
Mutation of the EPHA2 Tyrosine-Kinase Domain Dysregulates Cell Pattern Formation and Cytoskeletal Gene Expression in the Lens


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Mice and Lenses
2.2. Whole-Mount Imaging of tdT Labeled Lenses
2.3. Immunofluorescence Confocal Microscopy
2.4. Immunoblot and Immunoprecipitation Analyses
2.5. RNA Sequencing Analysis
3. Results
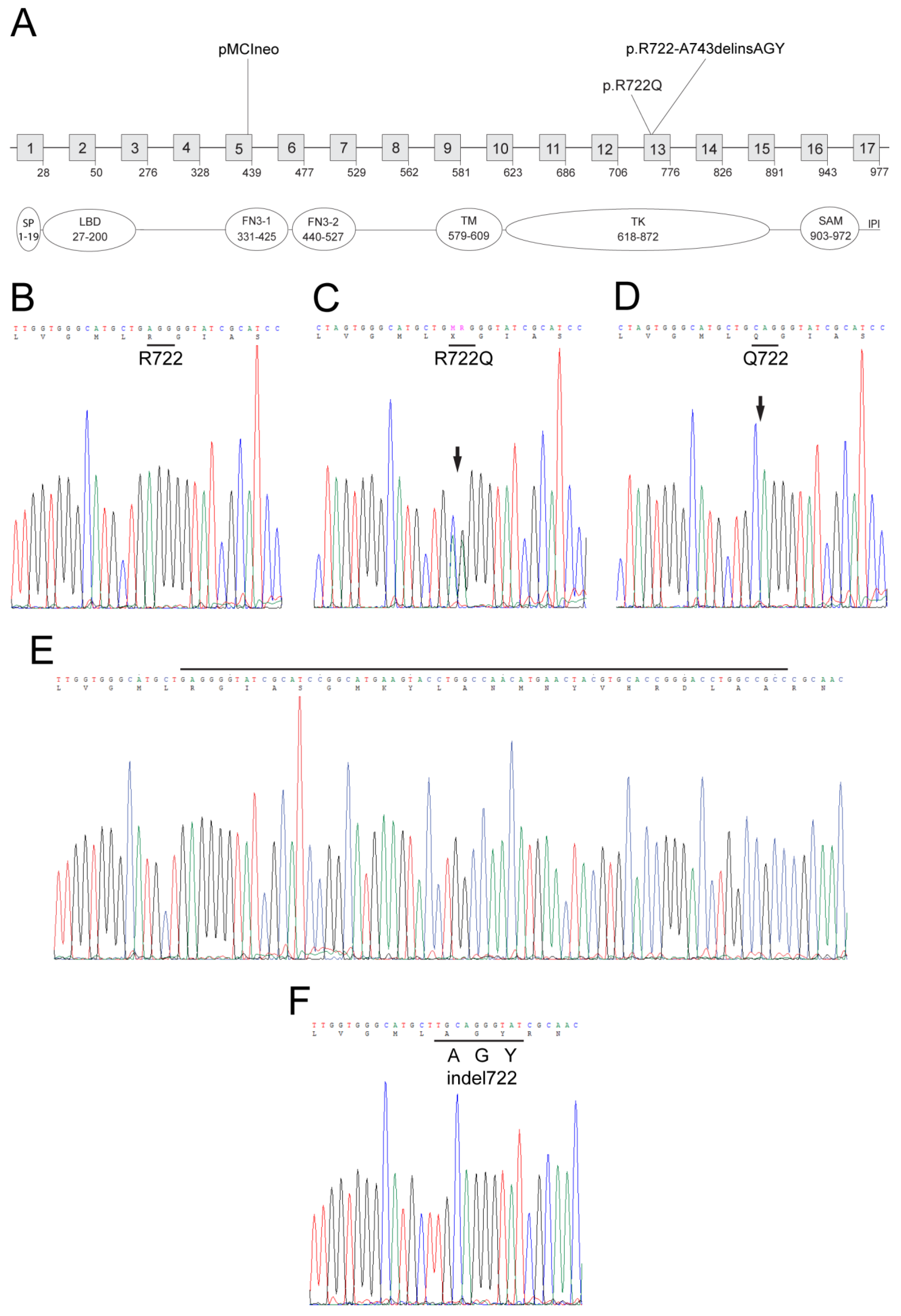
3.1. Epha2-Mutant Mice and Lenses
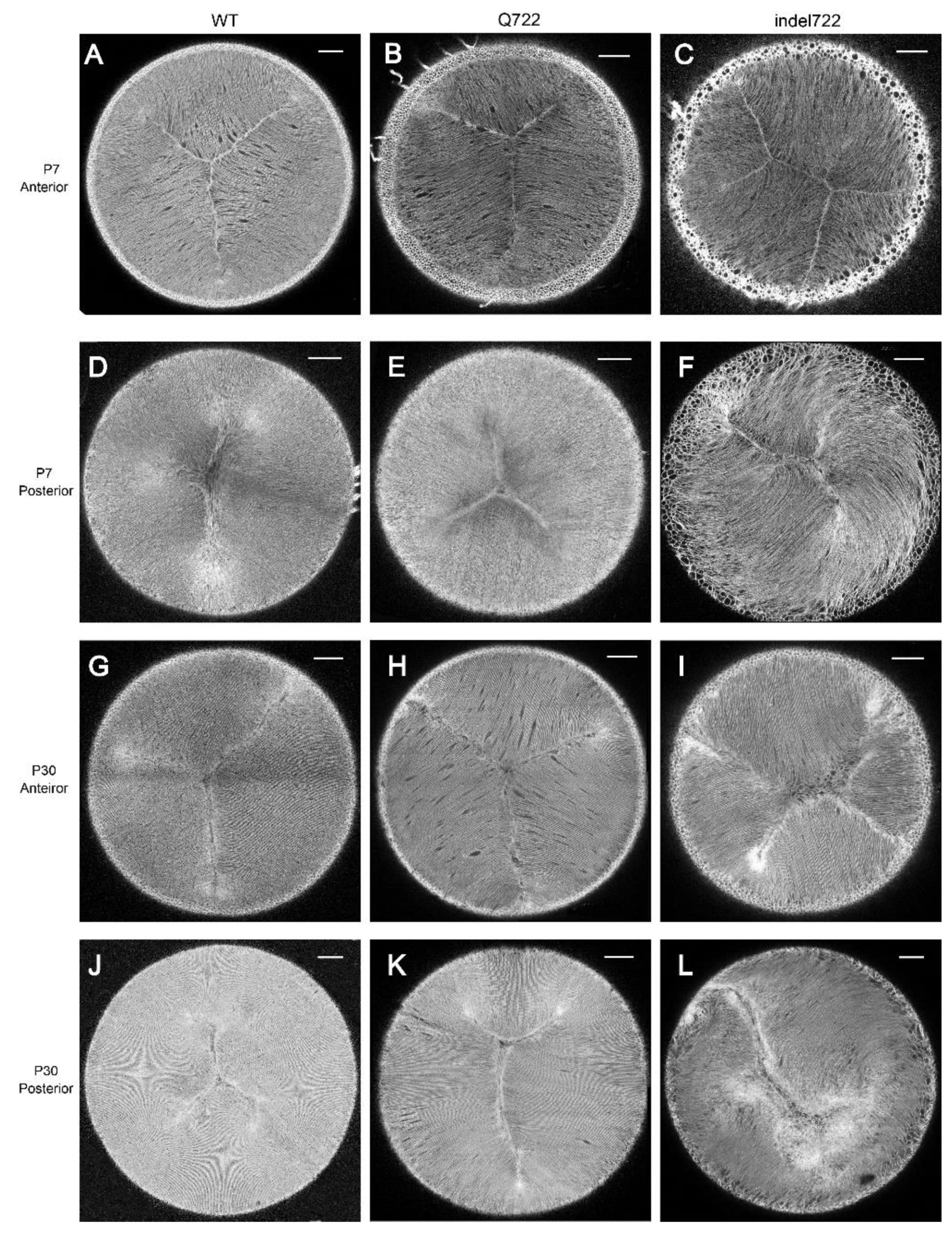
3.2. Lens Cell Alignment and Suture Formation in Epha2-Mutant Mice
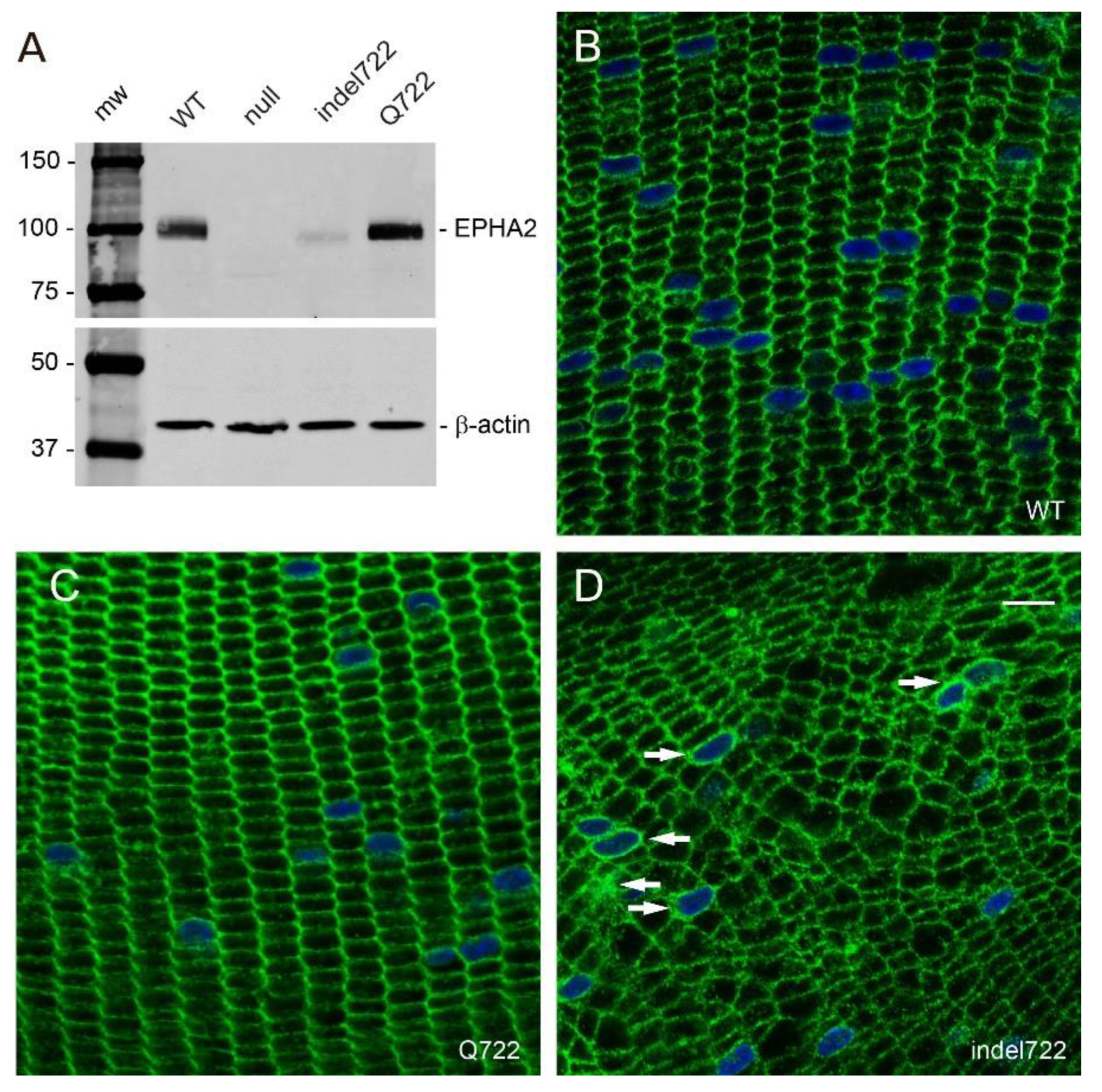
3.3. Expression and Distribution of EPHA2 Mutants in the Lens
3.4. EPHA2 Complex Formation and Phosphorylation in the Lens
3.5. Gene Expression Profiles of Epha2-Mutant and Epha2-Null Lenses
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hirai, H.; Maru, Y.; Hagiwara, K.; Nishida, J.; Takaku, F. A novel putative tyrosine kinase receptor encoded by the eph gene. Science 1987, 238, 1717–1720. [Google Scholar] [CrossRef]
- Lindberg, R.A.; Hunter, T. cDNA cloning and characterization of eck, an epithelial cell receptor protein-tyrosine kinase in the eph/elk family of protein kinases. Mol. Cell. Biol. 1990, 10, 6316–6324. [Google Scholar] [CrossRef]
- Lisabeth, E.M.; Falivelli, G. Pasquale EB. Eph receptor signaling and ephrins. Cold. Spring Harb. Perspect. Biol. 2013, 5, a009159. [Google Scholar] [CrossRef] [PubMed]
- Barquilla, A.; Pasquale, E.B. Eph receptors and ephrins: Therapeutic opportunities. Ann. Rev. Pharmacol. Toxicol. 2015, 55, 465–487. [Google Scholar] [CrossRef] [PubMed]
- Kania, A.; Klein, R. Mechanisms of ephrin-Eph signalling in development, physiology and disease. Nat. Rev. Mol. Cell. Biol. 2016, 17, 240–256. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.R.; Ahmed, F.; Paul, M.D.; Gedam, M.; Pasquale, E.B.; Hristova, K. The SAM domain inhibits EphA2 interactions in the plasma membrane. Biochim. Biophys. Acta. Mol. Cell. Res. 2017, 1864, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Hapiak, V.; Zheng, J.; Muller-Greven, J.; Bowman, D.; Lingerak, R.; Buck, M.; Wang, B.C.; Smith, A.W. A role of the SAM domain in EphA2 receptor activation. Sci. Rep. 2017, 7, 45084. [Google Scholar] [CrossRef] [PubMed]
- Miao, H.; Wang, B. EphA receptor signaling--complexity and emerging themes. Semin. Cell. Dev. Biol. 2012, 23, 16–25. [Google Scholar] [CrossRef]
- Xiao, T.; Xiao, Y.; Wang, W.; Tang, Y.Y.; Xiao, Z.; Su, M. Targeting EphA2 in cancer. J. Hematol. Oncol. 2020, 13, 114. [Google Scholar] [CrossRef]
- Miao, H.; Wang, B. Eph/ephrin signaling in epithelial development and homeostasis. Int. J. Biochem. Cell Biol. 2009, 41, 762–770. [Google Scholar] [CrossRef]
- London, M.; Gallo, E. The EphA2 and cancer connection: Potential for immune-based interventions. Mol. Biol. Rep. 2020, 47, 8037–8048. [Google Scholar] [CrossRef]
- Anderton, M.; van der Meulen, E.; Blumenthal, M.J.; Schafer, G. The Role of the Eph Receptor Family in Tumorigenesis. Cancers 2021, 13, 206. [Google Scholar] [CrossRef]
- Buraschi, S.; Neill, T.; Xu, S.Q.; Palladino, C.; Belfiore, A.; Iozzo, R.V.; Morrione, A. Progranulin/EphA2 axis: A novel oncogenic mechanism in bladder cancer. Matrix Biol. 2020, 93, 10–24. [Google Scholar] [CrossRef]
- Wang, J.; Zheng, X.; Peng, Q.; Zhang, X.; Qin, Z. Eph receptors: The bridge linking host and virus. Cell. Mol. Life Sci. 2020, 77, 2355–2365. [Google Scholar] [CrossRef]
- Swidergall, M.; Solis, N.V.; Wang, Z.; Phan, Q.T.; Marshall, M.E.; Lionakis, M.S.; Pearlman, E.; Filler, S.G. EphA2 Is a Neutrophil Receptor for Candida albicans that Stimulates Antifungal Activity during Oropharyngeal Infection. Cell. Rep. 2019, 28, 423–433.e5. [Google Scholar] [CrossRef] [PubMed]
- Darling, T.K.; Mimche, P.N.; Bray, C.; Umaru, B.; Brady, L.M.; Stone, C.; Eboumbou Moukoko, C.E.; Lane, T.E.; Ayong, L.S.; Lamb, T.L. EphA2 contributes to disruption of the blood-brain barrier in cerebral malaria. PLoS Pathog. 2020, 16, e1008261. [Google Scholar] [CrossRef] [PubMed]
- GBD 2019 Blindness and Vision Impairment Collaborators; Vision Loss Expert Group of the Global Burden of Disease Study. Causes of blindness and vision impairment in 2020 and trends over 30 years, and prevalence of avoidable blindness in relation to VISION 2020: The Right to Sight: An analysis for the Global Burden of Disease Study. Lancet Glob. Health 2021, 9, e144–e160. [Google Scholar] [CrossRef]
- Shiels, A.; Hejtmancik, J.F. Inherited cataracts: Genetic mechanisms and pathways new and old. Exp. Eye. Res. 2021, 209, 108662. [Google Scholar] [CrossRef] [PubMed]
- Shiels, A.; Bennett, T.M.; Knopf, H.L.; Maraini, G.; Li, A.; Jiao, X.; Hejtmancik, J.F. The EPHA2 gene is associated with cataracts linked to chromosome 1p. Mol. Vis. 2008, 14, 2042–2055. [Google Scholar]
- Jun, G.; Guo, H.; Klein, B.E.; Klein, R.; Wang, J.J.; Mitchell, P.; Miao, H.; Lee, K.E.; Joshi, T.; Buck, M.; et al. EPHA2 is associated with age-related cortical cataract in mice and humans. PLoS Genet. 2009, 5, e1000584. [Google Scholar] [CrossRef]
- Bennett, T.M.; M’Hamdi, O.; Hejtmancik, J.F.; Shiels, A. Germ-line and somatic EPHA2 coding variants in lens aging and cataract. PLoS ONE 2017, 12, e0189881. [Google Scholar]
- Beebe, D.C.; Coats, J.M. The lens organizes the anterior segment: Specification of neural crest cell differentiation in the avian eye. Dev. Biol. 2000, 220, 424–431. [Google Scholar] [CrossRef] [PubMed]
- Iribarren, R. Crystalline lens and refractive development. Prog. Retin. Eye Res. 2015, 47, 86–106. [Google Scholar] [CrossRef] [PubMed]
- Bassnett, S.; Shi, Y.; Vrensen, G.F. Biological glass: Structural determinants of eye lens transparency. Philos. Trans. R. Soc. B Biol. Sci. 2011, 366, 1250–1264. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, P.J.; Grey, A.C.; Maceo Heilman, B.; Lim, J.C.; Vaghefi, E. The physiological optics of the lens. Prog. Retin. Eye. Res. 2017, 56, e1–e24. [Google Scholar] [CrossRef] [PubMed]
- Cvekl, A.; Ashery-Padan, R. The cellular and molecular mechanisms of vertebrate lens development. Development 2014, 141, 4432–4447. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.; Nowak, R.B.; Fowler, V.M. The lens actin filament cytoskeleton: Diverse structures for complex functions. Exp. Eye Res. 2017, 156, 58–71. [Google Scholar] [CrossRef]
- Cvekl, A.; Zhang, X. Signaling and Gene Regulatory Networks in Mammalian Lens Development. Trends Genet. 2017, 33, 677–702. [Google Scholar] [CrossRef]
- Morishita, H.; Eguchi, T.; Tsukamoto, S.; Sakamaki, Y.; Takahashi, S.; Saito, C.; Koyama-Honda, I.; Mizushima, N. Organelle degradation in the lens by PLAAT phospholipases. Nature 2021, 592, 634–638. [Google Scholar] [CrossRef]
- Bassnett, S.; Wilmarth, P.A.; David, L.L. The membrane proteome of the mouse lens fiber cell. Mol. Vis. 2009, 15, 2448–2463. [Google Scholar]
- Cheng, C.; Gong, X. Diverse roles of Eph/ephrin signaling in the mouse lens. PLoS ONE 2011, 6, e28147. [Google Scholar] [CrossRef]
- Cheng, C.; Ansari, M.M.; Cooper, J.A.; Gong, X. EphA2 and Src regulate equatorial cell morphogenesis during lens development. Development 2013, 140, 4237–4245. [Google Scholar] [CrossRef]
- Cheng, C.; Fowler, V.M.; Gong, X. EphA2 and ephrin-A5 are not a receptor-ligand pair in the ocular lens. Exp. Eye Res. 2017, 162, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; de Maria, A.; Bennett, T.; Shiels, A.; Bassnett, S. A role for epha2 in cell migration and refractive organization of the ocular lens. Investig. Ophthalmol. Vis. Sci. 2012, 53, 551–559. [Google Scholar] [CrossRef]
- Zhou, Y.; Shiels, A. Epha2 and Efna5 participate in lens cell pattern-formation. Differentiation 2018, 102, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Dave, A.; Craig, J.E.; Skrzypiec, K.; Quinn, S.; Barnes, M.; Di Girolamo, N.; Mackey, D.A.; Burdon, K.P.; de Iongh, R.U.; Sharma, S. Epha2 genotype influences ultraviolet radiation induced cataract in mice. Exp. Eye Res. 2019, 188, 107806. [Google Scholar] [CrossRef] [PubMed]
- Brantley-Sieders, D.M.; Caughron, J.; Hicks, D.; Pozzi, A.; Ruiz, J.C.; Chen, J. EphA2 receptor tyrosine kinase regulates endothelial cell migration and vascular assembly through phosphoinositide 3-kinase-mediated Rac1 GTPase activation. J. Cell Sci. 2004, 117, 2037–2049. [Google Scholar] [CrossRef] [PubMed]
- Muzumdar, M.D.; Tasic, B.; Miyamichi, K.; Li, L.; Luo, L. A global double-fluorescent Cre reporter mouse. Genesis 2007, 45, 593–605. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Bennett, T.M.; Shiels, A. Mutation of the TRPM3 cation channel underlies progressive cataract development and lens calcification associated with pro-fibrotic and immune cell responses. FASEB J. 2021, 35, e21288. [Google Scholar] [CrossRef]
- Simirskii, V.N.; Lee, R.S.; Wawrousek, E.F. Duncan MK. Inbred FVB/N mice are mutant at the cp49/Bfsp2 locus and lack beaded filament proteins in the lens. Investig. Ophthalmol. Vis. Sci. 2006, 47, 4931–4934. [Google Scholar] [CrossRef]
- Zhou, Y.; Bennett, T.M.; Shiels, A. A charged multivesicular body protein (CHMP4B) is required for lens growth and differentiation. Differentiation 2019, 109, 16–27. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Bennett, T.M.; Shiels, A. Lens ER-stress response during cataract development in Mip-mutant mice. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2016, 1862, 1433–1442. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M. Twelve years of SAMtools and BCFtools. Gigascience 2021, 10, giab008. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Gu, Z.; Eils, R.; Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef]
- Gene Ontology Consortium. The Gene Ontology resource: Enriching a GOld mine. Nucleic Acids Res. 2021, 49, D325–D334. [Google Scholar] [CrossRef]
- Sugiyama, Y.; Akimoto, K.; Robinson, M.L.; Ohno, S.; Quinlan, R.A. A cell polarity protein aPKClambda is required for eye lens formation and growth. Dev. Biol. 2009, 336, 246–256. [Google Scholar] [CrossRef][Green Version]
- Kuszak, J.R.; Zoltoski, R.K.; Sivertson, C. Fibre cell organization in crystalline lenses. Exp. Eye Res. 2004, 78, 673–687. [Google Scholar] [CrossRef]
- Kuszak, J.R.; Zoltoski, R.K. Tiedemann CE. Development of lens sutures. Int. J. Dev. Biol. 2004, 48, 889–902. [Google Scholar] [CrossRef]
- Cooper, M.A.; Son, A.I.; Komlos, D.; Sun, Y.; Kleiman, N.J.; Zhou, R. Loss of ephrin-A5 function disrupts lens fiber cell packing and leads to cataract. Proc. Natl. Acad. Sci. USA 2008, 105, 16620–16625. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Shatadal, S.; Griep, A.E. Dlg-1 Interacts With and Regulates the Activities of Fibroblast Growth Factor Receptors and EphA2 in the Mouse Lens. Investig. Ophthalmol. Vis. Sci. 2016, 57, 707–718. [Google Scholar] [CrossRef] [PubMed]
- Fang, W.B.; Brantley-Sieders, D.M.; Hwang, Y.; Ham, A.J.; Chen, J. Identification and functional analysis of phosphorylated tyrosine residues within EphA2 receptor tyrosine kinase. J. Biol. Chem. 2008, 283, 16017–16026. [Google Scholar] [CrossRef]
- Wyatt, K.; Gao, C.; Tsai, J.Y.; Fariss, R.N.; Ray, S.; Wistow, G. A role for lengsin, a recruited enzyme, in terminal differentiation in the vertebrate lens. J. Biol. Chem. 2008, 283, 6607–6615. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Lerner, J.; Wyatt, M.K.; Cai, P.; Peterson, K.; Dong, L.; Wistow, G. The klotho-related protein KLPH (lctl) has preferred expression in lens and is essential for expression of clic5 and normal lens suture formation. Exp. Eye Res. 2018, 169, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Sakurai, H. Emerging and diverse functions of the EphA2 noncannonical pathway in cancer progression. Biol. Pharm. Bull. 2017, 40, 1616–1624. [Google Scholar] [CrossRef]
- Ma, X.; Ma, Z.; Jiao, X.; Hejtmancik, J.F. Functional non-coding polymorphism in an EPHA2 promoter PAX2 binding site modifies expression and alters the MAPK and AKT pathways. Sci. Rep. 2017, 7, 9992. [Google Scholar] [CrossRef]
- Zhang, T.; Hua, R.; Xiao, W.; Burdon, K.P.; Bhattacharya, S.S.; Craig, J.E.; Shang, D.; Zhao, X.; Mackey, D.A.; Moore, A.T.; et al. Mutations of the EPHA2 receptor tyrosine kinase gene cause autosomal dominant congenital cataract. Hum. Mutat. 2009, 30, E603–E611. [Google Scholar] [CrossRef]
- Park, J.E.; Son, A.I.; Hua, R.; Wang, L.; Zhang, X.; Zhou, R. Human cataract mutations in EPHA2 SAM domain alter receptor stability and function. PLoS ONE 2012, 7, e36564. [Google Scholar] [CrossRef]
- Dave, A.; Martin, S.; Kumar, R.; Craig, J.E.; Burdon, K.P.; Sharma, S. Epha2 mutations contribute to congenital cataract through diverse mechanisms. Mol. Vis. 2016, 22, 18–30. [Google Scholar]
- Patel, N.; Anand, D.; Monies, D.; Maddirevula, S.; Khan, A.O.; Algoufi, T.; Alowain, M.; Faqeih, M.; Alshammari, M.; Qudair, A.; et al. Novel phenotypes and loci identified through clinical genomics approaches to pediatric cataract. Hum. Genet. 2017, 136, 205–225. [Google Scholar] [CrossRef] [PubMed]
- Zhai, Y.; Zhu, S.; Li, J.; Yao, K. A novel human congenital cataract mutation in EPHA2 kinase domain (p.G668D) alters receptor stability and function. Investig. Ophthalmol. Vis. Sci. 2019, 60, 4717–4726. [Google Scholar] [CrossRef]
- Bian, T.; Zheng, M.; Jiang, D.; Liu, J.; Sun, H.; Li, X.; Liu, L.; Zhang, J.; Liu, Y. Prognostic biomarker TUBA1C is correlated to immune cell infiltration in the tumor microenvironment of lung adenocarcinoma. Cancer Cell Int. 2021, 21, 144. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Hu, W.; Xu, R.; Jin, J.; Szulc, Z.M.; Zhang, G.; Galadari, S.H.; Obeid, L.M.; Mao, C. Alkaline ceramidase 2 regulates beta1 integrin maturation and cell adhesion. FASEB J. 2009, 23, 656–666. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Panda, D. A centrosomal protein STARD9 promotes microtubule stability and regulates spindle microtubule dynamics. Cell Cycle 2018, 17, 2052–2068. [Google Scholar] [CrossRef] [PubMed]
- Grati, M.; Chakchouk, I.; Ma, Q.; Bensaid, M.; Desmidt, A.; Turki, N.; Yan, D.; Baanannou, A.; Mittal, R.; Driss, N.; et al. A missense mutation in DCDC2 causes human recessive deafness DFNB66, likely by interfering with sensory hair cell and supporting cell cilia length regulation. Hum. Mol. Genet. 2015, 24, 2482–2491. [Google Scholar] [CrossRef]
- Hu, Y.H.; Zhang, Y.; Jiang, L.Q.; Wang, S.; Lei, C.Q.; Sun, M.S.; Shu, H.B.; Liu, Y. WDFY1 mediates TLR3/4 signaling by recruiting TRIF. EMBO Rep. 2015, 16, 447–455. [Google Scholar] [CrossRef]
- Shu, D.Y.; Wojciechowski, M.C.; Lovicu, F.J. Bone morphogenetic protein-7 suppresses TGFbeta2-induced epithelial-mesenchymal transition in the lens: Implications for cataract prevention. Investig. Ophthalmol. Vis. Sci. 2017, 58, 781–796. [Google Scholar] [CrossRef]
- Harding, R.L.; Howley, S.; Baker, L.J.; Murphy, T.R.; Archer, W.E.; Wistow, G.; Hyde, D.R. Lengsin expression and function during zebrafish lens formation. Exp. Eye Res. 2008, 86, 807–818. [Google Scholar] [CrossRef][Green Version]
- Seco, C.Z.; Oonk, A.M.; Dominguez-Ruiz, M.; Draaisma, J.M.; Gandia, M.; Oostrik, J.; Neveling, K.; Kunst, H.P.M.; Hoefsloot, L.H.; del Castillo, I.; et al. Progressive hearing loss and vestibular dysfunction caused by a homozygous nonsense mutation in CLIC5. Eur. J. Hum. Genet. 2015, 23, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Wonkam-Tingang, E.; Schrauwen, I.; Esoh, K.K.; Bharadwaj, T.; Nouel-Saied, L.M.; Acharya, A.; Nasir, A.; Adadey, S.M.; Mowla, S.; Leal, S.M.; et al. Bi-allelic novel variants in CLIC5 identified in a Cameroonian multiplex family with non-syndromic hearing impairment. Genes 2020, 11, 1249. [Google Scholar] [CrossRef] [PubMed]
- Salles, F.T.; Andrade, L.R.; Tanda, S.; Grati, M.; Plona, K.L.; Gagnon, L.H.; Johnson, K.R.; Kachar, B.; Berryman, M.A. CLIC5 stabilizes membrane-actin filament linkages at the base of hair cell stereocilia in a molecular complex with radixin, taperin, and myosin VI. Cytoskeleton 2014, 71, 61–78. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, Y.; Bennett, T.M.; Ruzycki, P.A.; Shiels, A. Mutation of the EPHA2 Tyrosine-Kinase Domain Dysregulates Cell Pattern Formation and Cytoskeletal Gene Expression in the Lens. Cells 2021, 10, 2606. https://doi.org/10.3390/cells10102606
Zhou Y, Bennett TM, Ruzycki PA, Shiels A. Mutation of the EPHA2 Tyrosine-Kinase Domain Dysregulates Cell Pattern Formation and Cytoskeletal Gene Expression in the Lens. Cells. 2021; 10(10):2606. https://doi.org/10.3390/cells10102606
Chicago/Turabian StyleZhou, Yuefang, Thomas M. Bennett, Philip A. Ruzycki, and Alan Shiels. 2021. "Mutation of the EPHA2 Tyrosine-Kinase Domain Dysregulates Cell Pattern Formation and Cytoskeletal Gene Expression in the Lens" Cells 10, no. 10: 2606. https://doi.org/10.3390/cells10102606
APA StyleZhou, Y., Bennett, T. M., Ruzycki, P. A., & Shiels, A. (2021). Mutation of the EPHA2 Tyrosine-Kinase Domain Dysregulates Cell Pattern Formation and Cytoskeletal Gene Expression in the Lens. Cells, 10(10), 2606. https://doi.org/10.3390/cells10102606