
Epigenetic DNA Modifications Upregulate SPRY2 in Human Colorectal Cancers

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Human Tissue Collection
2.2. Isolation of Coloncytes from Colonic Mucosa
2.3. Cell Culture
2.4. Western Blotting
2.5. Combined Bisulfite Restriction Analysis
- Region #1: Promoter: putative CTCF binding site [2 CpGs] HpyCH4IV
- Forward primer: (5′→3′)TTAAAGTTTGGGTGTGTAAGGTTAG;
- Reverse primer: (5′→3′)AAAACCAACAAAACACAAATATCTC;
- Region #2: Promoter: upstream of the TSS [1 CpG] HpyCH4IV
- Forward primer: (5′→3′)TTAGAGATGGTAAGGGAAGTGGTAT;
- Reverse primer: (5′→3′)AACCAAAAAAACCCAACTATTTTAC;
- Region #3: Promoter: closest to the TSS [3 CpGs] BstUI
- Forward primer: (5′→3′)GTAGGAGGTATTTAAAAGGGTAAT;
- Reverse primer: (5′→3′)AAAAAAAATTCCCTAAATCAACTC;
- Region #4: Intragenic CpG island: putative CTCF binding site [4 CpGs] HpyCH4IV
- Forward primer: (5′→3′)TTGTTAGGTTTTTTGTAAAGTTTTT;
- Reverse primer: (5′→3′)CAAATTACAAACAATACCCCCTTC;
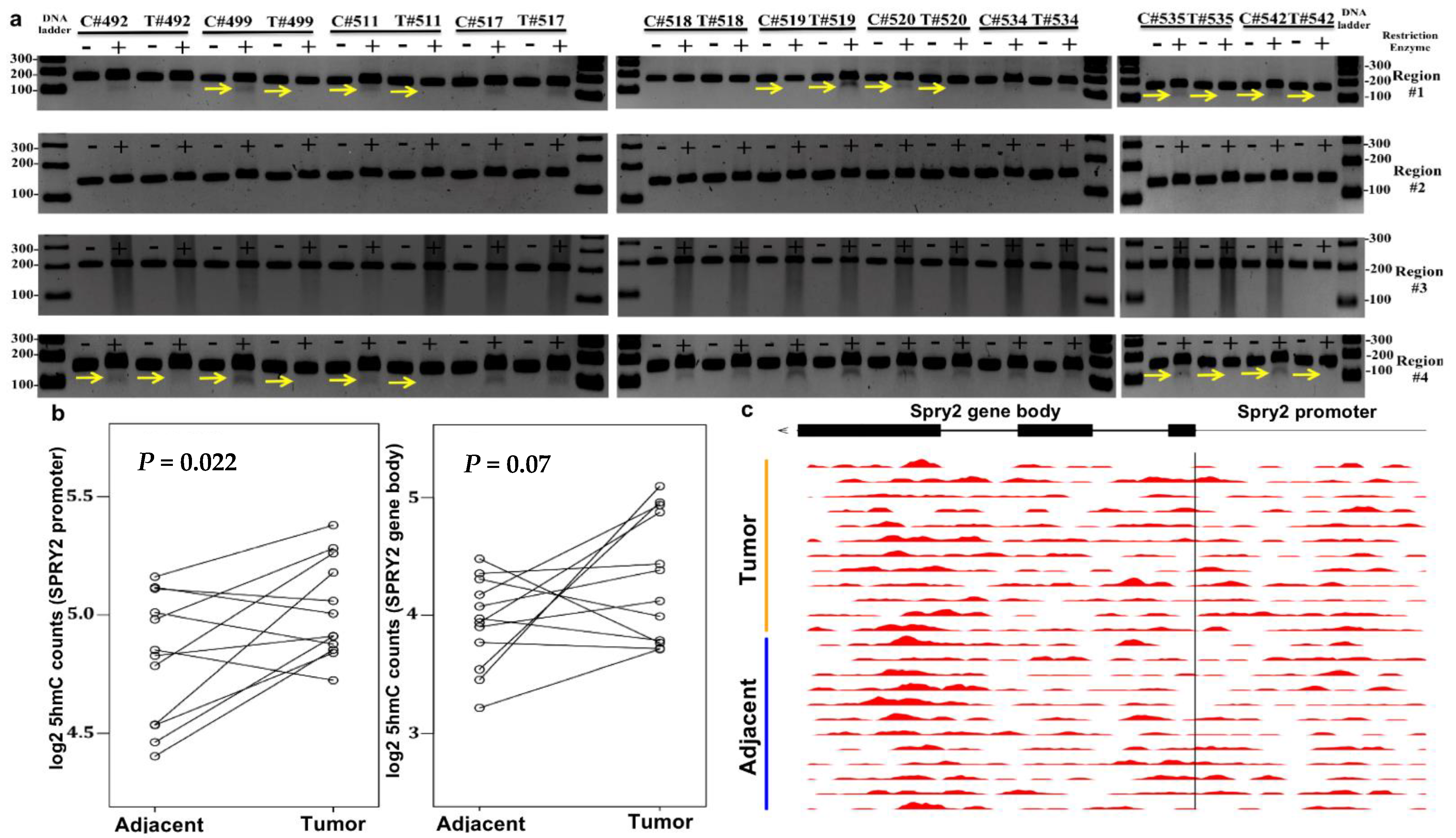
2.6. 5hmC-Modified Locations in the Gene Body
2.7. Bioinformatics Analysis
3. Results
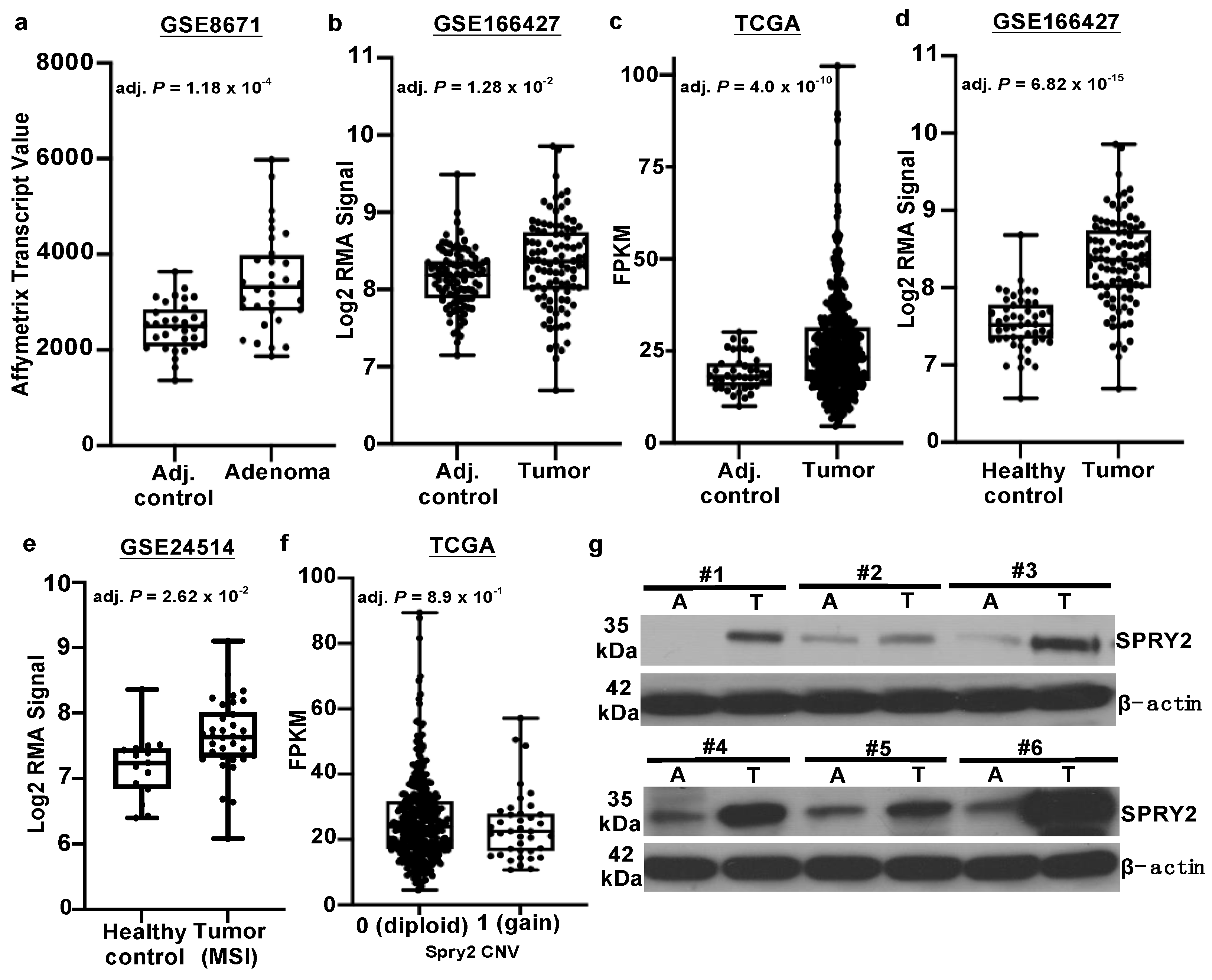
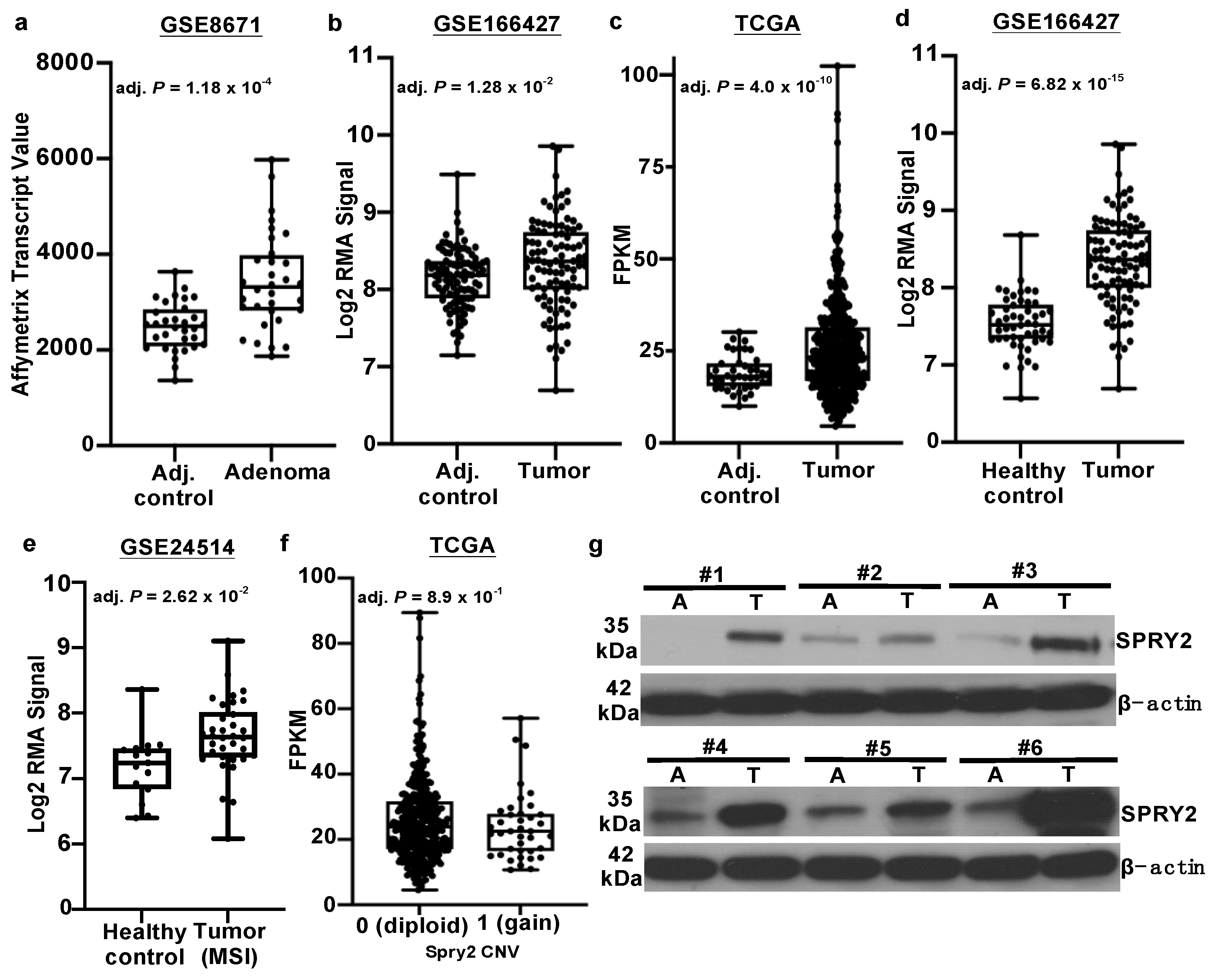
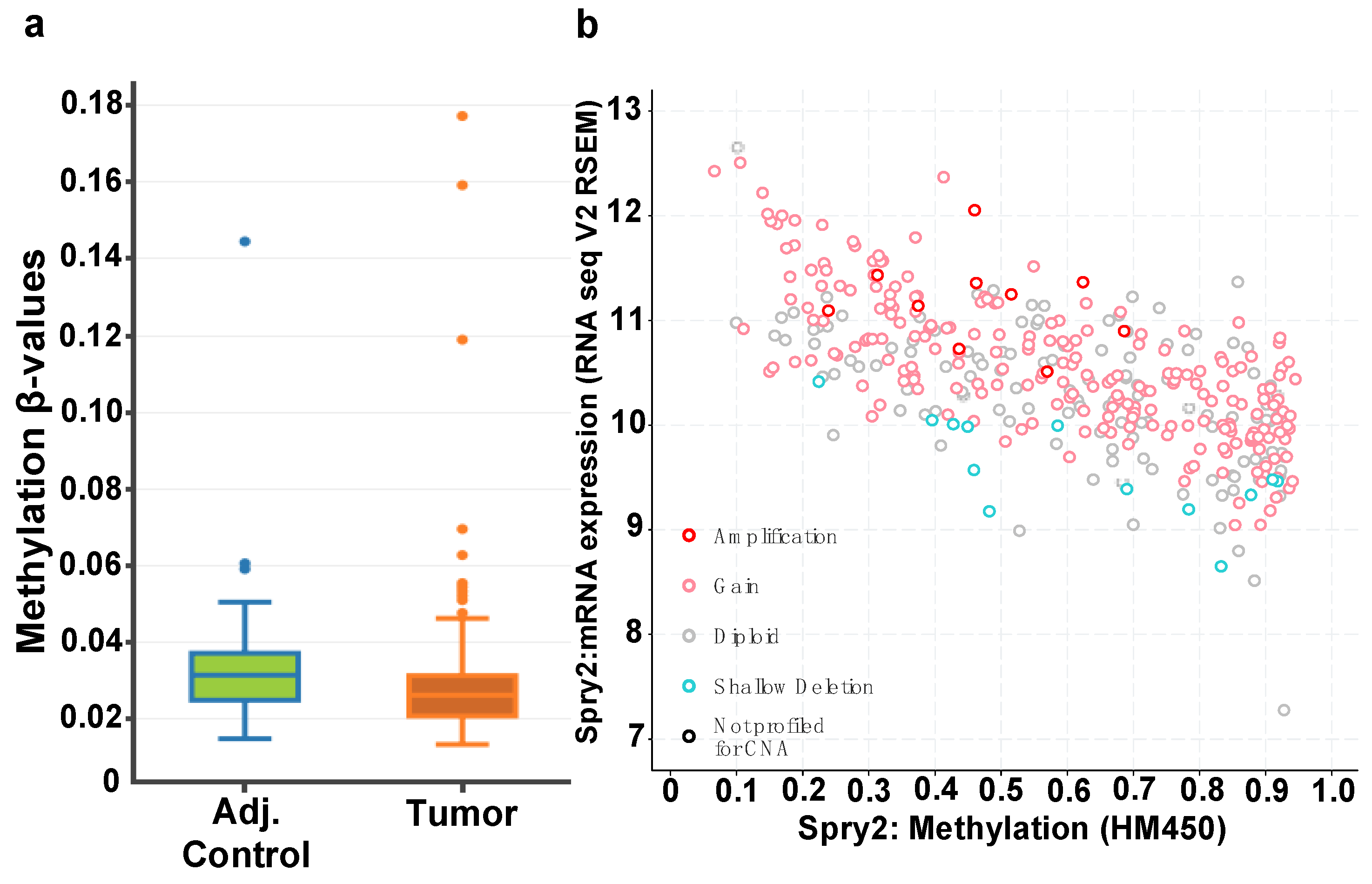
3.1. SPRY2 Is Upregulated in Adenomas and Colorectal Cancers
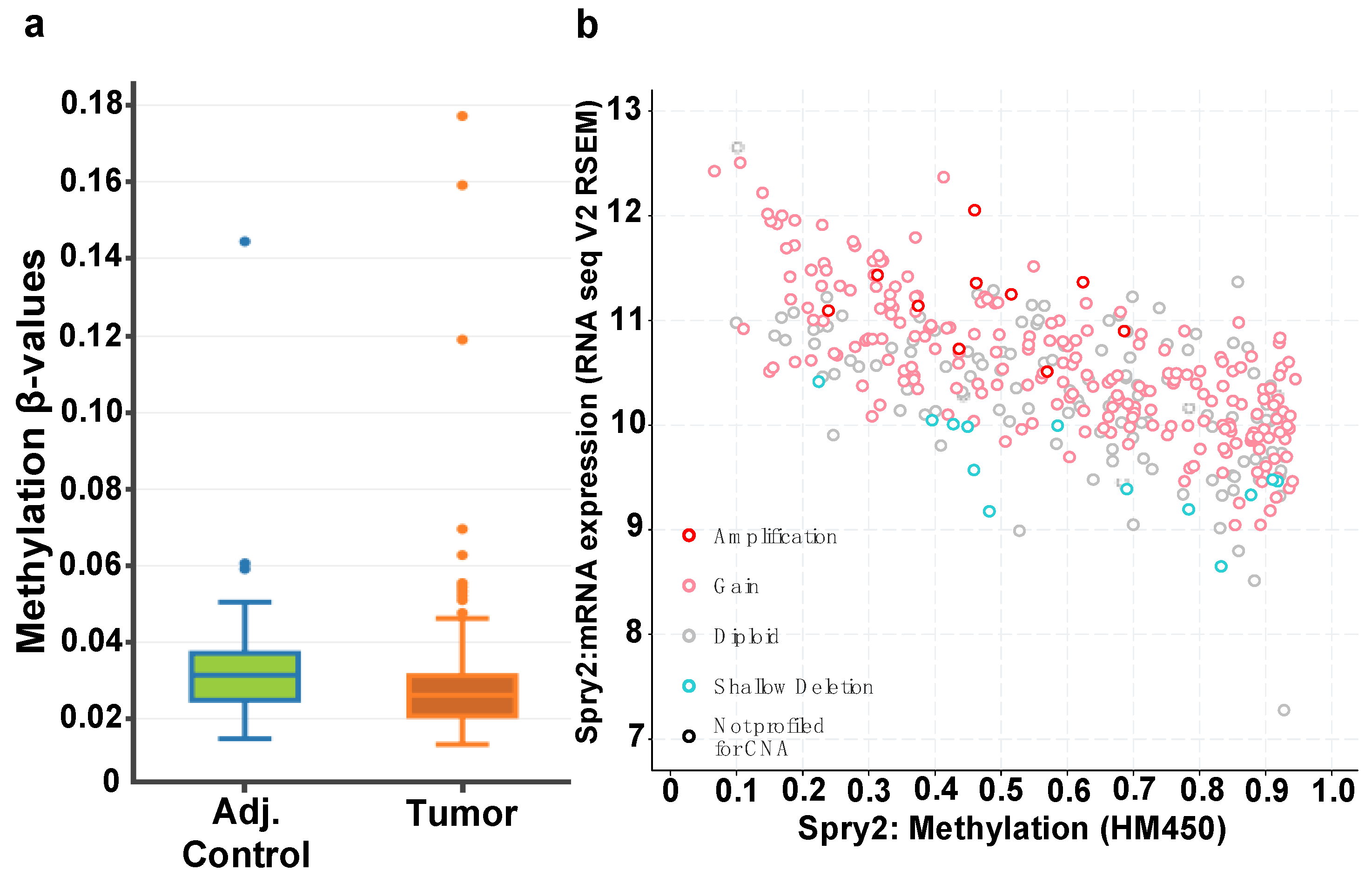
3.2. CTCF Binding Sites within Transcriptional Regulatory Regions of SPRY2 Are Differentially Methylated in CRC
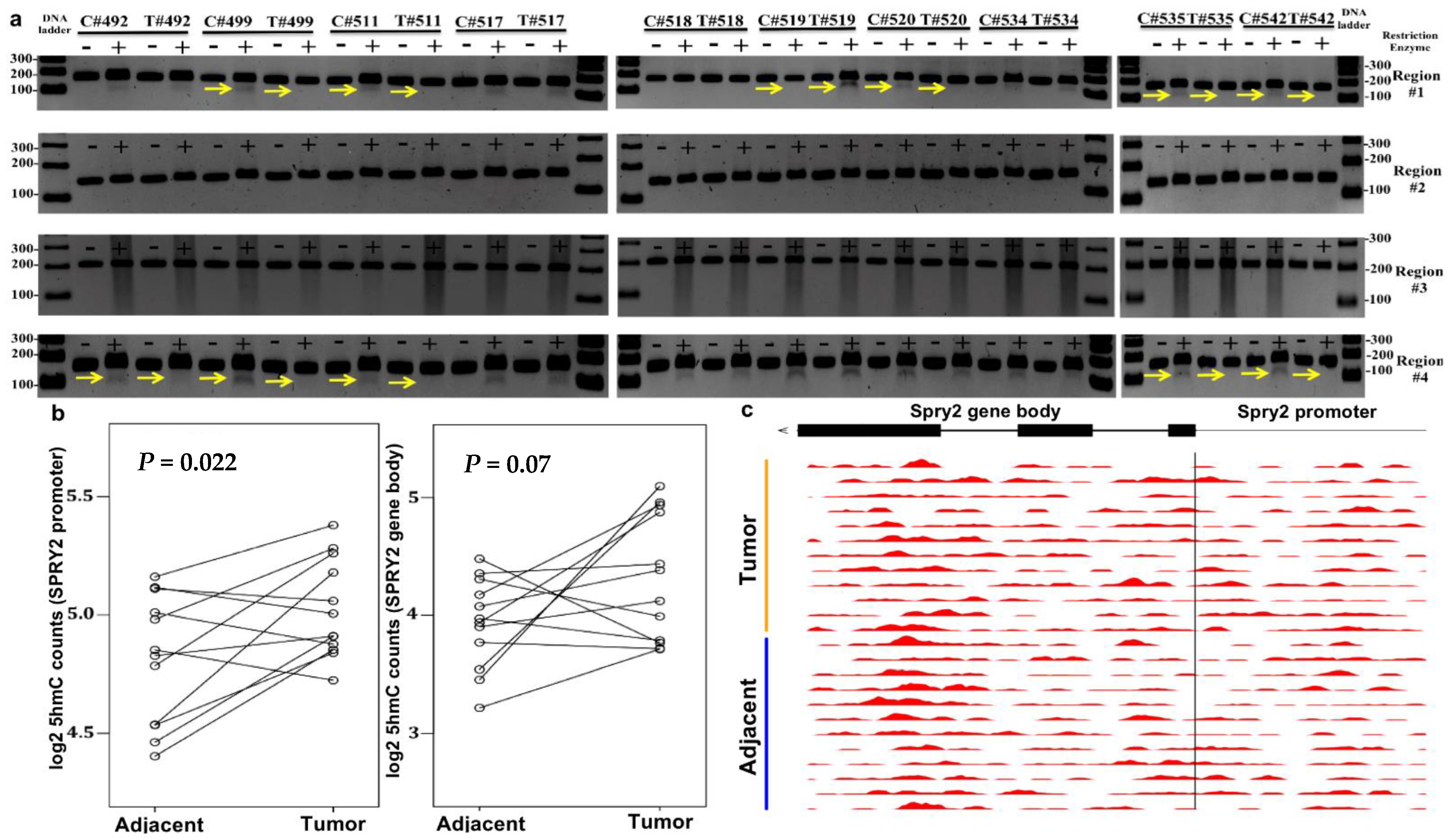
3.3. Increased SPRY2 5hmC Deposition Occurs in CRC
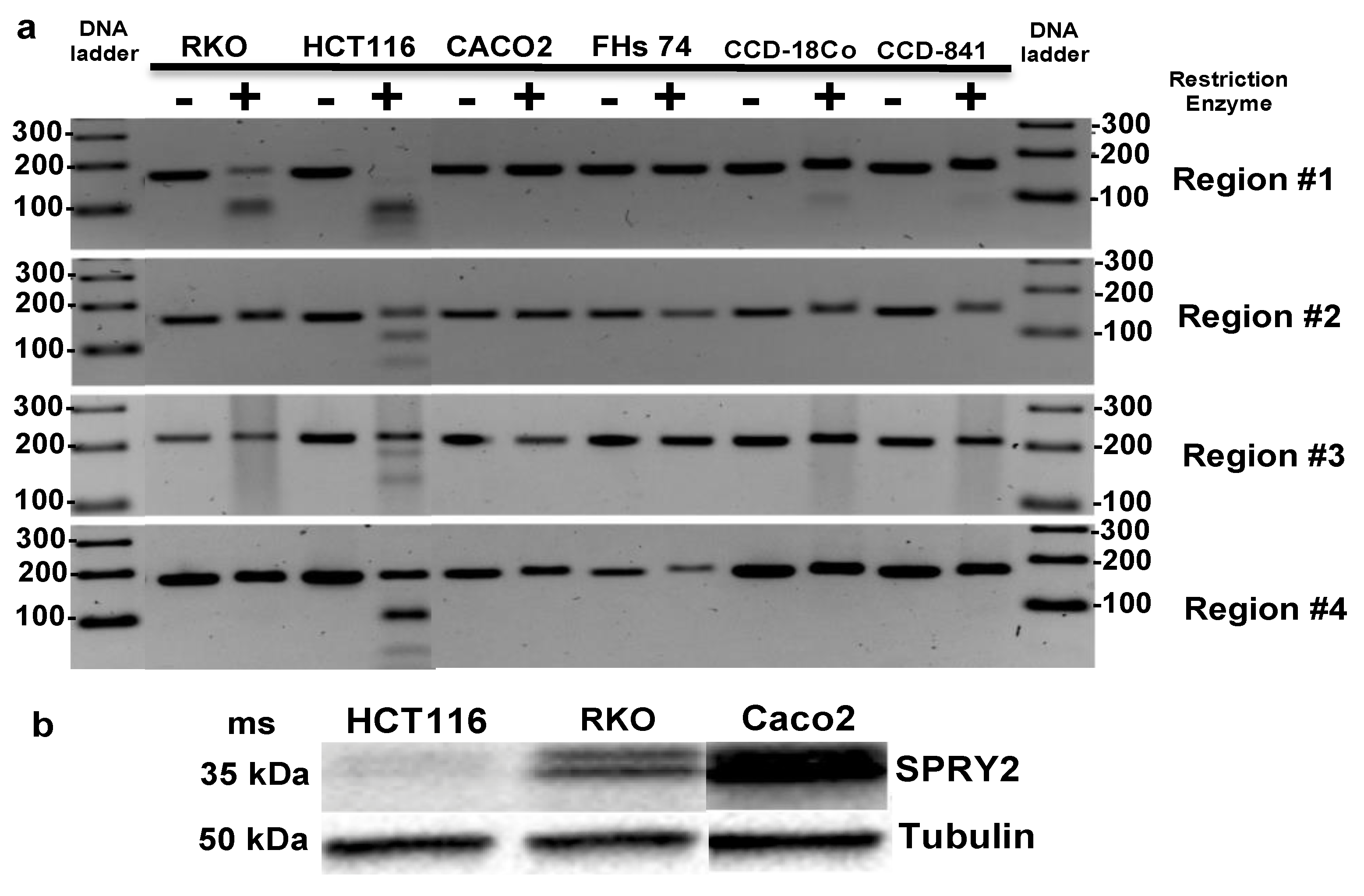
3.4. SPRY2 Is Differentially Methylated in CRC Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hacohen, N.; Kramer, S.; Sutherland, D.; Hiromi, Y.; Krasnow, M.A. Sprouty Encodes a Novel Antagonist of FGF Signaling that Patterns Apical Branching of the Drosophila Airways. Cell 1998, 92, 253–263. [Google Scholar] [CrossRef] [Green Version]
- Minowada, G.; Jarvis, L.A.; Chi, C.L.; Neubuser, A.; Sun, X.; Hacohen, N.; Krasnow, M.A.; Martin, G.R. Vertebrate Sprouty genes are induced by FGF signaling and can cause chondrodysplasia when overexpressed. Development 1999, 126, 4465. [Google Scholar] [CrossRef] [PubMed]
- Impagnatiello, M.-A.; Weitzer, S.; Gannon, G.; Compagni, A.; Cotten, M.; Christofori, G. Mammalian Sprouty-1 and -2 Are Membrane-Anchored Phosphoprotein Inhibitors of Growth Factor Signaling in Endothelial Cells. J. Cell Biol. 2001, 152, 1087–1098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gross, I.; Bassit, B.; Benezra, M.; Licht, J.D. Mammalian Sprouty Proteins Inhibit Cell Growth and Differentiation by Preventing Ras Activation. J. Biol. Chem. 2001, 276, 46460–46468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, J.; Yusoff, P.; Wong, E.S.M.; Chandramouli, S.; Lao, D.-H.; Fong, C.W.; Guy, G.R. The Cysteine-Rich Sprouty Translocation Domain Targets Mitogen-Activated Protein Kinase Inhibitory Proteins to Phosphatidylinositol 4,5-Bisphosphate in Plasma Membranes. Mol. Cell. Biol. 2002, 22, 7953–7966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubin, C.; Litvak, V.; Medvedovsky, H.; Zwang, Y.; Lev, S.; Yarden, Y. Sprouty Fine-Tunes EGF Signaling through Interlinked Positive and Negative Feedback Loops. Curr. Biol. 2003, 13, 297–307. [Google Scholar] [CrossRef] [Green Version]
- Shukla, A.; Rai, K.; Shukla, V.; Chaturvedi, N.K.; Bociek, R.G.; Pirruccello, S.J.; Band, H.; Lu, R.; Joshi, S.S. Sprouty 2: A novel attenuator of B-cell receptor and MAPK-Erk signaling in CLL. Blood 2016, 127, 2310–2321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sutterlüty, H.; Mayer, C.-E.; Setinek, U.; Attems, J.; Ovtcharov, S.; Mikula, M.; Mikulits, W.; Micksche, M.; Berger, W. Down-Regulation of Sprouty2 in Non–Small Cell Lung Cancer Contributes to Tumor Malignancy via Extracellular Signal-Regulated Kinase Pathway-Dependent and -Independent Mechanisms. Mol. Cancer Res. 2007, 5, 509–520. [Google Scholar] [CrossRef] [Green Version]
- Fong, C.W.; Chua, M.S.; McKie, A.B.; Ling, S.H.M.; Mason, V.; Li, R.; Yusoff, P.; Lo, T.L.; Leung, H.Y.; So, S.K.S.; et al. Sprouty 2, an Inhibitor of Mitogen-Activated Protein Kinase Signaling, Is Down-Regulated in Hepatocellular Carcinoma. Cancer Res. 2006, 66, 2048–2058. [Google Scholar] [CrossRef] [Green Version]
- Lo, T.L.; Yusoff, P.; Fong, C.W.; Guo, K.; McCaw, B.J.; Phillips, W.; Yang, H.; Wong, E.S.M.; Leong, H.F.; Zeng, Q.; et al. The Ras/Mitogen-Activated Protein Kinase Pathway Inhibitor and Likely Tumor Suppressor Proteins, Sprouty 1 and Sprouty 2 Are Deregulated in Breast Cancer. Cancer Res. 2004, 64, 6127–6136. [Google Scholar] [CrossRef] [Green Version]
- Fritzsche, S.; Kenzelmann, M.; Hoffmann, M.J.; Müller, M.; Engers, R.; Gröne, H.-J.; Schulz, W. Concomitant down-regulation of SPRY1 and SPRY2 in prostate carcinoma. Endocrine-Related Cancer 2006, 13, 839–849. [Google Scholar] [CrossRef] [Green Version]
- Park, J.-W.; Wollmann, G.; Urbiola, C.; Fogli, B.; Florio, T.; Geley, S.; Klimaschewski, L. Sprouty2 enhances the tumorigenic potential of glioblastoma cells. Neuro-Oncology 2018, 20, 1044–1054. [Google Scholar] [CrossRef] [Green Version]
- Holgren, C.; Dougherty, U.; Edwin, F.; Cerasi, D.; Taylor, I.; Fichera, A.; Joseph, L.; Bissonnette, M.; Khare, S. Sprouty-2 controls c-Met expression and metastatic potential of colon cancer cells: Sprouty/c-Met upregulation in human colonic adenocarcinomas. Oncogene 2010, 29, 5241–5253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Dougherty, U.; Bissonnette, M.; Khare, S. Role of Sprouty Proteins in Inflammatory Bowel Disease. Gastroenterology 2017, 152, S1037. [Google Scholar] [CrossRef]
- Zhang, Q.; Wei, T.; Shim, K.; Wright, K.; Xu, K.; Palka-Hamblin, H.L.; Jurkevich, A.; Khare, S. Atypical role of sprouty in colorectal cancer: Sprouty repression inhibits epithelial–mesenchymal transition. Oncogene 2016, 35, 3151–3162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schumacher, M.A.; Hsieh, J.J.; Liu, C.Y.; Appel, K.L.; Waddell, A.; Almohazey, D.; Katada, K.; Bernard, J.K.; Bucar, E.B.; Gadeock, S.; et al. Sprouty2 limits intestinal tuft and goblet cell numbers through GSK3β-mediated restriction of epithelial IL-33. Nat. Commun. 2021, 12, 836. [Google Scholar] [CrossRef] [PubMed]
- Ordóñez-Morán, P.; Irmisch, A.; Barbáchano, A.; Chicote, I.; Tenbaum, S.; Landolfi, S.; Tabernero, J.; Huelsken, J.; Munoz, A.; Palmer, H. SPROUTY2 is a β-catenin and FOXO3a target gene indicative of poor prognosis in colon cancer. Oncogene 2013, 33, 1975–1985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, Y.H.; Wu, C.L.; Tsao, C.J.; Chang, J.G.; Lu, P.J.; Yeh, K.T.; Uen, Y.-H.; Lee, J.-C.; Shiau, A.L. Deregulated expression of sprouty2 and microRNA-21 in human colon cancer: Correlation with the clinical stage of the disease. Cancer Biol. Ther. 2011, 11, 111–121. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.A.; Ladu, S.; Evert, M.; Dombrowski, F.; De Murtas, V.; Chen, X.; Calvisi, D.F. Synergistic role of sprouty2 inactivation and c-Met up-regulation in mouse and human hepatocarcinogenesis. Hepatology 2010, 52, 506–517. [Google Scholar] [CrossRef]
- Frank, M.J.; Dawson, D.W.; Bensinger, S.J.; Hong, J.S.; Knosp, W.M.; Xu, L.; Balatoni, C.E.; Allen, E.L.; Shen, R.R.; Bar-Sagi, D.; et al. Expression of sprouty2 inhibits B-cell proliferation and is epigenetically silenced in mouse and human B-cell lymphomas. Blood 2009, 113, 2478–2487. [Google Scholar] [CrossRef] [Green Version]
- Velasco, A.; Pallares, J.; Santacana, M.; Gatius, S.; Fernandez, M.; Domingo, M.; Valls, J.; Yeramian, A.; Encinas, M.; Dolcet, X.; et al. Promoter hypermethylation and expression of sprouty 2 in endometrial carcinoma. Hum. Pathol. 2010, 42, 185–193. [Google Scholar] [CrossRef]
- McKie, A.B.; Douglas, D.A.; Olijslagers, S.; Graham, J.; Omar, M.M.; Heer, R.; Gnanapragasam, V.J.; Robson, C.N.; Leung, H.Y. Epigenetic inactivation of the human sprouty2 (hSPRY2) homologue in prostate cancer. Oncogene 2005, 24, 2166–2174. [Google Scholar] [CrossRef]
- Rabaneda, L.G.; Geribaldi-Doldán, N.; Murillo-Carretero, M.; Carrasco, M.; Martínez-Salas, J.M.; Verástegui, C.; Castro, C. Altered regulation of the Spry2/Dyrk1A/PP2A triad by homocysteine impairs neural progenitor cell proliferation. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2016, 1863, 3015–3026. [Google Scholar] [CrossRef]
- Mustafi, R.; Dougherty, U.; Mustafi, D.; Ayaloglu-Butun, F.; Fletcher, M.; Adhikari, S.; Sadiq, F.; Meckel, K.; Haider, H.I.; Khalil, A.; et al. ADAM17 is a Tumor Promoter and Therapeutic Target in Western Diet–associated Colon Cancer. Clin. Cancer Res. 2016, 23, 549–561. [Google Scholar] [CrossRef] [Green Version]
- Seidelin, J.B.; Horn, T. Nielsen OH. Simple and efficient method for isolation and cultivation of endoscopically obtained human colonocytes. Am. J. Physiol.-Gastrointest. Liver Physiol. 2003, 285, G1122–G1128. [Google Scholar] [CrossRef] [Green Version]
- Khalil, H.; Nie, W.; Edwards, R.A.; Yoo, J. Isolation of Primary Myofibroblasts from Mouse and Human Colon Tissue. JoVE (J. Vis. Exp.) 2013, 80, e50611. [Google Scholar] [CrossRef] [Green Version]
- Khare, S.H.; Wilson, D.M.; Tien, X.Y.; Dudeja, P.K.; Wali, R.K.; Sitrin, M.D.; Brasitus, T.A. 1,25-Dihydroxycholecalciferol rapidly activates rat colonic particulate guanylate cyclase via a protein kinase C-dependent mechanism. Endocrinology 1993, 133, 2213–2219. [Google Scholar] [CrossRef] [PubMed]
- Li, L.-C.; Dahiya, R. MethPrimer: Designing primers for methylation PCRs. Bioinformatics 2002, 18, 1427–1431. [Google Scholar] [CrossRef] [Green Version]
- Xiong, Z.; Laird, P.W. COBRA: A sensitive and quantitative DNA methylation assay. Nucleic Acids Res. 1997, 25, 2532–2534. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Zhang, X.; Lu, X.; You, L.; Song, Y.; Luo, Z.; Zhang, J.; Nie, J.; Zheng, W.; Xu, D.; et al. 5-Hydroxymethylcytosine signatures in circulating cell-free DNA as diagnostic biomarkers for human cancers. Cell Res. 2017, 27, 1243–1257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomczak, K.; Czerwińska, P.; Wiznerowicz, M. The Cancer Genome Atlas (TCGA): An immeasurable source of knowledge. Contemp. Oncol. 2015, 19, A68–A77. [Google Scholar] [CrossRef]
- Sabates-Bellver, J.; Van der Flier, L.G.; de Palo, M.; Cattaneo, E.; Maake, C.; Rehrauer, H.; Laczko, E.; Kurowski, M.A.; Bujnicki, J.M.; Menigatti, M.; et al. Transcriptome profile of human colorectal adenomas. Mol. Cancer Res. 2007, 5, 1263–1275. [Google Scholar] [CrossRef] [Green Version]
- Seaborn: Statistical Data Visualization-Seaborn 0.9.0 Documentation, Seaborn.Pydata.org. 2018. Available online: http://seaborn.pydata.org/ (accessed on 15 April 2021).
- Rosenbloom, K.R.; Sloan, C.A.; Malladi, V.; Dreszer, T.R.; Learned, K.; Kirkup, V.M.; Wong, M.C.; Maddren, M.; Fang, R.; Heitner, S.G.; et al. ENCODE Data in the UCSC Genome Browser: Year 5 update. Nucleic Acids Res. 2012, 41, D56–D63. [Google Scholar] [CrossRef]
- Colaprico, A.; Silva, T.C.; Olsen, C.; Garofano, L.; Cava, C.; Garolini, D.; Sabedot, T.S.; Malta, T.M.; Pagnotta, S.M.; Castiglioni, I.; et al. TCGAbiolinks: An R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 2015, 44, e71. [Google Scholar] [CrossRef] [PubMed]
- Mermel, C.H.; Schumacher, S.E.; Hill, B.; Meyerson, M.L.; Beroukhim, R.; Getz, G. GISTIC2. 0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 2011, 12, R41. [Google Scholar] [CrossRef] [Green Version]
- Ding, W.; Chen, J.; Feng, G.; Chen, G.; Wu, J.; Guo, Y.; Ni, X.; Shi, T. DNMIVD: DNA methylation interactive visualization database. Nucleic Acids Res. 2019, 48, D856–D862. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, I.-H.; Chen, Y.-F.; Hsu, M.-T. Correlated 5-Hydroxymethylcytosine (5hmC) and Gene Expression Profiles Underpin Gene and Organ-Specific Epigenetic Regulation in Adult Mouse Brain and Liver. PLoS ONE 2017, 12, e0170779. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; D’Alessio, A.C.; Ito, S.; Wang, Z.; Cui, K.; Zhao, K.; Sun, Y.E.; Zhang, Y. Genome-wide analysis of 5-hydroxymethylcytosine distribution reveals its dual function in transcriptional regulation in mouse embryonic stem cells. Genes Dev. 2011, 25, 679–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madrid, A.; Chopra, P.; Alisch, R.S. Species-Specific 5 mC and 5 hmC Genomic Landscapes Indicate Epigenetic Contribution to Human Brain Evolution. Front. Mol. Neurosci. 2018, 11, 39. [Google Scholar] [CrossRef]
- Cai, J.; Chen, L.; Zhang, Z.; Zhang, X.; Lu, X.; Liu, W.; Shi, G.; Ge, Y.; Gao, P.; Yang, Y.; et al. Genome-wide mapping of 5-hydroxymethylcytosines in circulating cell-free DNA as a non-invasive approach for early detection of hepatocellular carcinoma. Gut 2019, 68, 2195–2205. [Google Scholar] [CrossRef] [Green Version]
- Barbáchano, A.; Fernández-Barral, A.; Pereira, F.P.; Segura, M.F.; Ordóñez-Morán, P.; Pau, E.C.D.S.; Sancho, J.M.G.; Hanniford, D.; Martínez, N.; Costales, A.; et al. SPROUTY-2 represses the epithelial phenotype of colon carcinoma cells via upregulation of ZEB1 mediated by ETS1 and miR-200/miR-150. Oncogene 2015, 35, 2991–3003. [Google Scholar] [CrossRef] [Green Version]
- Ahsan, S.; Raabe, E.H.; Haffner, M.C.; Vaghasia, A.; Warren, K.E.; Quezado, M.; Ballester, L.Y.; Nazarian, Y.; Eberhart, C.G.; Rodriguez, F.J. Increased 5-hydroxymethylcytosine and decreased 5-methylcytosine are indicators of global epigenetic dysregulation in diffuse intrinsic pontine glioma. Acta Neuropathol. Commun. 2014, 2, 59. [Google Scholar] [CrossRef] [Green Version]
- Vető, B.; Szabó, P.; Bacquet, C.; Apró, A.; Hathy, E.; Kiss, J.; Réthelyi, J.M.; Szeri, F.; Szüts, D.; Arányi, T. Inhibition of DNA methyltransferase leads to increased genomic 5-hydroxymethylcytosine levels in hematopoietic cells. FEBS Open Bio 2018, 8, 584–592. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.; Zhang, J.; Guo, Z.; Ma, Q.; Xu, Z.; Zhou, Y.; Xu, Z.; Li, Z.; Liu, Y.; Ye, X.; et al. Loss of 5-hydroxymethylcytosine is linked to gene body hypermethylation in kidney cancer. Cell Res. 2016, 26, 103–118. [Google Scholar] [CrossRef]
- Kubo, N.; Ishii, H.; Xiong, X.; Bianco, S.; Meitinger, F.; Hu, R.; Hocker, J.D.; Conte, M.; Gorkin, D.; Yu, M.; et al. CTCF Promotes Long-range Enhancer-promoter Interactions and Lineage-specific Gene Expression in Mammalian Cells. BioRxiv 2020. [Google Scholar] [CrossRef]
- Kentepozidou, E.; Aitken, S.J.; Feig, C.; Stefflova, K.; Ibarra-Soria, X.; Odom, D.T.; Roller, M.; Flicek, P. Clustered CTCF binding is an evolutionary mechanism to maintain topologically associating domains. Genome Biol. 2020, 21, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arpanahi, A.; Brinkworth, M.; Iles, D.; Krawetz, S.A.; Paradowska, A.; Platts, A.; Saida, M.; Steger, K.; Tedder, P.; Miller, D. Endonuclease-sensitive regions of human spermatozoal chromatin are highly enriched in promoter and CTCF binding sequences. Genome Res. 2009, 19, 1338–1349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, C.; Wang, Z.; Han, C.; Safgren, S.L.; Helmin, K.A.; Adelman, E.R.; Serafin, V.; Basso, G.; Eagen, K.P.; Gaspar-Maia, A.; et al. Cancer-specific CTCF binding facilitates oncogenic transcriptional dysregulation. Genome Biol. 2020, 21, 247. [Google Scholar] [CrossRef]
- Soto-Reyes, E.; González-Barrios, R.; Cisneros-Soberanis, F.; Herrera-Goepfert, R.; Pérez, V.; Cantú, D.; Prada, D.; Castro, C.; Recillas-Targa, F.; Herrera, L.A. Disruption of CTCF at the miR-125b1 locus in gynecological cancers. BMC Cancer 2012, 12, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, L.; Shen, H.; Laird, P.W.; Farnham, P.J.; Berman, B.P. Inferring regulatory element landscapes and transcription factor networks from cancer methylomes. Genome Biol. 2015, 16, 105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, I.J.; Yu, Y.S.; Mustafa, B.; Park, J.Y.; Seo, Y.B.; Kim, G.-D.; Kim, J.; Kim, C.M.; Noh, H.D.; Hong, S.-M.; et al. A Nine-Gene Signature for Predicting the Response to Preoperative Chemoradiotherapy in Patients with Locally Advanced Rectal Cancer. Cancers 2020, 12, 800. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.H.; Tsao, C.J.; Wu, C.L.; Chang, J.G.; Lu, P.J.; Yeh, K.T.; Shieh, G.-S.; Shiau, A.-L.; Lee, J.C. Sprouty2 protein enhances the response to gefitinib through epidermal growth factor receptor in colon cancer cells. Cancer Sci. 2010, 101, 2033–2038. [Google Scholar] [CrossRef]
- Song, L.; Jia, J.; Peng, X.; Xiao, W.; Li, Y. The performance of the SEPT9 gene methylation assay and a comparison with other CRC screening tests: A meta-analysis. Sci. Rep. 2017, 7, 3032. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Caco2 Cells | RKO Cells | HCT116 Cells | |
|---|---|---|---|
| SPRY2 protein expression | High | Medium | Low |
| 5mC presence region #1 | No | Yes | Yes |
| 5mC presence region #2 | No | No | Yes |
| 5mC presence region #3 | No | No | Yes |
| 5mC presence region #4 | No | No | Yes |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stuckel, A.J.; Zeng, S.; Lyu, Z.; Zhang, W.; Zhang, X.; Dougherty, U.; Mustafi, R.; Zhang, Q.; Joshi, T.; Bissonnette, M.; et al. Epigenetic DNA Modifications Upregulate SPRY2 in Human Colorectal Cancers. Cells 2021, 10, 2632. https://doi.org/10.3390/cells10102632
Stuckel AJ, Zeng S, Lyu Z, Zhang W, Zhang X, Dougherty U, Mustafi R, Zhang Q, Joshi T, Bissonnette M, et al. Epigenetic DNA Modifications Upregulate SPRY2 in Human Colorectal Cancers. Cells. 2021; 10(10):2632. https://doi.org/10.3390/cells10102632
Chicago/Turabian StyleStuckel, Alexei J., Shuai Zeng, Zhen Lyu, Wei Zhang, Xu Zhang, Urszula Dougherty, Reba Mustafi, Qiong Zhang, Trupti Joshi, Marc Bissonnette, and et al. 2021. "Epigenetic DNA Modifications Upregulate SPRY2 in Human Colorectal Cancers" Cells 10, no. 10: 2632. https://doi.org/10.3390/cells10102632
APA StyleStuckel, A. J., Zeng, S., Lyu, Z., Zhang, W., Zhang, X., Dougherty, U., Mustafi, R., Zhang, Q., Joshi, T., Bissonnette, M., Choudhury, S. R., & Khare, S. (2021). Epigenetic DNA Modifications Upregulate SPRY2 in Human Colorectal Cancers. Cells, 10(10), 2632. https://doi.org/10.3390/cells10102632