson, W.G.; Blier, L.; Sarrazin, J.-F.; Thibault, B.; Rivard, L.; Gula, L.; Leong-Sit, P.; et al. Ventricular Tachycardia Ablation versus Escalation of Antiarrhythmic Drugs. N. Engl. J. Med. 2016, 375, 111–121. [Google Scholar] [CrossRef]
- Sadek, M.M.; Schaller, R.D.; Supple, G.E.; Frankel, D.S.; Riley, M.P.; Hutchinson, M.D.; Garcia, F.C.; Lin, D.; Dixit, S.; Zado, E.S.; et al. Ventricular Tachycardia Ablation—The Right Approach for the Right Patient. Arrhythmia Electrophysiol. Rev. 2014, 3, 161–167. [Google Scholar] [CrossRef]
- Baldinger, S.H.; Stevenson, W.G.; John, R.M. Ablation of ischemic ventricular tachycardia: Evidence, techniques, results, and future directions. Curr. Opin. Cardiol. 2016, 31, 29–36. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Vaseghi, M.; Barwad, P.; Corrales, F.J.M.; Tandri, H.; Mathuria, N.; Shah, R.; Sorg, J.M.; Gima, J.; Mandal, K.; Morales, L.C.S.; et al. Cardiac Sympathetic Denervation for Refractory Ventricular Arrhythmias. J. Am. Coll. Cardiol. 2017, 69, 3070–3080. [Google Scholar] [CrossRef] [PubMed]
- Meng, L.; Tseng, C.-H.; Shivkumar, K.; Ajijola, O. Efficacy of Stellate Ganglion Blockade in Managing Electrical Storm. A Systematic Review. JACC Clin. Electrophysiol. 2017, 3, 942–949. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G. Regulation of the Inflammatory Response in Cardiac Repair. Circ. Res. 2012, 110, 159–173. [Google Scholar] [CrossRef] [PubMed]
- Amoni, M.; Claus, P.; Dries, E.; Nagaraju, C.; De Buck, S.; Vandenberk, B.; Ingelaere, S.; Vermoortele, D.; Roderick, H.L.; Sipido, K.R.; et al. Discrete sites of frequent premature ventricular complexes cluster within the infarct border zone and coincide with high frequency of delayed afterdepolarizations under adrenergic stimulation. Heart Rhythm. 2021, 7, 67. [Google Scholar] [CrossRef]
- Daseke, M.J.; Chalise, U.; Becirovic-Agic, M.; Salomon, J.D.; Cook, L.M.; Case, A.J.; Lindsey, M.L. Neutrophil signaling during myocardial infarction wound repair. Cell. Signal. 2021, 77, 109816. [Google Scholar] [CrossRef]
- Ma, Y.; Mouton, A.J.; Lindsey, M.L. Cardiac macrophage biology in the steady-state heart, the aging heart, and following myocardial infarction. Transl. Res. 2018, 191, 15–28. [Google Scholar] [CrossRef]
- Rossello, X.; Yellon, D.M. The RISK pathway and beyond. Basic Res. Cardiol. 2018, 113, 1–5. [Google Scholar] [CrossRef]
- Hadebe, N.; Cour, M.; Lecour, S. The SAFE pathway for cardioprotection: Is this a promising target? Basic Res. Cardiol. 2018, 113, 9. [Google Scholar] [CrossRef]
- Opie, L.H.; Commerford, P.J.; Gersh, B.J.; Pfeffer, M.A. Controversies in ventricular remodelling. Lancet 2006, 367, 356–367. [Google Scholar] [CrossRef]
- Ashikaga, H.; Mickelsen, S.R.; Ennis, D.B.; Rodriguez, I.; Kellman, P.; Wen, H.; McVeigh, E.R. Electromechanical analysis of infarct border zone in chronic myocardial infarction. Am. J. Physiol. Circ. Physiol. 2005, 289, H1099–H1105. [Google Scholar] [CrossRef] [PubMed]
- Jackson, B.M.; Gorman, J.H.; Salgo, I.S.; Moainie, S.L.; Plappert, T.; John-Sutton, M.S.; Edmunds, L.H.; Gorman, R.C. Border zone geometry increases wall stress after myocardial infarction: Contrast echocardiographic assessment. Am. J. Physiol. Circ. Physiol. 2003, 284, H475–H479. [Google Scholar] [CrossRef] [PubMed]
- Frisk, M.; Ruud, M.; Espe, E.K.S.; Aronsen, J.M.; Røe, Å.T.; Zhang, L.; Norseng, P.A.; Sejersted, O.M.; Christensen, G.A.; Sjaastad, I.; et al. Elevated ventricular wall stress disrupts cardiomyocyte t-tubule structure and calcium homeostasis. Cardiovasc. Res. 2016, 112, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Galan, D.T.; Bito, V.; Claus, P.; Holemans, P.; Abi-Char, J.; Nagaraju, C.K.; Dries, E.; Vermeulen, K.; Ventura-Clapier, R.; Sipido, K.R.; et al. Reduced mitochondrial respiration in the ischemic as well as in the remote nonischemic region in postmyocardial infarction remodeling. Am. J. Physiol. Circ. Physiol. 2016, 311, H1075–H1090. [Google Scholar] [CrossRef] [PubMed]
- Nagaraju, C.K.; Dries, E.; Popovic, N.; Singh, A.; Haemers, P.; Roderick, H.; Claus, P.; Sipido, K.R.; Driesen, R.B. Global fibroblast activation throughout the left ventricle but localized fibrosis after myocardial infarction. Sci. Rep. 2017, 7, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Cox, M.M.; Berman, I.; Myerburg, R.J.; Smets, M.J.; Kozlovskis, P.L. Morphometric mapping of regional myocyte diameters after healing of myocardial infarction in cats. J. Mol. Cell. Cardiol. 1991, 23, 127–135. [Google Scholar] [CrossRef]
- Yokoyama, T.; Lee, J.-K.; Miwa, K.; Opthof, T.; Tomoyama, S.; Nakanishi, H.; Yoshida, A.; Yasui, H.; Iida, T.; Miyagawa, S.; et al. Quantification of sympathetic hyperinnervation and denervation after myocardial infarction by three-dimensional assessment of the cardiac sympathetic network in cleared transparent murine hearts. PLoS ONE 2017, 12, e0182072. [Google Scholar] [CrossRef]
- Matsunari, I.; Schricke, U.; Bengel, F.M.; Haase, H.-U.; Barthel, P.; Schmidt, G.; Nekolla, S.G.; Schoemig, A.; Schwaiger, M. Extent of cardiac sympathetic neuronal damage is determined by the area of ischemia in patients with acute coronary syndromes. Circulation 2000, 101, 2579–2585. [Google Scholar] [CrossRef]
- Cao, J.-M.; Fishbein, M.C.; Han, J.B.; Lai, W.W.; Lai, A.C.; Wu, T.-J.; Czer, L.; Wolf, P.L.; Denton, T.A.; Shintaku, I.P.; et al. Relationship between regional cardiac hyperinnervation and ventricular arrhythmia. Circulation 2000, 101, 1960–1969. [Google Scholar] [CrossRef]
- Rajendran, P.S.; Nakamura, K.; Ajijola, O.; Vaseghi, M.; Armour, J.A.; Ardell, J.L.; Shivkumar, K. Myocardial infarction induces structural and functional remodelling of the intrinsic cardiac nervous system. J. Physiol. 2015, 594, 321–341. [Google Scholar] [CrossRef]
- Gardner, R.T.; Wang, L.; Lang, B.T.; Cregg, J.M.; Dunbar, C.L.; Woodward, W.R.; Silver, J.; Ripplinger, C.M.; Habecker, B.A. Targeting protein tyrosine phosphatase σ after myocardial infarction restores cardiac sympathetic innervation and prevents arrhythmias. Nat. Commun. 2015, 6, 6235. [Google Scholar] [CrossRef]
- Zhou, S.; Chen, L.S.; Miyauchi, Y.; Miyauchi, M.; Kar, S.; Kangavari, S.; Fishbein, M.C.; Sharifi, B.; Chen, P.-S. Mechanisms of Cardiac Nerve Sprouting After Myocardial Infarction in Dogs. Circ. Res. 2004, 95, 76–83. [Google Scholar] [CrossRef]
- Habecker, B.A.; Anderson, M.E.; Birren, S.J.; Fukuda, K.; Herring, N.; Hoover, D.B.; Kanazawa, H.; Paterson, D.J.; Ripplinger, C. Molecular and cellular neurocardiology: Development, and cellular and molecular adaptations to heart disease. J. Physiol. 2016, 594, 3853–3875. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Lee, P.; Mirams, G.R.; Sarathchandra, P.; Borg, T.K.; Gavaghan, D.J.; Kohl, P.; Bollensdorff, C. Cardiac tissue slices: Preparation, handling, and successful optical mapping. Am. J. Physiol. Circ. Physiol. 2015, 308, H1112–H1125. [Google Scholar] [CrossRef] [PubMed]
- Ajijola, O.; Yagishita, D.; Patel, K.J.; Vaseghi, M.; Zhou, W.; Yamakawa, K.; So, E.; Lux, R.L.; Mahajan, A.; Shivkumar, K. Focal myocardial infarction induces global remodeling of cardiac sympathetic innervation: Neural remodeling in a spatial context. Am. J. Physiol. Circ. Physiol. 2013, 305, H1031–H1040. [Google Scholar] [CrossRef] [PubMed]
- Ajijola, O.A.; Yagishita, D.; Reddy, N.K.; Yamakawa, K.; Vaseghi, M.; Downs, A.M.; Hoover, D.B.; Ardell, J.L.; Shivkumar, K. Remodeling of stellate ganglion neurons after spatially targeted myocardial infarction: Neuropeptide and morphologic changes. Heart Rhythm. 2015, 12, 1027–1035. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Kobayashi, K.; Joung, B.; Piccirillo, G.; Maruyama, M.; Vinters, H.V.; March, K.; Lin, S.-F.; Shen, C.; Fishbein, M.C.; et al. Electroanatomic Remodeling of the Left Stellate Ganglion After Myocardial Infarction. J. Am. Coll. Cardiol. 2012, 59, 954–961. [Google Scholar] [CrossRef]
- Padro, T.; Manfrini, O.; Bugiardini, R.; Canty, J.; Cenko, E.; De Luca, G.; Duncker, D.J.; Eringa, E.C.; Koller, A.; Tousoulis, D.; et al. ESC Working Group on Coronary Pathophysiology and Microcirculation position paper on ‘coronary microvascular dysfunction in cardiovascular disease′. Cardiovasc. Res. 2020, 116, 741–755. [Google Scholar] [CrossRef]
- Robbers, L.F.; Eerenberg, E.S.; Teunissen, P.F.; Jansen, M.F.; Hollander, M.R.; Horrevoets, A.J.; Knaapen, P.; Nijveldt, R.; Heymans, M.W.; Levi, M.M.; et al. Magnetic resonance imaging-defined areas of microvascular obstruction after acute myocardial infarction represent microvascular destruction and haemorrhage. Eur. Heart J. 2013, 34, 2346–2353. [Google Scholar] [CrossRef]
- van der Burg, A.E.B.; Bax, J.J.; Boersma, E.; Pauwels, E.K.; van der Wall, E.E.; Schalij, M.J. Impact of Viability, Ischemia, Scar Tissue, and Revascularization on Outcome After Aborted Sudden Death. Circulation 2003, 108, 1954–1959. [Google Scholar] [CrossRef]
- Travin, M.I.; Dessouki, A.; Cameron, T.; Helle, G.V. Use of exercise technetiumtium-99m sestamibi SPECT imagina to detect residual ischemia and for risk stratification after acute myocardial infarction. Am. J. Cardiol. 1995, 75, 665–669. [Google Scholar] [CrossRef]
- Wu, X.; Reboll, M.R.; Korf-Klingebiel, M.; Wollert, K.C. Angiogenesis after acute myocardial infarction. Cardiovasc. Res. 2021, 117, 1257–1273. [Google Scholar] [CrossRef] [PubMed]
- Merkus, D.; Muller-Delp, J.; Heaps, C.L. Coronary microvascular adaptations distal to epicardial artery stenosis. Am. J. Physiol. Circ. Physiol. 2021, 320, H2351–H2370. [Google Scholar] [CrossRef] [PubMed]
- Kalkman, E.A.; Van Haren, P.; Saxena, P.R.; Schoemaker, R.G. Regionally Different Vascular Response to Vasoactive Substances in the Remodelled Infarcted Rat Heart; Aberrant Vasculature in the Infarct Scar. J. Mol. Cell. Cardiol. 1997, 29, 1487–1497. [Google Scholar] [CrossRef] [PubMed]
- Sellke, F.W.; Kagaya, Y.; Johnson, R.G.; Shafique, T.; Schoen, F.J.; Grossman, W.; Weintraub, R.M. Endothelial modulation of porcine coronary microcirculation perfused via immature collaterals. Am. J. Physiol. Circ. Physiol. 1992, 262, H1669–H1675. [Google Scholar] [CrossRef] [PubMed]
- Gorenek, B.; Cengiz, O.; Kudaiberdieva, G.; Durak, I.; Dogan, V.; Yasar, B.; Birdane, A.; Cavusoglu, Y.; Ata, N. Mode of onset of polymorphic ventricular tachycardia in acute myocardial infarction. Can. J. Cardiol. 2010, 26, e254–e257. [Google Scholar] [CrossRef]
- Lerma, C.; Gorelick, A.; Ghanem, R.N.; Glass, L.; Huikuri, H.V. Patterns of ectopy leading to increased risk of fatal or near-fatal cardiac arrhythmia in patients with depressed left ventricular function after an acute myocardial infarction. EP Eur. 2013, 15, 1304–1312. [Google Scholar] [CrossRef]
- Janse, M.J.; van Capelle, F.J.; Morsink, H.; Kléber, A.G.; Wilms-Schopman, F.; Cardinal, R.; D’Alnoncourt, C.N.; Durrer, D. Flow of "injury" current and patterns of excitation during early ventricular arrhythmias in acute regional myocardial ischemia in isolated porcine and canine hearts. Evidence for two different arrhythmogenic mechanisms. Circ. Res. 1980, 47, 151–165. [Google Scholar] [CrossRef]
- Szumowski, L.; Sanders, P.; Walczak, F.; Hocini, M.; Jaïs, P.; Kepski, R.; Szufladowicz, E.; Urbanek, P.; Derejko, P.; Bodalski, R.; et al. Mapping and ablation of polymorphic ventricular tachycardia after myocardial infarction. J. Am. Coll. Cardiol. 2004, 44, 1700–1706. [Google Scholar] [CrossRef]
- Xing, D.; Martins, J.B. Triggered activity due to delayed afterdepolarizations in sites of focal origin of ischemic ventricular tachycardia. Am. J. Physiol. Circ. Physiol. 2004, 287, H2078–H2084. [Google Scholar] [CrossRef] [PubMed]
- Volders, P.G.; Vos, M.A.; Szabo, B.; Sipido, K.R.; De Groot, S.; Gorgels, A.P.; Wellens, H.J.; Lazzara, R. Progress in the understanding of cardiac early afterdepolarizations and torsades de pointes: Time to revise current concepts. Cardiovasc. Res. 2000, 46, 376–392. [Google Scholar] [CrossRef]
- Wit, A.L. Afterdepolarizations and triggered activity as a mechanism for clinical arrhythmias. Pacing Clin. Electrophysiol. 2018, 41, 883–896. [Google Scholar] [CrossRef] [PubMed]
- Janse, M.J.; Wit, A.L. Electrophysiological mechanisms of ventricular arrhythmias resulting from myocardial ischemia and infarction. Physiol. Rev. 1989, 69, 1049–1169. [Google Scholar] [CrossRef]
- Hegyi, B.; Bossuyt, J.; Griffiths, L.G.; Shimkunas, R.; Coulibaly, Z.; Jian, Z.; Grimsrud, K.N.; Sondergaard, C.S.; Ginsburg, K.S.; Chiamvimonvat, N.; et al. Complex electrophysiological remodeling in postinfarction ischemic heart failure. Proc. Natl. Acad. Sci. USA 2018, 115, E3036–E3044. [Google Scholar] [CrossRef]
- Dries, E.; Amoni, M.; Vandenberk, B.; Johnson, D.M.; Gilbert, G.; Nagaraju, C.K.; Puertas, R.D.; Abdesselem, M.; Santiago, D.J.; Roderick, H.L.; et al. Altered adrenergic response in myocytes bordering a chronic myocardial infarction underlies in vivo triggered activity and repolarization instability. J. Physiol. 2020, 598, 2875–2895. [Google Scholar] [CrossRef] [PubMed]
- Trayanova, N.A.; Doshi, A.N.; Prakosa, A. How personalized heart modeling can help treatment of lethal arrhythmias: A focus on ventricular tachycardia ablation strategies in post-infarction patients. WIREs Syst. Biol. Med. 2020, 12, e1477. [Google Scholar] [CrossRef]
- de Barros, B.G.; dos Santos, R.W.; Lobosco, M.; Alonso, S. Simulation of Ectopic Pacemakers in the Heart: Multiple Ectopic Beats Generated by Reentry inside Fibrotic Regions. BioMed Res. Int. 2015, 2015, 1–18. [Google Scholar] [CrossRef]
- Oliveira, R.S.; Alonso, S.; Campos, F.O.; Rocha, B.M.; Fernandes, J.F.; Kuehne, T.; Dos Santos, R.W. Ectopic beats arise from micro-reentries near infarct regions in simulations of a patient-specific heart model. Sci. Rep. 2018, 8, 1–14. [Google Scholar] [CrossRef]
- Haemers, P.; Sutherland, G.R.; Cikes, M.; Jakus, N.; Holemans, P.; Sipido, K.R.; Willems, R.; Claus, P. Further insights into blood pressure induced premature beats: Transient depolarizations are associated with fast myocardial deformation upon pressure decline. Heart Rhythm. 2015, 12, 2305–2315. [Google Scholar] [CrossRef]
- Johnson, D.M.; Antoons, G. Arrhythmogenic Mechanisms in Heart Failure: Linking β-Adrenergic Stimulation, Stretch, and Calcium. Front. Physiol. 2018, 9, 1453. [Google Scholar] [CrossRef]
- Ravens, U. Mechano-electric feedback and arrhythmias. Prog. Biophys. Mol. Biol. 2003, 82, 255–266. [Google Scholar] [CrossRef]
- Taggart, P.; Sutton, P.M. Cardiac mechano-electric feedback in man: Clinical relevance. Prog. Biophys. Mol. Biol. 1999, 71, 139–154. [Google Scholar] [CrossRef]
- De Bakker, J.M.; Van Capelle, F.J.; Janse, M.J.; Tasseron, S.; Vermeulen, J.T.; De Jonge, N.; Lahpor, J.R. Slow conduction in the infarcted human heart. ’Zigzag’ course of activation. Circulation 1993, 88, 915–926. [Google Scholar] [CrossRef]
- Ciaccio, E.J.; Coromilas, J.; Wit, A.L.; Peters, N.S.; Garan, H. Formation of Functional Conduction Block During the Onset of Reentrant Ventricular Tachycardia. Circ. Arrhythmia Electrophysiol. 2016, 9, e004462. [Google Scholar] [CrossRef] [PubMed]
- Baba, S.; Dun, W.; Cabo, C.; Boyden, P.A. Remodeling in cells from different regions of the reentrant circuit during ventricular tachycardia. Circulation 2005, 112, 2386–2396. [Google Scholar] [CrossRef] [PubMed]
- Restivo, M.; Gough, W.B.; El-Sherif, N. Ventricular arrhythmias in the subacute myocardial infarction period. High-resolution activation and refractory patterns of reentrant rhythms. Circ. Res. 1990, 66, 1310–1327. [Google Scholar] [CrossRef]
- Qin, D.; Zhang, Z.-H.; Caref, E.B.; Boutjdir, M.; Jain, P.; El-Sherif, N. Cellular and Ionic Basis of Arrhythmias in Postinfarction Remodeled Ventricular Myocardium. Circ. Res. 1996, 79, 461–473. [Google Scholar] [CrossRef] [PubMed]
- Orini, M.; Taggart, P.; Bhuva, A.; Roberts, N.; Di Salvo, C.; Yates, M.; Badiani, S.; Van Duijvenboden, S.; Lloyd, G.; Smith, A.; et al. Direct in vivo assessment of global and regional mechanoelectric feedback in the intact human heart. Heart Rhythm. 2021, 18, 1406–1413. [Google Scholar] [CrossRef] [PubMed]
- Orini, M.; Taggart, P.; Hayward, M.; Lambiase, P.D. Spatiotemporal characterization of the transition from sinus rhythm to ventricular fibrillation during an acute ischemic event in the intact human heart by whole-heart sock-mapping. Heart Rhythm Case Rep. 2017, 3, 259–263. [Google Scholar] [CrossRef] [PubMed]
- Haugaa, K.; Edvardsen, T.; Amlie, J.P. Prediction of Life-Threatening Arrhythmias—Still an Unresolved Problem. Cardiology 2011, 118, 129–137. [Google Scholar] [CrossRef]
- León, D.G.; López-Yunta, M.; Alfonso-Almazán, J.M.; Marina-Breysse, M.; Quintanilla, J.G.; Sánchez-González, J.; Galán-Arriola, C.; Castro-Núñez, F.; González-Ferrer, J.J.; Ibáñez, B.; et al. Three-dimensional cardiac fibre disorganization as a novel parameter for ventricular arrhythmia stratification after myocardial infarction. EP Eur. 2019, 21, 822–832. [Google Scholar] [CrossRef] [PubMed]
- Pashakhanloo, F.; Herzka, D.A.; Halperin, H.; McVeigh, E.R.; Trayanova, N.A. Role of 3-Dimensional Architecture of Scar and Surviving Tissue in Ventricular Tachycardia. Circ. Arrhythmia Electrophysiol. 2018, 11, e006131. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Chen, S.; Ernst, S.; Guzman, C.E.; Han, S.; Kalarus, Z.; Labadet, C.; Lin, Y.; Lo, L.; Nogami, A.; et al. 2019 APHRS expert consensus statement on three-dimensional mapping systems for tachycardia developed in collaboration with HRS, EHRA, and LAHRS. J. Arrhythmia 2020, 36, 215–270. [Google Scholar] [CrossRef] [PubMed]
- Gepstein, L.; Hayam, G.; Ben-Haim, S.A. A novel method for nonfluoroscopic catheter-based electroanatomical mapping of the heart. In vitro and in vivo accuracy results. Circulation 1997, 95, 1611–1622. [Google Scholar] [CrossRef]
- Wolf, M.; Sacher, F.; Cochet, H.; Kitamura, T.; Takigawa, M.; Yamashita, S.; Vlachos, K.; Cheniti, G.; Frontera, A.; Martin, R.; et al. Long-Term Outcome of Substrate Modification in Ablation of Post–Myocardial Infarction Ventricular Tachycardia. Circ. Arrhythmia Electrophysiol. 2018, 11, e005635. [Google Scholar] [CrossRef]
- Briceño, D.F.; Romero, J.; Villablanca, P.A.; Londoño, A.; Diaz, J.C.; Maraj, I.; Batul, S.A.; Madan, N.; Patel, J.; Jagannath, A.; et al. Long-term outcomes of different ablation strategies for ventricular tachycardia in patients with structural heart disease: Systematic review and meta-analysis. EP Eur. 2017, 20, 104–115. [Google Scholar] [CrossRef]
- Bogun, F.; Crawford, T.; Chalfoun, N.; Kühne, M.; Sarrazin, J.-F.; Wells, D.; Good, E.; Jongnarangsin, K.; Oral, H.; Chugh, A.; et al. Relationship of frequent postinfarction premature ventricular complexes to the reentry circuit of scar-related ventricular tachycardia. Heart Rhythm. 2008, 5, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Marrouche, N.F.; Verma, A.; Wazni, O.; Schweikert, R.; Martin, D.O.; Saliba, W.; Kilicaslan, F.; Cummings, J.; Burkhardt, J.; Bhargava, M.; et al. Mode of initiation and ablation of ventricular fibrillation storms in patients with ischemic cardiomyopathy. J. Am. Coll. Cardiol. 2004, 43, 1715–1720. [Google Scholar] [CrossRef] [PubMed]
- Okada, D.R.; Miller, J.; Chrispin, J.; Prakosa, A.; Trayanova, N.; Jones, S.; Maggioni, M.; Wu, K.C. Substrate Spatial Complexity Analysis for the Prediction of Ventricular Arrhythmias in Patients with Ischemic Cardiomyopathy. Circ. Arrhythmia Electrophysiol. 2020, 13, e007975. [Google Scholar] [CrossRef] [PubMed]
- Prakosa, A.; Arevalo, H.J.; Deng, D.; Boyle, P.M.; Nikolov, P.P.; Ashikaga, H.; Blauer, J.J.E.; Ghafoori, E.; Park, C.J.; Blake, R.C.; et al. Personalized virtual-heart technology for guiding the ablation of infarct-related ventricular tachycardia. Nat. Biomed. Eng. 2018, 2, 732–740. [Google Scholar] [CrossRef]
- Sharma, A.; Wong, D.; Weidlich, G.; Fogarty, T.; Jack, A.; Sumanaweera, T.; Maguire, P. Noninvasive stereotactic radiosurgery (CyberHeart) for creation of ablation lesions in the atrium. Heart Rhythm. 2010, 7, 802–810. [Google Scholar] [CrossRef] [PubMed]
- Lindsey, M.L.; Bolli, R.; Canty, J.M.; Du, X.-J.; Frangogiannis, N.; Frantz, S.; Gourdie, R.G.; Holmes, J.; Jones, S.P.; Kloner, R.A.; et al. Guidelines for experimental models of myocardial ischemia and infarction. Am. J. Physiol. Circ. Physiol. 2018, 314, H812–H838. [Google Scholar] [CrossRef]
- Anversa, P.; Beghi, C.; Kikkawa, Y.; Olivetti, G. Myocardial response to infarction in the rat. Morphometric measurement of infarct size and myocyte cellular hypertrophy. Am. J. Pathol. 1985, 118, 484–492. [Google Scholar] [PubMed]
- Anversa, P.; Olivetti, G.; Capasso, J.M. Cellular basis of ventricular remodeling after myocardial infarction. Am. J. Cardiol. 1991, 68, 7–16. [Google Scholar] [CrossRef]
- Heusch, G.; Skyschally, A.; Schulz, R. The in-situ pig heart with regional ischemia/reperfusion—Ready for translation. J. Mol. Cell. Cardiol. 2011, 50, 951–963. [Google Scholar] [CrossRef]
- Canty, J.M., Jr.; Suzuki, G.; Banas, M.D.; Verheyen, F.; Borgers, M.; Fallavollita, J.A. Hibernating Myocardium. Circ. Res. 2004, 94, 1142–1149. [Google Scholar] [CrossRef]
- Wu, M.; D’Hooge, J.; Ganame, J.; Ferferieva, V.; Sipido, K.R.; Maes, F.; Dymarkowski, S.; Bogaert, J.; Rademakers, F.E.; Claus, P. Non-invasive characterization of the area-at-risk using magnetic resonance imaging in chronic ischaemia. Cardiovasc. Res. 2011, 89, 166–174. [Google Scholar] [CrossRef] [PubMed]
- Wolf, E.; Kemter, E.; Klymiuk, N.; Reichart, B. Genetically modified pigs as donors of cells, tissues, and organs for xenotransplantation. Anim. Front. 2019, 9, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Längin, M.; Mayr, T.; Reichart, B.; Michel, S.; Buchholz, S.; Guethoff, S.; Dashkevich, A.; Baehr, A.; Egerer, S.; Bauer, A.; et al. Consistent success in life-supporting porcine cardiac xenotransplantation. Nature 2018, 564, 430–433. [Google Scholar] [CrossRef] [PubMed]
- DeSantiago, J.; Ai, X.; Islam, M.; Acuna, G.; Ziolo, M.T.; Bers, D.; Pogwizd, S.M. Arrhythmogenic Effects of β 2 -Adrenergic Stimulation in the Failing Heart Are Attributable to Enhanced Sarcoplasmic Reticulum Ca Load. Circ. Res. 2008, 102, 1389–1397. [Google Scholar] [CrossRef]
- Currie, S.; Quinn, F.R.; Sayeed, R.; Duncan, A.M.; Kettlewell, S.; Smith, G.L. Selective down-regulation of sub-endocardial ryanodine receptor expression in a rabbit model of left ventricular dysfunction. J. Mol. Cell. Cardiol. 2005, 39, 309–317. [Google Scholar] [CrossRef]
- De Villiers, C.; Riley, P.R. Mouse models of myocardial infarction: Comparing permanent ligation and ischaemia-reperfusion. Dis. Model. Mech. 2020, 13, 046565. [Google Scholar] [CrossRef]
- Dewald, O.; Ren, G.; Duerr, G.D.; Zoerlein, M.; Klemm, C.; Gersch, C.; Tincey, S.; Michael, L.H.; Entman, M.L.; Frangogiannis, N. Of Mice and Dogs: Species-Specific Differences in the Inflammatory Response Following Myocardial Infarction. Am. J. Pathol. 2004, 164, 665–677. [Google Scholar] [CrossRef]
- Fu, X.; Khalil, H.; Kanisicak, O.; Boyer, J.G.; Vagnozzi, R.J.; Maliken, B.D.; Sargent, M.A.; Prasad, V.; Valiente-Alandi, I.; Blaxall, B.C.; et al. Specialized fibroblast differentiated states underlie scar formation in the infarcted mouse heart. J. Clin. Investig. 2018, 128, 2127–2143. [Google Scholar] [CrossRef]
- Humeres, C.; Frangogiannis, N.G. Fibroblasts in the Infarcted, Remodeling, and Failing Heart. JACC Basic Transl. Sci. 2019, 4, 449–467. [Google Scholar] [CrossRef] [PubMed]
- Houser, S.R.; Margulies, K.B.; Murphy, A.M.; Spinale, F.G.; Francis, G.S.; Prabhu, S.D.; Rockman, H.A.; Kass, D.A.; Molkentin, J.; Sussman, M.A.; et al. Animal Models of Heart Failure. Circ. Res. 2012, 111, 131–150. [Google Scholar] [CrossRef] [PubMed]
- Sala, L.; Hegyi, B.; Bartolucci, C.; Altomare, C.; Rocchetti, M.; Váczi, K.; Mostacciuolo, G.; Szentandrássy, N.; Severi, S.; Nánási, P.P.; et al. Action potential contour contributes to species differences in repolarization response to β-adrenergic stimulation. EP Eur. 2018, 20, 1543–1552. [Google Scholar] [CrossRef] [PubMed]
- de Boer, T.; Stengl, M. Action potential contour and inter-species differences. EP Eur. 2017, 20, 1395–1396. [Google Scholar] [CrossRef] [PubMed]
- O’Hara, T.; Rudy, Y. Quantitative comparison of cardiac ventricular myocyte electrophysiology and response to drugs in human and nonhuman species. Am. J. Physiol. Circ. Physiol. 2012, 302, H1023–H1030. [Google Scholar] [CrossRef] [PubMed]
- Pinto, J.M.B. Electrical remodeling in ischemia and infarction. Cardiovasc. Res. 1999, 42, 284–297. [Google Scholar] [CrossRef]
- Nattel, S. Electrical coupling between cardiomyocytes and fibroblasts: Experimental testing of a challenging and important concept. Cardiovasc. Res. 2018, 114, 349–352. [Google Scholar] [CrossRef] [PubMed]
- Tomaselli, G.F. Electrophysiological remodeling in hypertrophy and heart failure. Cardiovasc. Res. 1999, 42, 270–283. [Google Scholar] [CrossRef]
- Litwin, S.E.; Bridge, J.H. Enhanced Na(+)-Ca2+ exchange in the infarcted heart. Implications for excitation-contraction coupling. Circ. Res. 1997, 81, 1083–1093. [Google Scholar] [CrossRef]
- Wit, A.L.; Peters, N.S. The role of gap junctions in the arrhythmias of ischemia and infarction. Heart Rhythm. 2012, 9, 308–311. [Google Scholar] [CrossRef]
- Wong, S.S.; Bassett, A.L.; Cameron, J.S.; Epstein, K.; Kozlovskis, P.; Myerburg, R.J. Dissimilarities in the electrophysiological abnormalities of lateral border and central infarct zone cells after healing of myocardial infarction in cats. Circ. Res. 1982, 51, 486–493. [Google Scholar] [CrossRef]
- Boyden, P.A.; Albala, A.; Dresdner, K.P., Jr. Electrophysiology and ultrastructure of canine subendocardial Purkinje cells isolated from control and 24-hour infarcted hearts. Circ. Res. 1989, 65, 955–970. [Google Scholar] [CrossRef]
- Ursell, P.C.; Gardner, P.I.; Albala, A.; Fenoglio, J.J., Jr.; Wit, A.L. Structural and electrophysiological changes in the epicardial border zone of canine myocardial infarcts during infarct healing. Circ. Res. 1985, 56, 436–451. [Google Scholar] [CrossRef]
- Haissaguerre, M.; Vigmond, E.; Stuyvers, B.; Hocini, M.; Bernus, O. Ventricular arrhythmias and the His–Purkinje system. Nat. Rev. Cardiol. 2016, 13, 155–166. [Google Scholar] [CrossRef]
- Cabo, C.; Boyden, P.A. Electrical remodeling of the epicardial border zone in the canine infarcted heart: A computational analysis. Am. J. Physiol. Circ. Physiol. 2003, 284, H372–H384. [Google Scholar] [CrossRef]
- Myerburg, R.J.; Epstein, K.; Gaide, M.S.; Wong, S.S.; Castellanos, A.; Gelband, H.; Bassett, A.L. Electrophysiologic consequences of experimental acute ischemia superimposed on healed myocardial infarction in cats. Am. J. Cardiol. 1982, 49, 323–330. [Google Scholar] [CrossRef]
- Huang, B.; Qin, D.; El-Sherif, N. Spatial alterations of Kv channels expression and K(+) currents in post-MI remodeled rat heart. Cardiovasc. Res. 2001, 52, 246–254. [Google Scholar] [CrossRef]
- Schwartz, P.J.; Vanoli, E.; Stramba-Badiale, M.; De Ferrari, G.M.; Billman, G.E.; Foreman, R.D. Autonomic mechanisms and sudden death. New insights from analysis of baroreceptor reflexes in conscious dogs with and without a myocardial infarction. Circulation 1988, 78, 969–979. [Google Scholar] [CrossRef]
- Myles, R.C.; Burton, F.L.; Cobbe, S.M.; Smith, G.L. Alternans of action potential duration and amplitude in rabbits with left ventricular dysfunction following myocardial infarction. J. Mol. Cell. Cardiol. 2011, 50, 510–521. [Google Scholar] [CrossRef]
- Tsujii, E.; Tanaka, H.; Oyamada, M.; Fujita, K.; Hamamoto, T.; Takamatsu, T. In situ visualization of the intracellular Ca2+ dynamics at the border of the acute myocardial infarct. Mol. Cell. Biochem. 2003, 248, 135–139. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; van Dessel, P.; Lopshire, J.C.; Groh, W.J.; Miller, J.; Wu, J.; Zipes, D. Optical mapping of the functional reentrant circuit of ventricular tachycardia in acute myocardial infarction. Heart Rhythm. 2004, 1, 451–459. [Google Scholar] [CrossRef] [PubMed]
- Cabo, C.; Yao, J.; Boyden, P.A.; Chen, S.; Hussain, W.; Duffy, H.S.; Ciaccio, E.J.; Peters, N.S.; Wit, A.L. Heterogeneous gap junction remodeling in reentrant circuits in the epicardial border zone of the healing canine infarct. Cardiovasc. Res. 2006, 72, 241–249. [Google Scholar] [CrossRef] [PubMed]
- Hund, T.J.; Decker, K.F.; Kanter, E.; Mohler, P.J.; Boyden, P.A.; Schuessler, R.B.; Yamada, K.A.; Rudy, Y. Role of activated CaMKII in abnormal calcium homeostasis and INa remodeling after myocardial infarction: Insights from mathematical modeling. J. Mol. Cell. Cardiol. 2008, 45, 420–428. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chou, C.-C.; Zhou, S.; Hayashi, H.; Nihei, M.; Liu, Y.-B.; Wen, M.-S.; Yeh, S.-J.; Fishbein, M.C.; Weiss, J.N.; Lin, S.-F.; et al. Remodelling of action potential and intracellular calcium cycling dynamics during subacute myocardial infarction promotes ventricular arrhythmias in Langendorff-perfused rabbit hearts. J. Physiol. 2007, 580, 895–906. [Google Scholar] [CrossRef] [PubMed]
- Mills, W.R.; Mal, N.; Forudi, F.; Popović, Z.B.; Penn, M.S.; Laurita, K.R. Optical mapping of late myocardial infarction in rats. Am. J. Physiol. Circ. Physiol. 2006, 290, H1298–H1306. [Google Scholar] [CrossRef] [PubMed]
- Pop, M.; Sermesant, M.; Liu, G.; Relan, J.; Mansi, T.; Soong, A.; Peyrat, J.-M.; Truong, M.V.; Fefer, P.; McVeigh, E.R.; et al. Construction of 3D MR image-based computer models of pathologic hearts, augmented with histology and optical fluorescence imaging to characterize action potential propagation. Med. Image Anal. 2012, 16, 505–523. [Google Scholar] [CrossRef]
- Pinali, C.; Malik, N.; Davenport, J.B.; Allan, L.J.; Murfitt, L.; Iqbal, M.M.; Boyett, M.R.; Wright, E.J.; Walker, R.; Zhang, Y.; et al. Post-Myocardial Infarction T-tubules Form Enlarged Branched Structures with Dysregulation of Junctophilin-2 and Bridging Integrator 1 (BIN-1). J. Am. Heart Assoc. 2017, 6, e004834. [Google Scholar] [CrossRef]
- Dun, W.; Baba, S.; Yagi, T.; Boyden, P.A. Dynamic remodeling of K+ and Ca2+ currents in cells that survived in the epicardial border zone of canine healed infarcted heart. Am. J. Physiol. Circ. Physiol. 2004, 287, H1046–H1054. [Google Scholar] [CrossRef]
- Kim, Y.-K.; Kim, S.-J.; Kramer, C.M.; Yatani, A.; Takagi, G.; Mankad, S.; Szigeti, G.P.; Singh, D.; Bishop, S.P.; Shannon, R.P.; et al. Altered Excitation–Contraction Coupling in Myocytes from Remodeled Myocardium after Chronic Myocardial Infarction. J. Mol. Cell. Cardiol. 2002, 34, 63–73. [Google Scholar] [CrossRef]
- Shimkunas, R.; Zhang, Z.; Wenk, J.F.; Soleimani, M.; Khazalpour, M.; Acevedo-Bolton, G.; Wang, G.; Saloner, D.; Mishra, R.; Wallace, A.W.; et al. Left Ventricular Myocardial Contractility Is Depressed in the Borderzone after Posterolateral Myocardial Infarction. Ann. Thorac. Surg. 2013, 95, 1619–1625. [Google Scholar] [CrossRef]
- Kimura, S.; Bassett, A.L.; Kohya, T.; Kozlovskis, P.L.; Myerburg, R.J. Simultaneous recording of action potentials from endocardium and epicardium during ischemia in the isolated cat ventricle: Relation of temporal electrophysiologic heterogeneities to arrhythmias. Circulation 1986, 74, 401–409. [Google Scholar] [CrossRef]
- Pinto, J.M.; Yuan, F.; Wasserlaue, B.J.; Bassett, A.L.; Myerburg, R.J. Regional Gradation of L-Type Calcium Currents in the Feline Heart with a Healed Myocardial Infarct. J. Cardiovasc. Electrophysiol. 1997, 8, 548–560. [Google Scholar] [CrossRef]
- Kimura, S.; Bassett, A.L.; Cameron, J.S.; Huikuri, H.; Kozlovskis, P.L.; Myerburg, R.J. Cellular electrophysiological changes during ischemia in isolated, coronary-perfused cat ventricle with healed myocardial infarction. Circulation 1988, 78, 401–406. [Google Scholar] [CrossRef]
- Weigand, K.; Witte, R.; Moukabary, T.; Chinyere, I.; Lancaster, J.; Pierce, M.K.; Goldman, S.; Juneman, E. In vivo Electrophysiological Study of Induced Ventricular Tachycardia in Intact Rat Model of Chronic Ischemic Heart Failure. IEEE Trans. Biomed. Eng. 2016, 64, 1393–1399. [Google Scholar] [CrossRef]
- Walker, N.L.; Burton, F.L.; Kettlewell, S.; Smith, G.L.; Cobbe, S.M. Mapping of Epicardial Activation in a Rabbit Model of Chronic Myocardial Infarction. J. Cardiovasc. Electrophysiol. 2007, 18, 862–868. [Google Scholar] [CrossRef] [PubMed]
- Dangman, K.H.; Danilo, P., Jr.; Hordof, A.J.; Mary-Rabine, L.; Reder, R.F.; Rosen, M.R. Electrophysiologic characteristics of human ventricular and Purkinje fibers. Circulation 1982, 65, 362–368. [Google Scholar] [CrossRef] [PubMed]
- Loennechen, J.P.; Wisløff, U.; Falck, G.; Ellingsen, Ø. Cardiomyocyte contractility and calcium handling partially recover after early deterioration during post-infarction failure in rat. Acta Physiol. Scand. 2002, 176, 17–26. [Google Scholar] [CrossRef]
- Kilic, A.; Li, T.; Nolan, T.D.C.; Nash, J.R.; Li, S.; Prastein, D.J.; Schwartzbauer, G.; Moainie, S.L.; Yankey, G.K.; Defilippi, C.; et al. Strain-related regional alterations of calcium-handling proteins in myocardial remodeling. J. Thorac. Cardiovasc. Surg. 2006, 132, 900–908. [Google Scholar] [CrossRef] [PubMed]
- Tomek, J.; Hao, G.; Tomkova, M.; Lewis, A.; Carr, C.; Paterson, D.J.; Rodriguez, B.; Bub, G.; Herring, N. β-Adrenergic Receptor Stimulation and Alternans in the Border Zone of a Healed Infarct: An ex vivo Study and Computational Investigation of Arrhythmogenesis. Front. Physiol. 2019, 10, 350. [Google Scholar] [CrossRef]
- Belevych, A.; Terentyev, D.; Terentyeva, R.; Nishijima, Y.; Sridhar, A.; Hamlin, R.L.; Carnes, C.; Györke, S. The relationship between arrhythmogenesis and impaired contractility in heart failure: Role of altered ryanodine receptor function. Cardiovasc. Res. 2011, 90, 493–502. [Google Scholar] [CrossRef]
- Belevych, A.E.; Terentyev, D.; Terentyeva, R.; Ho, H.-T.; Gyorke, I.; Bonilla, I.M.; Carnes, C.A.; Billman, G.E.; Györke, S. Shortened Ca2+ signaling refractoriness underlies cellular arrhythmogenesis in a postinfarction model of sudden cardiac death. Circ. Res. 2012, 110, 569–577. [Google Scholar] [CrossRef]
- Hirose, M.; Stuyvers, B.D.; Dun, W.; ter Keurs, H.E.; Boyden, P.A. Function of Ca 2+ Release Channels in Purkinje Cells That Survive in the Infarcted Canine Heart. Circ. Arrhythmia Electrophysiol. 2008, 1, 387–395. [Google Scholar] [CrossRef]
- Boyden, P.A.; Barbhaiya, C.; Lee, T.; Ter Keurs, H.E. Nonuniform Ca2+ transients in arrhythmogenic Purkinje cells that survive in the infarcted canine heart. Cardiovasc. Res. 2003, 57, 681–693. [Google Scholar] [CrossRef]
- Ter Keurs, H.E.D.J.; Boyden, P.A. Calcium and Arrhythmogenesis. Physiol. Rev. 2007, 87, 457–506. [Google Scholar] [CrossRef]
- Jalife, J. Modulated parasystole: Still relevant after all these years! Heart Rhythm. 2013, 10, 1441–1443. [Google Scholar] [CrossRef]
- Tallquist, M.D.; Molkentin, J.D. Redefining the identity of cardiac fibroblasts. Nat. Rev. Cardiol. 2017, 14, 484–491. [Google Scholar] [CrossRef]
- Ruiz-Villalba, A.; Romero, J.P.; Hernandez, S.C.; Vilas-Zornoza, A.; Fortelny, N.; Castro-Labrador, L.; Martin-Uriz, P.S.; Lorenzo-Vivas, E.; García-Olloqui, P.; Palacios, M.; et al. Single-Cell RNA-seq Analysis Reveals a Crucial Role for Collagen Triple Helix Repeat Containing 1 (CTHRC1) Cardiac Fibroblasts after Myocardial Infarction. Circulation 2020, 142, 1831–1847. [Google Scholar] [CrossRef]
- Nagaraju, C.K.; Dries, E.; Gilbert, G.; Abdesselem, M.; Wang, N.; Amoni, M.; Driesen, R.B.; Sipido, K.R. Myofibroblast modulation of cardiac myocyte structure and function. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef]
- Quinn, T.A.; Camelliti, P.; Rog-Zielinska, E.A.; Siedlecka, U.; Poggioli, T.; O’Toole, E.T.; Knopfel, T.; Kohl, P. Electrotonic coupling of excitable and nonexcitable cells in the heart revealed by optogenetics. Proc. Natl. Acad. Sci. USA 2016, 113, 14852–14857. [Google Scholar] [CrossRef] [PubMed]
- Schultz, F.; Swiatlowska, P.; Alvarez-Laviada, A.; Sanchez-Alonso, J.L.; Song, Q.; de Vries, A.A.F.; Pijnappels, D.A.; Ongstad, E.; Braga, V.M.M.; Entcheva, E.; et al. Cardiomyocyte–myofibroblast contact dynamism is modulated by connexin-43. FASEB J. 2019, 33, 10453–10468. [Google Scholar] [CrossRef] [PubMed]
- Rubart, M.; Tao, W.; Lu, X.-L.; Conway, S.J.; Reuter, S.P.; Lin, S.-F.; Soonpaa, M.H. Electrical coupling between ventricular myocytes and myofibroblasts in the infarcted mouse heart. Cardiovasc. Res. 2018, 114, 389–400. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.H. MicroRNA regulation of cardiac conduction and arrhythmias. Transl. Res. 2013, 161, 381–392. [Google Scholar] [CrossRef] [PubMed]
- Bang, C.; Batkai, S.; Dangwal, S.; Gupta, S.K.; Foinquinos, A.; Holzmann, A.; Just, A.; Remke, J.; Zimmer, K.; Zeug, A.; et al. Cardiac fibroblast–derived microRNA passenger strand-enriched exosomes mediate cardiomyocyte hypertrophy. J. Clin. Investig. 2014, 124, 2136–2146. [Google Scholar] [CrossRef] [PubMed]
- Shen, M.J.; Zipes, D.P. Role of the Autonomic Nervous System in Modulating Cardiac Arrhythmias. Circ. Res. 2014, 114, 1004–1021. [Google Scholar] [CrossRef] [PubMed]
- Vaseghi, M.; Shivkumar, K. The Role of the Autonomic Nervous System in Sudden Cardiac Death. Prog. Cardiovasc. Dis. 2008, 50, 404–419. [Google Scholar] [CrossRef] [PubMed]
- Gardner, R.T.; Ripplinger, C.; Myles, R.; Habecker, B.A. Molecular Mechanisms of Sympathetic Remodeling and Arrhythmias. Circ. Arrhythmia Electrophysiol. 2016, 9, e001359. [Google Scholar] [CrossRef]
- Vanoli, E.; De Ferrari, G.M.; Stramba-Badiale, M.; Hull, S.S., Jr.; Foreman, R.D.; Schwartz, P.J. Vagal stimulation and prevention of sudden death in conscious dogs with a healed myocardial infarction. Circ. Res. 1991, 68, 1471–1481. [Google Scholar] [CrossRef]
- Wang, L.; Olivas, A.; Stuart, S.D.F.; Tapa, S.; Blake, M.R.; Woodward, W.R.; Habecker, B.A.; Ripplinger, C.M. Cardiac sympathetic nerve transdifferentiation reduces action potential heterogeneity after myocardial infarction. Am. J. Physiol. Circ. Physiol. 2020, 318, H558–H565. [Google Scholar] [CrossRef]
- Jiang, H.; Lu, Z.; Yu, Y.; Zhao, D.; Yang, B.; Huang, C. Relationship between sympathetic nerve sprouting and repolarization dispersion at peri-infarct zone after myocardial infarction. Auton. Neurosci. 2007, 134, 18–25. [Google Scholar] [CrossRef]
- Yoshioka, K.; Gao, D.-W.; Chin, M.; Stillson, C.; Penades, E.; Lesh, M.; O’Connell, W.; Dae, M. Heterogeneous Sympathetic Innervation Influences Local Myocardial Repolarization in Normally Perfused Rabbit Hearts. Circulation 2000, 101, 1060–1066. [Google Scholar] [CrossRef]
- Vaseghi, M.; Lux, R.L.; Mahajan, A.; Shivkumar, K. Sympathetic stimulation increases dispersion of repolarization in humans with myocardial infarction. Am. J. Physiol. Circ. Physiol. 2012, 302, H1838–H1846. [Google Scholar] [CrossRef]
- Priori, S.G.; Mantica, M.; Schwartz, P.J. Delayed afterdepolarizations elicited in vivo by left stellate ganglion stimulation. Circulation 1988, 78, 178–185. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Sato, D.; Garfinkel, A.; Qu, Z.; Weiss, J.N. So Little Source, So Much Sink: Requirements for Afterdepolarizations to Propagate in Tissue. Biophys. J. 2010, 99, 1408–1415. [Google Scholar] [CrossRef]
- Dries, E.; Santiago, D.J.; Gilbert, G.; Lenaerts, I.; Vandenberk, B.; Nagaraju, C.K.; Johnson, D.M.; Holemans, P.; Roderick, H.; Macquaide, N.; et al. Hyperactive ryanodine receptors in human heart failure and ischaemic cardiomyopathy reside outside of couplons. Cardiovasc. Res. 2018, 114, 1512–1524. [Google Scholar] [CrossRef] [PubMed]
- Schobesberger, S.; Wright, P.; Tokar, S.; Bhargava, A.; Mansfield, C.; Glukhov, A.V.; Poulet, C.; Buzuk, A.; Monszpart, A.; Sikkel, M.; et al. T-tubule remodelling disturbs localized β2-adrenergic signalling in rat ventricular myocytes during the progression of heart failure. Cardiovasc. Res. 2017, 113, 770–782. [Google Scholar] [CrossRef] [PubMed]
- Vaseghi, M.; Gima, J.; Kanaan, C.; Ajijola, O.; Marmureanu, A.; Mahajan, A.; Shivkumar, K. Cardiac sympathetic denervation in patients with refractory ventricular arrhythmias or electrical storm: Intermediate and long-term follow-up. Heart Rhythm. 2014, 11, 360–366. [Google Scholar] [CrossRef] [PubMed]
- Marx, S.O.; Reiken, S.; Hisamatsu, Y.; Jayaraman, T.; Burkhoff, D.; Rosemblit, N.; Marks, A.R. PKA Phosphorylation Dissociates FKBP12.6 from the Calcium Release Channel (Ryanodine Receptor): Defective Regulation in Failing Hearts. Cell 2000, 101, 365–376. [Google Scholar] [CrossRef]
- Wehrens, X.; Lehnart, S.E.; Marks, A.R. Ryanodine Receptor-Targeted Anti-Arrhythmic Therapy. Ann. N. Y. Acad. Sci. 2005, 1047, 366–375. [Google Scholar] [CrossRef]
- Antoons, G.; Willems, R.; Sipido, K.R. Alternative strategies in arrhythmia therapy: Evaluation of Na/Ca exchange as an anti-arrhythmic target. Pharmacol. Ther. 2011, 134, 26–42. [Google Scholar] [CrossRef]
- Liu, N.; Napolitano, C.; Venetucci, L.A.; Priori, S.G. Flecainide and Antiarrhythmic Effects in a Mouse Model of Catecholaminergic Polymorphic Ventricular Tachycardia. Trends Cardiovasc. Med. 2012, 22, 35–39. [Google Scholar] [CrossRef]
- Kryshtal, D.O.; Blackwell, D.J.; Egly, C.L.; Smith, A.N.; Batiste, S.M.; Johnston, J.N.; Laver, D.R.; Knollmann, B.C. RYR2 Channel Inhibition Is the Principal Mechanism of Flecainide Action in CPVT. Circ. Res. 2021, 128, 321–331. [Google Scholar] [CrossRef]
- Zhou, Q.; Xiao, J.; Jiang, D.; Wang, R.; Vembaiyan, K.; Wang, A.; Smith, C.D.; Xie, C.; Chen, W.; Zhang, J.; et al. Carvedilol and its new analogs suppress arrhythmogenic store overload–induced Ca2+ release. Nat. Med. 2011, 17, 1003–1009. [Google Scholar] [CrossRef]
- Maruyama, M.; Xiao, J.; Zhou, Q.; Vembaiyan, K.; Chua, S.-K.; Der Lohe, M.R.-V.; Lin, S.-F.; Back, T.G.; Chen, S.R.W.; Chen, P.-S. Carvedilol analogue inhibits triggered activities evoked by both early and delayed afterdepolarizations. Heart Rhythm. 2012, 10, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Wang, Q.; Sibrian-Vazquez, M.; Klipp, R.C.; Reynolds, J.O.; Word, T.A.; Scott, L.; Salama, G.; Strongin, R.M.; Abramson, J.J.; et al. Treatment of catecholaminergic polymorphic ventricular tachycardia in mice using novel RyR2-modifying drugs. Int. J. Cardiol. 2017, 227, 668–673. [Google Scholar] [CrossRef] [PubMed]
- Pabel, S.; Mustroph, J.; Stehle, T.; Lebek, S.; Dybkova, N.; Keyser, A.; Rupprecht, L.; Wagner, S.; Neef, S.; Maier, L.S.; et al. Dantrolene reduces CaMKIIδC-mediated atrial arrhythmias. EP Eur. 2020, 22, 1111–1118. [Google Scholar] [CrossRef]
- Antoons, G.; Johnson, D.M.; Dries, E.; Santiago, D.J.; Ozdemir, S.; Lenaerts, I.; Beekman, J.D.; Houtman, M.J.; Sipido, K.R.; Vos, M.A. Calcium release near l-type calcium channels promotes beat-to-beat variability in ventricular myocytes from the chronic AV block dog. J. Mol. Cell. Cardiol. 2015, 89, 326–334. [Google Scholar] [CrossRef]
- Johnson, D.M.; Heijman, J.; Pollard, C.E.; Valentin, J.-P.; Crijns, H.J.; Abi-Gerges, N.; Volders, P.G. IKs restricts excessive beat-to-beat variability of repolarization during beta-adrenergic receptor stimulation. J. Mol. Cell. Cardiol. 2010, 48, 122–130. [Google Scholar] [CrossRef]
- Heijman, J.; Zaza, A.; Johnson, D.M.; Rudy, Y.; Peeters, R.; Volders, P.G.A.; Westra, R.L. Determinants of Beat-to-Beat Variability of Repolarization Duration in the Canine Ventricular Myocyte: A Computational Analysis. PLoS Comput. Biol. 2013, 9, e1003202. [Google Scholar] [CrossRef]
- Donahue, J.K. Gene Therapy for Post-Infarction Ventricular Tachycardia; Springer: New York, NY, USA, 2017; Volume 1521, pp. 307–321. [Google Scholar]
- Young, A.A.; Crossman, D.J.; Ruygrok, P.N.; Cannell, M.B. Mapping system for coregistration of cardiac mri and ex vivo tissue sampling. J. Magn. Reson. Imaging 2011, 34, 1065–1071. [Google Scholar] [CrossRef]
- Dauwe, D.; Nuyens, D.; De Buck, S.; Claus, P.; Gheysens, O.; Koole, M.; Coudyzer, W.; Driessche, N.V.; Janssens, L.; Ector, J.; et al. Three-dimensional rotational angiography fused with multimodal imaging modalities for targeted endomyocardial injections in the ischaemic heart. Eur. Heart J. Cardiovasc. Imaging 2014, 15, 900–907. [Google Scholar] [CrossRef]
- Sasano, T.; McDonald, A.D.; Kikuchi, K.; Donahue, J.K. Molecular ablation of ventricular tachycardia after myocardial infarction. Nat. Med. 2006, 12, 1256–1258. [Google Scholar] [CrossRef]
- Hesse, M.; Bednarz, R.; Carls, E.; Becker, C.; Bondareva, O.; Lother, A.; Geisen, C.; Dreßen, M.; Krane, M.; Roell, W.; et al. Proximity to injury, but neither number of nuclei nor ploidy define pathological adaptation and plasticity in cardiomyocytes. J. Mol. Cell. Cardiol. 2021, 152, 95–104. [Google Scholar] [CrossRef]
- Kuppe, C.; Flores, R.O.; Li, Z.; Hannani, M.; Tanevski, J.; Halder, M.; Cheng, M.; Ziegler, S.; Zhang, X.; Preisker, F.; et al. Spatial multi-omic map of human myocardial infarction. Bioxiv 2020. [Google Scholar] [CrossRef]
- Preissl, S.; Schwaderer, M.; Raulf, A.; Hesse, M.; Grüning, B.; Köbele, C.; Backofen, R.; Fleischmann, B.K.; Hein, L.; Gilsbach, R. Deciphering the Epigenetic Code of Cardiac Myocyte Transcription. Circ. Res. 2015, 117, 413–423. [Google Scholar] [CrossRef]
- Thienpont, B.; Aronsen, J.M.; Robinson, E.L.; Okkenhaug, H.; Loche, E.; Ferrini, A.; Brien, P.; Alkass, K.; Tomasso, A.; Agrawal, A.; et al. The H3K9 dimethyltransferases EHMT1/2 protect against pathological cardiac hypertrophy. J. Clin. Investig. 2016, 127, 335–348. [Google Scholar] [CrossRef]
- Kolesová, H.; Olejníčková, V.; Kvasilová, A.; Gregorovičová, M.; Sedmera, D. Tissue clearing and imaging methods for cardiovascular development. iScience 2021, 24, 102387. [Google Scholar] [CrossRef] [PubMed]
- Fischesser, D.M.; Meyer, E.C.; Sargent, M.; Molkentin, J.D. Refined CLARITY-Based Tissue Clearing for Three-Dimensional Fibroblast Organization in Healthy and Injured Mouse Hearts. J. Vis. Exp. 2021, 171, e62023. [Google Scholar] [CrossRef]
- Litviňuková, M.; Talavera-López, C.; Maatz, H.; Reichart, D.; Worth, C.L.; Lindberg, E.L.; Kanda, M.; Polanski, K.; Heinig, M.; Lee, M.; et al. Cells of the adult human heart. Nature 2020, 588, 466–472. [Google Scholar] [CrossRef] [PubMed]
- Tucker, N.R.; Chaffin, M.; Fleming, S.; Hall, A.; Parsons, V.A.; Bedi, K.C.; Akkad, A.-D.; Herndon, C.N.; Arduini, A.; Papangeli, I.; et al. Transcriptional and Cellular Diversity of the Human Heart. Circulation 2020, 142, 466–482. [Google Scholar] [CrossRef]
- Browaeys, R.; Saelens, W.; Saeys, Y. NicheNet: Modeling intercellular communication by linking ligands to target genes. Nat. Methods 2020, 17, 159–162. [Google Scholar] [CrossRef]
- Kelsey, G.; Stegle, O.; Reik, W. Single-cell epigenomics: Recording the past and predicting the future. Science 2017, 358, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Watson, S.; Scigliano, M.; Bardi, I.; Ascione, R.; Terracciano, C.M.; Perbellini, F. Preparation of viable adult ventricular myocardial slices from large and small mammals. Nat. Protoc. 2017, 12, 2623–2639. [Google Scholar] [CrossRef]
- Schneider-Warme, F.; Johnston, C.M.; Kohl, P. Organotypic myocardial slices as model system to study heterocellular interactions. Cardiovasc. Res. 2017, 114, 3–6. [Google Scholar] [CrossRef] [PubMed]
- Hulsmans, M.; Clauss, S.; Xiao, L.; Aguirre, A.D.; King, K.R.; Hanley, A.; Hucker, W.J.; Wülfers, E.M.; Seemann, G.; Courties, G.; et al. Macrophages Facilitate Electrical Conduction in the Heart. Cell 2017, 169, 510–522. [Google Scholar] [CrossRef] [PubMed]
- Simon-Chica, A.; Fernández, M.C.; Wülfers, E.M.; Lother, A.; Hilgendorf, I.; Seemann, G.; Ravens, U.; Kohl, P.; Schneider-Warme, F. Novel insights into the electrophysiology of murine cardiac macrophages: Relevance of voltage-gated potassium channels. Cardiovasc. Res. 2021. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
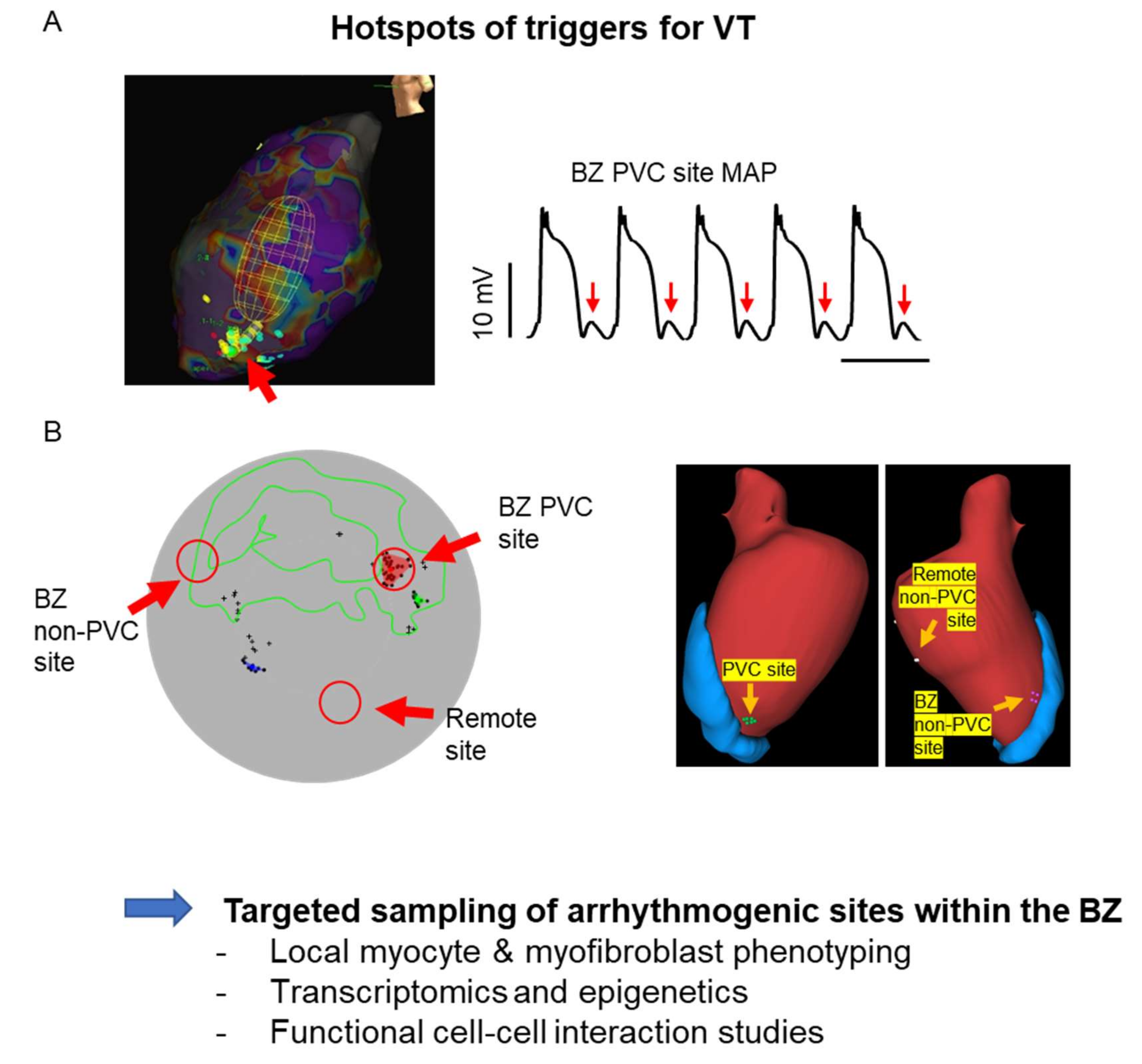{kind=link}
| Study | Species | MI Stage | Disease Model | Regions | Preparation | Observations |
|---|---|---|---|---|---|---|
| Tsujii et al., 2003 [148] | Rat | Acute (2 h) | LAD ligation | BZ (epi) vs. remote (epi) | tissue LV (optical mapping) | Ca2+ waves in BZ, uniform synchronous CaT in remote. |
| Takahashi et al., 2004 [149] | Dog | Acute (3–4 h) | Ligation side branch of LCX (ex vivo) | BZ (epi) vs. remote (epi) | tissue LV (optical mapping) | ↓ APD90, ↓ CV, ↓ APA, ↓ diastolic potential in BZ vs. remote. |
| Baba et al., 2005 [99] | Dog | Intermediate (5 d) | LAD ligation | central vs. outer reentry path (cBZ vs. oBZ) | single myocytes LV (whole-cell patch-clamp) | ↓ INa, ↓ ICaL, ↓ Ito in cBZ and oBZ. |
| Cabo et al., 2006 [150] | Dog | Intermediate (5 d) | LAD ligation | different regions of reentry path within BZ (epi): central vs. outer reentry path (cBZ vs. oBZ) | single myocytes and tissue LV (electrogram) | ↓ CV longitudinal and transverse vs. normal hearts, ↓ longitudinal CV in cBZ vs. oBZ myocytes, transverse CV unchanged in cBZ vs. oBZ myocytes. ↑ Cx43 laterisation in cBZ vs. oBZ myocytes |
| Hund T et al., 2008 [151] | Dog | Intermediate (5 d) | LAD ligation (2h) + reperfusion In silico model | BZ (epi) vs. remote (epi) | in silico | ↓ CaT amplitude, ↓ Vmax in BZ vs. remote with hyperactive CaMKII ↑ P-CaMKII in BZ vs. remote ↑ P-CaMKII at intercalated disk in BZ vs. control |
| Chou et al., 2007 [152] | Rabbit | Intermediate (7 d) | LCX ligation | BZ (epi) vs. remote (epi) | tissue LV (optical mapping) | ↑ extrasystoles in BZ, steeper ADP restitution in BZ, ↑ pacing-induced Ca2+ alternans in BZ vs. remote |
| Mills et al., 2006 [153] | Rat | Intermediate (7 d) | LAD ligation | BZ (epi) vs. remote (epi) | tissue LV (optical mapping) | APD90 = in BZ vs. remote, ↓ CV in BZ vs. remote. |
| Pop et al., 2012 [154] | Pig | Chronic (4 w) | Balloon occlusion in LAD or LCX (90 min) + reperfusion | BZ (epi) vs. remote (epi) | tissue LV (optical mapping) | ↓ APD90 in BZ vs. remote |
| Pinali et al., 2017 [155] | Pig | Chronic (4 w) | Microbead embolisation in LAD side branch | BZ vs. remote | tissue LV sampling | Cav1.2 =, BIN1 =, ↓JP2 in BZ vs. remote ↓ TT in BZ and remote vs. control, ↑ cell capacitance in BZ and remote vs. control. |
| Dun et al., 2004 [156] | Dog | Intermediate (14 d), Chronic (8 w) | LAD ligation | BZ (epi) vs. remote (epi) | single myocytes LV (whole-cell patch-clamp) | 14d: ↓ ICaL in BZ vs. remote, ↑ ISO effect in remote (presence of regional heterogeneity in adrenergic response); ↓ Ito in BZ vs. remote 8w: ↓ ICaL in BZ and remote, no ISO effect in BZ and remote (absence of regional heterogeneity in adrenergic response); Ito = in BZ vs. remote cell capacitance = in BZ vs. remote |
| Dries and Amoni et al., 2020 [89] | Pig | Chronic (6 w) | Copper-coated stent in LAD | BZ (mid) vs. remote (mid) | single myocytes LV (whole-cell patch-clamp) | ↑ DADs and spontaneous AP in BZ vs. remote, ↑ BVR in BZ vs. remote (with adrenergic signalling). Gene expression ↑ NPPA in BZ vs. remote. ↑ cell width, = cell length, = TTs in BZ vs. remote. |
| Kim et al., 2002 [157] | Sheep | Chronic (8 w) | LAD ligation | BZ (endo) vs. remote (endo) | single myocytes LV (whole-cell patch-clamp) | ↓ ICaL, ↓ CaT amplitude, ↑ CaT relaxation time, ↓ contraction in BZ vs. remote. ↓ SERCA in BZ vs. remote. ↑ cell length, ↑ cell width, ↑ cell capacitance in BZ vs. remote. |
| Shimkunas et al., 2013 [158] | Sheep | Chronic (17 w) | LCX ligation | BZ (epi) vs. remote (epi) | tissue LV (force measurements) | ↓ force development in BZ vs. remote |
| Wong et al. 1982 [139] | Cat | Chronic (2–7 months) | Ligation side branches of LAD and LCX | BZ (endo) vs. remote (endo) | tissue LV (microelectrode) | ↓ APD90, ↓ RMP (depolarised), ↓ Vmax in BZ vs. remote |
| Kimura et al. 1986 [159] | Cat | Chronic (2–6 months) | Ligation side branches of LAD and LCX | BZ (endo) vs. remote (endo) | tissue LV (ion-sensitive microelectrodes) | ↓ [K+], ↑ [Na+] in BZ vs. remote |
| Pinto et al. 1997 [160] | Cat | Chronic (>2 months) | Ligation side branches of LAD | BZ (endo) vs. remote (endo) | single myocytes LV (whole-cell patch-clamp) | ↓ ICaL in BZ and remote vs. control, ↓ APD in BZ, ↑ ADP in remote ↑ cell capacitance in remote vs. BZ/control |
| Kimura et al. 1988 [161] | Cat | Chronic (>2 months) | Ligation side branches of LAD and LCX | BZ (endo) vs. remote (endo) | tissue LV (microelectrode) | RMP =, APA =, APD90 =, APD50 = in BZ vs. remote |
| Weigand et al., 2016 [162] | Rat | Chronic (6 w) | LAD ligation | BZ (epi) vs. remote (epi) | whole heart (in vivo LV mapping) | ↓ MAPA, ↑ heterogeneity of repolarisation, ↓ Vmax, MAPD = in BZ vs. remote |
| Walker et al., 2007 [163] | Rabbit | Chronic (8 w) | LCX ligation | BZ (epi) vs. remote (epi) | tissue LV (optical mapping) | ↓ CV in BZ vs. remote |
| Dangman et al. 1982 [164] | Human | Chronic (end-stage HF) | - | BZ (endo) vs. remote (endo) | tissue LV (microelectrode) | ADP50 =, ADP100 =, Vmax =, RMP =, APA = in BZ vs. remote |
| Heygi et al., 2018 [88] | Pig | Chronic (5 months) | Microbead embolisation in LAD side branch | BZ vs. remote | single myocytes LV (whole-cell patch-clamp) | ↓APD95 in BZ, ↑ APD95 in remote, CaT amplitude/relaxation =, INa =, ↓ ICaL, ↓IK1, IKr =, INCX =, IKs =, ↑ DAD/AP frequency in BZ vs. remote ↓ cell shortening in BZ/remote vs. control, = cell shortening in BZ vs. remote |
| Loennechen et al., 2002 [165] | Rat | Chronic (56 d) | LAD ligation | Remote vs. sham, Remote vs. BZ | single myocytes LV | ↑ diastolic and systolic [Ca2+] in remote MI vs. sham; = diastolic and systolic [Ca2+] in remote vs. BZ ↑ cell length, ↑ cell width in remote vs. sham; = cell length, = cell width in remote vs. BZ cell shortening = in remote vs. BZ |
| Kilic et al., 2006 [166] | Sheep | Chronic (8 w) | LAD ligation | BZ vs. remote | whole heart (in vivo LV echo) | ↓ SERCA, ↓ PLB in peri-infarct vs. remote (correlated with regional strain on echo) |
| Tomek et al., 2019 [167] | Rat | Chronic (8 w) | Antero-apical cryo-infarction | BZ (epi) vs. remote (epi) | tissue LV (optical mapping) | ↑ alternans at longer cycle length in BZ vs. remote at baseline; ↓ alternans at longer cycle length in BZ vs. remote during adrenergic signalling |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amoni, M.; Dries, E.; Ingelaere, S.; Vermoortele, D.; Roderick, H.L.; Claus, P.; Willems, R.; Sipido, K.R. Ventricular Arrhythmias in Ischemic Cardiomyopathy—New Avenues for Mechanism-Guided Treatment. Cells 2021, 10, 2629. https://doi.org/10.3390/cells10102629
Amoni M, Dries E, Ingelaere S, Vermoortele D, Roderick HL, Claus P, Willems R, Sipido KR. Ventricular Arrhythmias in Ischemic Cardiomyopathy—New Avenues for Mechanism-Guided Treatment. Cells. 2021; 10(10):2629. https://doi.org/10.3390/cells10102629
Chicago/Turabian StyleAmoni, Matthew, Eef Dries, Sebastian Ingelaere, Dylan Vermoortele, H. Llewelyn Roderick, Piet Claus, Rik Willems, and Karin R. Sipido. 2021. "Ventricular Arrhythmias in Ischemic Cardiomyopathy—New Avenues for Mechanism-Guided Treatment" Cells 10, no. 10: 2629. https://doi.org/10.3390/cells10102629
APA StyleAmoni, M., Dries, E., Ingelaere, S., Vermoortele, D., Roderick, H. L., Claus, P., Willems, R., & Sipido, K. R. (2021). Ventricular Arrhythmias in Ischemic Cardiomyopathy—New Avenues for Mechanism-Guided Treatment. Cells, 10(10), 2629. https://doi.org/10.3390/cells10102629