
The Genetic Makeup of Myeloproliferative Neoplasms: Role of Germline Variants in Defining Disease Risk, Phenotypic Diversity and Outcome


Abstract
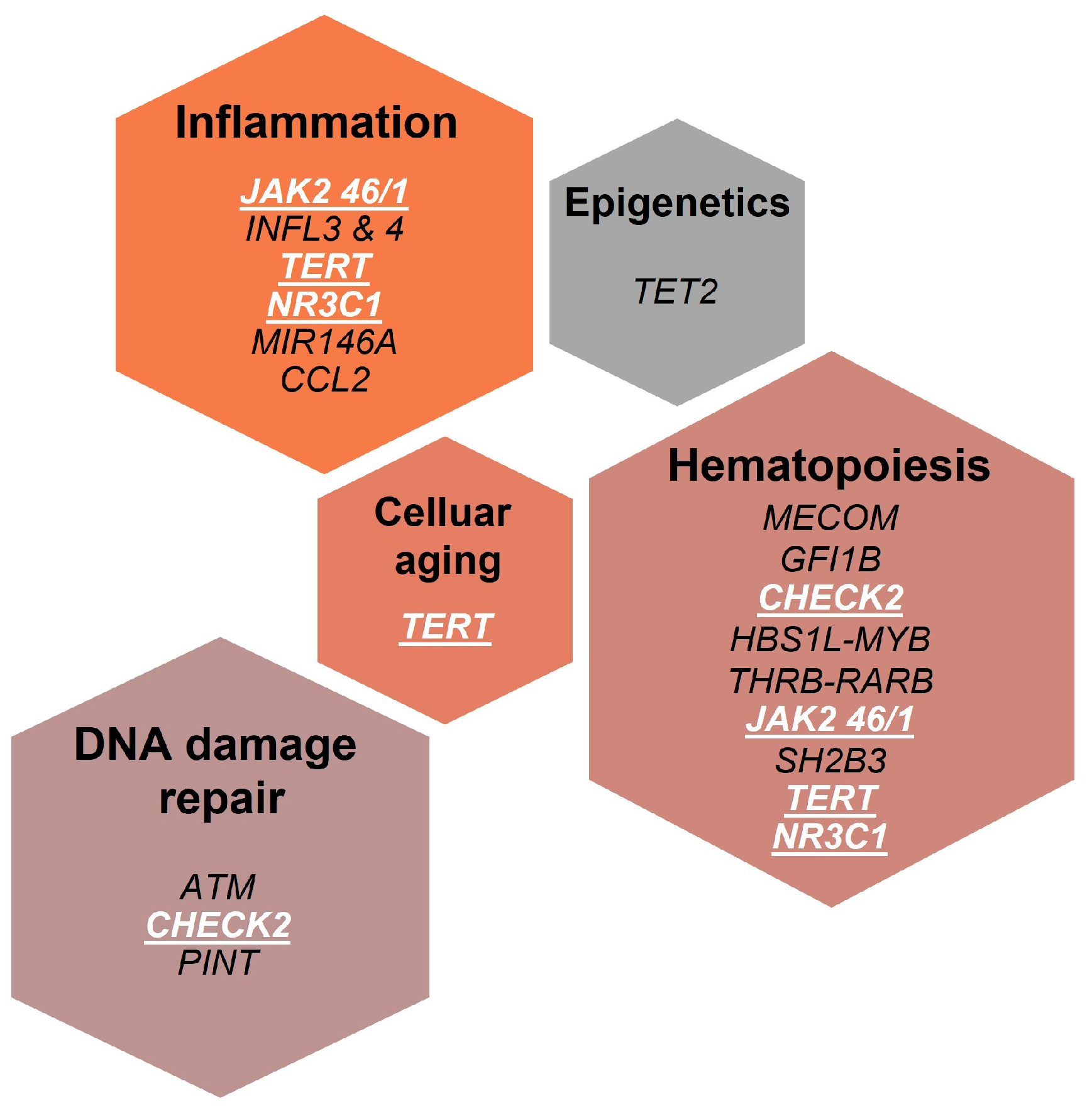
:1. Introduction
2. Host Genetic Variants Associated to Familial MPNs
3. Host Genetic Variants Associated to Increased MPN Risk in General Population
3.1. The JAK2 46/1 Haplotype
3.2. Telomere Reverse Transcriptase Gene (TERT) Polymorphisms
3.3. Polymorphisms in3q.26 (MECOM and HBS1L-MYB)
3.4. GFI1B and CHEK2 Polymorphisms
4. Host Genetic Variants Modulating Disease Phenotype and/or Outcome
4.1. The rs6198 SNP of the Glucocorticoid Gene
4.2. The rs1024611 SNP of CCL2
4.3. The rs2431697 of MIR146A
5. Host Genetic Variants Affecting Therapy Response
6. Host Genetic Determinants of Clonal Hematopoiesis of Indeterminate Potential (CHIP)
7. Conclusions and Perspectives
- (1)
- Hematopoiesis defines the tightly regulated process of formation of blood and immune cells. To generate these cells, HSCs give rise—throughout individuals’ life span—to an array of committed progenitors, which proliferate extensively and then differentiate into mature cells. Recent advances in genomics, such as accurate deep sequencing and novel methods of cell tracking, revolutionized the concept of hematopoiesis from a process made of discrete, punctuated phenotypic changes to a “continuum model”, typified by a continuous process of differentiation with blurred demarcation between different stages [82,83]. Genetic studies revealed, also, how mechanisms underlying hematopoiesis are modulated by genetic variations present throughout the population. The importance of these host genetic variations is highlighted by the fact that clinically measured hematopoietic traits typically show extensive interindividual variability and are highly heritable, which means that a relevant part of the observed phenotype variations can be attributed to genetic factors [82,84].
- (2)
- During genome duplication, cells may experience different exogenous and endogenous replication stresses, hampering the progression of DNA replication. Replication stress is a phenomenon exacerbated in cancer cells because of the loss of DNA repair genes or the activation of oncogenic pathways [85]. To counteract replication stress, cells are equipped with DNA damage response, an extensive network of signaling pathways accounting for recognition of DNA damage, DNA remodeling and repair, DNA damage bypass during replication, cell cycle control, and cell fate decisions in response to DNA alterations [86]. More than 450 genes are involved in this network. In addition to MPNs, a variety of polymorphisms in DDR genes have been associated with increased risk of developing acute myeloid leukemia [87] and breast cancer [88].
- (3)
- Inflammation refers to a host defense mechanism orchestrated by the immune system in response to harmful stimuli, such as pathogens, damaged cells, toxic compounds, or irradiation [89]. Cytokines are key mediators of the inflammatory response, by promoting the recruitment and activation of immune cells. After the human leucocyte antigen (HLA), chemokine genes are probably one of the most polymorphic sets of genes in the immune system, with remarkable effects on the immune response. A number of functionally relevant cytokine SNPs have been found repeatedly associated with disease of different etiologies but sharing a common pathogenetic aspect such as chronic inflammation [67].
Author Contributions
Funding
Conflicts of Interest
References
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef]
- Greenfield, G.; McMullin, M.F.; Mills, K. Molecular pathogenesis of the myeloproliferative neoplasms. J. Hematol. Oncol. 2021, 14, 103. [Google Scholar] [CrossRef] [PubMed]
- Grabek, J.; Straube, J.; Bywater, M.; Lane, S.W. MPN: The Molecular Drivers of Disease Initiation, Progression and Transformation and their Effect on Treatment. Cells 2020, 9, 1901. [Google Scholar] [CrossRef]
- Guglielmelli, P.; Lasho, T.L.; Rotunno, G.; Mudireddy, M.; Mannarelli, C.; Nicolosi, M.; Pacilli, A.; Pardanani, A.; Rumi, E.; Rosti, V.; et al. MIPSS70: Mutation-Enhanced International Prognostic Score System for Transplantation-Age Patients With Primary Myelofibrosis. J. Clin. Oncol. 2018, 36, 310–318. [Google Scholar] [CrossRef]
- Tefferi, A.; Guglielmelli, P.; Lasho, T.L.; Gangat, N.; Ketterling, R.P.; Pardanani, A.; Vannucchi, A.M. MIPSS70+ Version 2.0: Mutation and Karyotype-Enhanced International Prognostic Scoring System for Primary Myelofibrosis. J. Clin. Oncol. 2018, 36, 1769–1770. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Guglielmelli, P.; Nicolosi, M.; Mannelli, F.; Mudireddy, M.; Bartalucci, N.; Finke, C.M.; Lasho, T.L.; Hanson, C.A.; Ketterling, R.P.; et al. GIPSS: Genetically inspired prognostic scoring system for primary myelofibrosis. Leukemia 2018, 32, 1631–1642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMullin, M.F.; Anderson, L.A. Aetiology of Myeloproliferative Neoplasms. Cancers 2020, 12, 1810. [Google Scholar] [CrossRef]
- Tashi, T.; Swierczek, S.; Prchal, J.T. Familial MPN Predisposition. Curr. Hematol. Malig. Rep. 2017, 12, 442–447. [Google Scholar] [CrossRef]
- Kilpivaara, O.; Mukherjee, S.; Schram, A.M.; Wadleigh, M.; Mullally, A.; Ebert, B.L.; Bass, A.; Marubayashi, S.; Heguy, A.; Garcia-Manero, G.; et al. A germline JAK2 SNP is associated with predisposition to the development of JAK2(V617F)-positive myeloproliferative neoplasms. Nat. Genet. 2009, 41, 455–459. [Google Scholar] [CrossRef] [Green Version]
- Oddsson, A.; Kristinsson, S.Y.; Helgason, H.; Gudbjartsson, D.F.; Masson, G.; Sigurdsson, A.; Jonasdottir, A.; Jonasdottir, A.; Steingrimsdottir, H.; Vidarsson, B.; et al. The germline sequence variant rs2736100_C in TERT associates with myeloproliferative neoplasms. Leukemia 2014, 28, 1371–1374. [Google Scholar] [CrossRef] [Green Version]
- Hinds, D.A.; Barnholt, K.E.; Mesa, R.A.; Kiefer, A.K.; Do, C.B.; Eriksson, N.; Mountain, J.L.; Francke, U.; Tung, J.Y.; Nguyen, H.M.; et al. Germ line variants predispose to both JAK2 V617F clonal hematopoiesis and myeloproliferative neoplasms. Blood 2016, 128, 1121–1128. [Google Scholar] [CrossRef] [Green Version]
- Tapper, W.; Jones, A.V.; Kralovics, R.; Harutyunyan, A.S.; Zoi, K.; Leung, W.; Godfrey, A.L.; Guglielmelli, P.; Callaway, A.; Ward, D.; et al. Genetic variation at MECOM, TERT, JAK2 and HBS1L-MYB predisposes to myeloproliferative neoplasms. Nat. Commun. 2015, 6, 6691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bao, E.L.; Nandakumar, S.K.; Liao, X.; Bick, A.G.; Karjalainen, J.; Tabaka, M.; Gan, O.I.; Havulinna, A.S.; Kiiskinen, T.T.J.; Lareau, C.A.; et al. Inherited myeloproliferative neoplasm risk affects haematopoietic stem cells. Nature 2020, 586, 769–775. [Google Scholar] [CrossRef]
- Saliba, J.; Saint-Martin, C.; Di Stefano, A.; Lenglet, G.; Marty, C.; Keren, B.; Pasquier, F.; Valle, V.D.; Secardin, L.; Leroy, G.; et al. Germline duplication of ATG2B and GSKIP predisposes to familial myeloid malignancies. Nat. Genet. 2015, 47, 1131–1140. [Google Scholar] [CrossRef] [PubMed]
- Harutyunyan, A.S.; Giambruno, R.; Krendl, C.; Stukalov, A.; Klampfl, T.; Berg, T.; Chen, D.; Milosevic Feenstra, J.D.; Jager, R.; Gisslinger, B.; et al. Germline RBBP6 mutations in familial myeloproliferative neoplasms. Blood 2016, 127, 362–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirvonen, E.A.M.; Pitkanen, E.; Hemminki, K.; Aaltonen, L.A.; Kilpivaara, O. Whole-exome sequencing identifies novel candidate predisposition genes for familial polycythemia vera. Hum. Genom. 2017, 11, 6. [Google Scholar] [CrossRef] [PubMed]
- Anelli, L.; Zagaria, A.; Specchia, G.; Albano, F. The JAK2 GGCC (46/1) Haplotype in Myeloproliferative Neoplasms: Causal or Random? Int. J. Mol. Sci. 2018, 19, 1152. [Google Scholar] [CrossRef] [Green Version]
- Trifa, A.P.; Banescu, C.; Bojan, A.S.; Voina, C.M.; Popa, S.; Visan, S.; Ciubean, A.D.; Tripon, F.; Dima, D.; Popov, V.M.; et al. MECOM, HBS1L-MYB, THRB-RARB, JAK2, and TERT polymorphisms defining the genetic predisposition to myeloproliferative neoplasms: A study on 939 patients. Am. J. Hematol. 2018, 93, 100–106. [Google Scholar] [CrossRef] [Green Version]
- Nahajevszky, S.; Andrikovics, H.; Batai, A.; Adam, E.; Bors, A.; Csomor, J.; Gopcsa, L.; Koszarska, M.; Kozma, A.; Lovas, N.; et al. The prognostic impact of germline 46/1 haplotype of Janus kinase 2 in cytogenetically normal acute myeloid leukemia. Haematologica 2011, 96, 1613–1618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hermouet, S.; Vilaine, M. The JAK2 46/1 haplotype: A marker of inappropriate myelomonocytic response to cytokine stimulation, leading to increased risk of inflammation, myeloid neoplasm, and impaired defense against infection? Haematologica 2011, 96, 1575–1579. [Google Scholar] [CrossRef] [PubMed]
- Shay, J.W.; Wright, W.E. Telomerase: A target for cancer therapeutics. Cancer Cell 2002, 2, 257–265. [Google Scholar] [CrossRef] [Green Version]
- Blasco, M.A. Telomeres and human disease: Ageing, cancer and beyond. Nat. Rev. Genet. 2005, 6, 611–622. [Google Scholar] [CrossRef]
- Savage, S.A.; Alter, B.P. Dyskeratosis congenita. Hematol. Oncol. Clin. N. Am. 2009, 23, 215–231. [Google Scholar] [CrossRef]
- Maciejewski, J.P.; Selleri, C.; Sato, T.; Anderson, S.; Young, N.S. A severe and consistent deficit in marrow and circulating primitive hematopoietic cells (long-term culture-initiating cells) in acquired aplastic anemia. Blood 1996, 88, 1983–1991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldman, F.D.; Aubert, G.; Klingelhutz, A.J.; Hills, M.; Cooper, S.R.; Hamilton, W.S.; Schlueter, A.J.; Lambie, K.; Eaves, C.J.; Lansdorp, P.M. Characterization of primitive hematopoietic cells from patients with dyskeratosis congenita. Blood 2008, 111, 4523–4531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, P.; Gu, A.; Ji, G.; Zhao, L.; Zhao, P.; Lu, A. The TERT rs2736100 polymorphism and cancer risk: A meta-analysis based on 25 case-control studies. BMC Cancer 2012, 12, 7. [Google Scholar] [CrossRef] [PubMed]
- Mocellin, S.; Verdi, D.; Pooley, K.A.; Landi, M.T.; Egan, K.M.; Baird, D.M.; Prescott, J.; De Vivo, I.; Nitti, D. Telomerase reverse transcriptase locus polymorphisms and cancer risk: A field synopsis and meta-analysis. J. Natl. Cancer Inst. 2012, 104, 840–854. [Google Scholar] [CrossRef]
- Thompson, C.A.H.; Wong, J.M.Y. Non-canonical Functions of Telomerase Reverse Transcriptase: Emerging Roles and Biological Relevance. Curr. Top. Med. Chem. 2020, 20, 498–507. [Google Scholar] [CrossRef]
- Wang, F.; Fu, P.; Pang, Y.; Liu, C.; Shao, Z.; Zhu, J.; Li, J.; Wang, T.; Zhang, X.; Liu, J. TERT rs2736100T/G polymorphism upregulates interleukin 6 expression in non-small cell lung cancer especially in adenocarcinoma. Tumour Biol. 2014, 35, 4667–4672. [Google Scholar] [CrossRef]
- Masselli, E.; Pozzi, G.; Gobbi, G.; Merighi, S.; Gessi, S.; Vitale, M.; Carubbi, C. Cytokine Profiling in Myeloproliferative Neoplasms: Overview on Phenotype Correlation, Outcome Prediction, and Role of Genetic Variants. Cells 2020, 9, 2136. [Google Scholar] [CrossRef]
- Telomeres Mendelian Randomization, C.; Haycock, P.C.; Burgess, S.; Nounu, A.; Zheng, J.; Okoli, G.N.; Bowden, J.; Wade, K.H.; Timpson, N.J.; Evans, D.M.; et al. Association Between Telomere Length and Risk of Cancer and Non-Neoplastic Diseases: A Mendelian Randomization Study. JAMA Oncol. 2017, 3, 636–651. [Google Scholar] [CrossRef] [Green Version]
- Mangino, M.; Hwang, S.J.; Spector, T.D.; Hunt, S.C.; Kimura, M.; Fitzpatrick, A.L.; Christiansen, L.; Petersen, I.; Elbers, C.C.; Harris, T.; et al. Genome-wide meta-analysis points to CTC1 and ZNF676 as genes regulating telomere homeostasis in humans. Hum. Mol. Genet. 2012, 21, 5385–5394. [Google Scholar] [CrossRef] [Green Version]
- Codd, V.; Nelson, C.P.; Albrecht, E.; Mangino, M.; Deelen, J.; Buxton, J.L.; Hottenga, J.J.; Fischer, K.; Esko, T.; Surakka, I.; et al. Identification of seven loci affecting mean telomere length and their association with disease. Nat. Genet. 2013, 45, 422–427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levy, D.; Neuhausen, S.L.; Hunt, S.C.; Kimura, M.; Hwang, S.J.; Chen, W.; Bis, J.C.; Fitzpatrick, A.L.; Smith, E.; Johnson, A.D.; et al. Genome-wide association identifies OBFC1 as a locus involved in human leukocyte telomere biology. Proc. Natl. Acad. Sci. USA 2010, 107, 9293–9298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giaccherini, M.; Macauda, A.; Sgherza, N.; Sainz, J.; Gemignani, F.; Maldonado, J.M.S.; Jurado, M.; Tavano, F.; Mazur, G.; Jerez, A.; et al. Genetic polymorphisms associated with telomere length and risk of developing myeloproliferative neoplasms. Blood Cancer J. 2020, 10, 89. [Google Scholar] [CrossRef] [PubMed]
- Kustikova, O.S.; Schwarzer, A.; Stahlhut, M.; Brugman, M.H.; Neumann, T.; Yang, M.; Li, Z.; Schambach, A.; Heinz, N.; Gerdes, S.; et al. Activation of Evi1 inhibits cell cycle progression and differentiation of hematopoietic progenitor cells. Leukemia 2013, 27, 1127–1138. [Google Scholar] [CrossRef] [PubMed]
- Germeshausen, M.; Ancliff, P.; Estrada, J.; Metzler, M.; Ponstingl, E.; Rutschle, H.; Schwabe, D.; Scott, R.H.; Unal, S.; Wawer, A.; et al. MECOM-associated syndrome: A heterogeneous inherited bone marrow failure syndrome with amegakaryocytic thrombocytopenia. Blood Adv. 2018, 2, 586–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamazaki, H.; Suzuki, M.; Otsuki, A.; Shimizu, R.; Bresnick, E.H.; Engel, J.D.; Yamamoto, M. A remote GATA2 hematopoietic enhancer drives leukemogenesis in inv(3)(q21;q26) by activating EVI1 expression. Cancer Cell 2014, 25, 415–427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groschel, S.; Sanders, M.A.; Hoogenboezem, R.; de Wit, E.; Bouwman, B.A.M.; Erpelinck, C.; van der Velden, V.H.J.; Havermans, M.; Avellino, R.; van Lom, K.; et al. A single oncogenic enhancer rearrangement causes concomitant EVI1 and GATA2 deregulation in leukemia. Cell 2014, 157, 369–381. [Google Scholar] [CrossRef] [Green Version]
- Qayyum, R.; Snively, B.M.; Ziv, E.; Nalls, M.A.; Liu, Y.; Tang, W.; Yanek, L.R.; Lange, L.; Evans, M.K.; Ganesh, S.; et al. A meta-analysis and genome-wide association study of platelet count and mean platelet volume in african americans. PLoS Genet. 2012, 8, e1002491. [Google Scholar] [CrossRef] [Green Version]
- Menzel, S.; Jiang, J.; Silver, N.; Gallagher, J.; Cunningham, J.; Surdulescu, G.; Lathrop, M.; Farrall, M.; Spector, T.D.; Thein, S.L. The HBS1L-MYB intergenic region on chromosome 6q23.3 influences erythrocyte, platelet, and monocyte counts in humans. Blood 2007, 110, 3624–3626. [Google Scholar] [CrossRef] [Green Version]
- Ganesh, S.K.; Zakai, N.A.; van Rooij, F.J.; Soranzo, N.; Smith, A.V.; Nalls, M.A.; Chen, M.H.; Kottgen, A.; Glazer, N.L.; Dehghan, A.; et al. Multiple loci influence erythrocyte phenotypes in the CHARGE Consortium. Nat. Genet. 2009, 41, 1191–1198. [Google Scholar] [CrossRef] [Green Version]
- Soranzo, N.; Spector, T.D.; Mangino, M.; Kuhnel, B.; Rendon, A.; Teumer, A.; Willenborg, C.; Wright, B.; Chen, L.; Li, M.; et al. A genome-wide meta-analysis identifies 22 loci associated with eight hematological parameters in the HaemGen consortium. Nat. Genet. 2009, 41, 1182–1190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira, M.A.; Hottenga, J.J.; Warrington, N.M.; Medland, S.E.; Willemsen, G.; Lawrence, R.W.; Gordon, S.; de Geus, E.J.; Henders, A.K.; Smit, J.H.; et al. Sequence variants in three loci influence monocyte counts and erythrocyte volume. Am. J. Hum. Genet. 2009, 85, 745–749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thein, S.L.; Menzel, S.; Peng, X.; Best, S.; Jiang, J.; Close, J.; Silver, N.; Gerovasilli, A.; Ping, C.; Yamaguchi, M.; et al. Intergenic variants of HBS1L-MYB are responsible for a major quantitative trait locus on chromosome 6q23 influencing fetal hemoglobin levels in adults. Proc. Natl. Acad. Sci. USA 2007, 104, 11346–11351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uda, M.; Galanello, R.; Sanna, S.; Lettre, G.; Sankaran, V.G.; Chen, W.; Usala, G.; Busonero, F.; Maschio, A.; Albai, G.; et al. Genome-wide association study shows BCL11A associated with persistent fetal hemoglobin and amelioration of the phenotype of beta-thalassemia. Proc. Natl. Acad. Sci. USA 2008, 105, 1620–1625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lettre, G.; Sankaran, V.G.; Bezerra, M.A.; Araujo, A.S.; Uda, M.; Sanna, S.; Cao, A.; Schlessinger, D.; Costa, F.F.; Hirschhorn, J.N.; et al. DNA polymorphisms at the BCL11A, HBS1L-MYB, and beta-globin loci associate with fetal hemoglobin levels and pain crises in sickle cell disease. Proc. Natl. Acad. Sci. USA 2008, 105, 11869–11874. [Google Scholar] [CrossRef] [Green Version]
- Stadhouders, R.; Aktuna, S.; Thongjuea, S.; Aghajanirefah, A.; Pourfarzad, F.; van Ijcken, W.; Lenhard, B.; Rooks, H.; Best, S.; Menzel, S.; et al. HBS1L-MYB intergenic variants modulate fetal hemoglobin via long-range MYB enhancers. J. Clin. Investig. 2014, 124, 1699–1710. [Google Scholar] [CrossRef] [Green Version]
- Sankaran, V.G.; Menne, T.F.; Scepanovic, D.; Vergilio, J.A.; Ji, P.; Kim, J.; Thiru, P.; Orkin, S.H.; Lander, E.S.; Lodish, H.F. MicroRNA-15a and -16-1 act via MYB to elevate fetal hemoglobin expression in human trisomy 13. Proc. Natl. Acad. Sci. USA 2011, 108, 1519–1524. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Guo, S.; Ebert, B.L.; Zhang, H.; Peng, X.; Bosco, J.; Pretz, J.; Schlanger, R.; Wang, J.Y.; Mak, R.H.; et al. MicroRNA-mediated control of cell fate in megakaryocyte-erythrocyte progenitors. Dev. Cell 2008, 14, 843–853. [Google Scholar] [CrossRef] [Green Version]
- Garcia, P.; Clarke, M.; Vegiopoulos, A.; Berlanga, O.; Camelo, A.; Lorvellec, M.; Frampton, J. Reduced c-Myb activity compromises HSCs and leads to a myeloproliferation with a novel stem cell basis. EMBO J. 2009, 28, 1492–1504. [Google Scholar] [CrossRef]
- Pierini, T.; Di Giacomo, D.; Pierini, V.; Gorello, P.; Barba, G.; Lema Fernandez, A.G.; Pellanera, F.; Iannotti, T.; Falzetti, F.; La Starza, R.; et al. MYB deregulation from a EWSR1-MYB fusion at leukemic evolution of a JAK2 (V617F) positive primary myelofibrosis. Mol. Cytogenet. 2016, 9, 68. [Google Scholar] [CrossRef] [Green Version]
- Beauchemin, H.; Shooshtharizadeh, P.; Pinder, J.; Dellaire, G.; Moroy, T. Dominant negative Gfi1b mutations cause moderate thrombocytopenia and an impaired stress thrombopoiesis associated with mild erythropoietic abnormalities in mice. Haematologica 2020, 105, 2457–2470. [Google Scholar] [CrossRef] [Green Version]
- Rudd, M.F.; Sellick, G.S.; Webb, E.L.; Catovsky, D.; Houlston, R.S. Variants in the ATM-BRCA2-CHEK2 axis predispose to chronic lymphocytic leukemia. Blood 2006, 108, 638–644. [Google Scholar] [CrossRef]
- Tefferi, A.; Lasho, T.L.; Patnaik, M.M.; Finke, C.M.; Hussein, K.; Hogan, W.J.; Elliott, M.A.; Litzow, M.R.; Hanson, C.A.; Pardanani, A. JAK2 germline genetic variation affects disease susceptibility in primary myelofibrosis regardless of V617F mutational status: Nullizygosity for the JAK2 46/1 haplotype is associated with inferior survival. Leukemia 2010, 24, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Lasho, T.L.; Mudireddy, M.; Finke, C.M.; Hanson, C.A.; Ketterling, R.P.; Gangat, N.; Pardanani, A. The germline JAK2 GGCC (46/1) haplotype and survival among 414 molecularly-annotated patients with primary myelofibrosis. Am. J. Hematol. 2019, 94, 299–305. [Google Scholar] [CrossRef] [PubMed]
- Varricchio, L.; Masselli, E.; Alfani, E.; Battistini, A.; Migliaccio, G.; Vannucchi, A.M.; Zhang, W.; Rondelli, D.; Godbold, J.; Ghinassi, B.; et al. The dominant negative beta isoform of the glucocorticoid receptor is uniquely expressed in erythroid cells expanded from polycythemia vera patients. Blood 2011, 118, 425–436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poletto, V.; Rosti, V.; Villani, L.; Catarsi, P.; Carolei, A.; Campanelli, R.; Massa, M.; Martinetti, M.; Viarengo, G.; Malovini, A.; et al. A3669G polymorphism of glucocorticoid receptor is a susceptibility allele for primary myelofibrosis and contributes to phenotypic diversity and blast transformation. Blood 2012, 120, 3112–3117. [Google Scholar] [CrossRef] [Green Version]
- Masselli, E.; Carubbi, C.; Cambo, B.; Pozzi, G.; Gobbi, G.; Mirandola, P.; Follini, E.; Pagliaro, L.; Di Marcantonio, D.; Bonatti, F.; et al. The -2518 A/G polymorphism of the monocyte chemoattractant protein-1 as a candidate genetic predisposition factor for secondary myelofibrosis and biomarker of disease severity. Leukemia 2018, 32, 2266–2270. [Google Scholar] [CrossRef] [Green Version]
- Masselli, E.; Carubbi, C.; Pozzi, G.; Percesepe, A.; Campanelli, R.; Villani, L.; Gobbi, G.; Bonomini, S.; Roti, G.; Rosti, V.; et al. Impact of the rs1024611 Polymorphism of CCL2 on the Pathophysiology and Outcome of Primary Myelofibrosis. Cancers 2021, 13, 2552. [Google Scholar] [CrossRef] [PubMed]
- Ferrer-Marin, F.; Arroyo, A.B.; Bellosillo, B.; Cuenca, E.J.; Zamora, L.; Hernandez-Rivas, J.M.; Hernandez-Boluda, J.C.; Fernandez-Rodriguez, C.; Luno, E.; Garcia Hernandez, C.; et al. miR-146a rs2431697 identifies myeloproliferative neoplasm patients with higher secondary myelofibrosis progression risk. Leukemia 2020, 34, 2648–2659. [Google Scholar] [CrossRef]
- Zhou, J.; Cidlowski, J.A. The human glucocorticoid receptor: One gene, multiple proteins and diverse responses. Steroids 2005, 70, 407–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Derijk, R.H.; Schaaf, M.J.; Turner, G.; Datson, N.A.; Vreugdenhil, E.; Cidlowski, J.; de Kloet, E.R.; Emery, P.; Sternberg, E.M.; Detera-Wadleigh, S.D. A human glucocorticoid receptor gene variant that increases the stability of the glucocorticoid receptor beta-isoform mRNA is associated with rheumatoid arthritis. J. Rheumatol. 2001, 28, 2383–2388. [Google Scholar]
- Varricchio, L.; Migliaccio, A.R. The role of glucocorticoid receptor (GR) polymorphisms in human erythropoiesis. Am. J. Blood Res. 2014, 4, 53–72. [Google Scholar] [PubMed]
- Rovin, B.H.; Lu, L.; Saxena, R. A novel polymorphism in the MCP-1 gene regulatory region that influences MCP-1 expression. Biochem. Biophys. Res. Commun. 1999, 259, 344–348. [Google Scholar] [CrossRef] [PubMed]
- Pham, M.H.; Bonello, G.B.; Castiblanco, J.; Le, T.; Sigala, J.; He, W.; Mummidi, S. The rs1024611 regulatory region polymorphism is associated with CCL2 allelic expression imbalance. PLoS ONE 2012, 7, e49498. [Google Scholar] [CrossRef]
- Colobran, R.; Pujol-Borrell, R.; Armengol, M.P.; Juan, M. The chemokine network. II. On how polymorphisms and alternative splicing increase the number of molecular species and configure intricate patterns of disease susceptibility. Clin. Exp. Immunol. 2007, 150, 1–12. [Google Scholar] [CrossRef]
- McDermott, D.H.; Yang, Q.; Kathiresan, S.; Cupples, L.A.; Massaro, J.M.; Keaney, J.F., Jr.; Larson, M.G.; Vasan, R.S.; Hirschhorn, J.N.; O’Donnell, C.J.; et al. CCL2 polymorphisms are associated with serum monocyte chemoattractant protein-1 levels and myocardial infarction in the Framingham Heart Study. Circulation 2005, 112, 1113–1120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Yin, S.; Zheng, L.; Tang, W.; Kang, M.; Wei, W.; Sui, K. Relationship between the Monocyte Chemo-attractant Protein-1 gene rs1024611 A>G Polymorphism and Cancer Susceptibility: A Meta-analysis Involving 14,617 Subjects. Immunol. Investig. 2021, 50, 461–477. [Google Scholar] [CrossRef] [PubMed]
- Fisher, D.A.C.; Fowles, J.S.; Zhou, A.; Oh, S.T. Inflammatory Pathophysiology as a Contributor to Myeloproliferative Neoplasms. Front. Immunol. 2021, 12, 683401. [Google Scholar] [CrossRef] [PubMed]
- Karin, M.; Greten, F.R. NF-kappaB: Linking inflammation and immunity to cancer development and progression. Nat. Rev. Immunol. 2005, 5, 749–759. [Google Scholar] [CrossRef] [PubMed]
- Melgarejo, E.; Medina, M.A.; Sanchez-Jimenez, F.; Urdiales, J.L. Monocyte chemoattractant protein-1: A key mediator in inflammatory processes. Int. J. Biochem. Cell Biol. 2009, 41, 998–1001. [Google Scholar] [CrossRef] [PubMed]
- Boldin, M.P.; Taganov, K.D.; Rao, D.S.; Yang, L.; Zhao, J.L.; Kalwani, M.; Garcia-Flores, Y.; Luong, M.; Devrekanli, A.; Xu, J.; et al. miR-146a is a significant brake on autoimmunity, myeloproliferation, and cancer in mice. J. Exp. Med. 2011, 208, 1189–1201. [Google Scholar] [CrossRef]
- Prokunina-Olsson, L. Genetics of the Human Interferon Lambda Region. J. Interferon Cytokine Res. 2019, 39, 599–608. [Google Scholar] [CrossRef] [Green Version]
- Hasselbalch, H.C.; Holmstrom, M.O. Perspectives on interferon-alpha in the treatment of polycythemia vera and related myeloproliferative neoplasms: Minimal residual disease and cure? Semin. Immunopathol. 2019, 41, 5–19. [Google Scholar] [CrossRef] [Green Version]
- Lindgren, M.; Samuelsson, J.; Nilsson, L.; Knutsen, H.; Ghanima, W.; Westin, J.; Johansson, P.L.; Andreasson, B. Genetic variation in IL28B (IFNL3) and response to interferon-alpha treatment in myeloproliferative neoplasms. Eur. J. Haematol. 2018, 100, 419–425. [Google Scholar] [CrossRef]
- Jager, R.; Gisslinger, H.; Fuchs, E.; Bogner, E.; Milosevic Feenstra, J.D.; Weinzierl, J.; Schischlik, F.; Gisslinger, B.; Schalling, M.; Zorer, M.; et al. Germline genetic factors influence the outcome of interferon-alpha therapy in polycythemia vera. Blood 2021, 137, 387–391. [Google Scholar] [CrossRef]
- Kohnke, T.; Majeti, R. Clonal hematopoiesis: From mechanisms to clinical intervention. Cancer Discov. 2021. [Google Scholar] [CrossRef]
- Zink, F.; Stacey, S.N.; Norddahl, G.L.; Frigge, M.L.; Magnusson, O.T.; Jonsdottir, I.; Thorgeirsson, T.E.; Sigurdsson, A.; Gudjonsson, S.A.; Gudmundsson, J.; et al. Clonal hematopoiesis, with and without candidate driver mutations, is common in the elderly. Blood 2017, 130, 742–752. [Google Scholar] [CrossRef] [Green Version]
- Bick, A.G.; Weinstock, J.S.; Nandakumar, S.K.; Fulco, C.P.; Bao, E.L.; Zekavat, S.M.; Szeto, M.D.; Liao, X.; Leventhal, M.J.; Nasser, J.; et al. Inherited causes of clonal haematopoiesis in 97,691 whole genomes. Nature 2020, 586, 763–768. [Google Scholar] [CrossRef]
- Vannucchi, A.M.; Guglielmelli, P. The JAK2 46/1 (GGCC) MPN-predisposing haplotype: A risky haplotype, after all. Am. J. Hematol. 2019, 94, 283–285. [Google Scholar] [CrossRef] [Green Version]
- Liggett, L.A.; Sankaran, V.G. Unraveling Hematopoiesis through the Lens of Genomics. Cell 2020, 182, 1384–1400. [Google Scholar] [CrossRef]
- Laurenti, E.; Gottgens, B. From haematopoietic stem cells to complex differentiation landscapes. Nature 2018, 553, 418–426. [Google Scholar] [CrossRef]
- Bao, E.L.; Cheng, A.N.; Sankaran, V.G. The genetics of human hematopoiesis and its disruption in disease. EMBO Mol. Med. 2019, 11, e10316. [Google Scholar] [CrossRef]
- Yoshida, K.; Fujita, M. DNA damage responses that enhance resilience to replication stress. Cell Mol. Life Sci. 2021. [Google Scholar] [CrossRef]
- McPherson, K.S.; Korzhnev, D.M. Targeting protein-protein interactions in the DNA damage response pathways for cancer chemotherapy. RSC Chem. Biol. 2021, 2, 1167–1195. [Google Scholar] [CrossRef]
- Esposito, M.T.; So, C.W. DNA damage accumulation and repair defects in acute myeloid leukemia: Implications for pathogenesis, disease progression, and chemotherapy resistance. Chromosoma 2014, 123, 545–561. [Google Scholar] [CrossRef] [PubMed]
- Mehmood, A.; Kayani, M.A.; Ahmed, M.W.; Nisar, A.; Mahjabeen, I. Association between single nucleotide polymorphisms of DNA damage response pathway genes and increased risk in breast cancer. Future Oncol. 2020, 16, 1977–1995. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Deng, H.; Cui, H.; Fang, J.; Zuo, Z.; Deng, J.; Li, Y.; Wang, X.; Zhao, L. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget 2018, 9, 7204–7218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramamoorthy, S.; Cidlowski, J.A. Exploring the molecular mechanisms of glucocorticoid receptor action from sensitivity to resistance. Endocr. Dev. 2013, 24, 41–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
| SNPs | Gene Function (Relative to Hematopoiesis) | Associated Driver Mutations | Associated MPN Phenotype | Ref. |
|---|---|---|---|---|
| JAK246/1 haplotype | Hematopoiesis, cytokine receptor signaling | All (>JAK2V617F) | All (>PV and PMF) | [9,10,11,12,13] |
| TERT | Telomere length | All | All | |
| rs2736100 | [10,12] | |||
| rs7705526 | [11,13] | |||
| rs2853677 | [11,13] | |||
| MECOM | HSC maintenance, differentiation | JAK2V617F and CALR type 1/type 1-like | PV MF and ET (only in presence of CALR) | |
| rs2201862 | [12] | |||
| rs3851397 | [11] | |||
| rs9847631 | [13] | |||
| HBS1L-MYB rs9376092 | Peripheral blood cell counts, fetal hemoglobin levels | none | ET (only in presence of JAK2V617F) | [11,12] |
| GFI1B | HSC quiescence, erythroid and megakaryocytic differentiation | n/a | n/a | |
| rs621940 | [11] | |||
| rs1633768 | [13] | |||
| rs524137 | [13] | |||
| CHEK2 | DNA damage response | n/a | n/a | |
| rs555607708 | [11] | |||
| rs17879961 | [13] | |||
| SH2B3 rs7310615 | Negative regulation of normal hematopoiesis | n/a | n/a | [11,13] |
| ATM rs1800057 | DNA damage response | n/a | n/a | [11,13] |
| TET2 | HSC self-renewal, commitment, terminal differentiation of monocytes | n/a | n/a | |
| rs1548483 | [11] | |||
| rs62329718 | [13] | |||
| PINT rs58270997 | DNA damage response, hematopoietic stem cell maintenance, and differentiation (via PRC2) | n/a | n/a | [11,13] |
| THRB-RARB rs4858647 | unknown | none | PMF | [12] |
| GATA2 rs9864772 | HSC activity and self-renewal, myeloid and myelo-erythroid differentiation, erythroid precursors maintenance | n/a | n/a | [13] |
| SCHIP1 rs77249081 | unknown | n/a | n/a | [13] |
| KPNA4 rs74676712 | unknown | n/a | n/a | [13] |
| NUDT3 rs116466979 | unknown | n/a | n/a | [13] |
| MKLN1 rs61471615 | unknown | n/a | n/a | [13] |
| MRPS31 rs8002412 | unknown | n/a | n/a | [13] |
| ZNF521 rs9946154 | HSC differentiation and B-lymphoid cell development | n/a | n/a | [13] |
| RUNX1 rs55857134 | Differentiation of megakaryocytes and lymphocytes | n/a | n/a | [13] |
| SNPs | Gene Function (Relative to Hematopoiesis) | Detection Methods | Allele Variant | MPN Cohort | Disease Subtype Associations | Disease Phenotype Associations | Ref. |
|---|---|---|---|---|---|---|---|
| JAK2 46/1 haplotype rs12343867 (T/C) | Hematopoiesis, cytokine receptor signaling | RT-PCR | T allele (wild type) | 130 PMF | n/e | ↓ OS | [55] |
| RT-PCR | T allele (wild type) | 414 PMF | n/e | ↓ OS | [56] | ||
| NR3C1 rs6198 (A/G) | Immune response regulation, erythrocytosis | PCR-SSCP + sequencing | G-allele | 57 MPNs 22 CTRLs | PV | n/e | [57] |
| HRM analysis + sequencing | G-allele (homozygous) | 499 PMF 2948 CTRLs | PMF | ↑ CD34+ cells, splenomegaly, ↑WBC, ↓ LFS * | [58] | ||
| CCL2rs1024611 (A/G) | Chemokine production | RT-PCR | G-allele | 177 MPNs 149 CTRLs | sMF | ↓ Hb, ↑ IPSS, ↑ blasts, ↑ fibrosis | [59] |
| RT-PCR | G-allele (homozygous) | 773 PMF 323 CTRLs | PMF in males | ↓ OS | [60] | ||
| MIR146A rs2431697 (C/T) | NF-κB signaling modulation | RT-PCR | T-allele (homozygous) | 967 MPNs 600 CTRLs | sMF | ↓ MF-free survival in PV and ET | [61] |
| SNPs | Gene Function (Relative to Hematopoiesis) | Detection Methods | Allele Variant | MPN Cohort | Type of Therapy | Response Assessment | Association(s) | Ref. |
|---|---|---|---|---|---|---|---|---|
| INFL4 rs12979860 (T/C) | Cytokine production | RT-PCR | C/C | 100 MPNs | Inf α-2b, peg α-2b peg α-2a | HR | Higher HR rate in PVs | [76] |
| RT-PCR | C/C | 122 PVs | ropeg α-2b | HR, MR | Higher MR rate | [77] | ||
| INFL4 rs368234815 (G/TT) | RT-PCR | TT/TT | 122 PVs | ropeg α-2b | HR, MR | Higher MR rate | [77] | |
| INFL4 rs8099917 (T/G) | RT-PCR | T/T |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Masselli, E.; Pozzi, G.; Carubbi, C.; Vitale, M. The Genetic Makeup of Myeloproliferative Neoplasms: Role of Germline Variants in Defining Disease Risk, Phenotypic Diversity and Outcome. Cells 2021, 10, 2597. https://doi.org/10.3390/cells10102597
Masselli E, Pozzi G, Carubbi C, Vitale M. The Genetic Makeup of Myeloproliferative Neoplasms: Role of Germline Variants in Defining Disease Risk, Phenotypic Diversity and Outcome. Cells. 2021; 10(10):2597. https://doi.org/10.3390/cells10102597
Chicago/Turabian StyleMasselli, Elena, Giulia Pozzi, Cecilia Carubbi, and Marco Vitale. 2021. "The Genetic Makeup of Myeloproliferative Neoplasms: Role of Germline Variants in Defining Disease Risk, Phenotypic Diversity and Outcome" Cells 10, no. 10: 2597. https://doi.org/10.3390/cells10102597
APA StyleMasselli, E., Pozzi, G., Carubbi, C., & Vitale, M. (2021). The Genetic Makeup of Myeloproliferative Neoplasms: Role of Germline Variants in Defining Disease Risk, Phenotypic Diversity and Outcome. Cells, 10(10), 2597. https://doi.org/10.3390/cells10102597