
A Novel Calpain Inhibitor Compound Has Protective Effects on a Zebrafish Model of Spinocerebellar Ataxia Type 3

 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Transgenic SCA3 Zebrafish
2.2. Calpain Inhibitor Testing
2.3. Zebrafish Swimming Analysis
2.4. Flow Cytometric Analysis of Insoluble Ataxin-3
2.5. Western Blotting
2.6. Data Analysis
3. Results
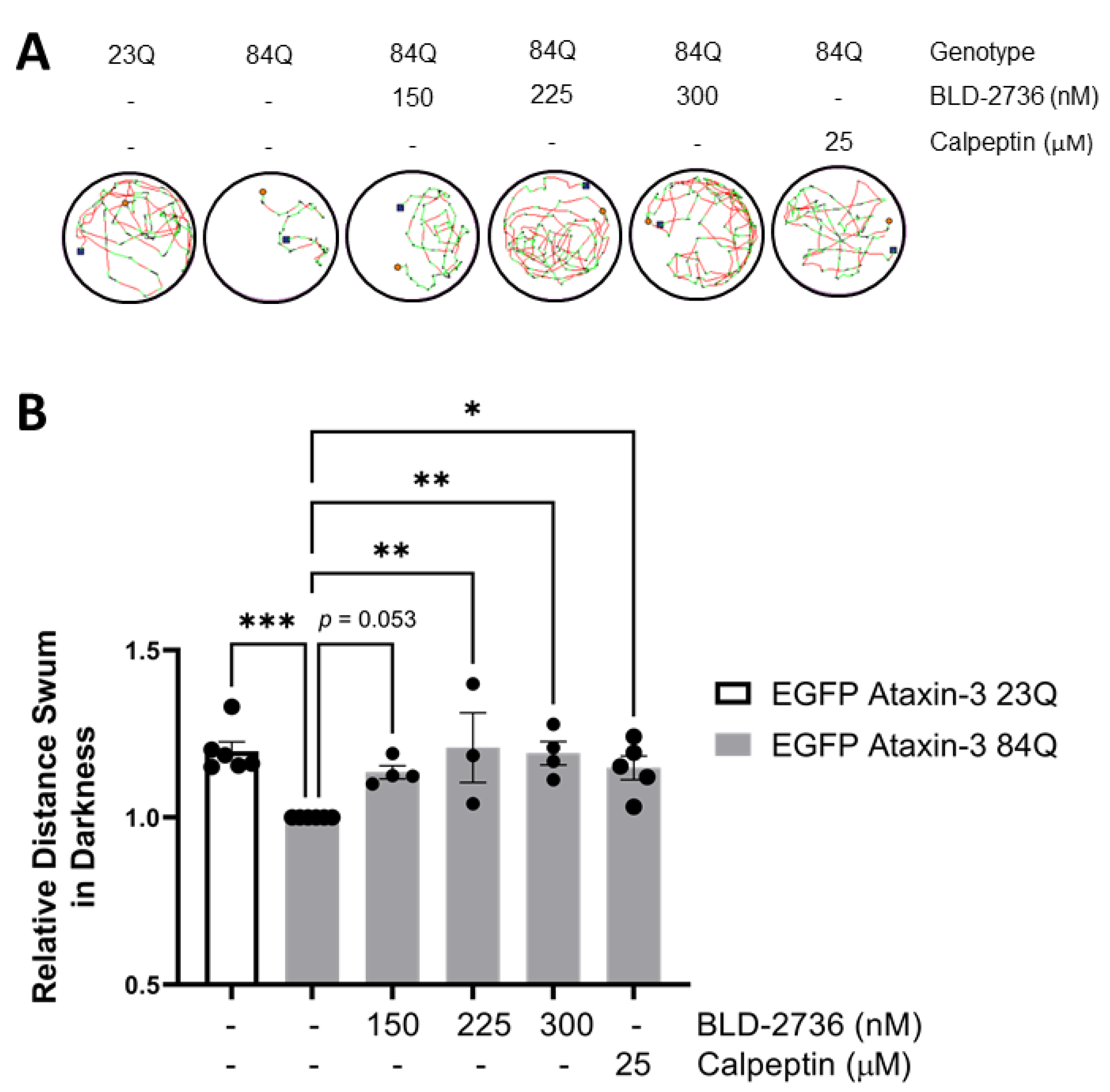
3.1. Treatment with BLD-2736 Improves Swimming of SCA3 Zebrafish Larvae
3.2. Treatment with BLD-2736 Decreases the Presence of Protein Aggregates and PolyQ Expanded Ataxin-3 in SCA3 Zebrafish
3.3. Delayed BLD-2736 Treatment Can Remove Formed Ataxin-3 Aggregates
3.4. Treatment with BLD-2736 Increased Synthesis of LC3II and Autophagic Activity
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- do Carmo Costa, M.; Paulson, H.L. Toward Understanding Machado-Joseph Disease. Prog. Neurobiol. 2012, 97, 239–257. [Google Scholar] [CrossRef] [Green Version]
- Durr, A.; Stevanin, G.; Cancel, G.; Duyckaerts, C.; Abbas, N.; Didierjean, O.; Chneiweiss, H.; Benomar, A.; Lyon-Caen, O.; Julien, J.; et al. Spinocerebellar ataxia 3 and Machado-Joseph disease: Clinical, molecular, and neuropathological features. Ann. Neurol. 1996, 39, 490–499. [Google Scholar] [CrossRef] [PubMed]
- Ranum, L.P.; Lundgren, J.K.; Schut, L.J.; Ahrens, M.J.; Perlman, S.; Aita, J.; Bird, T.D.; Gomez, C.; Orr, H.T. Spinocerebellar ataxia type 1 and Machado-Joseph disease: Incidence of CAG expansions among adult-onset ataxia patients from 311 families with dominant, recessive, or sporadic ataxia. Am. J. Hum. Genet. 1995, 57, 603–608. [Google Scholar] [PubMed]
- Schöls, L.; Vieira-Saecker, A.M.; Schöls, S.; Przuntek, H.; Epplen, J.T.; Riess, O. Trinucleotide expansion within the MJD1 gene presents clinically as spinocerebellar ataxia and occurs most frequently in German SCA patients. Hum. Mol. Genet. 1995, 4, 1001–1005. [Google Scholar] [CrossRef] [PubMed]
- Bettencourt, C.; Santos, C.; Montiel, R.; Kay, T.; Vasconcelos, J.; Maciel, P.; Lima, M. The (CAG)n tract of Machado–Joseph Disease gene (ATXN3): A comparison between DNA and mRNA in patients and controls. Eur. J. Hum. Genet. 2010, 18, 621–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gan, S.R.; Shi, S.S.; Wu, J.J.; Wang, N.; Zhao, G.X.; Weng, S.T.; Murong, S.X.; Lu, C.Z.; Wu, Z.Y. High frequency of Machado-Joseph disease identified in Southeastern Chinese kindreds with spinocerebellar ataxia. BMC Med. Genet. 2010, 11, 47. [Google Scholar] [CrossRef] [Green Version]
- Burt, T.; Currie, B.; Kilburn, C.; Lethlean, A.K.; Dempsey, K.; Blair, I.; Cohen, A.; Nicholson, G. Machado-Joseph disease in east Arnhem Land, Australia: Chromosome 14q32.1 expanded repeat confirmed in four families. Neurology 1996, 46, 1118–1122. [Google Scholar] [CrossRef] [PubMed]
- Durr, A. Autosomal dominant cerebellar ataxias: Polyglutamine expansions and beyond. Lancet Neurol. 2010, 9, 885–894. [Google Scholar] [CrossRef]
- Kawaguchi, Y.; Okamoto, T.; Taniwaki, M.; Aizawa, M.; Inoue, M.; Katayama, S.; Kawakami, H.; Nakamura, S.; Nishimura, M.; Akiguchi, I.; et al. CAG expansions in a novel gene for Machado-Joseph disease at chromosome 14q32.1. Nat. Genet. 1994, 8, 221–228. [Google Scholar] [CrossRef]
- Takiyama, Y.; Nishizawa, M.; Tanaka, H.; Kawashima, S.; Sakamoto, H.; Karube, Y.; Shimazaki, H.; Soutome, M.; Endo, K.; Ohta, S.; et al. The gene for Machado-Joseph disease maps to human chromosome 14q. Nat. Genet. 1993, 4, 300–304. [Google Scholar] [CrossRef]
- Matsumura, R.; Takayanagi, T.; Murata, K.; Futamura, N.; Hirano, M.; Ueno, S. Relationship of (CAG)nC configuration to repeat instability of the Machado-Joseph disease gene. Hum. Genet. 1996, 98, 643–645. [Google Scholar] [CrossRef] [PubMed]
- Abe, Y.; Tanaka, F.; Matsumoto, M.; Doyu, M.; Hirayama, M.; Kachi, T.; Sobue, G. CAG repeat number correlates with the rate of brainstem and cerebellar atrophy in Machado-Joseph disease. Neurology 1998, 51, 882. [Google Scholar] [CrossRef] [PubMed]
- Maciel, P.; Gaspar, C.; DeStefano, A.L.; Silveira, I.; Coutinho, P.; Radvany, J.; Dawson, D.M.; Sudarsky, L.; Guimarães, J.; Loureiro, J.E.; et al. Correlation between CAG repeat length and clinical features in Machado-Joseph disease. Am. J. Hum. Genet. 1995, 57, 54–61. [Google Scholar]
- Li, F.; Macfarlan, T.; Pittman, R.N.; Chakravarti, D. Ataxin-3 is a histone-binding protein with two independent transcriptional corepressor activities. J. Biol. Chem. 2002, 277, 45004–45012. [Google Scholar] [CrossRef] [Green Version]
- Evert, B.O.; Araujo, J.; Vieira-Saecker, A.M.; de Vos, R.A.; Harendza, S.; Klockgether, T.; Wüllner, U. Ataxin-3 represses transcription via chromatin binding, interaction with histone deacetylase 3, and histone deacetylation. J. Neurosci. 2006, 26, 11474–11486. [Google Scholar] [CrossRef]
- Evers, M.M.; Toonen, L.J.A.; van Roon-Mom, W.M.C. Ataxin-3 protein and RNA toxicity in spinocerebellar ataxia type 3: Current insights and emerging therapeutic strategies. Mol. Neurobiol. 2014, 49, 1513–1531. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Tang, T.S.; Tu, H.; Nelson, O.; Pook, M.; Hammer, R.; Nukina, N.; Bezprozvanny, I. Deranged Calcium Signaling and Neurodegeneration in Spinocerebellar Ataxia Type 3. J. Neurosci. 2008, 28, 12713. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Li, J.J.; Liu, S.; Zhao, J.; Jiang, Y.J.; Song, A.X.; Hu, H.Y. Aggregation of polyglutamine-expanded ataxin-3 sequesters its specific interacting partners into inclusions: Implication in a loss-of-function pathology. Sci. Rep. 2014, 4, 6410. [Google Scholar] [CrossRef] [Green Version]
- Rüb, U.; Schöls, L.; Paulson, H.; Auburger, G.; Kermer, P.; Jen, J.C.; Seidel, K.; Korf, H.W.; Deller, T. Clinical features, neurogenetics and neuropathology of the polyglutamine spinocerebellar ataxias type 1, 2, 3, 6 and 7. Prog. Neurobiol. 2013, 104, 38–66. [Google Scholar] [CrossRef] [PubMed]
- Rüb, U.; Brunt, E.R.; Deller, T. New insights into the pathoanatomy of spinocerebellar ataxia type 3 (Machado-Joseph disease). Curr. Opin. Neurol. 2008, 21, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Goti, D.; Katzen, S.M.; Mez, J.; Kurtis, N.; Kiluk, J.; Ben-Haïem, L.; Jenkins, N.A.; Copeland, N.G.; Kakizuka, A.; Sharp, A.H.; et al. A mutant ataxin-3 putative-cleavage fragment in brains of Machado-Joseph disease patients and transgenic mice is cytotoxic above a critical concentration. J. Neurosci. 2004, 24, 10266–10279. [Google Scholar] [CrossRef]
- Schmidt, T.; Landwehrmeyer, G.B.; Schmitt, I.; Trottier, Y.; Auburger, G.; Laccone, F.; Klockgether, T.; Völpel, M.; Epplen, J.T.; Schöls, L.; et al. An isoform of ataxin-3 accumulates in the nucleus of neuronal cells in affected brain regions of SCA3 patients. Brain Pathol. 1998, 8, 669–679. [Google Scholar] [CrossRef] [PubMed]
- Yamada, M.; Tsuji, S.; Takahashi, H. Pathology of CAG repeat diseases. Neuropathology 2000, 20, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Haacke, A.; Broadley, S.A.; Boteva, R.; Tzvetkov, N.; Hartl, F.U.; Breuer, P. Proteolytic cleavage of polyglutamine-expanded ataxin-3 is critical for aggregation and sequestration of non-expanded ataxin-3. Hum. Mol. Genet. 2006, 15, 555–568. [Google Scholar] [CrossRef] [PubMed]
- Ellisdon, A.M.; Thomas, B.; Bottomley, S.P. The Two-stage Pathway of Ataxin-3 Fibrillogenesis Involves a Polyglutamine-independent Step. J. Biol. Chem. 2006, 281, 16888–16896. [Google Scholar] [CrossRef] [Green Version]
- Bevivino, A.E.; Loll, P.J. An expanded glutamine repeat destabilizes native ataxin-3 structure and mediates formation of parallel β-fibrils. Proc. Natl. Acad. Sci. USA 2001, 98, 11955–11960. [Google Scholar] [CrossRef] [Green Version]
- Ikeda, H.; Yamaguchi, M.; Sugai, S.; Aze, Y.; Narumiya, S.; Kakizuka, A. Expanded polyglutamine in the Machado-Joseph disease protein induces cell death in vitro and in vivo. Nat. Genet. 1996, 13, 196–202. [Google Scholar] [CrossRef]
- Warrick, J.M.; Paulson, H.L.; Gray-Board, G.L.; Bui, Q.T.; Fischbeck, K.H.; Pittman, R.N.; Bonini, N.M. Expanded Polyglutamine Protein Forms Nuclear Inclusions and Causes Neural Degeneration in Drosophila. Cell 1998, 93, 939–949. [Google Scholar] [CrossRef] [Green Version]
- Morley, J.F.; Brignull, H.R.; Weyers, J.J.; Morimoto, R.I. The threshold for polyglutamine-expansion protein aggregation and cellular toxicity is dynamic and influenced by aging in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 2002, 99, 10417–10422. [Google Scholar] [CrossRef] [Green Version]
- Marsh, J.L.; Walker, H.; Theisen, H.; Zhu, Y.Z.; Fielder, T.; Purcell, J.; Thompson, L.M. Expanded polyglutamine peptides alone are intrinsically cytotoxic and cause neurodegeneration in Drosophila. Hum. Mol. Genet. 2000, 9, 13–25. [Google Scholar] [CrossRef] [Green Version]
- Schiffer, N.W.; Broadley, S.A.; Hirschberger, T.; Tavan, P.; Kretzschmar, H.A.; Giese, A.; Haass, C.; Hartl, F.U.; Schmid, B. Identification of anti-prion compounds as efficient inhibitors of polyglutamine protein aggregation in a zebrafish model. J. Biol. Chem. 2007, 282, 9195–9203. [Google Scholar] [CrossRef] [Green Version]
- Teixeira-Castro, A.; Ailion, M.; Jalles, A.; Brignull, H.R.; Vilaça, J.L.; Dias, N.; Rodrigues, P.; Oliveira, J.F.; Neves-Carvalho, A.; Morimoto, R.I.; et al. Neuron-specific proteotoxicity of mutant ataxin-3 in C. elegans: Rescue by the DAF-16 and HSF-1 pathways. Hum. Mol. Genet. 2011, 20, 2996–3009. [Google Scholar] [CrossRef] [Green Version]
- Graham, R.K.; Deng, Y.; Slow, E.J.; Haigh, B.; Bissada, N.; Lu, G.; Pearson, J.; Shehadeh, J.; Bertram, L.; Murphy, Z.; et al. Cleavage at the caspase-6 site is required for neuronal dysfunction and degeneration due to mutant huntingtin. Cell 2006, 125, 1179–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wellington, C.L.; Hayden, M.R. Of molecular interactions, mice and mechanisms: New insights into Huntington’s disease. Curr. Opin. Neurol. 1997, 10, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, A.J.; Neves-Carvalho, A.; Teixeira-Castro, A.; Rokka, A.; Corthals, G.; Logarinho, E.; Maciel, P. Absence of ataxin-3 leads to enhanced stress response in C. elegans. PLoS ONE 2011, 6, e18512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamashita, T.; Hideyama, T.; Hachiga, K.; Teramoto, S.; Takano, J.; Iwata, N.; Saido, T.C.; Kwak, S. A role for calpain-dependent cleavage of TDP-43 in amyotrophic lateral sclerosis pathology. Nat. Commun. 2012, 3, 1307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hübener, J.; Weber, J.J.; Richter, C.; Honold, L.; Weiss, A.; Murad, F.; Breuer, P.; Wüllner, U.; Bellstedt, P.; Paquet-Durand, F.; et al. Calpain-mediated ataxin-3 cleavage in the molecular pathogenesis of spinocerebellar ataxia type 3 (SCA3). Hum. Mol. Genet. 2013, 22, 508–518. [Google Scholar] [CrossRef] [Green Version]
- Koch, P.; Breuer, P.; Peitz, M.; Jungverdorben, J.; Kesavan, J.; Poppe, D.; Doerr, J.; Ladewig, J.; Mertens, J.; Tüting, T.; et al. Excitation-induced ataxin-3 aggregation in neurons from patients with Machado-Joseph disease. Nature 2011, 480, 543–546. [Google Scholar] [CrossRef]
- Bezprozvanny, I. Calcium signaling and neurodegenerative diseases. Trends Mol. Med. 2009, 15, 89–100. [Google Scholar] [CrossRef] [Green Version]
- Weber, J.J.; Haas, E.; Maringer, Y.; Hauser, S.; Casadei, N.L.; Chishti, A.H.; Riess, O.; Hübener-Schmid, J. Calpain-1 ablation partially rescues disease-associated hallmarks in models of Machado-Joseph disease. Hum. Mol. Genet. 2020, 29, 892–906. [Google Scholar] [CrossRef]
- Haacke, A.; Hartl, F.U.; Breuer, P. Calpain inhibition is sufficient to suppress aggregation of polyglutamine-expanded ataxin-3. J. Biol. Chem. 2007, 282, 18851–18856. [Google Scholar] [CrossRef] [Green Version]
- Simões, A.T.; Gonçalves, N.; Koeppen, A.; Déglon, N.; Kügler, S.; Duarte, C.B.; Pereira de Almeida, L. Calpastatin-mediated inhibition of calpains in the mouse brain prevents mutant ataxin 3 proteolysis, nuclear localization and aggregation, relieving Machado-Joseph disease. Brain 2012, 135, 2428–2439. [Google Scholar] [CrossRef]
- Watchon, M.; Yuan, K.C.; Mackovski, N.; Svahn, A.J.; Cole, N.J.; Goldsbury, C.; Rinkwitz, S.; Becker, T.S.; Nicholson, G.A.; Laird, A.S. Calpain Inhibition Is Protective in Machado-Joseph Disease Zebrafish Due to Induction of Autophagy. J. Neurosci. Off. J. Soc. Neurosci. 2017, 37, 7782–7794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simões, A.T.; Gonçalves, N.; Nobre, R.J.; Duarte, C.B.; Pereira de Almeida, L. Calpain inhibition reduces ataxin-3 cleavage alleviating neuropathology and motor impairments in mouse models of Machado-Joseph disease. Hum. Mol. Genet. 2014, 23, 4932–4944. [Google Scholar] [CrossRef] [PubMed]
- Robinson, K.J.; Tym, M.C.; Hogan, A.; Watchon, M.; Yuan, K.C.; Plenderleith, S.K.; Don, E.K.; Laird, A.S. Flow cytometry allows rapid detection of protein aggregates in cellular and zebrafish models of spinocerebellar ataxia 3. Dis. Models Mech. 2021. accepted. [Google Scholar]
- Menzies, F.M.; Garcia-Arencibia, M.; Imarisio, S.; O’Sullivan, N.C.; Ricketts, T.; Kent, B.A.; Rao, M.V.; Lam, W.; Green-Thompson, Z.W.; Nixon, R.A.; et al. Calpain inhibition mediates autophagy-dependent protection against polyglutamine toxicity. Cell Death Differ. 2015, 22, 433–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de la Fuente, S.; Sansa, A.; Hidalgo, I.; Vivancos, N.; Romero-Guevara, R.; Garcera, A.; Soler, R.M. Calpain system is altered in survival motor neuron-reduced cells from in vitro and in vivo spinal muscular atrophy models. Cell Death Dis. 2020, 11, 487. [Google Scholar] [CrossRef] [PubMed]
- Ong, S.B.; Lee, W.H.; Shao, N.Y.; Ismail, N.I.; Katwadi, K.; Lim, M.M.; Kwek, X.Y.; Michel, N.A.; Li, J.; Newson, J.; et al. Calpain Inhibition Restores Autophagy and Prevents Mitochondrial Fragmentation in a Human iPSC Model of Diabetic Endotheliopathy. Stem Cell Rep. 2019, 12, 597–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuro, M.; Yoshizawa, K.; Uehara, N.; Miki, H.; Takahashi, K.; Tsubura, A. Calpain Inhibition Restores Basal Autophagy and Suppresses MNU-induced Photoreceptor Cell Death in Mice. In Vivo 2011, 25, 617. [Google Scholar]
- Russo, R.; Berliocchi, L.; Adornetto, A.; Varano, G.P.; Cavaliere, F.; Nucci, C.; Rotiroti, D.; Morrone, L.A.; Bagetta, G.; Corasaniti, M.T. Calpain-mediated cleavage of Beclin-1 and autophagy deregulation following retinal ischemic injury in vivo. Cell Death Dis. 2011, 2, e144. [Google Scholar] [CrossRef] [Green Version]
- Watchon, M.; Luu, L.; Robinson, K.J.; Yuan, K.C.; De Luca, A.; Suddull, H.J.; Guillemin, G.; Cole, N.J.; Nicholson, G.A.; Chung, R.S.; et al. Sodium valproate increases activity of the sirtuin pathway resulting in beneficial effects for spinocerebellar ataxia-3 in vivo. Mol. Brain 2021, 14, 128. [Google Scholar] [CrossRef] [PubMed]
- Paquet, D.; Bhat, R.; Sydow, A.; Mandelkow, E.M.; Berg, S.; Hellberg, S.; Fälting, J.; Distel, M.; Köster, R.W.; Schmid, B.; et al. A zebrafish model of tauopathy allows in vivo imaging of neuronal cell death and drug evaluation. J. Clin. Investig. 2009, 119, 1382–1395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chai, Y.; Shao, J.; Miller, V.M.; Williams, A.; Paulson, H.L. Live-cell imaging reveals divergent intracellular dynamics of polyglutamine disease proteins and supports a sequestration model of pathogenesis. Proc. Natl. Acad. Sci. USA 2002, 99, 9310–9315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whiten, D.R.; San Gil, R.; McAlary, L.; Yerbury, J.J.; Ecroyd, H.; Wilson, M.R. Rapid flow cytometric measurement of protein inclusions and nuclear trafficking. Sci. Rep. 2016, 6, 31138. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Abdel-Aziz, A.K.; Abdelfatah, S.; Abdellatif, M.; Abdoli, A.; Abel, S.; Abeliovich, H.; Abildgaard, M.H.; Abudu, Y.P.; Acevedo-Arozena, A.; et al. Guidelines for the Use and Interpretation of Assays for Monitoring Autophagy, (4th edition). Autophagy 2021, 17, 1–382. [Google Scholar] [CrossRef]
- Weber, J.J.; Golla, M.; Guaitoli, G.; Wanichawan, P.; Hayer, S.N.; Hauser, S.; Krahl, A.C.; Nagel, M.; Samer, S.; Aronica, E.; et al. A combinatorial approach to identify calpain cleavage sites in the Machado-Joseph disease protein ataxin-3. Brain 2017, 140, 1280–1299. [Google Scholar] [CrossRef]
- Weber, J.J.; Pereira Sena, P.; Singer, E.; Nguyen, H.P. Killing Two Angry Birds with One Stone: Autophagy Activation by Inhibiting Calpains in Neurodegenerative Diseases and Beyond. BioMed Res. Int. 2019, 2019, 4741252. [Google Scholar] [CrossRef] [Green Version]
- Williams, A.; Sarkar, S.; Cuddon, P.; Ttofi, E.K.; Saiki, S.; Siddiqi, F.H.; Jahreiss, L.; Fleming, A.; Pask, D.; Goldsmith, P.; et al. Novel targets for Huntington’s disease in an mTOR-independent autophagy pathway. Nat. Chem. Biol. 2008, 4, 295–305. [Google Scholar] [CrossRef] [Green Version]
- Xia, H.G.; Zhang, L.; Chen, G.; Zhang, T.; Liu, J.; Jin, M.; Ma, X.; Ma, D.; Yuan, J. Control of basal autophagy by calpain1 mediated cleavage of ATG5. Autophagy 2010, 6, 61–66. [Google Scholar] [CrossRef] [Green Version]
- Yousefi, S.; Perozzo, R.; Schmid, I.; Ziemiecki, A.; Schaffner, T.; Scapozza, L.; Brunner, T.; Simon, H.U. Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis. Nat. Cell. Biol. 2006, 8, 1124–1132. [Google Scholar] [CrossRef]
- Chinskey, N.D.; Zheng, Q.-D.; Zacks, D.N. Control of photoreceptor autophagy after retinal detachment: The switch from survival to death. Investig. Ophthalmol. Vis. Sci. 2014, 55, 688–695. [Google Scholar] [CrossRef] [Green Version]
- Nascimento-Ferreira, I.; Santos-Ferreira, T.; Sousa-Ferreira, L.; Auregan, G.; Onofre, I.; Alves, S.; Dufour, N.; Colomer Gould, V.F.; Koeppen, A.; Déglon, N.; et al. Overexpression of the autophagic beclin-1 protein clears mutant ataxin-3 and alleviates Machado–Joseph disease. Brain 2011, 134, 1400–1415. [Google Scholar] [CrossRef] [PubMed]
- Onofre, I.; Mendonça, N.; Lopes, S.; Nobre, R.; de Melo, J.B.; Carreira, I.M.; Januário, C.; Gonçalves, A.F.; de Almeida, L.P. Fibroblasts of Machado Joseph Disease patients reveal autophagy impairment. Sci. Rep. 2016, 6, 28220. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.Y.; Wang, S.C.; Lei, M.; Wang, Z.; Xiong, K. Regulatory role of calpain in neuronal death. Neural Regen. Res. 2018, 13, 556–562. [Google Scholar] [PubMed]
- Ashkenazi, A.; Bento, C.F.; Ricketts, T.; Vicinanza, M.; Siddiqi, F.; Pavel, M.; Squitieri, F.; Hardenberg, M.C.; Imarisio, S.; Menzies, F.M.; et al. Polyglutamine tracts regulate beclin 1-dependent autophagy. Nature 2017, 545, 108–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsujinaka, T.; Kajiwara, Y.; Kambayashi, J.; Sakon, M.; Higuchi, N.; Tanaka, T.; Mori, T. Synthesis of a new cell penetrating calpain inhibitor (calpeptin). Biochem. Biophys. Res. Commun. 1988, 153, 1201–1208. [Google Scholar] [CrossRef]
- Vosler, P.S.; Brennan, C.S.; Chen, J. Calpain-Mediated Signaling Mechanisms in Neuronal Injury and Neurodegeneration. Mol. Neurobiol. 2008, 38, 78–100. [Google Scholar] [CrossRef] [Green Version]
- Bartus, R.T. The Calpain Hypothesis of Neurodegeneration: Evidence for a Common Cytotoxic Pathway. Neuroscientist 1997, 3, 314–327. [Google Scholar] [CrossRef]
- Yoshikawa, Y.; Mukai, H.; Hino, F.; Asada, K.; Kato, I. Isolation of Two Novel Genes, Down-regulated in Gastric Cancer. Jpn. J. Cancer Res. 2000, 91, 459–463. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.; Martin, S.G.; Patel, P.M.; Green, A.R.; Rakha, E.A.; Ellis, I.O.; Storr, S.J. Low calpain-9 is associated with adverse disease-specific survival following endocrine therapy in breast cancer. BMC Cancer 2014, 14, 995. [Google Scholar] [CrossRef] [Green Version]
- Peng, P.; Wu, W.; Zhao, J.; Song, S.; Wang, X.; Jia, D.; Shao, M.; Zhang, M.; Li, L.; Wang, L.; et al. Decreased expression of Calpain-9 predicts unfavorable prognosis in patients with gastric cancer. Sci. Rep. 2016, 6, 29604. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Robinson, K.J.; Yuan, K.; Plenderleith, S.K.; Watchon, M.; Laird, A.S. A Novel Calpain Inhibitor Compound Has Protective Effects on a Zebrafish Model of Spinocerebellar Ataxia Type 3. Cells 2021, 10, 2592. https://doi.org/10.3390/cells10102592
Robinson KJ, Yuan K, Plenderleith SK, Watchon M, Laird AS. A Novel Calpain Inhibitor Compound Has Protective Effects on a Zebrafish Model of Spinocerebellar Ataxia Type 3. Cells. 2021; 10(10):2592. https://doi.org/10.3390/cells10102592
Chicago/Turabian StyleRobinson, Katherine J., Kristy Yuan, Stuart K. Plenderleith, Maxinne Watchon, and Angela S. Laird. 2021. "A Novel Calpain Inhibitor Compound Has Protective Effects on a Zebrafish Model of Spinocerebellar Ataxia Type 3" Cells 10, no. 10: 2592. https://doi.org/10.3390/cells10102592