
Lipid Profiling of Alzheimer’s Disease Brain Highlights Enrichment in Glycerol(phospho)lipid, and Sphingolipid Metabolism


 , ,
, ,  ,
,  and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Tissue Samples
2.2. Chemicals and Internal Standards
2.3. Sample Preparation
2.4. LC-MS/MS Analysis
2.5. Statistical Analysis
2.5.1. Data Quality Control
2.5.2. Univariate and One-Way ANOVA Analysis
2.5.3. Machine Learning Models
3. Results
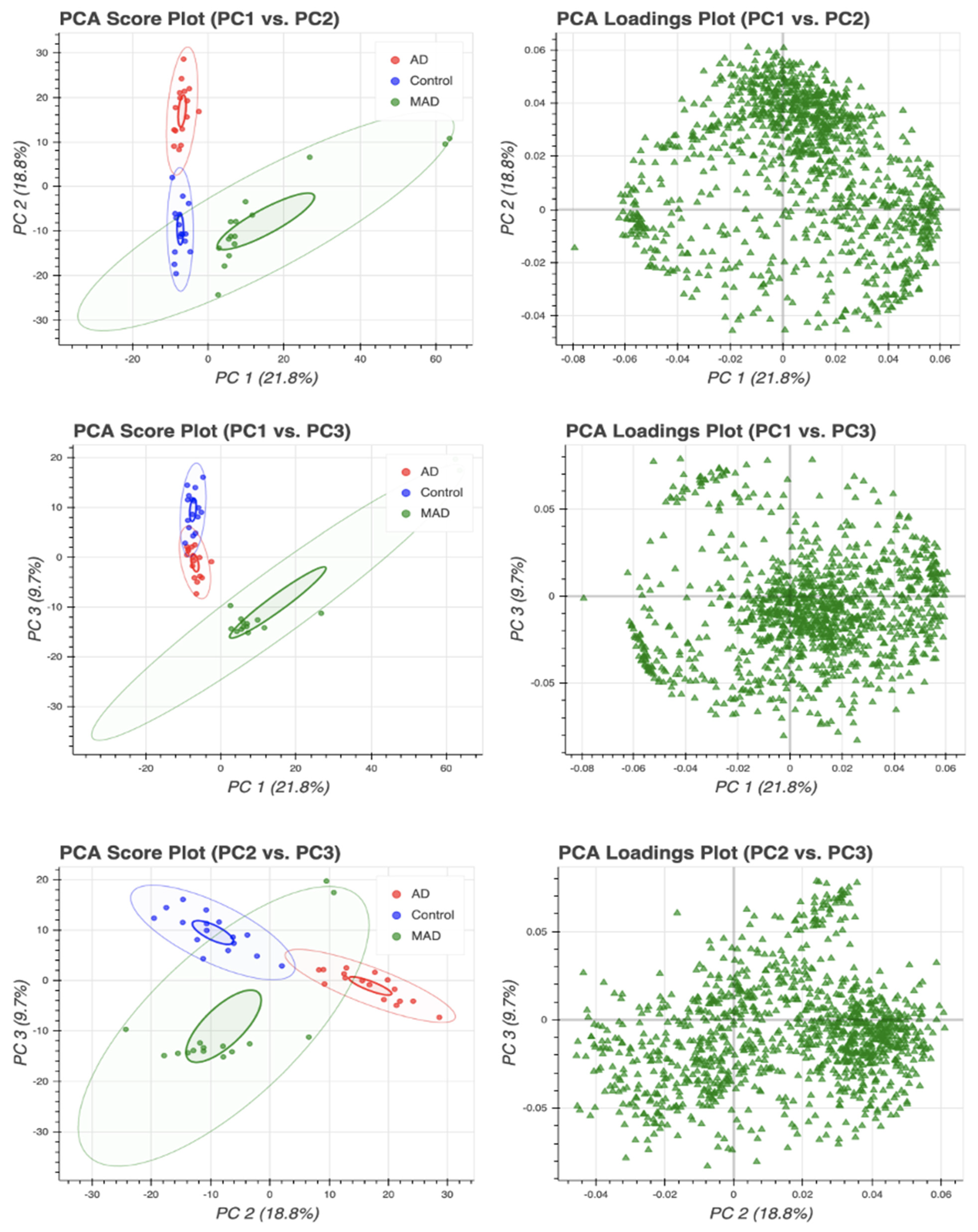
3.1. Principal Component Analysis (PCA)
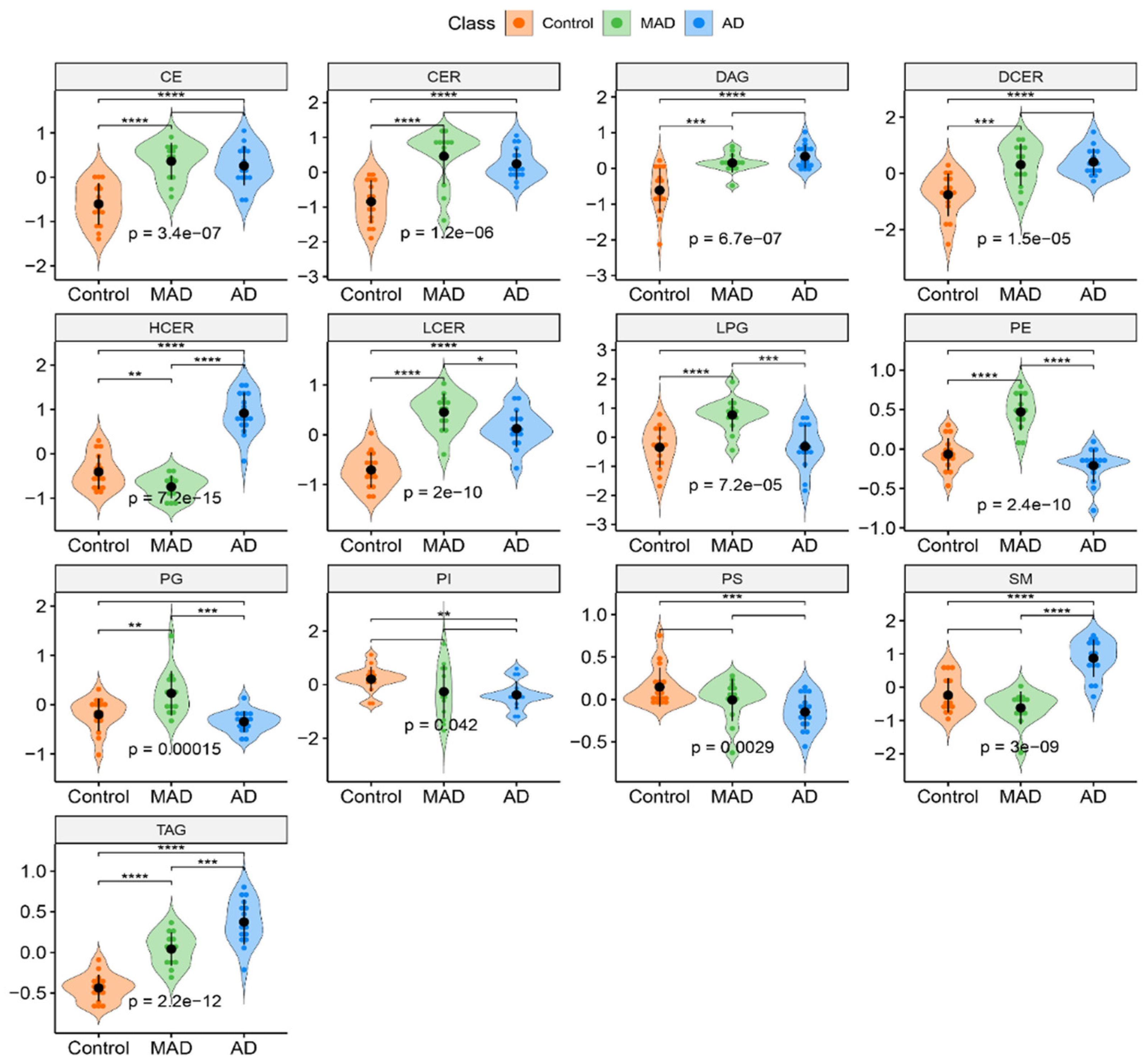
3.2. Overall Lipid Subclass Changes between the Groups
3.3. Diagnostic Performance of the Machine Learning Model
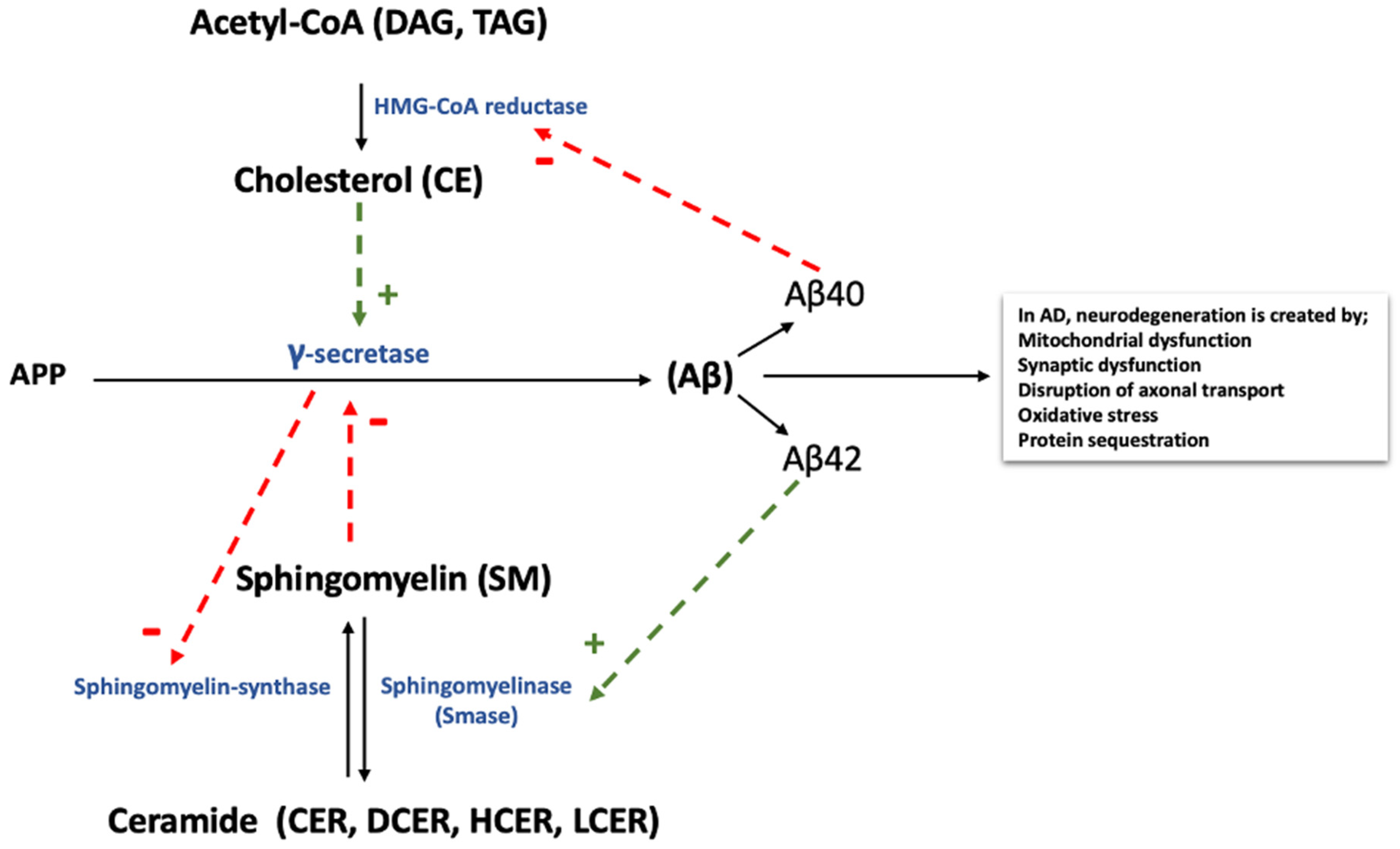
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Livingston, G.; Sommerlad, A.; Orgeta, V.; Costafreda, S.G.; Huntley, J.; Ames, D.; Ballard, C.; Banerjee, S.; Burns, A.; Cohen-Mansfield, J.; et al. Dementia prevention, intervention, and care. Lancet 2017, 390, 2673–2734. [Google Scholar] [CrossRef] [Green Version]
- Hebert, L.E.; Weuve, J.; Scherr, P.A.; Evans, D.A. Alzheimer disease in the United States (2010–2050) estimated using the 2010 census. Neurology 2013, 80, 1778–1783. [Google Scholar] [CrossRef] [Green Version]
- 2020 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2020, 16, 391–460. [CrossRef]
- McKhann, G.; Drachman, D.; Folstein, M.; Katzman, R.; Price, D.; Stadlan, E.M. Clinical diagnosis of Alzheimer’s disease: Report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology 1984, 34, 939–944. [Google Scholar] [CrossRef] [Green Version]
- Karran, E.; Mercken, M.; De Strooper, B. The amyloid cascade hypothesis for Alzheimer’s disease: An appraisal for the development of therapeutics. Nat. Rev. Drug Discov. 2011, 10, 698–712. [Google Scholar] [CrossRef]
- Savva, G.M.; Wharton, S.B.; Ince, P.G.; Forster, G.; Matthews, F.E.; Brayne, C. Age, neuropathology, and dementia. N. Engl. J. Med. 2009, 360, 2302–2309. [Google Scholar] [CrossRef] [PubMed]
- Shaw, L.M.; Vanderstichele, H.; Knapik-Czajka, M.; Clark, C.M.; Aisen, P.S.; Petersen, R.C.; Blennow, K.; Soares, H.; Simon, A.; Lewczuk, P.; et al. Cerebrospinal fluid biomarker signature in Alzheimer’s disease neuroimaging initiative subjects. Ann. Neurol. 2009, 65, 403–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, X.; Nasaruddin, M.B.; Elliott, C.T.; McGuinness, B.; Passmore, A.P.; Kehoe, P.G.; Hölscher, C.; McClean, P.L.; Graham, S.F.; Green, B.D. Alzheimer’s disease-like pathology has transient effects on the brain and blood metabolome. Neurobiol. Aging 2016, 38, 151–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKhann, G.M.; Knopman, D.S.; Chertkow, H.; Hyman, B.T.; Jack, C.R., Jr.; Kawas, C.H.; Klunk, W.E.; Koroshetz, W.J.; Manly, J.J.; Mayeux, R.; et al. The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2011, 7, 263–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jack, C.R., Jr.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2018, 14, 535–562. [Google Scholar] [CrossRef] [PubMed]
- Jack, C.R.; Wiste, H.J.; Weigand, S.D.; Therneau, T.M.; Lowe, V.J.; Knopman, D.S.; Botha, H.; Graff-Radford, J.; Jones, D.T.; Ferman, T.J.; et al. Predicting future rates of tau accumulation on PET. Brain 2020, 143, 3136–3150. [Google Scholar] [CrossRef]
- Wood, P.L. Lipidomics of Alzheimer’s disease: Current status. Alzheimer’s Res. Ther. 2012, 4, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Köfeler, H.C.; Fauland, A.; Rechberger, G.N.; Trötzmüller, M. Mass spectrometry based lipidomics: An overview of technological platforms. Metabolites 2012, 2, 19–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, M.W.; Braidy, N.; Poljak, A.; Sachdev, P.S. The application of lipidomics to biomarker research and pathomechanisms in Alzheimer’s disease. Curr. Opin. Psychiatry 2017, 30, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Nasaruddin, M.L.; Hölscher, C.; Kehoe, P.; Graham, S.F.; Green, B.D. Wide-ranging alterations in the brain fatty acid complement of subjects with late Alzheimer’s disease as detected by GC-MS. Am. J. Transl. Res. 2016, 8, 154–165. [Google Scholar] [PubMed]
- Nasaruddin, M.L.; Pan, X.; McGuinness, B.; Passmore, P.; Kehoe, P.G.; Hölscher, C.; Graham, S.F.; Green, B.D. Evidence That Parietal Lobe Fatty Acids May Be More Profoundly Affected in Moderate Alzheimer’s Disease (AD) Pathology Than in Severe AD Pathology. Metabolites 2018, 8, 69. [Google Scholar] [CrossRef] [Green Version]
- Zhang, A.H.; Ma, Z.M.; Kong, L.; Gao, H.L.; Sun, H.; Wang, X.Q.; Yu, J.B.; Han, Y.; Yan, G.L.; Wang, X.J. High-throughput lipidomics analysis to discover lipid biomarkers and profiles as potential targets for evaluating efficacy of Kai-Xin-San against APP/PS1 transgenic mice based on UPLC-Q/TOF-MS. Biomed. Chromatogr. BMC 2020, 34, e4724. [Google Scholar] [CrossRef]
- Barupal, D.K.; Baillie, R.; Fan, S.; Saykin, A.J.; Meikle, P.J.; Arnold, M.; Nho, K.; Fiehn, O.; Kaddurah-Daouk, R.; Alzheimer Disease Metabolomics Consortium. Sets of coregulated serum lipids are associated with Alzheimer’s disease pathophysiology. Alzheimer’s Dement. 2019, 11, 619–627. [Google Scholar] [CrossRef]
- Wood, P.L.; Barnette, B.L.; Kaye, J.A.; Quinn, J.F.; Woltjer, R.L. Non-targeted lipidomics of CSF and frontal cortex grey and white matter in control, mild cognitive impairment, and Alzheimer’s disease subjects. Acta Neuropsychiatr. 2015, 27, 270–278. [Google Scholar] [CrossRef]
- McKeith, I.G.; Dickson, D.W.; Lowe, J.; Emre, M.; O’Brien, J.T.; Feldman, H.; Cummings, J.; Duda, J.E.; Lippa, C.; Perry, E.K.; et al. Diagnosis and management of dementia with Lewy bodies: Third report of the DLB Consortium. Neurology 2005, 65, 1863–1872. [Google Scholar] [CrossRef] [Green Version]
- Montine, T.J.; Phelps, C.H.; Beach, T.G.; Bigio, E.H.; Cairns, N.J.; Dickson, D.W.; Duyckaerts, C.; Frosch, M.P.; Masliah, E.; Mirra, S.S.; et al. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: A practical approach. Acta Neuropathol. 2012, 123, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Angarita-Zapata, J.S.; Masegosa, A.D.; Triguero, I. General-Purpose Automated Machine Learning for Transportation: A Case Study of Auto-Sklearn for Traffic Forecasting; Springer: Cham, Switzerland, 2020; pp. 728–744. [Google Scholar]
- René de Cotret, L.P.; Otto, M.R.; Stern, M.J.; Siwick, B.J. An open-source software ecosystem for the interactive exploration of ultrafast electron scattering data. Adv. Struct. Chem. Imaging 2018, 4, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parsons, H.; Viant, M. Variance stabilising transformations for NMR metabolomics data. BMC Syst. Biol. 2007, 1, 22. [Google Scholar] [CrossRef] [Green Version]
- Lalwani, A.M.; Yilmaz, A.; Bisgin, H.; Ugur, Z.; Akyol, S.; Graham, S.F. The Biochemical Profile of Post-Mortem Brain from People Who Suffered from Epilepsy Reveals Novel Insights into the Etiopathogenesis of the Disease. Metabolites 2020, 10, 261. [Google Scholar] [CrossRef] [PubMed]
- Graham, S.F.; Turkoglu, O.; Yilmaz, A.; Ustun, I.; Ugur, Z.; Bjorndhal, T.; Han, B.; Mandal, R.; Wishart, D.; Bahado-Singh, R.O. Targeted metabolomics highlights perturbed metabolism in the brain of autism spectrum disorder sufferers. Metabolomics 2020, 16, 59. [Google Scholar] [CrossRef] [PubMed]
- Varma, V.R.; Oommen, A.M.; Varma, S.; Casanova, R.; An, Y.; Andrews, R.M.; O’Brien, R.; Pletnikova, O.; Troncoso, J.C.; Toledo, J.; et al. Brain and blood metabolite signatures of pathology and progression in Alzheimer disease: A targeted metabolomics study. PLoS Med. 2018, 15, e1002482. [Google Scholar] [CrossRef] [PubMed]
- Tajima, Y.; Ishikawa, M.; Maekawa, K.; Murayama, M.; Senoo, Y.; Nishimaki-Mogami, T.; Nakanishi, H.; Ikeda, K.; Arita, M.; Taguchi, R.; et al. Lipidomic analysis of brain tissues and plasma in a mouse model expressing mutated human amyloid precursor protein/tau for Alzheimer’s disease. Lipids Health Dis. 2013, 12, 68. [Google Scholar] [CrossRef] [Green Version]
- Dart, C. Lipid microdomains and the regulation of ion channel function. J. Physiol. 2010, 588, 3169–3178. [Google Scholar] [CrossRef] [PubMed]
- Schneider, A.; Schulz-Schaeffer, W.; Hartmann, T.; Schulz, J.B.; Simons, M. Cholesterol depletion reduces aggregation of amyloid-beta peptide in hippocampal neurons. Neurobiol. Dis. 2006, 23, 573–577. [Google Scholar] [CrossRef]
- Xiong, H.; Callaghan, D.; Jones, A.; Walker, D.G.; Lue, L.F.; Beach, T.G.; Sue, L.I.; Woulfe, J.; Xu, H.; Stanimirovic, D.B.; et al. Cholesterol retention in Alzheimer’s brain is responsible for high beta- and gamma-secretase activities and Abeta production. Neurobiol. Dis. 2008, 29, 422–437. [Google Scholar] [CrossRef] [Green Version]
- Grimm, M.O.; Mett, J.; Grimm, H.S.; Hartmann, T. APP Function and Lipids: A Bidirectional Link. Front. Mol. Neurosci. 2017, 10, 63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corder, E.H.; Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.E.; Gaskell, P.C.; Small, G.W.; Roses, A.D.; Haines, J.L.; Pericak-Vance, M.A. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 1993, 261, 921–923. [Google Scholar] [CrossRef] [PubMed]
- Martins, I.J.; Berger, T.; Sharman, M.J.; Verdile, G.; Fuller, S.J.; Martins, R.N. Cholesterol metabolism and transport in the pathogenesis of Alzheimer’s disease. J. Neurochem. 2009, 111, 1275–1308. [Google Scholar] [CrossRef] [PubMed]
- Zhao, N.; Liu, C.C.; Qiao, W.; Bu, G. Apolipoprotein E, Receptors, and Modulation of Alzheimer’s Disease. Biol. Psychiatry 2018, 83, 347–357. [Google Scholar] [CrossRef]
- Sato, N.; Morishita, R. The roles of lipid and glucose metabolism in modulation of β-amyloid, tau, and neurodegeneration in the pathogenesis of Alzheimer disease. Front. Aging Neurosci. 2015, 7, 199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bandaru, V.V.R.; Troncoso, J.; Wheeler, D.; Pletnikova, O.; Wang, J.; Conant, K.; Haughey, N.J. ApoE4 disrupts sterol and sphingolipid metabolism in Alzheimer’s but not normal brain. Neurobiol. Aging 2009, 30, 591–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Gaamouch, F.; Jing, P.; Xia, J.; Cai, D. Alzheimer’s Disease Risk Genes and Lipid Regulators. J. Alzheimer’s Dis. JAD 2016, 53, 15–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frisardi, V.; Panza, F.; Seripa, D.; Farooqui, T.; Farooqui, A.A. Glycerophospholipids and glycerophospholipid-derived lipid mediators: A complex meshwork in Alzheimer’s disease pathology. Prog. Lipid Res. 2011, 50, 313–330. [Google Scholar] [CrossRef] [PubMed]
- Shindou, H.; Shimizu, T. Acyl-CoA:lysophospholipid acyltransferases. J. Biol. Chem. 2009, 284, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Cutler, R.G.; Kelly, J.; Storie, K.; Pedersen, W.A.; Tammara, A.; Hatanpaa, K.; Troncoso, J.C.; Mattson, M.P. Involvement of oxidative stress-induced abnormalities in ceramide and cholesterol metabolism in brain aging and Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2004, 101, 2070–2075. [Google Scholar] [CrossRef] [Green Version]
- González-Domínguez, R.; García-Barrera, T.; Gómez-Ariza, J.L. Combination of metabolomic and phospholipid-profiling approaches for the study of Alzheimer’s disease. J. Proteom. 2014, 104, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Nitsch, R.; Pittas, A.; Blusztajn, J.K.; Slack, B.E.; Growdon, J.H.; Wurtman, R.J. Alterations of phospholipid metabolites in postmortem brain from patients with Alzheimer’s disease. Ann. N. Y. Acad. Sci. 1991, 640, 110–113. [Google Scholar] [CrossRef]
- Mulder, C.; Wahlund, L.O.; Teerlink, T.; Blomberg, M.; Veerhuis, R.; van Kamp, G.J.; Scheltens, P.; Scheffer, P.G. Decreased lysophosphatidylcholine/phosphatidylcholine ratio in cerebrospinal fluid in Alzheimer’s disease. J. Neural. Transm. 2003, 110, 949–955. [Google Scholar] [CrossRef] [PubMed]
- Igarashi, M.; Ma, K.; Gao, F.; Kim, H.W.; Rapoport, S.I.; Rao, J.S. Disturbed choline plasmalogen and phospholipid fatty acid concentrations in Alzheimer’s disease prefrontal cortex. J. Alzheimer’s Dis. JAD 2011, 24, 507–517. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.Y.; Tan, M.S.; Yu, J.T.; Tan, L. Role of pro-inflammatory cytokines released from microglia in Alzheimer’s disease. Ann. Transl. Med. 2015, 3, 136. [Google Scholar] [CrossRef] [PubMed]
- Wong, M.W.; Braidy, N.; Poljak, A.; Pickford, R.; Thambisetty, M.; Sachdev, P.S. Dysregulation of lipids in Alzheimer’s disease and their role as potential biomarkers. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2017, 13, 810–827. [Google Scholar] [CrossRef] [PubMed]
- Mielke, M.M.; Bandaru, V.V.R.; Haughey, N.J.; Xia, J.; Fried, L.P.; Yasar, S.; Albert, M.; Varma, V.; Harris, G.; Schneider, E.B.; et al. Serum ceramides increase the risk of Alzheimer disease: The Women’s Health and Aging Study II. Neurology 2012, 79, 633–641. [Google Scholar] [CrossRef] [Green Version]
- Han, X.; Holtzman, D.M.; McKeel, D.W., Jr. Plasmalogen deficiency in early Alzheimer’s disease subjects and in animal models: Molecular characterization using electrospray ionization mass spectrometry. J. Neurochem. 2001, 77, 1168–1180. [Google Scholar] [CrossRef]
- Posse de Chaves, E.; Sipione, S. Sphingolipids and gangliosides of the nervous system in membrane function and dysfunction. FEBS Lett. 2010, 584, 1748–1759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarasiuk, J.; Kapica-Topczewska, K.; Kułakowska, A.; Halicka, D.; Drozdowski, W.; Kornhuber, J.; Lewczuk, P. Increased concentration of the CSF Tau protein and its phosphorylated form in the late juvenile metachromatic leukodystrophy form: A case report. J. Neural. Transm. 2012, 119, 759–762. [Google Scholar] [CrossRef] [PubMed]
- Satoi, H.; Tomimoto, H.; Ohtani, R.; Kitano, T.; Kondo, T.; Watanabe, M.; Oka, N.; Akiguchi, I.; Furuya, S.; Hirabayashi, Y.; et al. Astroglial expression of ceramide in Alzheimer’s disease brains: A role during neuronal apoptosis. Neuroscience 2005, 130, 657–666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, X.M.; Holtzman, D.W.; McKeel, D.W., Jr.; Kelley, J.; Morris, J.C. Substantial sulfatide deficiency and ceramide elevation in very early Alzheimer’s disease: Potential role in disease pathogenesis. J. Neurochem. 2002, 82, 809–818. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Huang, Y.; Li, B.; Gong, C.X.; Schuchman, E.H. Deregulation of sphingolipid metabolism in Alzheimer’s disease. Neurobiol. Aging 2010, 31, 398–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patil, S.; Melrose, J.; Chan, C. Involvement of astroglial ceramide in palmitic acid-induced Alzheimer-like changes in primary neurons. Eur. J. Neurosci. 2007, 26, 2131–2141. [Google Scholar] [CrossRef] [PubMed]
- Mielke, M.M.; Haughey, N.J.; Bandaru, V.V.; Weinberg, D.D.; Darby, E.; Zaidi, N.; Pavlik, V.; Doody, R.S.; Lyketsos, C.G. Plasma sphingomyelins are associated with cognitive progression in Alzheimer’s disease. J. Alzheimer’s Dis. JAD 2011, 27, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Jazvinšćak Jembrek, M.; Hof, P.R.; Šimić, G. Ceramides in Alzheimer’s Disease: Key Mediators of Neuronal Apoptosis Induced by Oxidative Stress and Aβ Accumulation. Oxid. Med. Cell Longev. 2015, 2015, 346783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mullen, T.D.; Obeid, L.M. Ceramide and apoptosis: Exploring the enigmatic connections between sphingolipid metabolism and programmed cell death. Anti-Cancer Agents Med. Chem. 2012, 12, 340–363. [Google Scholar] [CrossRef]
- Katsel, P.; Li, C.; Haroutunian, V. Gene expression alterations in the sphingolipid metabolism pathways during progression of dementia and Alzheimer’s disease: A shift toward ceramide accumulation at the earliest recognizable stages of Alzheimer’s disease? Neurochem. Res. 2007, 32, 845–856. [Google Scholar] [CrossRef] [PubMed]
- Grimm, M.O.; Grimm, H.S.; Pätzold, A.J.; Zinser, E.G.; Halonen, R.; Duering, M.; Tschäpe, J.A.; De Strooper, B.; Müller, U.; Shen, J.; et al. Regulation of cholesterol and sphingomyelin metabolism by amyloid-beta and presenilin. Nat. Cell Biol. 2005, 7, 1118–1123. [Google Scholar] [CrossRef] [PubMed]
- Malaplate-Armand, C.; Florent-Béchard, S.; Youssef, I.; Koziel, V.; Sponne, I.; Kriem, B.; Leininger-Muller, B.; Olivier, J.-L.; Oster, T.; Pillot, T. Soluble oligomers of amyloid-beta peptide induce neuronal apoptosis by activating a cPLA2-dependent sphingomyelinase-ceramide pathway. Neurobiol. Dis. 2006, 23, 178–189. [Google Scholar] [CrossRef]
- Kosicek, M.; Zetterberg, H.; Andreasen, N.; Peter-Katalinic, J.; Hecimovic, S. Elevated cerebrospinal fluid sphingomyelin levels in prodromal Alzheimer’s disease. Neurosci. Lett. 2012, 516, 302–305. [Google Scholar] [CrossRef] [PubMed]
- Yanagisawa, K. GM1 ganglioside and Alzheimer’s disease. Glycoconj. J. 2015, 32, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Sevigny, J.; Chiao, P.; Bussière, T.; Weinreb, P.H.; Williams, L.; Maier, M.; Dunstan, R.; Salloway, S.; Chen, T.; Ling, Y.; et al. The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature 2016, 537, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Schneider, L. A resurrection of aducanumab for Alzheimer’s disease. Lancet Neurol. 2020, 19, 111–112. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
| Controls | Mild AD | AD | p-Value | |
|---|---|---|---|---|
| n | 16 | 15 | 15 | |
| Age, mean (SD) | 79.12 (6.28) | 84.573 (8.03) | 81.33 (6.51) | 0.3306 |
| Gender | ||||
| Male | 8 | 8 | 7 | 0.8704 |
| Female | 8 | 7 | 8 | |
| PMI in hours (SD) | 49.93 (0.45) | 38.96 (0.48) | 42.07 (0.47) | 0.3826 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Akyol, S.; Ugur, Z.; Yilmaz, A.; Ustun, I.; Gorti, S.K.K.; Oh, K.; McGuinness, B.; Passmore, P.; Kehoe, P.G.; Maddens, M.E.; et al. Lipid Profiling of Alzheimer’s Disease Brain Highlights Enrichment in Glycerol(phospho)lipid, and Sphingolipid Metabolism. Cells 2021, 10, 2591. https://doi.org/10.3390/cells10102591
Akyol S, Ugur Z, Yilmaz A, Ustun I, Gorti SKK, Oh K, McGuinness B, Passmore P, Kehoe PG, Maddens ME, et al. Lipid Profiling of Alzheimer’s Disease Brain Highlights Enrichment in Glycerol(phospho)lipid, and Sphingolipid Metabolism. Cells. 2021; 10(10):2591. https://doi.org/10.3390/cells10102591
Chicago/Turabian StyleAkyol, Sumeyya, Zafer Ugur, Ali Yilmaz, Ilyas Ustun, Santosh Kapil Kumar Gorti, Kyungjoon Oh, Bernadette McGuinness, Peter Passmore, Patrick G. Kehoe, Michael E. Maddens, and et al. 2021. "Lipid Profiling of Alzheimer’s Disease Brain Highlights Enrichment in Glycerol(phospho)lipid, and Sphingolipid Metabolism" Cells 10, no. 10: 2591. https://doi.org/10.3390/cells10102591
APA StyleAkyol, S., Ugur, Z., Yilmaz, A., Ustun, I., Gorti, S. K. K., Oh, K., McGuinness, B., Passmore, P., Kehoe, P. G., Maddens, M. E., Green, B. D., & Graham, S. F. (2021). Lipid Profiling of Alzheimer’s Disease Brain Highlights Enrichment in Glycerol(phospho)lipid, and Sphingolipid Metabolism. Cells, 10(10), 2591. https://doi.org/10.3390/cells10102591