
Cochlear Inflammaging in Relation to Ion Channels and Mitochondrial Functions

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
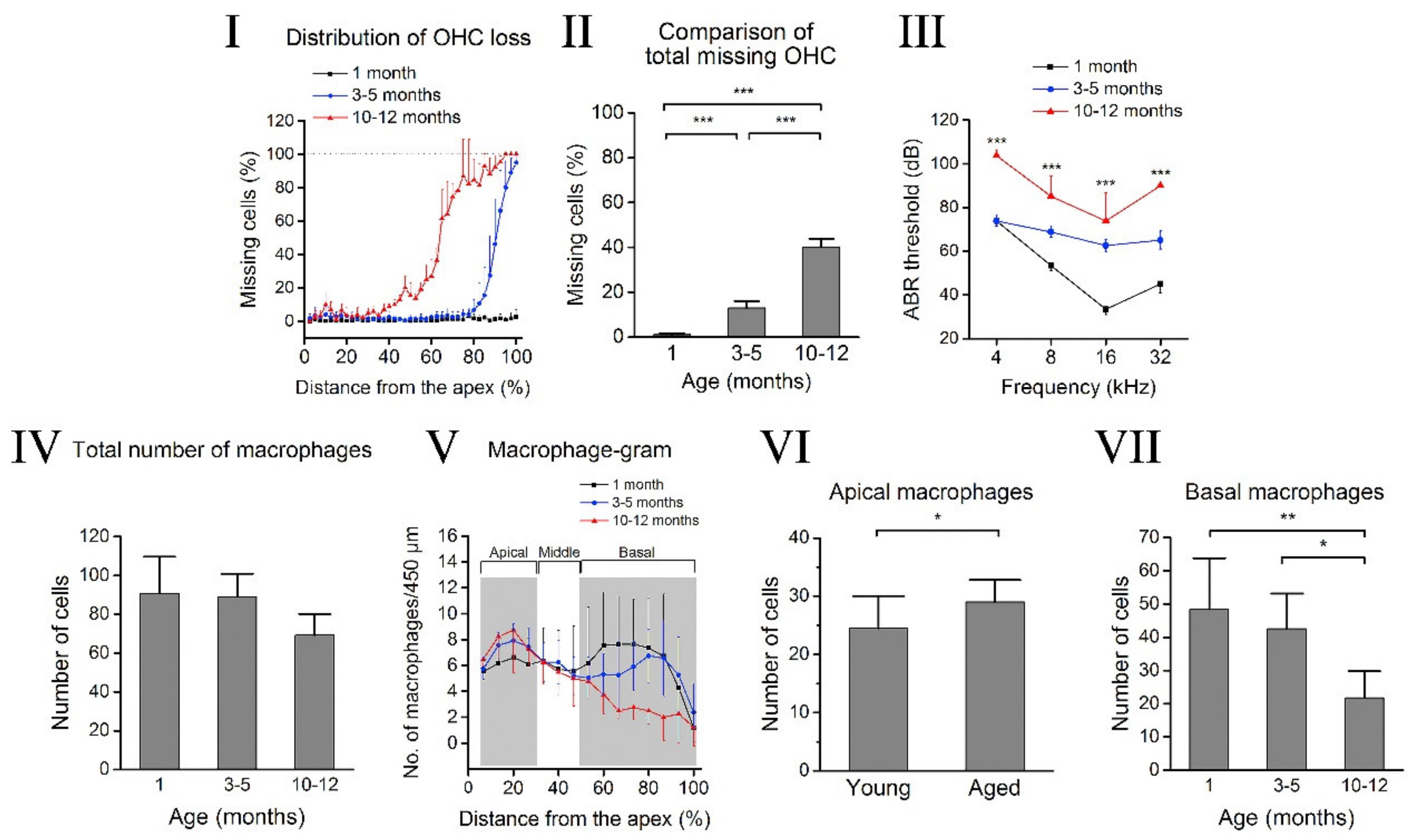
2. Cochlear Aging
3. Cochlear Inflammaging
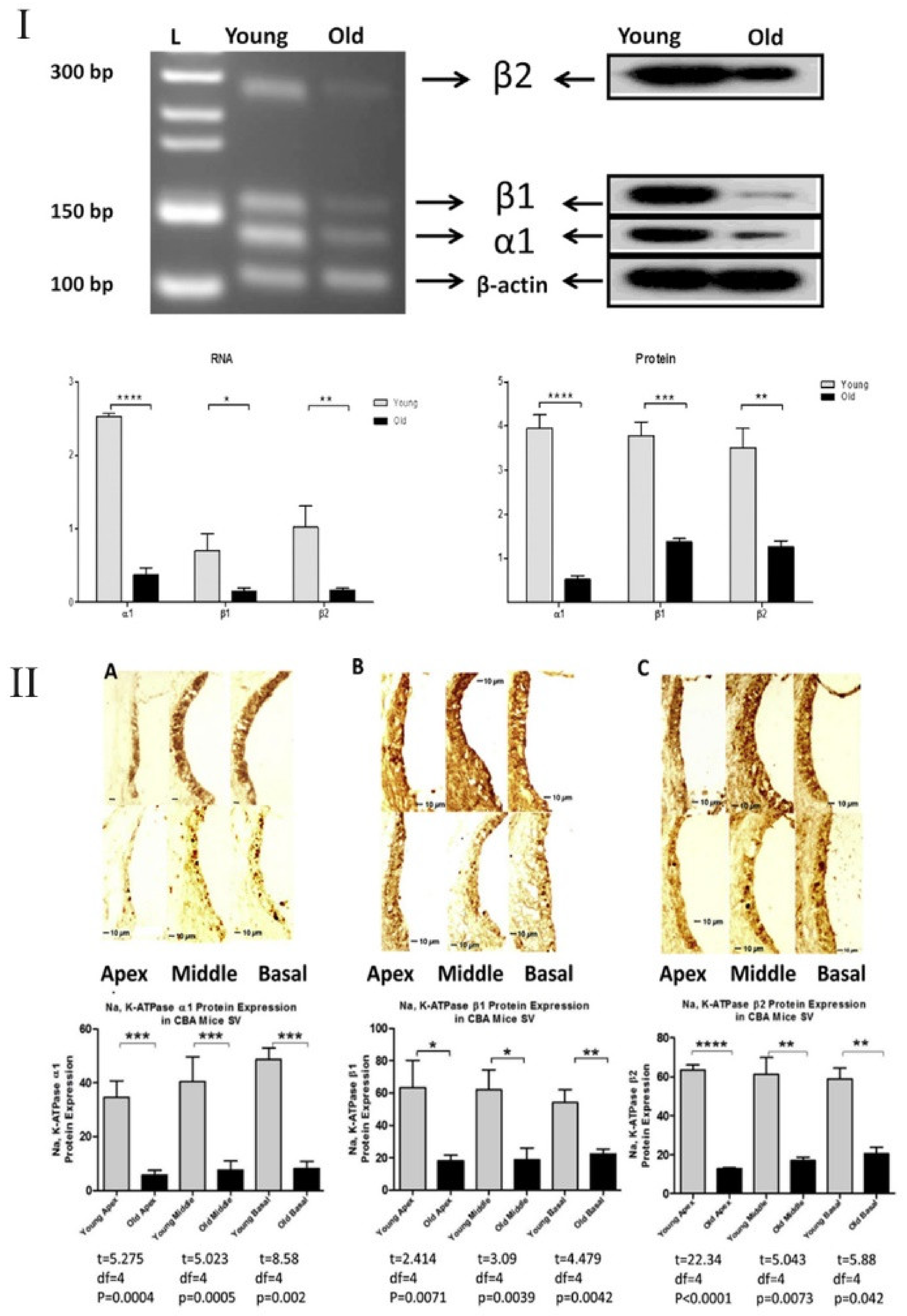
4. Cochlear Inflammaging and Ion Channels/Transporters
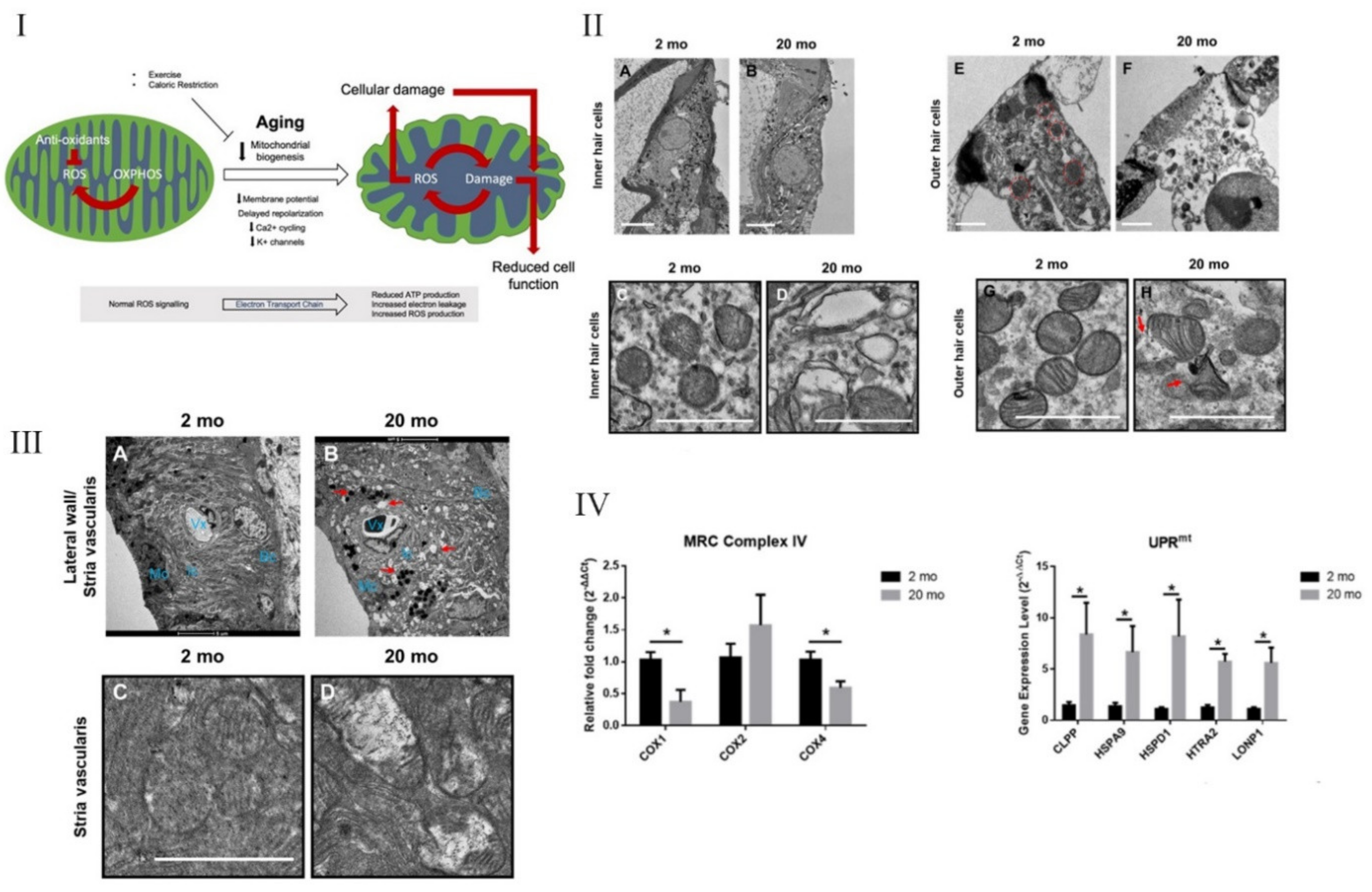
5. Necroptosis, Mitochondrial Dysfunction, and Inflammaging in the Inner Ear
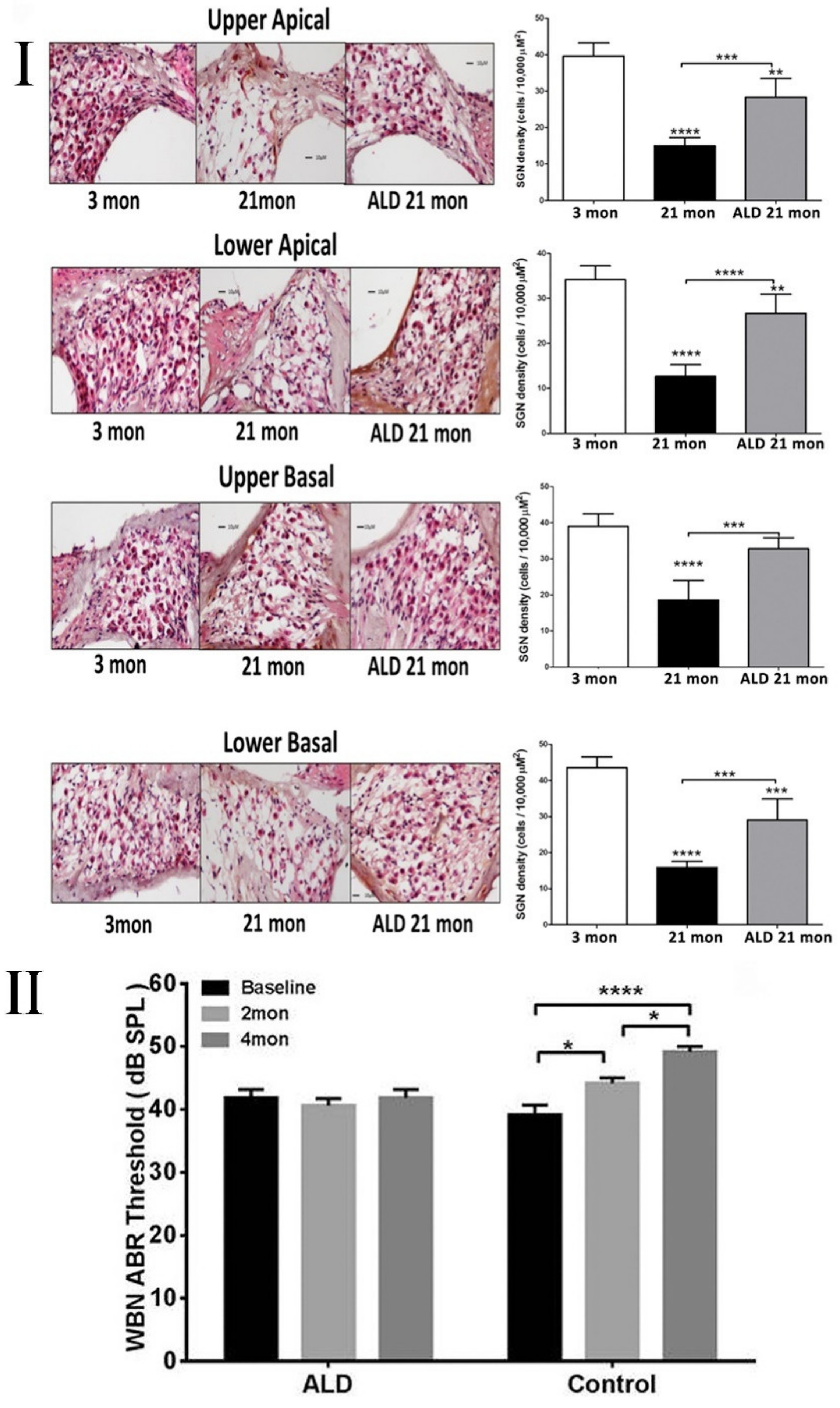
6. Intervention Strategies to Prevent Damage/Treat the Aging Cochlea
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Norman, R. The aging of the world’s population. In Geriatric Dermatology; CRC Press: Boca Raton, FL, USA, 2020; pp. 1–3. [Google Scholar]
- United Nations—Department of Economic and Social Affairs—Population Division (2020). World Population Ageing 2019; ST/ESA/SER.A/444; United Nations: New York, NY, USA, 2020. [Google Scholar]
- Mather, M.; Scommegna, P.; Kilduff, L. Fact Sheet: Aging in the United States. Available online: https://www.prb.org/resources/fact-sheet-aging-in-the-united-states/ (accessed on 6 June 2021).
- Ames, B.N.; Shigenaga, M.K.; Hagen, T.M. Oxidants, antioxidants, and the degenerative diseases of aging. Proc. Natl. Acad. Sci. USA 1993, 90, 7915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fulop, T.; Larbi, A.; Witkowski, J.M.; McElhaney, J.; Loeb, M.; Mitnitski, A.; Pawelec, G. Aging, frailty and age-related diseases. Biogerontology 2010, 11, 547–563. [Google Scholar] [CrossRef] [PubMed]
- Jenny, N.S. Inflammation in aging: Cause, effect, or both? Discov. Med. 2012, 13, 451–460. [Google Scholar] [PubMed]
- Singh, T.; Newman, A.B. Inflammatory markers in population studies of aging. Ageing Res. Rev. 2011, 10, 319–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedman, E.M.; Hayney, M.; Love, G.D.; Singer, B.H.; Ryff, C.D. Plasma interleukin-6 and soluble IL-6 receptors are associated with psychological well-being in aging women. Health Psychol. 2007, 26, 305–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maggio, M.; Guralnik, J.M.; Longo, D.L.; Ferrucci, L. Interleukin-6 in Aging and Chronic Disease: A Magnificent Pathway. J. Gerontol. Ser. A 2006, 61, 575–584. [Google Scholar] [CrossRef]
- Puzianowska-Kuźnicka, M.; Owczarz, M.; Wieczorowska-Tobis, K.; Nadrowski, P.; Chudek, J.; Slusarczyk, P.; Skalska, A.; Jonas, M.; Franek, E.; Mossakowska, M. Interleukin-6 and C-reactive protein, successful aging, and mortality: The PolSenior study. Immun. Ageing 2016, 13, 21. [Google Scholar] [CrossRef] [Green Version]
- Ruggiero, C.; Metter, E.J.; Cherubini, A.; Maggio, M.; Sen, R.; Najjar, S.S.; Windham, G.B.; Ble, A.; Senin, U.; Ferrucci, L. White blood cell count and mortality in the Baltimore Longitudinal Study of Aging. J. Am. Coll. Cardiol. 2007, 49, 1841–1850. [Google Scholar] [CrossRef] [Green Version]
- Taaffe, D.R.; Harris, T.B.; Ferrucci, L.; Rowe, J.; Seeman, T.E. Cross-sectional and Prospective Relationships of Interleukin-6 and C-Reactive Protein with Physical Performance in Elderly Persons: MacArthur Studies of Successful Aging. J. Gerontol. Ser. A 2000, 55, M709–M715. [Google Scholar] [CrossRef]
- Xing, Z.; Gauldie, J.; Cox, G.; Baumann, H.; Jordana, M.; Lei, X.F.; Achong, M.K. IL-6 is an antiinflammatory cytokine required for controlling local or systemic acute inflammatory responses. J. Clin. Investig. 1998, 101, 311–320. [Google Scholar] [CrossRef]
- Ferrucci, L.; Fabbri, E. Inflammageing: Chronic inflammation in ageing, cardiovascular disease, and frailty. Nat. Rev. Cardiol. 2018, 15, 505–522. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Bonafè, M.; Valensin, S.; Olivieri, F.; De Luca, M.; Ottaviani, E.; De Benedictis, G. Inflammaging: An evolutionary perspective on immunosenescence. Ann. N. Y. Acad. Sci. 2000, 908, 244–254. [Google Scholar] [CrossRef]
- Watson, N.; Ding, B.; Zhu, X.; Frisina, R.D. Chronic inflammation—Inflammaging—In the ageing cochlea: A novel target for future presbycusis therapy. Ageing Res. Rev. 2017, 40, 142–148. [Google Scholar] [CrossRef]
- Chung, H.Y.; Kim, H.J.; Kim, K.W.; Choi, J.S.; Yu, B.P. Molecular inflammation hypothesis of aging based on the anti-aging mechanism of calorie restriction. Microsc. Res. Tech. 2002, 59, 264–272. [Google Scholar] [CrossRef]
- Franceschi, C.; Campisi, J. Chronic Inflammation (Inflammaging) and Its Potential Contribution to Age-Associated Diseases. J. Gerontol. Ser. A 2014, 69, S4–S9. [Google Scholar] [CrossRef]
- Murakami, M.; Hirano, T. The molecular mechanisms of chronic inflammation development. Front. Immunol. 2012, 3, 323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanada, F.; Taniyama, Y.; Muratsu, J.; Otsu, R.; Shimizu, H.; Rakugi, H.; Morishita, R. Source of Chronic Inflammation in Aging. Front. Cardiovasc. Med. 2018, 5, 12. [Google Scholar] [CrossRef] [Green Version]
- Blake, G.J.; Ridker, P.M. Inflammatory bio-markers and cardiovascular risk prediction. J. Intern. Med. 2002, 252, 283–294. [Google Scholar] [CrossRef] [PubMed]
- Dandona, P.; Aljada, A.; Bandyopadhyay, A. Inflammation: The link between insulin resistance, obesity and diabetes. Trends Immunol. 2004, 25, 4–7. [Google Scholar] [CrossRef] [PubMed]
- Dorfmüller, P.; Perros, F.; Balabanian, K.; Humbert, M. Inflammation in pulmonary arterial hypertension. Eur. Respir. J. 2003, 22, 358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansson, G.K.; Hermansson, A. The immune system in atherosclerosis. Nat. Immunol. 2011, 12, 204–212. [Google Scholar] [CrossRef]
- Harrison, D.G.; Guzik, T.J.; Lob, H.E.; Madhur, M.S.; Marvar, P.J.; Thabet, S.R.; Vinh, A.; Weyand, C.M. Inflammation, Immunity, and Hypertension. Hypertension 2011, 57, 132–140. [Google Scholar] [CrossRef] [Green Version]
- Heppner, F.L.; Ransohoff, R.M.; Becher, B. Immune attack: The role of inflammation in Alzheimer disease. Nat. Rev. Neurosci. 2015, 16, 358. [Google Scholar] [CrossRef] [PubMed]
- Koenig, W.; Sund, M.; Fröhlich, M.; Fischer, H.-G.; Löwel, H.; Döring, A.; Hutchinson, W.L.; Pepys, M.B. C-Reactive Protein, a Sensitive Marker of Inflammation, Predicts Future Risk of Coronary Heart Disease in Initially Healthy Middle-Aged Men. Circulation 1999, 99, 237–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koziorowski, D.; Tomasiuk, R.; Szlufik, S.; Friedman, A. Inflammatory cytokines and NT-proCNP in Parkinson’s disease patients. Cytokine 2012, 60, 762–766. [Google Scholar] [CrossRef] [PubMed]
- Luft, V.C.; Schmidt, M.I.; Pankow, J.S.; Couper, D.; Ballantyne, C.M.; Young, J.H.; Duncan, B.B. Chronic inflammation role in the obesity-diabetes association: A case-cohort study. Diabetol. Metab. Syndr. 2013, 5, 31. [Google Scholar] [CrossRef] [Green Version]
- Savoia, C.; Schiffrin, E.L. Inflammation in hypertension. Curr. Opin. Nephrol. Hypertens. 2006, 15, 152–158. [Google Scholar] [CrossRef]
- Stenvinkel, P.; Heimbürger, O.; Paultre, F.; Diczfalusy, U.; Wang, T.; Berglund, L.; Jogestrand, T. Strong association between malnutrition, inflammation, and atherosclerosis in chronic renal failure. Kidney Int. 1999, 55, 1899–1911. [Google Scholar] [CrossRef] [Green Version]
- Walker, D.G.; Dalsing-Hernandez, J.E.; Campbell, N.A.; Lue, L.-F. Decreased expression of CD200 and CD200 receptor in Alzheimer’s disease: A potential mechanism leading to chronic inflammation. Exp. Neurol. 2009, 215, 5–19. [Google Scholar] [CrossRef] [Green Version]
- Candore, G.; Caruso, C.; Colonna-Romano, G. Inflammation, genetic background and longevity. Biogerontology 2010, 11, 565–573. [Google Scholar] [CrossRef] [Green Version]
- Lio, D.; Scola, L.; Crivello, A.; Colonna-Romano, G.; Candore, G.; Bonafé, M.; Cavallone, L.; Marchegiani, F.; Olivieri, F.; Franceschi, C.; et al. Inflammation, genetics, and longevity: Further studies on the protective effects in men of IL-10 −1082 promoter SNP and its interaction with TNF-α −308 promoter SNP. J. Med. Genet. 2003, 40, 296–299. [Google Scholar] [CrossRef] [Green Version]
- Xia, S.; Zhang, X.; Zheng, S.; Khanabdali, R.; Kalionis, B.; Wu, J.; Wan, W.; Tai, X. An Update on Inflamm-Aging: Mechanisms, Prevention, and Treatment. J. Immunol. Res. 2016, 2016, 8426874. [Google Scholar] [CrossRef]
- Dalton, D.S.; Cruickshanks, K.J.; Klein, B.E.K.; Klein, R.; Wiley, T.L.; Nondahl, D.M. The Impact of Hearing Loss on Quality of Life in Older Adults. Gerontology 2003, 43, 661–668. [Google Scholar] [CrossRef]
- Frisina, D.R.; Frisina, R.D. Speech recognition in noise and presbycusis: Relations to possible neural mechanisms. Hear. Res. 1997, 106, 95–104. [Google Scholar] [CrossRef]
- Frisina, R.D.; Williamson, T.T.; Bazard, P.; Zhu, X.; Dinga, B. 2.44—The Aging Cochlea. In The Senses: A Comprehensive Reference, 2nd ed.; Fritzsch, B., Ed.; Elsevier: Oxford, UK, 2020; pp. 871–883. [Google Scholar]
- Huang, Q.; Tang, J. Age-related hearing loss or presbycusis. Eur. Arch. Oto-Rhino-Laryngol. 2010, 267, 1179–1191. [Google Scholar] [CrossRef]
- Gates, G.A.; Cooper, J.C. Incidence of Hearing Decline in the Elderly. Acta Oto-Laryngol. 1991, 111, 240–248. [Google Scholar] [CrossRef] [PubMed]
- Gates, G.A.; Mills, J.H. Presbycusis. Lancet 2005, 366, 1111–1120. [Google Scholar] [CrossRef]
- Liu, X.; Yan, D. Ageing and hearing loss. J. Pathol. 2007, 211, 188–197. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Puel, J.-L. Presbycusis: An Update on Cochlear Mechanisms and Therapies. J. Clin. Med. 2020, 9, 218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuknecht, H.F. Presbycusis. Laryngoscope 1955, 65, 402–419. [Google Scholar] [CrossRef]
- Schuknecht, H.F. Further Observations on the Pathology of Presbycusis. Arch. Otolaryngol. 1964, 80, 369–382. [Google Scholar] [CrossRef]
- Schuknecht, H.F. Pathology of presbycusis. Geriatr. Otorhinolaryngol. 1989, 40–44. [Google Scholar]
- Schuknecht, H.F.; Gacek, M.R. Cochlear Pathology in Presbycusis. Ann. Otol. Rhinol. Laryngol. 1993, 102, 1–16. [Google Scholar] [CrossRef]
- Bazard, P.; Frisina, R.D.; Acosta, A.A.; Dasgupta, S.; Bauer, M.A.; Zhu, X.Z.; Ding, B. Roles of Key Ion Channels and Transport Proteins in Age-related Hearing Loss. Int. J. Mol. Sci. 2021, 22, 6158. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Li, Y.; Chen, L.; Zhang, Q.; Pan, N.; Nichols, D.H.; Zhang, W.J.; Fritzsch, B.; He, D.Z.Z. Organ of Corti and Stria Vascularis: Is there an Interdependence for Survival? PLoS ONE 2016, 11, e0168953. [Google Scholar] [CrossRef] [PubMed]
- Salt, A.N.; Mleichar, I.; Thalmann, R. Mechanisms of endocochlear potential generation by stria vascularis. Laryngoscope 1987, 97, 984–991. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, S.; Ando, M.; Kakigi, A. Mechanism Generating Endocochlear Potential: Role Played by Intermediate Cells in Stria Vascularis. Biophys. J. 2000, 79, 2572–2582. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Zhao, H.B. The role of an inwardly rectifying K+ channel (Kir4.1) in the inner ear and hearing loss. Neuroscience 2014, 265, 137–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, B.; Walton, J.P.; Zhu, X.; Frisina, R.D. Age-related changes in Na, K-ATPase expression, subunit isoform selection and assembly in the stria vascularis lateral wall of mouse cochlea. Hear. Res. 2018, 367, 59–73. [Google Scholar] [CrossRef] [PubMed]
- Mills, J.H.; Schmiedt, R.A.; Schulte, B.A.; Dubno, J.R. Age-Related Hearing Loss: A Loss of Voltage, Not Hair Cells. Semin. Hear. 2006, 27, 228–236. [Google Scholar] [CrossRef]
- Ohlemiller, K.K.; Frisina, R.D. Age-Related Hearing Loss and Its Cellular and Molecular Bases. In Auditory Trauma, Protection, and Repair; Schacht, J., Popper, A.N., Fay, R.R., Eds.; Springer: Boston, MA, USA, 2008; pp. 145–194. [Google Scholar]
- Pan, C.-c.; Chu, H.-q.; Lai, Y.-b.; Sun, Y.-b.; Du, Z.-h.; Liu, Y.; Chen, J.; Tong, T.; Chen, Q.-g.; Zhou, L.-q.; et al. Downregulation of inwardly rectifying potassium channel 5.1 expression in C57BL/6J cochlear lateral wall. J. Huazhong Univ. Sci. Technol. 2016, 36, 406–409. [Google Scholar] [CrossRef] [PubMed]
- Pauler, M.; Schuknecht, H.F.; White, J.A. Atrophy of the stria vascularis as a cause of sensorineural hearing loss. Laryngoscope 1988, 98, 754–759. [Google Scholar] [CrossRef] [PubMed]
- Schmiedt, R.A.; Lang, H.; Okamura, H.-o.; Schulte, B.A. Effects of Furosemide Applied Chronically to the Round Window: A Model of Metabolic Presbyacusis. J. Neurosci. 2002, 22, 9643–9650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuknecht, H.F. Pathology of the Ear; Harvard University Press: Cambridge, MA, USA, 1974; Volume 85, pp. 576–577. [Google Scholar]
- Schulte, B.A.; Schmiedt, R.A. Lateral wall Na, K-ATPase and endocochlear potentials decline with age in quiet-reared gerbils. Hear. Res. 1992, 61, 35–46. [Google Scholar] [CrossRef]
- Sewell, W.F. The effects of furosemide on the endocochlear potential and auditory-nerve fiber tuning curves in cats. Hear. Res. 1984, 14, 305–314. [Google Scholar] [CrossRef]
- Takeuchi, S.; Ando, M. Inwardly rectifying K+ currents in intermediate cells in the cochlea of gerbils: A possible contribution to the endocochlear potential. Neurosci. Lett. 1998, 247, 175–178. [Google Scholar] [CrossRef]
- Gratton, M.A.; Smyth, B.J.; Lam, C.F.; Boettcher, F.A.; Schmiedt, R.A. Decline in the endocochlear potential corresponds to decreased Na,K-ATPase activity in the lateral wall of quiet-aged gerbils. Hear. Res. 1997, 108, 9–16. [Google Scholar] [CrossRef]
- Carraro, M.; Harrison, R.V. Degeneration of stria vascularis in age-related hearing loss; a corrosion cast study in a mouse model. Acta Oto-Laryngol. 2016, 136, 385–390. [Google Scholar] [CrossRef]
- Fetoni, A.R.; Picciotti, P.M.; Paludetti, G.; Troiani, D. Pathogenesis of presbycusis in animal models: A review. Exp. Gerontol. 2011, 46, 413–425. [Google Scholar] [CrossRef]
- Gratton, M.A.; Schmiedt, R.A.; Schulte, B.A. Age-related decreases in endocochlear potential are associated with vascular abnormalities in the stria vascularis. Hear. Res. 1996, 102, 181–190. [Google Scholar] [CrossRef]
- Gratton, M.A.; Schulte, B.A. Alterations in microvasculature are associated with atrophy of the stria vascularis in quiet-aged gerbils. Hear. Res. 1995, 82, 44–52. [Google Scholar] [CrossRef]
- Kurata, N.; Schachern, P.A.; Paparella, M.M.; Cureoglu, S. Histopathologic Evaluation of Vascular Findings in the Cochlea in Patients with Presbycusis. JAMA Otolaryngol. Head Neck Surg. 2016, 142, 173–178. [Google Scholar] [CrossRef] [Green Version]
- Wu, P.Z.; Liberman, L.D.; Bennett, K.; de Gruttola, V.; O’Malley, J.T.; Liberman, M.C. Primary Neural Degeneration in the Human Cochlea: Evidence for Hidden Hearing Loss in the Aging Ear. Neuroscience 2019, 407, 8–20. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.-Y. Pathophysiology of Age-Related Hearing Loss (Peripheral and Central). Korean J. Audiol. 2013, 17, 45–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kujawa, S.G.; Liberman, M.C. Adding Insult to Injury: Cochlear Nerve Degeneration after “Temporary” Noise-Induced Hearing Loss. J. Neurosci. 2009, 29, 14077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parthasarathy, A.; Kujawa, S.G. Synaptopathy in the Aging Cochlea: Characterizing Early-Neural Deficits in Auditory Temporal Envelope Processing. J. Neurosci. 2018, 38, 7108–7119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sergeyenko, Y.; Lall, K.; Liberman, M.C.; Kujawa, S.G. Age-Related Cochlear Synaptopathy: An Early-Onset Contributor to Auditory Functional Decline. J. Neurosci. 2013, 33, 13686–13694. [Google Scholar] [CrossRef]
- Kujawa, S.G.; Liberman, M.C. Acceleration of Age-Related Hearing Loss by Early Noise Exposure: Evidence of a Misspent Youth. J. Neurosci. 2006, 26, 2115. [Google Scholar] [CrossRef]
- Verschuur, C.A.; Dowell, A.; Syddall, H.E.; Ntani, G.; Simmonds, S.J.; Baylis, D.; Gale, C.R.; Walsh, B.; Cooper, C.; Lord, J.M.; et al. Markers of inflammatory status are associated with hearing threshold in older people: Findings from the Hertfordshire ageing study. Age Ageing 2011, 41, 92–97. [Google Scholar] [CrossRef] [Green Version]
- Verschuur, C.; Agyemang-Prempeh, A.; Newman, T.A. Inflammation is associated with a worsening of presbycusis: Evidence from the MRC national study of hearing. Int. J. Audiol. 2014, 53, 469–475. [Google Scholar] [CrossRef]
- Lassale, C.; Vullo, P.; Cadar, D.; Batty, G.D.; Steptoe, A.; Zaninotto, P. Association of inflammatory markers with hearing impairment: The English Longitudinal Study of Ageing. Brain Behav. Immun. 2020, 83, 112–119. [Google Scholar] [CrossRef]
- Sardone, R.; Lampignano, L.; Guerra, V.; Zupo, R.; Donghia, R.; Castellana, F.; Battista, P.; Bortone, I.; Procino, F.; Castellana, M.; et al. Relationship between Inflammatory Food Consumption and Age-Related Hearing Loss in a Prospective Observational Cohort: Results from the Salus in Apulia Study. Nutrients 2020, 12, 426. [Google Scholar] [CrossRef] [Green Version]
- Rask-Andersen, H.; Stahle, J. Lymphocyte-Macrophage Activity in the Endolymphatic Sac. ORL J. Otorhinolaryngol. Relat. Spec. 1979, 41, 177–192. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Zhang, J.; Neng, L.; Shi, X. Characterization and Inflammatory Response of Perivascular-Resident Macrophage-Like Melanocytes in the Vestibular System. J. Assoc. Res. Otolaryngol. 2013, 14, 635–643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Dai, M.; Fridberger, A.; Hassan, A.; DeGagne, J.; Neng, L.; Zhang, F.; He, W.; Ren, T.; Trune, D.; et al. Perivascular-resident macrophage-like melanocytes in the inner ear are essential for the integrity of the intrastrial fluid–blood barrier. Proc. Natl. Acad. Sci. USA 2012, 109, 10388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frye, M.D.; Yang, W.; Zhang, C.; Xiong, B.; Hu, B.H. Dynamic activation of basilar membrane macrophages in response to chronic sensory cell degeneration in aging mouse cochleae. Hear. Res. 2017, 344, 125–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, X.; Qiu, S.; Zhuang, W.; Yuan, N.; Wang, C.; Zhang, S.; Sun, T.; Guo, W.; Gao, F.; Yang, S.; et al. NLRP3-inflammasomes are triggered by age-related hearing loss in the inner ear of mice. Am. J. Transl. Res. 2017, 9, 5611–5618. [Google Scholar] [PubMed]
- Menardo, J.; Tang, Y.; Ladrech, S.; Lenoir, M.; Casas, F.; Michel, C.; Bourien, J.; Ruel, J.; Rebillard, G.; Maurice, T.; et al. Oxidative Stress, Inflammation, and Autophagic Stress as the Key Mechanisms of Premature Age-Related Hearing Loss in SAMP8 Mouse Cochlea. Antioxid. Redox Signal. 2011, 16, 263–274. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.; Xiong, H.; Liu, Y.; Pang, J.; Lin, H.; Zhang, W.; Zheng, Y. Transcriptomic analysis highlights cochlear inflammation associated with age-related hearing loss in C57BL/6 mice using next generation sequencing. PeerJ 2020, 8, e9737. [Google Scholar] [CrossRef]
- Dhukhwa, A.; Bhatta, P.; Sheth, S.; Korrapati, K.; Tieu, C.; Mamillapalli, C.; Ramkumar, V.; Mukherjea, D. Targeting Inflammatory Processes Mediated by TRPVI and TNF-α for Treating Noise-Induced Hearing Loss. Front. Cell. Neurosci. 2019, 13, 444. [Google Scholar] [CrossRef] [Green Version]
- Fujioka, M.; Kanzaki, S.; Okano, H.J.; Masuda, M.; Ogawa, K.; Okano, H. Proinflammatory cytokines expression in noise-induced damaged cochlea. J. Neurosci. Res. 2006, 83, 575–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Umugire, A.; Lee, S.; Kim, D.; Choi, M.; Kim, H.-S.; Cho, H.-H. Avenanthramide-C prevents noise- and drug-induced hearing loss while protecting auditory hair cells from oxidative stress. Cell Death Discov. 2019, 5, 115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wakabayashi, K.; Fujioka, M.; Kanzaki, S.; Okano, H.J.; Shibata, S.; Yamashita, D.; Masuda, M.; Mihara, M.; Ohsugi, Y.; Ogawa, K.; et al. Blockade of interleukin-6 signaling suppressed cochlear inflammatory response and improved hearing impairment in noise-damaged mice cochlea. Neurosci. Res. 2010, 66, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Wong, A.K.O.; Beattie, K.A.; Bhargava, A.; Cheung, M.; Webber, C.E.; Chettle, D.R.; Papaioannou, A.; Adachi, J.D. Bone lead (Pb) content at the tibia is associated with thinner distal tibia cortices and lower volumetric bone density in postmenopausal women. Bone 2015, 79, 58–64. [Google Scholar] [CrossRef] [Green Version]
- Nakamoto, T.; Mikuriya, T.; Sugahara, K.; Hirose, Y.; Hashimoto, T.; Shimogori, H.; Takii, R.; Nakai, A.; Yamashita, H. Geranylgeranylacetone suppresses noise-induced expression of proinflammatory cytokines in the cochlea. Auris Nasus Larynx 2012, 39, 270–274. [Google Scholar] [CrossRef]
- Zhang, G.; Zheng, H.; Pyykko, I.; Zou, J. The TLR-4/NF-κB signaling pathway activation in cochlear inflammation of rats with noise-induced hearing loss. Hear. Res. 2019, 379, 59–68. [Google Scholar] [CrossRef]
- Celaya, A.M.; Rodríguez-de la Rosa, L.; Bermúdez-Muñoz, J.M.; Zubeldia, J.M.; Romá-Mateo, C.; Avendaño, C.; Pallardó, F.V.; Varela-Nieto, I. IGF-1 Haploinsufficiency Causes Age-Related Chronic Cochlear Inflammation and Increases Noise-Induced Hearing Loss. Cells 2021, 10, 1686. [Google Scholar] [CrossRef] [PubMed]
- Frye, M.D.; Ryan, A.F.; Kurabi, A. Inflammation associated with noise-induced hearing loss. J. Acoust. Soc. Am. 2019, 146, 4020–4032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paciello, F.; Di Pino, A.; Rolesi, R.; Troiani, D.; Paludetti, G.; Grassi, C.; Fetoni, A.R. Anti-oxidant and anti-inflammatory effects of caffeic acid: In vivo evidences in a model of noise-induced hearing loss. Food Chem. Toxicol. 2020, 143, 111555. [Google Scholar] [CrossRef]
- Shih, C.-P.; Chen, H.-C.; Lin, Y.-C.; Chen, H.-K.; Wang, H.; Kuo, C.-Y.; Lin, Y.-Y.; Wang, C.-H. Middle-ear dexamethasone delivery via ultrasound microbubbles attenuates noise-induced hearing loss. Laryngoscope 2019, 129, 1907–1914. [Google Scholar] [CrossRef]
- Shih, C.-P.; Kuo, C.-Y.; Lin, Y.-Y.; Lin, Y.-C.; Chen, H.-K.; Wang, H.; Chen, H.-C.; Wang, C.-H. Inhibition of Cochlear HMGB1 Expression Attenuates Oxidative Stress and Inflammation in an Experimental Murine Model of Noise-Induced Hearing Loss. Cells 2021, 10, 810. [Google Scholar] [CrossRef] [PubMed]
- Marzetti, E.; Hwang, J.C.Y.; Lees, H.A.; Wohlgemuth, S.E.; Dupont-Versteegden, E.E.; Carter, C.S.; Bernabei, R.; Leeuwenburgh, C. Mitochondrial death effectors: Relevance to sarcopenia and disuse muscle atrophy. Biochim. Biophys. Acta Gen. Subj. 2010, 1800, 235–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shanmugam, M.K.; Sethi, G. Role of Epigenetics in Inflammation-Associated Diseases. In Epigenetics: Development and Disease; Kundu, T.K., Ed.; Springer: Dordrecht, The Netherlands, 2013; pp. 627–657. [Google Scholar]
- Sohal, R.S.; Orr, W.C. The redox stress hypothesis of aging. Free Radic. Biol. Med. 2012, 52, 539–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarantino, G.; Savastano, S.; Colao, A. Hepatic steatosis, low-grade chronic inflammation and hormone/growth factor/adipokine imbalance. World J. Gastroenterol. 2010, 16, 4773. [Google Scholar] [CrossRef]
- Cunningham, J.N., Jr.; Carter, N.W.; Rector, F.C., Jr.; Seldin, D.W. Resting transmembrane potential difference of skeletal muscle in normal subjects and severely ill patients. J. Clin. Investig. 1971, 50, 49–59. [Google Scholar] [CrossRef] [Green Version]
- Eisenhut, M.; Southern, K. Positive sweat test following meningococcal septicaemia. Acta Paediatr. Nurtur. Child 2002, 91, 361–362. [Google Scholar] [CrossRef]
- Welt, L.G. Membrane transport defect: The sick cell. Trans. Assoc. Am. Phys. 1967, 80, 217–226. [Google Scholar]
- Welt, L.G.; Sachs, J.R.; McManus, T.J. An ion transport defect in erythrocytes from uremic patients. Trans. Assoc. Am. Phys. 1964, 77, 169–181. [Google Scholar]
- Clausen, M.V.; Hilbers, F.; Poulsen, H. The Structure and Function of the Na,K-ATPase Isoforms in Health and Disease. Front. Physiol. 2017, 8, 371. [Google Scholar] [CrossRef]
- Erichsen, S.; Zuo, J.; Curtis, L.; Rarey, K.; Hultcrantz, M. Na,K-ATPase α- and β-isoforms in the developing cochlea of the mouse. Hear. Res. 1996, 100, 143–149. [Google Scholar] [CrossRef]
- Watabe, T.; Xu, M.; Watanabe, M.; Nabekura, J.; Higuchi, T.; Hori, K.; Sato, M.P.; Nin, F.; Hibino, H.; Ogawa, K.; et al. Time-controllable Nkcc1 knockdown replicates reversible hearing loss in postnatal mice. Sci. Rep. 2017, 7, 13605. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Yan, Y.; Liu, L.; Xie, Z.; Malhotra, D.; Joe, B.; Shapiro, J.I. Impairment of Na/K-ATPase Signaling in Renal Proximal Tubule Contributes to Dahl Salt-sensitive Hypertension. J. Biol. Chem. 2011, 286, 22806–22813. [Google Scholar] [CrossRef] [Green Version]
- Delpire, E.; Lu, J.; England, R.; Dull, C.; Thorne, T. Deafness and imbalance associated with inactivation of the secretory Na-K-2Cl co-transporter. Nat. Genet. 1999, 22, 192–195. [Google Scholar] [CrossRef] [PubMed]
- Halonen, J.; Hinton, A.S.; Frisina, R.D.; Ding, B.; Zhu, X.; Walton, J.P. Long-term treatment with aldosterone slows the progression of age-related hearing loss. Hear. Res. 2016, 336, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Chu, H.; Chen, J.; Zhou, L.; Chen, Q.; Yu, Y.; Wu, Z.; Wang, S.; Lai, Y.; Pan, C.; et al. Age-related change in the expression of NKCC1 in the cochlear lateral wall of C57BL/6J mice. Acta Oto-Laryngol. 2014, 134, 1047–1051. [Google Scholar] [CrossRef] [PubMed]
- Frisina, R.D.; Ding, B.; Zhu, X.; Walton, J.P. Age-related hearing loss: Prevention of threshold declines, cell loss and apoptosis in spiral ganglion neurons. Aging 2016, 8, 2081–2099. [Google Scholar] [CrossRef] [Green Version]
- Diaz, R.C.; Vazquez, A.E.; Dou, H.; Wei, D.; Cardell, E.L.; Lingrel, J.; Shull, G.E.; Doyle, K.J.; Yamoah, E.N. Conservation of Hearing by Simultaneous Mutation of Na,K-ATPase and NKCC1. J. Assoc. Res. Otolaryngol. 2007, 8, 422–434. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, C.; Höcherl, K.; Schweda, F.; Kurtz, A.; Bucher, M. Regulation of Renal Sodium Transporters during Severe Inflammation. J. Am. Soc. Nephrol. 2007, 18, 1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bone, R.C. The Pathogenesis of Sepsis. Ann. Intern. Med. 1991, 115, 457–469. [Google Scholar] [CrossRef] [PubMed]
- Allgayer, H.; Kruis, W.; Paumgartner, G.; Wiebecke, B.; Brown, L.; Erdmann, E. Inverse relationship between colonic (Na+ + K+)-ATPase activity and degree of mucosal inflammation in inflammatory bowel disease. Dig. Dis. Sci. 1988, 33, 417–422. [Google Scholar] [CrossRef]
- Yan, Y.; Shapiro, A.P.; Haller, S.; Katragadda, V.; Liu, L.; Tian, J.; Basrur, V.; Malhotra, D.; Xie, Z.-j.; Abraham, N.G.; et al. Involvement of Reactive Oxygen Species in a Feed-forward Mechanism of Na/K-ATPase-mediated Signaling Transduction. J. Biol. Chem. 2013, 288, 34249–34258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sodhi, K.; Nichols, A.; Mallick, A.; Klug, R.L.; Liu, J.; Wang, X.; Srikanthan, K.; Goguet-Rubio, P.; Nawab, A.; Pratt, R.; et al. The Na/K-ATPase Oxidant Amplification Loop Regulates Aging. Sci. Rep. 2018, 8, 9721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Tian, J.; Chaudhry, M.; Maxwell, K.; Yan, Y.; Wang, X.; Shah, P.T.; Khawaja, A.A.; Martin, R.; Robinette, T.J.; et al. Attenuation of Na/K-ATPase Mediated Oxidant Amplification with pNaKtide Ameliorates Experimental Uremic Cardiomyopathy. Sci. Rep. 2016, 6, 34592. [Google Scholar] [CrossRef] [PubMed]
- Sodhi, K.; Maxwell, K.; Yan, Y.; Liu, J.; Chaudhry, M.A.; Getty, M.; Xie, Z.; Abraham, N.G.; Shapiro, J.I. pNaKtide inhibits Na/K-ATPase reactive oxygen species amplification and attenuates adipogenesis. Sci. Adv. 2015, 1, e1500781. [Google Scholar] [CrossRef] [Green Version]
- Qu, X.; Zhang, Z.; Hu, W.; Lou, M.; Zhai, B.; Mei, S.; Hu, Z.; Zhang, L.; Liu, D.; Liu, Z.; et al. Attenuation of the Na/K-ATPase/Src/ROS amplification signaling pathway by astaxanthin ameliorates myocardial cell oxidative stress injury. Mol. Med. Rep. 2020, 22, 5125–5134. [Google Scholar] [CrossRef]
- Srikanthan, K.; Shapiro, J.I.; Sodhi, K. The Role of Na/K-ATPase Signaling in Oxidative Stress Related to Obesity and Cardiovascular Disease. Molecules 2016, 21, 1172. [Google Scholar] [CrossRef] [Green Version]
- Xie, Z.; Cai, T. Na+-K+—ATPase-mediated signal transduction: From protein interaction to cellular function. Mol. Interv. 2003, 3, 157–168. [Google Scholar] [CrossRef]
- Tian, J.; Liu, J.; Garlid, K.D.; Shapiro, J.I.; Xie, Z. Involvement of mitogen-activated protein kinases and reactive oxygen species in the inotropic action of ouabain on cardiac myocytes. A potential role for mitochondrial K(ATP) channels. Mol Cell Biochem. 2003, 242, 181–187. [Google Scholar] [CrossRef]
- Liu, J.; Tian, J.; Haas, M.; Shapiro, J.I.; Askari, A.; Xie, Z. Ouabain Interaction with Cardiac Na+/K+-ATPase Initiates Signal Cascades Independent of Changes in Intracellular Na+ and Ca2+ Concentrations. J. Biol. Chem. 2000, 275, 27838–27844. [Google Scholar] [CrossRef]
- Mittal, M.; Siddiqui, M.R.; Tran, K.; Reddy, S.P.; Malik, A.B. Reactive Oxygen Species in Inflammation and Tissue Injury. Antioxid. Redox Signal. 2013, 20, 1126–1167. [Google Scholar] [CrossRef] [Green Version]
- Bertelsen, L.S.; Eckmann, L.; Barrett, K.E. Prolonged interferon-γ exposure decreases ion transport, NKCC1, and Na+-K+-ATPase expression in human intestinal xenografts in vivo. Am. J. Physiol.-Gastrointest. Liver Physiol. 2004, 286, G157–G165. [Google Scholar] [CrossRef]
- King, S.J.; Bunz, M.; Chappell, A.; Scharl, M.; Docherty, M.; Jung, B.; Lytle, C.; McCole, D.F. AMPK mediates inhibition of electrolyte transport and NKCC1 activity by reactive oxygen species. Am. J. Physiol.-Gastrointest. Liver Physiol. 2019, 317, G171–G181. [Google Scholar] [CrossRef] [PubMed]
- Koumangoye, R.; Omer, S.; Kabeer, M.H.; Delpire, E. Novel Human NKCC1 Mutations Cause Defects in Goblet Cell Mucus Secretion and Chronic Inflammation. Cell. Mol. Gastroenterol. Hepatol. 2020, 9, 239–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, Y.; Wu, M.; Shen, J.; Tang, J.; Li, J.; Xu, J.; Dang, B.; Chen, G. Inhibition of the NKCC1/NF-κB Signaling Pathway Decreases Inflammation and Improves Brain Edema and Nerve Cell Apoptosis in an SBI Rat Model. Front. Mol. Neurosci. 2021, 14, 1993. [Google Scholar] [CrossRef]
- Shen, C.-H.; Lin, J.-Y.; Chang, Y.-L.; Wu, S.-Y.; Peng, C.-K.; Wu, C.-P.; Huang, K.-L. Inhibition of NKCC1 Modulates Alveolar Fluid Clearance and Inflammation in Ischemia-Reperfusion Lung Injury via TRAF6-Mediated Pathways. Front. Immunol. 2018, 9, 2049. [Google Scholar] [CrossRef] [PubMed]
- Ando, M.; Takeuchi, S. Immunological identification of an inward rectifier K+ channel (Kir4.1) in the intermediate cell (melanocyte) of the cochlear stria vascularis of gerbils and rats. Cell Tissue Res. 1999, 298, 179–183. [Google Scholar] [CrossRef]
- Brew, H.M.; Forsythe, I.D. Systematic variation of potassium current amplitudes across the tonotopic axis of the rat medial nucleus of the trapezoid body. Hear. Res. 2005, 206, 116–132. [Google Scholar] [CrossRef]
- Rusznák, Z.; Bakondi, G.; Pocsai, K.; Pór, A.; Kosztka, L.; Pál, B.; Nagy, D.; Szucs, G. Voltage-gated potassium channel (Kv) subunits expressed in the rat cochlear nucleus. J. Histochem. Cytochem. Off. J. Histochem. Soc. 2008, 56, 443–465. [Google Scholar] [CrossRef] [Green Version]
- Takeuchi, S.; Ando, M.; Sato, T.; Kakigi, A. Three-dimensional and ultrastructural relationships between intermediate cells and capillaries in the gerbil stria vascularis. Hear. Res. 2001, 155, 103–112. [Google Scholar] [CrossRef]
- Kuo, M.M.-C.; Haynes, W.J.; Loukin, S.H.; Kung, C.; Saimi, Y. Prokaryotic K+ channels: From crystal structures to diversity. FEMS Microbiol. Rev. 2005, 29, 961–985. [Google Scholar] [CrossRef] [Green Version]
- Eisenhut, M.; Wallace, H. Ion channels in inflammation. Pflügers Arch. Eur. J. Physiol. 2011, 461, 401–421. [Google Scholar] [CrossRef]
- Xie, L.-H.; John, S.A.; Ribalet, B.; Weiss, J.N. Activation of inwardly rectifying potassium (Kir) channels by phosphatidylinosital-4,5-bisphosphate (PIP2): Interaction with other regulatory ligands. Prog. Biophys. Mol. Biol. 2007, 94, 320–335. [Google Scholar] [CrossRef]
- Krawczyk, C.M.; Holowka, T.; Sun, J.; Blagih, J.; Amiel, E.; DeBerardinis, R.J.; Cross, J.R.; Jung, E.; Thompson, C.B.; Jones, R.G.; et al. Toll-like receptor–induced changes in glycolytic metabolism regulate dendritic cell activation. Blood 2010, 115, 4742–4749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajamäki, K.; Nordström, T.; Nurmi, K.; Åkerman, K.E.O.; Kovanen, P.T.; Öörni, K.; Eklund, K.K. Extracellular Acidosis Is a Novel Danger Signal Alerting Innate Immunity via the NLRP3 Inflammasome. J. Biol. Chem. 2013, 288, 13410–13419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roiniotis, J.; Dinh, H.; Masendycz, P.; Turner, A.; Elsegood, C.L.; Scholz, G.M.; Hamilton, J.A. Hypoxia Prolongs Monocyte/Macrophage Survival and Enhanced Glycolysis Is Associated with Their Maturation under Aerobic Conditions. J. Immunol. 2009, 182, 7974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, A.C.Y.; Ryan, A.F. Mechanisms of sensorineural cell damage, death and survival in the cochlea. Front. Aging Neurosci. 2015, 7, 58. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Garagnani, P.; Parini, P.; Giuliani, C.; Santoro, A. Inflammaging: A new immune–metabolic viewpoint for age-related diseases. Nat. Rev. Endocrinol. 2018, 14, 576–590. [Google Scholar] [CrossRef] [PubMed]
- Hirayama, D.; Iida, T.; Nakase, H. The Phagocytic Function of Macrophage-Enforcing Innate Immunity and Tissue Homeostasis. Int. J. Mol. Sci. 2017, 19, 92. [Google Scholar] [CrossRef] [Green Version]
- Van Horssen, J.; van Schaik, P.; Witte, M. Inflammation and mitochondrial dysfunction: A vicious circle in neurodegenerative disorders? Neurosci. Lett. 2019, 710, 132931. [Google Scholar] [CrossRef]
- Kastl, L.; Sauer, S.W.; Ruppert, T.; Beissbarth, T.; Becker, M.S.; Süss, D.; Krammer, P.H.; Gülow, K. TNF-α mediates mitochondrial uncoupling and enhances ROS-dependent cell migration via NF-κB activation in liver cells. FEBS Lett. 2014, 588, 175–183. [Google Scholar] [CrossRef] [Green Version]
- Samavati, L.; Lee, I.; Mathes, I.; Lottspeich, F.; Hüttemann, M. Tumor Necrosis Factor α Inhibits Oxidative Phosphorylation through Tyrosine Phosphorylation at Subunit I of Cytochrome c Oxidase. J. Biol. Chem. 2008, 283, 21134–21144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lassmann, H.; van Horssen, J. Oxidative stress and its impact on neurons and glia in multiple sclerosis lesions. Biochim. Biophys. Acta Mol. Basis Dis. 2016, 1862, 506–510. [Google Scholar] [CrossRef]
- Bonora, M.; Pinton, P. Mitochondrial DNA keeps you young. Cell Death Dis. 2018, 9, 1–3. [Google Scholar] [CrossRef]
- Jang, J.Y.; Blum, A.; Liu, J.; Finkel, T. The role of mitochondria in aging. J. Clin. Investig. 2018, 128, 3662–3670. [Google Scholar] [CrossRef] [Green Version]
- Jara, C.; Torres, A.K.; Olesen, M.A.; Tapia-Rojas, C. Mitochondrial Dysfunction as a Key Event during Aging: From Synaptic Failure to Memory Loss. In Mitochondria and Brain Disorders; IntechOpen: London, UK, 2019. [Google Scholar]
- Salminen, A.; Kaarniranta, K.; Kauppinen, A. Inflammaging: Disturbed interplay between autophagy and inflammasomes. Aging 2012, 4, 166–175. [Google Scholar] [CrossRef] [Green Version]
- Shokolenko, I.N.; Wilson, G.L.; Alexeyev, M.F. Aging: A mitochondrial DNA perspective, critical analysis and an update. World J. Exp. Med. 2014, 4, 46–57. [Google Scholar] [CrossRef]
- Wu, L.; Sun, Y.; Hu, Y.-J.; Yang, Y.; Yao, L.-L.; Zhou, X.-X.; Wang, H.; Zhang, R.; Huang, X.; Kong, W.-J. Increased p66Shc in the inner ear of D-galactose-induced aging mice with accumulation of mitochondrial DNA 3873-bp deletion: p66Shc and mtDNA damage in the inner ear during aging. PLoS ONE 2012, 7, e50483. [Google Scholar] [CrossRef] [Green Version]
- Frisina, R.D. Age-related hearing loss: Ear and brain mechanisms. Ann. N. Y. Acad. Sci. 2009, 1170, 708–717. [Google Scholar] [CrossRef]
- Melgar-Rojas, P.; Alvarado, J.C.; Fuentes-Santamaría, V.; Juiz, J.M. Cellular Mechanisms of Age-Related Hearing Loss. In Free Radicals in ENT Pathology; Miller, J., Le Prell, C.G., Rybak, L., Eds.; Springer: Cham, Switzerland, 2015; pp. 305–333. [Google Scholar]
- Xiong, H.; Chen, S.; Lai, L.; Yang, H.; Xu, Y.; Pang, J.; Su, Z.; Lin, H.; Zheng, Y. Modulation of miR-34a/SIRT1 signaling protects cochlear hair cells against oxidative stress and delays age-related hearing loss through coordinated regulation of mitophagy and mitochondrial biogenesis. Neurobiol. Aging 2019, 79, 30–42. [Google Scholar] [CrossRef]
- Lyu, A.-R.; Kim, T.H.; Park, S.J.; Shin, S.-A.; Jeong, S.-H.; Yu, Y.; Huh, Y.H.; Je, A.R.; Park, M.J.; Park, Y.-H. Mitochondrial Damage and Necroptosis in Aging Cochlea. Int. J. Mol. Sci. 2020, 21, 2505. [Google Scholar] [CrossRef] [Green Version]
- Ribeiro, D.; Castro, I.; Fahimi, H.D.; Schrader, M. Peroxisome morphology in pathology. Histol. Histopatholol. 2012, 27, 661–676. [Google Scholar]
- Seo, Y.J.; Ju, H.M.; Lee, S.H.; Kwak, S.H.; Kang, M.J.; Yoon, J.-H.; Kim, C.-H.; Cho, H.-J. Damage of Inner Ear Sensory Hair Cells via Mitochondrial Loss in a Murine Model of Sleep Apnea with Chronic Intermittent Hypoxia. Sleep 2017, 40, zsx106. [Google Scholar] [CrossRef] [PubMed]
- Strickland, M.; Yacoubi-Loueslati, B.; Bouhaouala-Zahar, B.; Pender, S.L.F.; Larbi, A. Relationships Between Ion Channels, Mitochondrial Functions and Inflammation in Human Aging. Front. Physiol. 2019, 10, 158. [Google Scholar] [CrossRef] [Green Version]
- Qian, S.; Fan, J.; Billiar, T.R.; Scott, M.J. Inflammasome and Autophagy Regulation: A Two-way Street. Mol. Med. 2017, 23, 188–195. [Google Scholar]
- Bruunsgaard, H.; Andersen-Ranberg, K.; Jeune, B.; Pedersen, A.N.; Skinhøj, P.; Pedersen, B.K. A high plasma concentration of TNF-alpha is associated with dementia in centenarians. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 1999, 54, M357–M364. [Google Scholar] [CrossRef] [Green Version]
- Holmes, C.; Cunningham, C.; Zotova, E.; Woolford, J.; Dean, C.; Kerr, S.; Culliford, D.; Perry, V.H. Systemic inflammation and disease progression in Alzheimer disease. Neurology 2009, 73, 768–774. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, S.; Kawamoto, S.; Ohtani, N.; Hara, E. Impact of senescence-associated secretory phenotype and its potential as a therapeutic target for senescence-associated diseases. Cancer Sci. 2017, 108, 563–569. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Nagasu, H.; Murakami, T.; Hoang, H.; Broderick, L.; Hoffman, H.M.; Horng, T. Inflammasome activation leads to Caspase-1-dependent mitochondrial damage and block of mitophagy. Proc. Natl. Acad. Sci. USA 2014, 111, 15514–15519. [Google Scholar] [CrossRef] [Green Version]
- Nakanishi, H.; Kawashima, Y.; Kurima, K.; Chae, J.J.; Ross, A.M.; Pinto-Patarroyo, G.; Patel, S.K.; Muskett, J.A.; Ratay, J.S.; Chattaraj, P.; et al. NLRP3 mutation and cochlear autoinflammation cause syndromic and nonsyndromic hearing loss DFNA34 responsive to anakinra therapy. Proc. Natl. Acad. Sci. USA 2017, 114, E7766. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, H.; Kawashima, Y.; Kurima, K.; Muskett, J.A.; Kim, H.J.; Brewer, C.C.; Griffith, A.J. Gradual Symmetric Progression of DFNA34 Hearing Loss Caused by an NLRP3 Mutation and Cochlear Autoinflammation. Otol. Neurotol. 2018, 39, e181–e185. [Google Scholar] [CrossRef]
- Philips Jennifer, A. NLRP3, keep it down so we can hear. Sci. Transl. Med. 2017, 9, eaao6126. [Google Scholar] [CrossRef]
- Rao, V.; Kaja, S.; Gentile, S. Ion Channels in Aging and Aging-Related Diseases. In Molecular Mechanisms of the Aging Process and Rejuvenation; IntechOpen: London, UK, 2016. [Google Scholar]
- Jahangir, A.; Ozcan, C.; Holmuhamedov, E.L.; Terzic, A. Increased calcium vulnerability of senescent cardiac mitochondria: Protective role for a mitochondrial potassium channel opener. Mech. Ageing Dev. 2001, 122, 1073–1086. [Google Scholar] [CrossRef]
- Connors, L.H.; Sam, F.; Skinner, M.; Salinaro, F.; Sun, F.; Ruberg, F.L.; Berk, J.L.; Seldin, D.C. Heart Failure Due to Age-Related Cardiac Amyloid Disease Associated with Wild-Type Transthyretin: A Prospective, Observational Cohort Study. Circulation 2016, 133, 282–290. [Google Scholar] [CrossRef] [Green Version]
- O’Rourke, B. Mitochondrial Ion Channels. Annu. Rev. Physiol. 2007, 69, 19–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bazard, P.; Frisina, R.D.; Walton, J.P.; Bhethanabotla, V.R. Nanoparticle-based Plasmonic Transduction for Modulation of Electrically Excitable Cells. Sci. Rep. 2017, 7, 7803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Damnjanovic, R.; Bazard, P.; Frisina, R.D.; Bhethanabotla, V.R. Hybrid Electro-Plasmonic Neural Stimulation with Visible-Light-Sensitive Gold Nanoparticles. ACS Nano 2020, 14, 10917–10928. [Google Scholar] [CrossRef] [PubMed]
- Dillon, H. Hearing Aids; Hodder Arnold: London, UK, 2008. [Google Scholar]
- Huber, M.; Burger, T.; Illg, A.; Kunze, S.; Giourgas, A.; Braun, L.; Kröger, S.; Nickisch, A.; Rasp, G.; Becker, A. Mental health problems in adolescents with cochlear implants: Peer problems persist after controlling for additional handicaps. Front. Psychol. 2015, 6, 953. [Google Scholar] [CrossRef] [Green Version]
- Plath, P. Problems in fitting hearing aids in the elderly. Acta Oto-Laryngol. 1991, 111, 278–280. [Google Scholar] [CrossRef] [PubMed]
- Schorr, E.A.; Roth, F.P.; Fox, N.A. Quality of Life for Children with Cochlear Implants: Perceived Benefits and Problems and the Perception of Single Words and Emotional Sounds. J. Speech Lang. Hear. Res. 2009, 52, 141–152. [Google Scholar] [CrossRef] [Green Version]
- Haake, S.M.; Dinh, C.T.; Chen, S.; Eshraghi, A.A.; Van De Water, T.R. Dexamethasone protects auditory hair cells against TNFα-initiated apoptosis via activation of PI3K/Akt and NFκB signaling. Hear. Res. 2009, 255, 22–32. [Google Scholar] [CrossRef]
- Rath, P.C.; Aggarwal, B.B. TNF-Induced Signaling in Apoptosis. J. Clin. Immunol. 1999, 19, 350–364. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.-W.; Luo, Y.; Wu, Z.; Tao, Z.; Jones, R.O.; Zhao, H.-B. Paradoxical Enhancement of Active Cochlear Mechanics in Long-Term Administration of Salicylate. J. Neurophysiol. 2005, 93, 2053–2061. [Google Scholar] [CrossRef]
- Yang, K.; Huang, Z.-W.; Liu, Z.-Q.; Xiao, B.-K.; Peng, J.-H. Long-term administration of salicylate enhances prestin expression in rat cochlea. Int. J. Audiol. 2009, 48, 18–23. [Google Scholar] [CrossRef] [PubMed]
- Yu, N.; Zhu, M.L.; Johnson, B.; Liu, Y.P.; Jones, R.O.; Zhao, H.B. Prestin up-regulation in chronic salicylate (aspirin) administration: An implication of functional dependence of prestin expression. Cell. Mol. Life Sci. 2008, 65, 2407–2418. [Google Scholar] [CrossRef] [Green Version]
- Lowthian, J.A.; Britt, C.J.; Rance, G.; Lin, F.R.; Woods, R.L.; Wolfe, R.; Nelson, M.R.; Dillon, H.A.; Ward, S.; Reid, C.M.; et al. Slowing the progression of age-related hearing loss: Rationale and study design of the ASPIRIN in HEARING, retinal vessels imaging and neurocognition in older generations (ASPREE-HEARING) trial. Contemp. Clin. Trials 2016, 46, 60–66. [Google Scholar] [CrossRef]
- Coleman, J.; Huang, X.; Liu, J.; Kopke, R.; Jackson, R. Dosing study on the effectiveness of salicylate/N-acetylcysteine for prevention of noise-induced hearing loss. Noise Health 2010, 12, 159–165. [Google Scholar]
- Gao, X.-r.; Adhikari, C.M.; Peng, L.-y.; Guo, X.-g.; Zhai, Y.-s.; He, X.-y.; Zhang, L.-Y.; Lin, J.; Zuo, Z.-y. Efficacy of different doses of aspirin in decreasing blood levels of inflammatory markers in patients with cardiovascular metabolic syndrome. J. Pharm. Pharmacol. 2010, 61, 1505–1510. [Google Scholar] [CrossRef]
- Yamashita, D.; Jiang, H.Y.; Le Prell, C.G.; Schacht, J.; Miller, J.M. Post-exposure treatment attenuates noise-induced hearing loss. Neuroscience 2005, 134, 633–642. [Google Scholar] [CrossRef]
- Kalinec, G.M.; Lomberk, G.; Urrutia, R.A.; Kalinec, F. Resolution of Cochlear Inflammation: Novel Target for Preventing or Ameliorating Drug-, Noise- and Age-related Hearing Loss. Front. Cell. Neurosci. 2017, 11, 192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, T.; Stables, M.; Hobbs, A.; de Souza, P.; Colville-Nash, P.; Warner, T.; Newson, J.; Bellingan, G.; Gilroy, D.W. Effects of Low-Dose Aspirin on Acute Inflammatory Responses in Humans. J. Immunol. 2009, 183, 2089. [Google Scholar] [CrossRef]
- Ferrucci, L.; Guralnik, J.M. Inflammation, hormones, and body composition at a crossroad. Am. J. Med. 2003, 115, 501–502. [Google Scholar] [CrossRef]
- Mancini, A.; Di Segni, C.; Raimondo, S.; Olivieri, G.; Silvestrini, A.; Meucci, E.; Currò, D. Thyroid hormones, oxidative stress, and inflammation. Mediat. Inflamm. 2016, 2016, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Forrest, D.; Erway, L.C.; Ng, L.; Altschuler, R.; Curran, T. Thyroid hormone receptor β is essential for development of auditory function. Nat. Genet. 1996, 13, 354. [Google Scholar] [CrossRef]
- Ng, L.; Cordas, E.; Wu, X.; Vella, K.R.; Hollenberg, A.N.; Forrest, D. Age-related hearing loss and degeneration of cochlear hair cells in mice lacking thyroid hormone receptor β1. Endocrinology 2015, 156, 3853–3865. [Google Scholar] [CrossRef]
- Sharlin, D.S.; Ng, L.; Verrey, F.; Visser, T.J.; Liu, Y.; Olszewski, R.T.; Hoa, M.; Heuer, H.; Forrest, D. Deafness and loss of cochlear hair cells in the absence of thyroid hormone transporters Slc16a2 (Mct8) and Slc16a10 (Mct10). Sci. Rep. 2018, 8, 4403. [Google Scholar] [CrossRef]
- Song, L.; McGee, J.; Walsh, E.J. The influence of thyroid hormone deficiency on the development of cochlear nonlinearities. J. Assoc. Res. Otolaryngol. 2008, 9, 464–476. [Google Scholar] [CrossRef] [Green Version]
- Hussein, M.M.; Asal, S.I.; Salem, T.M.; Mohammed, A.M. The effect of L-thyroxine hormone therapy on hearing loss in hypothyroid patients. Egypt. J. Otolaryngol. 2017, 33, 637. [Google Scholar] [CrossRef]
- Iwai, K.; Nakagawa, T.; Endo, T.; Matsuoka, Y.; Kita, T.; Kim, T.S.; Tabata, Y.; Ito, J. Cochlear Protection by Local Insulin-Like Growth Factor-1 Application Using Biodegradable Hydrogel. Laryngoscope 2006, 116, 529–533. [Google Scholar] [CrossRef] [PubMed]
- Muus, J.S.; Weir, F.W.; Kreicher, K.L.; Bowlby, D.A.; Discolo, C.M.; Meyer, T.A. Hearing loss in children with growth hormone deficiency. Int. J. Pediatr. Otorhinolaryngol. 2017, 100, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Yamahara, K.; Yamamoto, N.; Nakagawa, T.; Ito, J. Insulin-like growth factor 1: A novel treatment for the protection or regeneration of cochlear hair cells. Hear. Res. 2015, 330, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, S.; Sugahara, K.; Hashimoto, M.; Hirose, Y.; Shimogori, H.; Yamashita, H. The minimum peptides of IGF-1 and substance P protect vestibular hair cells against neomycin ototoxicity. Acta Oto-Laryngol. 2015, 135, 411–415. [Google Scholar] [CrossRef]
- Attias, J.; Zarchi, O.; Nageris, B.I.; Laron, Z. Cochlear hearing loss in patients with Laron syndrome. Eur. Arch. Oto-Rhino-Laryngol. 2012, 269, 461–466. [Google Scholar] [CrossRef]
- Ding, B.; Frisina, R.D.; Zhu, X.; Sakai, Y.; Sokolowski, B.; Walton, J.P. Direct control of Na+-K+-2Cl−-cotransport protein (NKCC1) expression with aldosterone. Am. J. Physiol. Cell Physiol. 2014, 306, C66–C75. [Google Scholar] [CrossRef] [Green Version]
- Bazard, P.; Ding, B.; Chittam, H.K.; Zhu, X.; Parks, T.A.; Taylor-Clark, T.E.; Bhethanabotla, V.R.; Frisina, R.D.; Walton, J.P. Aldosterone up-regulates voltage-gated potassium currents and NKCC1 protein membrane fractions. Sci. Rep. 2020, 10, 15604. [Google Scholar] [CrossRef]
- Frisina, R.D.; Bazard, P.; Bauer, M.; Pineros, J.; Zhu, X.; Ding, B. Translational implications of the interactions between hormones and age-related hearing loss. Hear. Res. 2021, 402, 108093. [Google Scholar] [CrossRef]
- Williamson, T.T.; Ding, B.; Zhu, X.; Frisina, R.D. Hormone replacement therapy attenuates hearing loss: Mechanisms involving estrogen and the IGF-1 pathway. Aging Cell 2019, 18, e12939. [Google Scholar] [CrossRef] [PubMed]
- Guimaraes, P.; Frisina, S.T.; Mapes, F.; Tadros, S.F.; Frisina, D.R.; Frisina, R.D. Progestin negatively affects hearing in aged women. Proc. Natl. Acad. Sci. USA 2006, 103, 14246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Böttger, E.C.; Schacht, J. The mitochondrion: A perpetrator of acquired hearing loss. Hear. Res. 2013, 303, 12–19. [Google Scholar] [CrossRef] [Green Version]
- Lenaz, G.; Baracca, A.; Fato, R.; Genova, M.L.; Solaini, G. New Insights into Structure and Function of Mitochondria and Their Role in Aging and Disease. Antioxid. Redox Signal. 2006, 8, 417–437. [Google Scholar] [CrossRef] [PubMed]
- Seidman, M.D.; Khan, M.J.; Tang, W.X.; Quirk, W.S. Influence of lecithin on mitochondrial DNA and age-related hearing loss. Otolaryngol. Head Neck Surg. 2002, 127, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Le, T.; Keithley, E.M. Effects of antioxidants on the aging inner ear. Hear. Res. 2007, 226, 194–202. [Google Scholar] [CrossRef] [PubMed]
- Someya, S.; Xu, J.; Kondo, K.; Ding, D.; Salvi, R.J.; Yamasoba, T.; Rabinovitch, P.S.; Weindruch, R.; Leeuwenburgh, C.; Tanokura, M. Age-related hearing loss in C57BL/6J mice is mediated by Bak-dependent mitochondrial apoptosis. Proc. Natl. Acad. Sci. USA 2009, 106, 19432–19437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seidman, M.D. Effects of dietary restriction and antioxidants on presbyacusis. Laryngoscope 2000, 110, 727–738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takumida, M.; Anniko, M. Radical scavengers: A remedy for presbyacusis. A pilot study. Acta Oto-Laryngol. 2005, 125, 1290–1295. [Google Scholar] [CrossRef] [PubMed]
- Takumida, M.; Anniko, M. Radical scavengers for elderly patients with age-related hearing loss. Acta Oto-Laryngol. 2009, 129, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Polanski, J.; Cruz, O. Evaluation of antioxidant treatment in presbyacusis: Prospective, placebo-controlled, double-blind, randomised trial. J. Laryngol. Otol. 2013, 127, 134–141. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bazard, P.; Pineros, J.; Frisina, R.D.; Bauer, M.A.; Acosta, A.A.; Paganella, L.R.; Borakiewicz, D.; Thivierge, M.; Mannering, F.L.; Zhu, X.; et al. Cochlear Inflammaging in Relation to Ion Channels and Mitochondrial Functions. Cells 2021, 10, 2761. https://doi.org/10.3390/cells10102761
Bazard P, Pineros J, Frisina RD, Bauer MA, Acosta AA, Paganella LR, Borakiewicz D, Thivierge M, Mannering FL, Zhu X, et al. Cochlear Inflammaging in Relation to Ion Channels and Mitochondrial Functions. Cells. 2021; 10(10):2761. https://doi.org/10.3390/cells10102761
Chicago/Turabian StyleBazard, Parveen, Jennifer Pineros, Robert D. Frisina, Mark A. Bauer, Alejandro A. Acosta, Lauren R. Paganella, Dominika Borakiewicz, Mark Thivierge, Freyda L. Mannering, Xiaoxia Zhu, and et al. 2021. "Cochlear Inflammaging in Relation to Ion Channels and Mitochondrial Functions" Cells 10, no. 10: 2761. https://doi.org/10.3390/cells10102761