
Lymphatic Clearance of Immune Cells in Cardiovascular Disease

 , ,
, ,  ,
,  and
and 
Abstract
1. Introduction
2. Immune Cells in the Heart
2.1. Neutrophils: The First Cells to Arrive at the Site of Infarction
2.2. Monocyte/Macrophages: Cells Involved in Both Inflammation and Its Resolution
2.3. Dendritic Cells: Regulators of Immune Tolerance during MI
2.4. T-Lymphocytes: CD4+ Helper T Cells
2.5. T-Lymphocytes: CD8+ Cytotoxic T Cells
2.6. B-Lymphocytes
3. Cardiac Lymphatic Network
3.1. Lymphatic Development in the Embryo
3.2. Cardiac Lymphatic Remodelling Following Myocardial Infarction
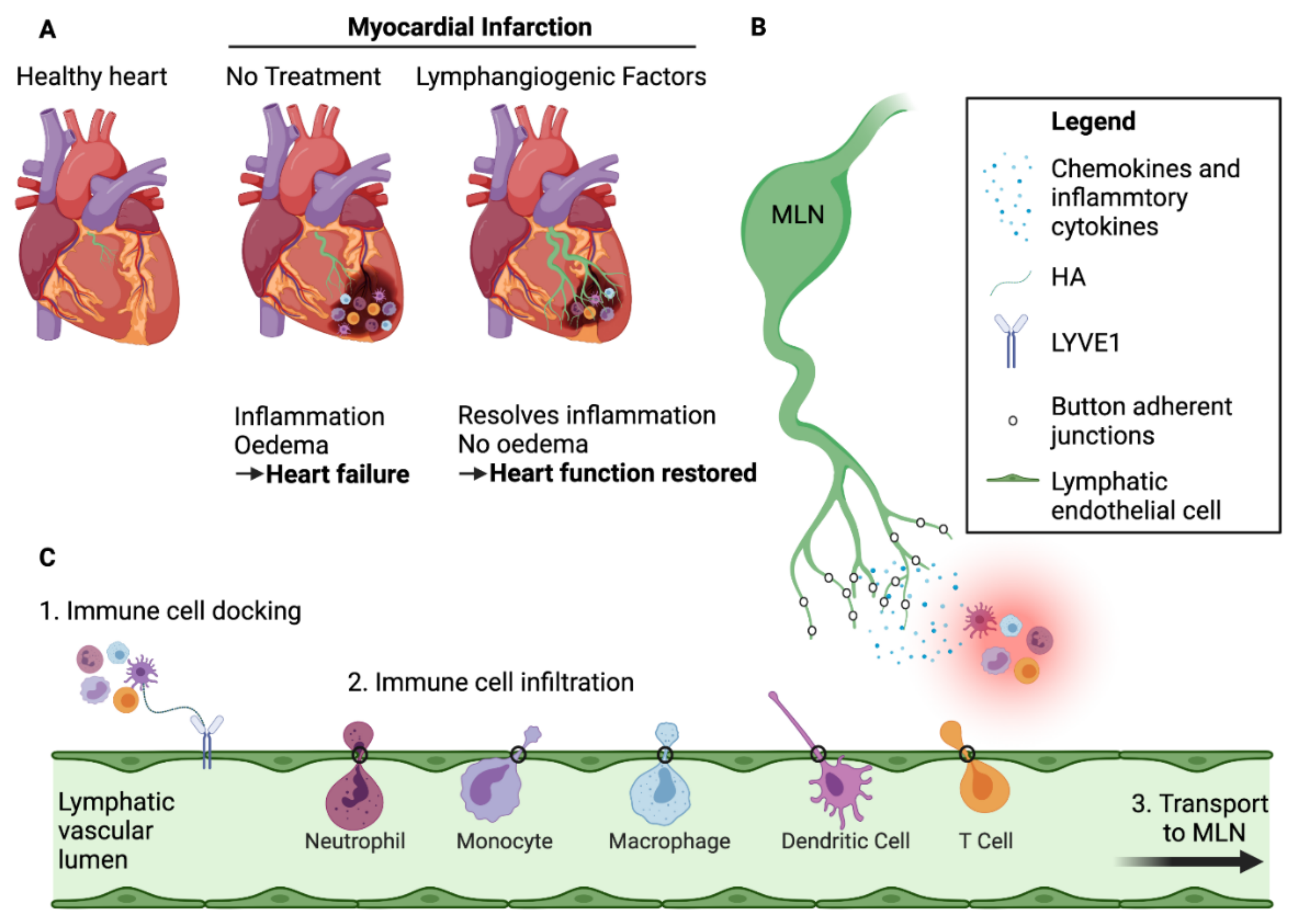
3.3. Exit of Immune Cells from the Infarcted Heart—The Role of Cardiac Lymphatics
3.4. Mechanisms of Immune Cell Exit via the Cardiac Lymphatics
4. Lymphangiogenic Therapy Post-MI
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Potente, M.; Mäkinen, T. Vascular heterogeneity and specialization in development and disease. Nat. Rev. Mol. Cell Biol. 2017, 18, 477–494. [Google Scholar] [CrossRef] [PubMed]
- Miyasaka, M.; Tanaka, T. Lymphocyte trafficking across high endothelial venules: Dogmas and enigmas. Nat. Rev. Immunol. 2004, 4, 360–370. [Google Scholar] [CrossRef] [PubMed]
- Gancz, D.; Perlmoter, G.; Yaniv, K. Formation and Growth of Cardiac Lymphatics during Embryonic Development, Heart Regeneration, and Disease. Cold Spring Harb. Perspect. Biol. 2019, 12, a037176. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Anzai, A.; Katsumata, Y.; Matsuhashi, T.; Ito, K.; Endo, J.; Yamamoto, T.; Takeshima, A.; Shinmura, K.; Shen, W.; et al. Temporal dynamics of cardiac immune cell accumulation following acute myocardial infarction. J. Mol. Cell Cardiol. 2013, 62, 24–35. [Google Scholar] [CrossRef]
- Rusinkevich, V.; Huang, Y.; Chen, Z.Y.; Qiang, W.; Wang, Y.G.; Shi, Y.F.; Yang, H.T. Temporal dynamics of immune response following prolonged myocardial ischemia/reperfusion with and without cyclosporine A. Acta Pharm. Sin 2019, 40, 1168–1183. [Google Scholar] [CrossRef]
- Daseke, M.J., 2nd; Chalise, U.; Becirovic-Agic, M.; Salomon, J.D.; Cook, L.M.; Case, A.J.; Lindsey, M.L. Neutrophil signaling during myocardial infarction wound repair. Cell Signal. 2021, 77, 109816. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Yabluchanskiy, A.; Iyer, R.P.; Cannon, P.L.; Flynn, E.R.; Jung, M.; Henry, J.; Cates, C.A.; Deleon-Pennell, K.Y.; Lindsey, M.L. Temporal neutrophil polarization following myocardial infarction. Cardiovasc. Res. 2016, 110, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Horckmans, M.; Ring, L.; Duchene, J.; Santovito, D.; Schloss, M.J.; Drechsler, M.; Weber, C.; Soehnlein, O.; Steffens, S. Neutrophils orchestrate post-myocardial infarction healing by polarizing macrophages towards a reparative phenotype. Eur. Heart J. 2017, 38, 187–197. [Google Scholar] [CrossRef]
- Dewald, O.; Zymek, P.; Winkelmann, K.; Koerting, A.; Ren, G.; Abou-Khamis, T.; Michael, L.H.; Rollins, B.J.; Entman, M.L.; Frangogiannis, N.G. CCL2/Monocyte Chemoattractant Protein-1 regulates inflammatory responses critical to healing myocardial infarcts. Circ. Res. 2005, 96, 881–889. [Google Scholar] [CrossRef]
- Nahrendorf, M.; Swirski, F.K.; Aikawa, E.; Stangenberg, L.; Wurdinger, T.; Figueiredo, J.L.; Libby, P.; Weissleder, R.; Pittet, M.J. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J. Exp. Med. 2007, 204, 3037–3047. [Google Scholar] [CrossRef]
- Elkenhans, B.; Protti, A.; Shah, A.; Onthank, D.; Botnar, R. Visualization of elastin using cardiac magnetic resonance imaging after myocardial infarction as inflammatory response. Sci. Rep. 2021, 11, 1–9. [Google Scholar] [CrossRef]
- Ma, Y.; Mouton, A.J.; Lindsey, M.L. Cardiac macrophage biology in the steady-state heart, the aging heart, and following myocardial infarction. Transl. Res. 2018, 191, 15–28. [Google Scholar] [CrossRef] [PubMed]
- Bajpai, G.; Bredemeyer, A.; Li, W.; Zaitsev, K.; Koenig, A.L.; Lokshina, I.; Mohan, J.; Ivey, B.; Hsiao, H.M.; Weinheimer, C.; et al. Tissue Resident CCR2- and CCR2+ Cardiac Macrophages Differentially Orchestrate Monocyte Recruitment and Fate Specification Following Myocardial Injury. Circ. Res. 2019, 124, 263–278. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Hsiao, H.M.; Higashikubo, R.; Saunders, B.T.; Bharat, A.; Goldstein, D.R.; Krupnick, A.S.; Gelman, A.E.; Lavine, K.J.; Kreisel, D. Heart-resident CCR2(+) macrophages promote neutrophil extravasation through TLR9/MyD88/CXCL5 signaling. JCI Insight 2016, 1. [Google Scholar] [CrossRef]
- Dick, S.A.; Macklin, J.A.; Nejat, S.; Momen, A.; Clemente-Casares, X.; Althagafi, M.G.; Chen, J.; Kantores, C.; Hosseinzadeh, S.; Aronoff, L.; et al. Self-renewing resident cardiac macrophages limit adverse remodeling following myocardial infarction. Nat. Immunol. 2019, 20, 29–39. [Google Scholar] [CrossRef] [PubMed]
- O’Rourke, S.A.; Dunne, A.; Monaghan, M.G. The Role of Macrophages in the Infarcted Myocardium: Orchestrators of ECM Remodeling. Front. Cardiovasc. Med. 2019, 6, 101. [Google Scholar] [CrossRef]
- Van Amerongen, M.J.; Harmsen, M.C.; van Rooijen, N.; Petersen, A.H.; van Luyn, M.J. Macrophage depletion impairs wound healing and increases left ventricular remodeling after myocardial injury in mice. Am. J. Pathol. 2007, 170, 818–829. [Google Scholar] [CrossRef] [PubMed]
- Simoes, F.C.; Cahill, T.J.; Kenyon, A.; Gavriouchkina, D.; Vieira, J.M.; Sun, X.; Pezzolla, D.; Ravaud, C.; Masmanian, E.; Weinberger, M.; et al. Macrophages directly contribute collagen to scar formation during zebrafish heart regeneration and mouse heart repair. Nat. Commun. 2020, 11, 600. [Google Scholar] [CrossRef]
- Shiraishi, M.; Shintani, Y.; Shintani, Y.; Ishida, H.; Saba, R.; Yamaguchi, A.; Adachi, H.; Yashiro, K.; Suzuki, K. Alternatively activated macrophages determine repair of the infarcted adult murine heart. J. Clin. Invest. 2016, 126, 2151–2166. [Google Scholar] [CrossRef]
- Lee, J.S.; Jeong, S.J.; Kim, S.; Chalifour, L.; Yun, T.J.; Miah, M.A.; Li, B.; Majdoubi, A.; Sabourin, A.; Keler, T.; et al. Conventional Dendritic Cells Impair Recovery after Myocardial Infarction. J. Immunol. 2018, 201, 1784–1798. [Google Scholar] [CrossRef]
- Van der Borght, K.; Scott, C.L.; Nindl, V.; Bouche, A.; Martens, L.; Sichien, D.; Van Moorleghem, J.; Vanheerswynghels, M.; De Prijck, S.; Saeys, Y.; et al. Myocardial Infarction Primes Autoreactive T Cells through Activation of Dendritic Cells. Cell Rep. 2017, 18, 3005–3017. [Google Scholar] [CrossRef]
- Anzai, A.; Anzai, T.; Nagai, S.; Maekawa, Y.; Naito, K.; Kaneko, H.; Sugano, Y.; Takahashi, T.; Abe, H.; Mochizuki, S.; et al. Regulatory role of dendritic cells in postinfarction healing and left ventricular remodeling. Circulation 2012, 125, 1234–1245. [Google Scholar] [CrossRef]
- Choo, E.H.; Lee, J.H.; Park, E.H.; Park, H.E.; Jung, N.C.; Kim, T.H.; Koh, Y.S.; Kim, E.; Seung, K.B.; Park, C.; et al. Infarcted Myocardium-Primed Dendritic Cells Improve Remodeling and Cardiac Function After Myocardial Infarction by Modulating the Regulatory T Cell and Macrophage Polarization. Circulation 2017, 135, 1444–1457. [Google Scholar] [CrossRef] [PubMed]
- Zhu, R.; Sun, H.; Yu, K.; Zhong, Y.; Shi, H.; Wei, Y.; Su, X.; Xu, W.; Luo, Q.; Zhang, F.; et al. Interleukin-37 and Dendritic Cells Treated With Interleukin-37 Plus Troponin I Ameliorate Cardiac Remodeling After Myocardial Infarction. J. Am. Heart Assoc. 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, U.; Beyersdorf, N.; Weirather, J.; Podolskaya, A.; Bauersachs, J.; Ertl, G.; Kerkau, T.; Frantz, S. Activation of CD4+ T lymphocytes improves wound healing and survival after experimental myocardial infarction in mice. Circulation 2012, 125, 1652–1663. [Google Scholar] [CrossRef]
- Yang, Z.; Day, Y.J.; Toufektsian, M.C.; Xu, Y.; Ramos, S.I.; Marshall, M.A.; French, B.A.; Linden, J. Myocardial infarct-sparing effect of adenosine A2A receptor activation is due to its action on CD4+ T lymphocytes. Circulation 2006, 114, 2056–2064. [Google Scholar] [CrossRef]
- Weirather, J.; Hofmann, U.D.; Beyersdorf, N.; Ramos, G.C.; Vogel, B.; Frey, A.; Ertl, G.; Kerkau, T.; Frantz, S. Foxp3+ CD4+ T cells improve healing after myocardial infarction by modulating monocyte/macrophage differentiation. Circ. Res. 2014, 115, 55–67. [Google Scholar] [CrossRef]
- Xia, N.; Lu, Y.; Gu, M.; Li, N.; Liu, M.; Jiao, J.; Zhu, Z.; Li, J.; Li, D.; Tang, T.; et al. A Unique Population of Regulatory T Cells in Heart Potentiates Cardiac Protection From Myocardial Infarction. Circulation 2020, 142, 1956–1973. [Google Scholar] [CrossRef] [PubMed]
- Zacchigna, S.; Martinelli, V.; Moimas, S.; Colliva, A.; Anzini, M.; Nordio, A.; Costa, A.; Rehman, M.; Vodret, S.; Pierro, C.; et al. Paracrine effect of regulatory T cells promotes cardiomyocyte proliferation during pregnancy and after myocardial infarction. Nat. Commun. 2018, 9, 1–12. [Google Scholar] [CrossRef]
- Santos-Zas, I.; Lemarie, J.; Zlatanova, I.; Cachanado, M.; Seghezzi, J.C.; Benamer, H.; Goube, P.; Vandestienne, M.; Cohen, R.; Ezzo, M.; et al. Cytotoxic CD8(+) T cells promote granzyme B-dependent adverse post-ischemic cardiac remodeling. Nat. Commun. 2021, 12, 1–13. [Google Scholar] [CrossRef]
- Ilatovskaya, D.V.; Pitts, C.; Clayton, J.; Domondon, M.; Troncoso, M.; Pippin, S.; DeLeon-Pennell, K.Y. CD8(+) T-cells negatively regulate inflammation post-myocardial infarction. Am. J. Physiol. Heart Circ. Physiol. 2019, 317, H581–H596. [Google Scholar] [CrossRef] [PubMed]
- Zouggari, Y.; Ait-Oufella, H.; Bonnin, P.; Simon, T.; Sage, A.P.; Guerin, C.; Vilar, J.; Caligiuri, G.; Tsiantoulas, D.; Laurans, L.; et al. B lymphocytes trigger monocyte mobilization and impair heart function after acute myocardial infarction. Nat. Med. 2013, 19, 1273–1280. [Google Scholar] [CrossRef]
- Mo, F.; Luo, Y.; Yan, Y.; Li, J.; Lai, S.; Wu, W. Are activated B cells involved in the process of myocardial fibrosis after acute myocardial infarction? An in vivo experiment. BMC Cardiovasc. Disord. 2021, 21, 1–14. [Google Scholar] [CrossRef]
- Adamo, L.; Staloch, L.J.; Rocha-Resende, C.; Matkovich, S.J.; Jiang, W.; Bajpai, G.; Weinheimer, C.J.; Kovacs, A.; Schilling, J.D.; Barger, P.M.; et al. Modulation of subsets of cardiac B lymphocytes improves cardiac function after acute injury. JCI Insight 2018, 3. [Google Scholar] [CrossRef]
- Pittman, K.; Kubes, P. Damage-associated molecular patterns control neutrophil recruitment. J. Innate Immun. 2013, 5, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Arslan, F.; de Kleijn, D.P.; Pasterkamp, G. Innate immune signaling in cardiac ischemia. Nat. Rev. Cardiol 2011, 8, 292–300. [Google Scholar] [CrossRef]
- Prince, L.R.; Whyte, M.K.; Sabroe, I.; Parker, L.C. The role of TLRs in neutrophil activation. Curr. Opin. Pharmacol. 2011, 11, 397–403. [Google Scholar] [CrossRef]
- Chia, S.; Nagurney, J.T.; Brown, D.F.; Raffel, O.C.; Bamberg, F.; Senatore, F.; Wackers, F.J.; Jang, I.K. Association of leukocyte and neutrophil counts with infarct size, left ventricular function and outcomes after percutaneous coronary intervention for ST-elevation myocardial infarction. Am. J. Cardiol. 2009, 103, 333–337. [Google Scholar] [CrossRef] [PubMed]
- Honold, L.; Nahrendorf, M. Resident and Monocyte-Derived Macrophages in Cardiovascular Disease. Circ. Res. 2018, 122, 113–127. [Google Scholar] [CrossRef] [PubMed]
- Epelman, S.; Lavine, K.J.; Beaudin, A.E.; Sojka, D.K.; Carrero, J.A.; Calderon, B.; Brija, T.; Gautier, E.L.; Ivanov, S.; Satpathy, A.T.; et al. Embryonic and adult-derived resident cardiac macrophages are maintained through distinct mechanisms at steady state and during inflammation. Immunity 2014, 40, 91–104. [Google Scholar] [CrossRef]
- Gomez Perdiguero, E.; Klapproth, K.; Schulz, C.; Busch, K.; Azzoni, E.; Crozet, L.; Garner, H.; Trouillet, C.; de Bruijn, M.F.; Geissmann, F.; et al. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature 2015, 518, 547–551. [Google Scholar] [CrossRef] [PubMed]
- Heidt, T.; Courties, G.; Dutta, P.; Sager, H.B.; Sebas, M.; Iwamoto, Y.; Sun, Y.; Da Silva, N.; Panizzi, P.; van der Laan, A.M.; et al. Differential contribution of monocytes to heart macrophages in steady-state and after myocardial infarction. Circ. Res. 2014, 115, 284–295. [Google Scholar] [CrossRef] [PubMed]
- Leid, J.; Carrelha, J.; Boukarabila, H.; Epelman, S.; Jacobsen, S.E.; Lavine, K.J. Primitive Embryonic Macrophages are Required for Coronary Development and Maturation. Circ. Res. 2016, 118, 1498–1511. [Google Scholar] [CrossRef] [PubMed]
- Cahill, T.J.; Sun, X.; Ravaud, C.; Villa Del Campo, C.; Klaourakis, K.; Lupu, I.E.; Lord, A.M.; Browne, C.; Jacobsen, S.E.W.; Greaves, D.R.; et al. Tissue-resident macrophages regulate lymphatic vessel growth and patterning in the developing heart. Development 2021, 148. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G.; Mendoza, L.H.; Ren, G.; Akrivakis, S.; Jackson, P.L.; Michael, L.H.; Smith, C.W.; Entman, M.L. MCSF expression is induced in healing myocardial infarcts and may regulate monocyte and endothelial cell phenotype. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, H483–H492. [Google Scholar] [CrossRef]
- Peet, C.; Ivetic, A.; Bromage, D.I.; Shah, A.M. Cardiac monocytes and macrophages after myocardial infarction. Cardiovasc. Res. 2020, 116, 1101–1112. [Google Scholar] [CrossRef]
- Molenaar, B.; Timmer, L.T.; Droog, M.; Perini, I.; Versteeg, D.; Kooijman, L.; Monshouwer-Kloots, J.; de Ruiter, H.; Gladka, M.M.; van Rooij, E. Single-cell transcriptomics following ischemic injury identifies a role for B2M in cardiac repair. Commun. Biol. 2021, 4, 1–15. [Google Scholar] [CrossRef]
- Frangogiannis, N.G.; Ren, G.; Dewald, O.; Zymek, P.; Haudek, S.; Koerting, A.; Winkelmann, K.; Michael, L.H.; Lawler, J.; Entman, M.L. Critical role of endogenous thrombospondin-1 in preventing expansion of healing myocardial infarcts. Circulation 2005, 111, 2935–2942. [Google Scholar] [CrossRef]
- Dobaczewski, M.; Bujak, M.; Li, N.; Gonzalez-Quesada, C.; Mendoza, L.H.; Wang, X.F.; Frangogiannis, N.G. Smad3 signaling critically regulates fibroblast phenotype and function in healing myocardial infarction. Circ. Res. 2010, 107, 418–428. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; de Castro Bras, L.E.; Toba, H.; Iyer, R.P.; Hall, M.E.; Winniford, M.D.; Lange, R.A.; Tyagi, S.C.; Lindsey, M.L. Myofibroblasts and the extracellular matrix network in post-myocardial infarction cardiac remodeling. Pflug. Arch. 2014, 466, 1113–1127. [Google Scholar] [CrossRef]
- Satoh, T.; Kidoya, H.; Naito, H.; Yamamoto, M.; Takemura, N.; Nakagawa, K.; Yoshioka, Y.; Morii, E.; Takakura, N.; Takeuchi, O.; et al. Critical role of Trib1 in differentiation of tissue-resident M2-like macrophages. Nature 2013, 495, 524–528. [Google Scholar] [CrossRef] [PubMed]
- Al-Darraji, A.; Haydar, D.; Chelvarajan, L.; Tripathi, H.; Levitan, B.; Gao, E.; Venditto, V.J.; Gensel, J.C.; Feola, D.J.; Abdel-Latif, A. Azithromycin therapy reduces cardiac inflammation and mitigates adverse cardiac remodeling after myocardial infarction: Potential therapeutic targets in ischemic heart disease. PLoS ONE 2018, 13, e0200474. [Google Scholar] [CrossRef]
- Shintani, Y.; Ito, T.; Fields, L.; Shiraishi, M.; Ichihara, Y.; Sato, N.; Podaru, M.; Kainuma, S.; Tanaka, H.; Suzuki, K. IL-4 as a Repurposed Biological Drug for Myocardial Infarction through Augmentation of Reparative Cardiac Macrophages: Proof-of-Concept Data in Mice. Sci. Rep. 2017, 7, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Chen, B.; Zhao, J.; Peng, Z.; Xu, W.; Yu, G. Effect of intravenous transplantation of hUCB-MSCs on M1/M2 subtype conversion in monocyte/macrophages of AMI mice. Biomed. Pharmacother. 2019, 111, 624–630. [Google Scholar] [CrossRef] [PubMed]
- Deng, S.; Zhou, X.; Ge, Z.; Song, Y.; Wang, H.; Liu, X.; Zhang, D. Exosomes from adipose-derived mesenchymal stem cells ameliorate cardiac damage after myocardial infarction by activating S1P/SK1/S1PR1 signaling and promoting macrophage M2 polarization. Int. J. Biochem. Cell Biol. 2019, 114, 105564. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Nurakhayev, S.; Nurkesh, A.; Zharkinbekov, Z.; Saparov, A. Macrophage Polarization in Cardiac Tissue Repair Following Myocardial Infarction. Int. J. Mol. Sci. 2021, 22, 2715. [Google Scholar] [CrossRef]
- Dai, H.; Thomson, A.W.; Rogers, N.M. Dendritic Cells as Sensors, Mediators, and Regulators of Ischemic Injury. Front. Immunol. 2019, 10, 2418. [Google Scholar] [CrossRef]
- Forte, E.; Perkins, B.; Sintou, A.; Kalkat, H.S.; Papanikolaou, A.; Jenkins, C.; Alsubaie, M.; Chowdhury, R.A.; Duffy, T.M.; Skelly, D.A.; et al. Cross-Priming Dendritic Cells Exacerbate Immunopathology After Ischemic Tissue Damage in the Heart. Circulation 2021, 143, 821–836. [Google Scholar] [CrossRef]
- Satpathy, A.T.; Kc, W.; Albring, J.C.; Edelson, B.T.; Kretzer, N.M.; Bhattacharya, D.; Murphy, T.L.; Murphy, K.M. Zbtb46 expression distinguishes classical dendritic cells and their committed progenitors from other immune lineages. J. Exp. Med. 2012, 209, 1135–1152. [Google Scholar] [CrossRef]
- Dzionek, A.; Sohma, Y.; Nagafune, J.; Cella, M.; Colonna, M.; Facchetti, F.; Gunther, G.; Johnston, I.; Lanzavecchia, A.; Nagasaka, T.; et al. BDCA-2, a novel plasmacytoid dendritic cell-specific type II C-type lectin, mediates antigen capture and is a potent inhibitor of interferon alpha/beta induction. J. Exp. Med. 2001, 194, 1823–1834. [Google Scholar] [CrossRef]
- Nagai, T.; Honda, S.; Sugano, Y.; Matsuyama, T.A.; Ohta-Ogo, K.; Asaumi, Y.; Ikeda, Y.; Kusano, K.; Ishihara, M.; Yasuda, S.; et al. Decreased myocardial dendritic cells is associated with impaired reparative fibrosis and development of cardiac rupture after myocardial infarction in humans. J. Am. Heart Assoc. 2014, 3, e000839. [Google Scholar] [CrossRef]
- Yan, X.; Zhang, H.; Fan, Q.; Hu, J.; Tao, R.; Chen, Q.; Iwakura, Y.; Shen, W.; Lu, L.; Zhang, Q.; et al. Dectin-2 Deficiency Modulates Th1 Differentiation and Improves Wound Healing After Myocardial Infarction. Circ. Res. 2017, 120, 1116–1129. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Zong, W.; Zhang, M.; Tu, Y.; Zhou, Q.; Ni, M.; Li, Z.; Liu, H.; Zhang, J. Increased Ratio of Circulating T-Helper 1 to T-Helper 2 Cells and Severity of Coronary Artery Disease in Patients with Acute Myocardial Infarction: A Prospective Observational Study. Med. Sci. Monit. 2019, 25, 6034–6042. [Google Scholar] [CrossRef] [PubMed]
- Rudensky, A.Y. Regulatory T cells and Foxp3. Immunol. Rev. 2011, 241, 260–268. [Google Scholar] [CrossRef] [PubMed]
- De Villiers, C.; Riley, P.R. Mouse models of myocardial infarction: Comparing permanent ligation and ischaemia-reperfusion. Dis. Model. Mech. 2020, 13. [Google Scholar] [CrossRef]
- Tang, T.T.; Yuan, J.; Zhu, Z.F.; Zhang, W.C.; Xiao, H.; Xia, N.; Yan, X.X.; Nie, S.F.; Liu, J.; Zhou, S.F.; et al. Regulatory T cells ameliorate cardiac remodeling after myocardial infarction. Basic Res. Cardiol. 2012, 107, 232. [Google Scholar] [CrossRef]
- Jujo, K.; Ii, M.; Sekiguchi, H.; Klyachko, E.; Misener, S.; Tanaka, T.; Tongers, J.; Roncalli, J.; Renault, M.A.; Thorne, T.; et al. CXC-chemokine receptor 4 antagonist AMD3100 promotes cardiac functional recovery after ischemia/reperfusion injury via endothelial nitric oxide synthase-dependent mechanism. Circulation 2013, 127, 63–73. [Google Scholar] [CrossRef]
- Wang, Y.; Dembowsky, K.; Chevalier, E.; Stuve, P.; Korf-Klingebiel, M.; Lochner, M.; Napp, L.C.; Frank, H.; Brinkmann, E.; Kanwischer, A.; et al. C-X-C Motif Chemokine Receptor 4 Blockade Promotes Tissue Repair After Myocardial Infarction by Enhancing Regulatory T Cell Mobilization and Immune-Regulatory Function. Circulation 2019, 139, 1798–1812. [Google Scholar] [CrossRef]
- Piao, W.; Xiong, Y.; Li, L.; Saxena, V.; Smith, K.D.; Hippen, K.L.; Paluskievicz, C.; Willsonshirkey, M.; Blazar, B.R.; Abdi, R.; et al. Regulatory T Cells Condition Lymphatic Endothelia for Enhanced Transendothelial Migration. Cell Rep. 2020, 30, 1052.e1055–1062.e1055. [Google Scholar] [CrossRef]
- Suntharalingam, G.; Perry, M.R.; Ward, S.; Brett, S.J.; Castello-Cortes, A.; Brunner, M.D.; Panoskaltsis, N. Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. N. Engl. J. Med. 2006, 355, 1018–1028. [Google Scholar] [CrossRef]
- Klaourakis, K.; Vieira, J.M.; Riley, P.R. The evolving cardiac lymphatic vasculature in development, repair and regeneration. Nat. Rev. Cardiol. 2021, 18, 368–379. [Google Scholar] [CrossRef]
- Park, C.; Kim, T.M.; Malik, A.B. Transcriptional regulation of endothelial cell and vascular development. Circ. Res. 2013, 112, 1380–1400. [Google Scholar] [CrossRef]
- Wigle, J.T.; Oliver, G. Prox1 function is required for the development of the murine lymphatic system. Cell 1999, 98, 769–778. [Google Scholar] [CrossRef]
- Gale, N.W.; Thurston, G.; Hackett, S.F.; Renard, R.; Wang, Q.; McClain, J.; Martin, C.; Witte, C.; Witte, M.H.; Jackson, D.; et al. Angiopoietin-2 is required for postnatal angiogenesis and lymphatic patterning, and only the latter role is rescued by Angiopoietin-1. Dev. Cell 2002, 3, 411–423. [Google Scholar] [CrossRef]
- Harvey, N.L.; Srinivasan, R.S.; Dillard, M.E.; Johnson, N.C.; Witte, M.H.; Boyd, K.; Sleeman, M.W.; Oliver, G. Lymphatic vascular defects promoted by Prox1 haploinsufficiency cause adult-onset obesity. Nat. Genet. 2005, 37, 1072–1081. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.K.; Harvey, N.; Noh, Y.H.; Schacht, V.; Hirakawa, S.; Detmar, M.; Oliver, G. Prox1 is a master control gene in the program specifying lymphatic endothelial cell fate. Dev. Dyn. 2002, 225, 351–357. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Nguyen, V.P.; Petrova, T.V.; Cruz, M.; Alitalo, K.; Dumont, D.J. Embryonic vascular endothelial cells are malleable to reprogramming via Prox1 to a lymphatic gene signature. BMC Dev. Biol. 2010, 10, 1–10. [Google Scholar] [CrossRef]
- Karkkainen, M.J.; Haiko, P.; Sainio, K.; Partanen, J.; Taipale, J.; Petrova, T.V.; Jeltsch, M.; Jackson, D.G.; Talikka, M.; Rauvala, H.; et al. Vascular endothelial growth factor C is required for sprouting of the first lymphatic vessels from embryonic veins. Nat. Immunol. 2003, 5, 74–80. [Google Scholar] [CrossRef]
- Klotz, L.; Norman, S.; Vieira, J.M.; Masters, M.; Rohling, M.; Dube, K.N.; Bollini, S.; Matsuzaki, F.; Carr, C.A.; Riley, P.R. Cardiac lymphatics are heterogeneous in origin and respond to injury. Nature 2015, 522, 62–67. [Google Scholar] [CrossRef]
- Ulvmar, M.H.; Martinez-Corral, I.; Stanczuk, L.; Makinen, T. Pdgfrb-Cre targets lymphatic endothelial cells of both venous and non-venous origins. Genesis 2016, 54, 350–358. [Google Scholar] [CrossRef]
- Makinen, T.; Jussila, L.; Veikkola, T.; Karpanen, T.; Kettunen, M.I.; Pulkkanen, K.J.; Kauppinen, R.; Jackson, D.G.; Kubo, H.; Nishikawa, S.; et al. Inhibition of lymphangiogenesis with resulting lymphedema in transgenic mice expressing soluble VEGF receptor-3. Nat. Med. 2001, 7, 199–205. [Google Scholar] [CrossRef]
- Chen, H.I.; Sharma, B.; Akerberg, B.N.; Numi, H.J.; Kivela, R.; Saharinen, P.; Aghajanian, H.; McKay, A.S.; Bogard, P.E.; Chang, A.H.; et al. The sinus venosus contributes to coronary vasculature through VEGFC-stimulated angiogenesis. Development 2014, 141, 4500–4512. [Google Scholar] [CrossRef]
- Juszynski, M.; Ciszek, B.; Stachurska, E.; Jablonska, A.; Ratajska, A. Development of lymphatic vessels in mouse embryonic and early postnatal hearts. Dev. Dyn. 2008, 237, 2973–2986. [Google Scholar] [CrossRef] [PubMed]
- Bradham, R.R.; Parker, E.F. The cardiac lymphatics. Ann. Thorac. Surg. 1973, 15, 526–535. [Google Scholar] [CrossRef]
- Flaht-Zabost, A.; Gula, G.; Ciszek, B.; Czarnowska, E.; Jankowska-Steifer, E.; Madej, M.; Niderla-Bielińska, J.; Radomska-Leśniewska, D.; Ratajska, A. Cardiac Mouse Lymphatics: Developmental and Anatomical Update. Anat. Rec. 2014, 297, 1115–1130. [Google Scholar] [CrossRef] [PubMed]
- Ratajska, A.; Gula, G.; Flaht-Zabost, A.; Czarnowska, E.; Ciszek, B.; Jankowska-Steifer, E.; Niderla-Bielinska, J.; Radomska-Lesniewska, D. Comparative and Developmental Anatomy of Cardiac Lymphatics. Sci. World J. 2014, 2014. [Google Scholar] [CrossRef] [PubMed]
- Shore, L.R. The Lymphatic Drainage of the Human Heart. J. Anat. 1929, 63, 291–313. [Google Scholar]
- Shimada, T.; Zhang, L.; Abe, K.; Yamabe, M.; Miyamoto, T. Developmental morphology of blood and lymphatic capillary networks in mammalian hearts, with special reference to three-dimensional architecture. Ital. J. Anat. Embryol. 2001, 106, 203–211. [Google Scholar] [PubMed]
- Henri, O.; Pouehe, C.; Houssari, M.; Galas, L.; Nicol, L.; Edwards-Levy, F.; Henry, J.P.; Dumesnil, A.; Boukhalfa, I.; Banquet, S.; et al. Selective Stimulation of Cardiac Lymphangiogenesis Reduces Myocardial Edema and Fibrosis Leading to Improved Cardiac Function Following Myocardial Infarction. Circulation 2016, 133, 1484–1497. [Google Scholar] [CrossRef]
- Geissler, H.; Allen, S.; Davis, K.; Sauer, H.; Laine, G.; Kuhn-Régnier, F.; Dapunt, O.; Vivie, E.d.; Mehlhorn, U. Impact of cardiopulmonary bypass and cardioplegic arrest on myocardial efficiency. Crit. Care 1999, 3, 1–2. [Google Scholar] [CrossRef]
- Laine, G.A.; Allen, S.J. Left ventricular myocardial edema. Lymph flow, interstitial fibrosis, and cardiac function. Circ. Res. 1991, 68, 1713–1721. [Google Scholar] [CrossRef]
- Shimizu, Y.; Polavarapu, R.; Eskla, K.L.; Pantner, Y.; Nicholson, C.K.; Ishii, M.; Brunnhoelzl, D.; Mauria, R.; Husain, A.; Naqvi, N.; et al. Impact of Lymphangiogenesis on Cardiac Remodeling After Ischemia and Reperfusion Injury. J. Am. Heart Assoc. 2018, 7, e009565. [Google Scholar] [CrossRef]
- Kholova, I.; Dragneva, G.; Cermakova, P.; Laidinen, S.; Kaskenpaa, N.; Hazes, T.; Cermakova, E.; Steiner, I.; Yla-Herttuala, S. Lymphatic vasculature is increased in heart valves, ischaemic and inflamed hearts and in cholesterol-rich and calcified atherosclerotic lesions. Eur. J. Clin. Invest. 2011, 41, 487–497. [Google Scholar] [CrossRef]
- Geissler, H.J.; Dashkevich, A.; Fischer, U.M.; Fries, J.W.; Kuhn-Regnier, F.; Addicks, K.; Mehlhorn, U.; Bloch, W. First year changes of myocardial lymphatic endothelial markers in heart transplant recipients. Eur. J. Cardiothorac. Surg. 2006, 29, 767–771. [Google Scholar] [CrossRef] [PubMed]
- Rutanen, J.; Leppanen, P.; Tuomisto, T.T.; Rissanen, T.T.; Hiltunen, M.O.; Vajanto, I.; Niemi, M.; Hakkinen, T.; Karkola, K.; Stacker, S.A.; et al. Vascular endothelial growth factor-D expression in human atherosclerotic lesions. Cardiovasc. Res. 2003, 59, 971–979. [Google Scholar] [CrossRef]
- Dongaonkar, R.M.; Stewart, R.H.; Geissler, H.J.; Laine, G.A. Myocardial microvascular permeability, interstitial oedema, and compromised cardiac function. Cardiovasc. Res. 2010, 87, 331–339. [Google Scholar] [CrossRef]
- Vieira, J.M.; Norman, S.; Villa Del Campo, C.; Cahill, T.J.; Barnette, D.N.; Gunadasa-Rohling, M.; Johnson, L.A.; Greaves, D.R.; Carr, C.A.; Jackson, D.G.; et al. The cardiac lymphatic system stimulates resolution of inflammation following myocardial infarction. J. Clin. Invest. 2018, 128, 3402–3412. [Google Scholar] [CrossRef] [PubMed]
- Frantz, S.; Hofmann, U.; Fraccarollo, D.; Schafer, A.; Kranepuhl, S.; Hagedorn, I.; Nieswandt, B.; Nahrendorf, M.; Wagner, H.; Bayer, B.; et al. Monocytes/macrophages prevent healing defects and left ventricular thrombus formation after myocardial infarction. FASEB J. 2013, 27, 871–881. [Google Scholar] [CrossRef]
- Adamo, L.; Rocha-Resende, C.; Prabhu, S.D.; Mann, D.L. Reappraising the role of inflammation in heart failure. Nat. Rev. Cardiol. 2020, 17, 269–285. [Google Scholar] [CrossRef] [PubMed]
- Jantsch, J.; Binger, K.J.; Muller, D.N.; Titze, J. Macrophages in homeostatic immune function. Front. Physiol. 2014, 5, 146. [Google Scholar] [CrossRef]
- Kastenmuller, W.; Torabi-Parizi, P.; Subramanian, N.; Lammermann, T.; Germain, R.N. A spatially organized multicellular innate immune response in lymph nodes limits systemic pathogen spread. Cell 2012, 150, 1235–1248. [Google Scholar] [CrossRef] [PubMed]
- Von Andrian, U.H.; Mempel, T.R. Homing and cellular traffic in lymph nodes. Nat. Rev. Immunol 2003, 3, 867–878. [Google Scholar] [CrossRef]
- Bellingan, G.J.; Caldwell, H.; Howie, S.E.; Dransfield, I.; Haslett, C. In vivo fate of the inflammatory macrophage during the resolution of inflammation: Inflammatory macrophages do not die locally but emigrate to the draining lymph nodes. J. Immunol. 1996, 157, 2577–2585. [Google Scholar] [PubMed]
- Hou, Y.; Bock, F.; Hos, D.; Cursiefen, C. Lymphatic Trafficking in the Eye: Modulation of Lymphatic Trafficking to Promote Corneal Transplant Survival. Cells 2021, 10, 1661. [Google Scholar] [CrossRef] [PubMed]
- Bellingan, G.J.; Xu, P.; Cooksley, H.; Cauldwell, H.; Shock, A.; Bottoms, S.; Haslett, C.; Mutsaers, S.E.; Laurent, G.J. Adhesion molecule-dependent mechanisms regulate the rate of macrophage clearance during the resolution of peritoneal inflammation. J. Exp. Med. 2002, 196, 1515–1521. [Google Scholar] [CrossRef]
- Jackson, D.G. Leucocyte Trafficking via the Lymphatic Vasculature- Mechanisms and Consequences. Front. Immunol. 2019, 10, 471. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.P.; Platt, N.; Wykes, M.; Major, J.R.; Powell, T.J.; Jenkins, C.D.; MacPherson, G.G. A discrete subpopulation of dendritic cells transports apoptotic intestinal epithelial cells to T cell areas of mesenteric lymph nodes. J. Exp. Med. 2000, 191, 435–444. [Google Scholar] [CrossRef]
- Jakubzick, C.; Bogunovic, M.; Bonito, A.J.; Kuan, E.L.; Merad, M.; Randolph, G.J. Lymph-migrating, tissue-derived dendritic cells are minor constituents within steady-state lymph nodes. J. Exp. Med. 2008, 205, 2839–2850. [Google Scholar] [CrossRef]
- MacPherson, G.G.; Jenkins, C.D.; Stein, M.J.; Edwards, C. Endotoxin-mediated dendritic cell release from the intestine. Characterization of released dendritic cells and TNF dependence. J. Immunol. 1995, 154, 1317–1322. [Google Scholar]
- Gorlino, C.V.; Ranocchia, R.P.; Harman, M.F.; Garcia, I.A.; Crespo, M.I.; Moron, G.; Maletto, B.A.; Pistoresi-Palencia, M.C. Neutrophils exhibit differential requirements for homing molecules in their lymphatic and blood trafficking into draining lymph nodes. J. Immunol. 2014, 193, 1966–1974. [Google Scholar] [CrossRef]
- Chtanova, T.; Schaeffer, M.; Han, S.J.; van Dooren, G.G.; Nollmann, M.; Herzmark, P.; Chan, S.W.; Satija, H.; Camfield, K.; Aaron, H.; et al. Dynamics of neutrophil migration in lymph nodes during infection. Immunity 2008, 29, 487–496. [Google Scholar] [CrossRef]
- Beauvillain, C.; Cunin, P.; Doni, A.; Scotet, M.; Jaillon, S.; Loiry, M.L.; Magistrelli, G.; Masternak, K.; Chevailler, A.; Delneste, Y.; et al. CCR7 is involved in the migration of neutrophils to lymph nodes. Blood 2011, 117, 1196–1204. [Google Scholar] [CrossRef]
- Forster, R.; Braun, A.; Worbs, T. Lymph node homing of T cells and dendritic cells via afferent lymphatics. Trends Immunol. 2012, 33, 271–280. [Google Scholar] [CrossRef]
- Daroczy, J. The structure and dynamic function of the dermal lymphatic capillaries. Br. J. Dermatol. 1983, 109, 99–102. [Google Scholar]
- Schmid-Schonbein, G.W. The second valve system in lymphatics. Lymphat. Res. Biol. 2003, 1, 25–31. [Google Scholar] [CrossRef]
- Bazigou, E.; Wilson, J.T.; Moore, J.E., Jr. Primary and secondary lymphatic valve development: Molecular, functional and mechanical insights. Microvasc. Res. 2014, 96, 38–45. [Google Scholar] [CrossRef]
- Yao, L.C.; Baluk, P.; Srinivasan, R.S.; Oliver, G.; McDonald, D.M. Plasticity of button-like junctions in the endothelium of airway lymphatics in development and inflammation. Am. J. Pathol. 2012, 180, 2561–2575. [Google Scholar] [CrossRef] [PubMed]
- Baluk, P.; Fuxe, J.; Hashizume, H.; Romano, T.; Lashnits, E.; Butz, S.; Vestweber, D.; Corada, M.; Molendini, C.; Dejana, E.; et al. Functionally specialized junctions between endothelial cells of lymphatic vessels. J. Exp. Med. 2007, 204, 2349–2362. [Google Scholar] [CrossRef] [PubMed]
- Romani, N.; Ratzinger, G.; Pfaller, K.; Salvenmoser, W.; Stossel, H.; Koch, F.; Stoitzner, P. Migration of dendritic cells into lymphatics-the Langerhans cell example: Routes, regulation, and relevance. Int. Rev. Cytol. 2001, 207, 237–270. [Google Scholar] [CrossRef] [PubMed]
- Pflicke, H.; Sixt, M. Preformed portals facilitate dendritic cell entry into afferent lymphatic vessels. J. Exp. Med. 2009, 206, 2925–2935. [Google Scholar] [CrossRef]
- Scallan, J.P.; Zawieja, S.D.; Castorena-Gonzalez, J.A.; Davis, M.J. Lymphatic pumping: Mechanics, mechanisms and malfunction. J. Physiol. 2016, 594, 5749–5768. [Google Scholar] [CrossRef]
- Schineis, P.; Runge, P.; Halin, C. Cellular traffic through afferent lymphatic vessels. Vasc. Pharmacol. 2019, 112, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Ji, H.; Cao, R.; Yang, Y.; Zhang, Y.; Iwamoto, H.; Lim, S.; Nakamura, M.; Andersson, P.; Wang, J.; Sun, Y.; et al. TNFR1 mediates TNF-α-induced tumour lymphangiogenesis and metastasis by modulating VEGF-C-VEGFR3 signalling. Nat. Commun. 2014, 5, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Pang, I.K.; Ichinohe, T.; Iwasaki, A. IL-1R signaling in dendritic cells replaces pattern-recognition receptors in promoting CD8(+) T cell responses to influenza A virus. Nat. Immunol. 2013, 14, 246–253. [Google Scholar] [CrossRef] [PubMed]
- MartIn-Fontecha, A.; Sebastiani, S.; Hopken, U.E.; Uguccioni, M.; Lipp, M.; Lanzavecchia, A.; Sallusto, F. Regulation of dendritic cell migration to the draining lymph node: Impact on T lymphocyte traffic and priming. J. Exp. Med. 2003, 198, 615–621. [Google Scholar] [CrossRef]
- Ohl, L.; Mohaupt, M.; Czeloth, N.; Hintzen, G.; Kiafard, Z.; Zwirner, J.; Blankenstein, T.; Henning, G.; Forster, R. CCR7 governs skin dendritic cell migration under inflammatory and steady-state conditions. Immunity 2004, 21, 279–288. [Google Scholar] [CrossRef]
- Saeki, H.; Moore, A.M.; Brown, M.J.; Hwang, S.T. Cutting edge: Secondary lymphoid-tissue chemokine (SLC) and CC chemokine receptor 7 (CCR7) participate in the emigration pathway of mature dendritic cells from the skin to regional lymph nodes. J. Immunol. 1999, 162, 2472–2475. [Google Scholar]
- Clatworthy, M.R.; Aronin, C.E.; Mathews, R.J.; Morgan, N.Y.; Smith, K.G.; Germain, R.N. Immune complexes stimulate CCR7-dependent dendritic cell migration to lymph nodes. Nat. Med. 2014, 20, 1458–1463. [Google Scholar] [CrossRef]
- Haemmerle, M.; Keller, T.; Egger, G.; Schachner, H.; Steiner, C.W.; Stokic, D.; Neumayer, C.; Brown, M.K.; Kerjaschki, D.; Hantusch, B. Enhanced lymph vessel density, remodeling, and inflammation are reflected by gene expression signatures in dermal lymphatic endothelial cells in type 2 diabetes. Diabetes 2013, 62, 2509–2529. [Google Scholar] [CrossRef]
- Kiermaier, E.; Moussion, C.; Veldkamp, C.T.; Gerardy-Schahn, R.; de Vries, I.; Williams, L.G.; Chaffee, G.R.; Phillips, A.J.; Freiberger, F.; Imre, R.; et al. Polysialylation controls dendritic cell trafficking by regulating chemokine recognition. Science 2016, 351, 186–190. [Google Scholar] [CrossRef]
- Johnson, L.A.; Clasper, S.; Holt, A.P.; Lalor, P.F.; Baban, D.; Jackson, D.G. An inflammation-induced mechanism for leukocyte transmigration across lymphatic vessel endothelium. J. Exp. Med. 2006, 203, 2763–2777. [Google Scholar] [CrossRef]
- Johnson, L.A.; Banerji, S.; Lagerholm, B.C.; Jackson, D.G. Dendritic cell entry to lymphatic capillaries is orchestrated by CD44 and the hyaluronan glycocalyx. Life Sci. Alliance 2021, 4, e202000908. [Google Scholar] [CrossRef]
- Johnson, L.A.; Banerji, S.; Lawrance, W.; Gileadi, U.; Prota, G.; Holder, K.A.; Roshorm, Y.M.; Hanke, T.; Cerundolo, V.; Gale, N.W.; et al. Dendritic cells enter lymph vessels by hyaluronan-mediated docking to the endothelial receptor LYVE-1. Nat. Immunol. 2017, 18, 762–770. [Google Scholar] [CrossRef]
- Stanly, T.A.; Fritzsche, M.; Banerji, S.; Shrestha, D.; Schneider, F.; Eggeling, C.; Jackson, D.G. The cortical actin network regulates avidity-dependent binding of hyaluronan by the lymphatic vessel endothelial receptor LYVE-1. J. Biol. Chem. 2020, 295, 5036–5050. [Google Scholar] [CrossRef] [PubMed]
- Vigl, B.; Aebischer, D.; Nitschke, M.; Iolyeva, M.; Rothlin, T.; Antsiferova, O.; Halin, C. Tissue inflammation modulates gene expression of lymphatic endothelial cells and dendritic cell migration in a stimulus-dependent manner. Blood 2011, 118, 205–215. [Google Scholar] [CrossRef]
- Jackson, D.G. Hyaluronan in the lymphatics: The key role of the hyaluronan receptor LYVE-1 in leucocyte trafficking. Matrix Biol. 2019, 78–79, 219–235. [Google Scholar] [CrossRef] [PubMed]
- Arasa, J.; Collado-Diaz, V.; Kritikos, I.; Medina-Sanchez, J.D.; Friess, M.C.; Sigmund, E.C.; Schineis, P.; Hunter, M.C.; Tacconi, C.; Paterson, N.; et al. Upregulation of VCAM-1 in lymphatic collectors supports dendritic cell entry and rapid migration to lymph nodes in inflammation. J. Exp. Med. 2021, 218. [Google Scholar] [CrossRef] [PubMed]
- Rigby, D.A.; Ferguson, D.J.P.; Johnson, L.A.; Jackson, D.G. Neutrophils rapidly transit inflamed lymphatic vessel endothelium via integrin-dependent proteolysis and lipoxin-induced junctional retraction. J. Leukoc. Biol. 2015, 98, 897–912. [Google Scholar] [CrossRef] [PubMed]
- Hampton, H.R.; Bailey, J.; Tomura, M.; Brink, R.; Chtanova, T. Microbe-dependent lymphatic migration of neutrophils modulates lymphocyte proliferation in lymph nodes. Nat. Commun. 2015, 6, 7139. [Google Scholar] [CrossRef]
- Weber, M.; Hauschild, R.; Schwarz, J.; Moussion, C.; de Vries, I.; Legler, D.F.; Luther, S.A.; Bollenbach, T.; Sixt, M. Interstitial dendritic cell guidance by haptotactic chemokine gradients. Science 2013, 339, 328–332. [Google Scholar] [CrossRef]
- Tal, O.; Lim, H.Y.; Gurevich, I.; Milo, I.; Shipony, Z.; Ng, L.G.; Angeli, V.; Shakhar, G. DC mobilization from the skin requires docking to immobilized CCL21 on lymphatic endothelium and intralymphatic crawling. J. Exp. Med. 2011, 208, 2141–2153. [Google Scholar] [CrossRef] [PubMed]
- Vaahtomeri, K.; Brown, M.; Hauschild, R.; De Vries, I.; Leithner, A.F.; Mehling, M.; Kaufmann, W.A.; Sixt, M. Locally Triggered Release of the Chemokine CCL21 Promotes Dendritic Cell Transmigration across Lymphatic Endothelia. Cell Rep. 2017, 19, 902–909. [Google Scholar] [CrossRef]
- Tatin, F.; Renaud-Gabardos, E.; Godet, A.C.; Hantelys, F.; Pujol, F.; Morfoisse, F.; Calise, D.; Viars, F.; Valet, P.; Masri, B.; et al. Apelin modulates pathological remodeling of lymphatic endothelium after myocardial infarction. JCI Insight 2017, 2. [Google Scholar] [CrossRef]
- Ishikawa, Y.; Akishima-Fukasawa, Y.; Ito, K.; Akasaka, Y.; Tanaka, M.; Shimokawa, R.; Kimura-Matsumoto, M.; Morita, H.; Sato, S.; Kamata, I.; et al. Lymphangiogenesis in myocardial remodelling after infarction. Histopathology 2007, 51, 345–353. [Google Scholar] [CrossRef]
- Kataru, R.P.; Jung, K.; Jang, C.; Yang, H.; Schwendener, R.A.; Baik, J.E.; Han, S.H.; Alitalo, K.; Koh, G.Y. Critical role of CD11b+ macrophages and VEGF in inflammatory lymphangiogenesis, antigen clearance, and inflammation resolution. Blood 2009, 113, 5650–5659. [Google Scholar] [CrossRef]
- Tan, K.W.; Chong, S.Z.; Wong, F.H.; Evrard, M.; Tan, S.M.; Keeble, J.; Kemeny, D.M.; Ng, L.G.; Abastado, J.P.; Angeli, V. Neutrophils contribute to inflammatory lymphangiogenesis by increasing VEGF-A bioavailability and secreting VEGF-D. Blood 2013, 122, 3666–3677. [Google Scholar] [CrossRef] [PubMed]
- Taira, A.; Morishita, Y.; Arikawa, K.; Murata, K.; Hamada, Y.; Akita, H. Flow velocity of cardiac lymph and contractility of the heart: An experimental study. Ann. Thorac. Surg. 1977, 23, 230–234. [Google Scholar] [CrossRef]
- Houssari, M.; Dumesnil, A.; Tardif, V.; Kivela, R.; Pizzinat, N.; Boukhalfa, I.; Godefroy, D.; Schapman, D.; Hemanthakumar, K.A.; Bizou, M.; et al. Lymphatic and Immune Cell Cross-Talk Regulates Cardiac Recovery After Experimental Myocardial Infarction. Arter. Thromb. Vasc. Biol. 2020, 40, 1722–1737. [Google Scholar] [CrossRef] [PubMed]
- Veikkola, T.; Jussila, L.; Makinen, T.; Karpanen, T.; Jeltsch, M.; Petrova, T.V.; Kubo, H.; Thurston, G.; McDonald, D.M.; Achen, M.G.; et al. Signalling via vascular endothelial growth factor receptor-3 is sufficient for lymphangiogenesis in transgenic mice. EMBO J. 2001, 20, 1223–1231. [Google Scholar] [CrossRef]
- Hartikainen, J.; Hassinen, I.; Hedman, A.; Kivela, A.; Saraste, A.; Knuuti, J.; Husso, M.; Mussalo, H.; Hedman, M.; Rissanen, T.T.; et al. Adenoviral intramyocardial VEGF-DDeltaNDeltaC gene transfer increases myocardial perfusion reserve in refractory angina patients: A phase I/IIa study with 1-year follow-up. Eur. Heart J. 2017, 38, 2547–2555. [Google Scholar] [CrossRef]
- Trincot, C.E.; Xu, W.; Zhang, H.; Kulikauskas, M.R.; Caranasos, T.G.; Jensen, B.C.; Sabine, A.; Petrova, T.V.; Caron, K.M. Adrenomedullin Induces Cardiac Lymphangiogenesis After Myocardial Infarction and Regulates Cardiac Edema Via Connexin 43. Circ. Res. 2019, 124, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Yoon, C.M.; Hong, B.S.; Moon, H.G.; Lim, S.; Suh, P.G.; Kim, Y.K.; Chae, C.B.; Gho, Y.S. Sphingosine-1-phosphate promotes lymphangiogenesis by stimulating S1P1/Gi/PLC/Ca2+ signaling pathways. Blood 2008, 112, 1129–1138. [Google Scholar] [CrossRef]
- Pham, T.H.; Baluk, P.; Xu, Y.; Grigorova, I.; Bankovich, A.J.; Pappu, R.; Coughlin, S.R.; McDonald, D.M.; Schwab, S.R.; Cyster, J.G. Lymphatic endothelial cell sphingosine kinase activity is required for lymphocyte egress and lymphatic patterning. J. Exp. Med. 2010, 207, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Cyster, J.G.; Schwab, S.R. Sphingosine-1-phosphate and lymphocyte egress from lymphoid organs. Annu. Rev. Immunol. 2012, 30, 69–94. [Google Scholar] [CrossRef] [PubMed]
- Czeloth, N.; Bernhardt, G.; Hofmann, F.; Genth, H.; Forster, R. Sphingosine-1-phosphate mediates migration of mature dendritic cells. J. Immunol. 2005, 175, 2960–2967. [Google Scholar] [CrossRef]
- Zhang, H.F.; Wang, Y.L.; Tan, Y.Z.; Wang, H.J.; Tao, P.; Zhou, P. Enhancement of cardiac lymphangiogenesis by transplantation of CD34(+)VEGFR-3(+) endothelial progenitor cells and sustained release of VEGF-C. Basic Res. Cardiol. 2019, 114, 1–17. [Google Scholar] [CrossRef]
- Boopathy, A.V.; Davis, M.E. Self-assembling peptide-based delivery of therapeutics for myocardial infarction. Methods Mol. Biol. 2014, 1141, 159–164. [Google Scholar] [CrossRef]
- Iwamiya, T.; Segard, B.D.; Matsuoka, Y.; Imamura, T. Human cardiac fibroblasts expressing VCAM1 improve heart function in postinfarct heart failure rat models by stimulating lymphangiogenesis. PLoS ONE 2020, 15, e0237810. [Google Scholar] [CrossRef]
- Lee, E.; Pandey, N.B.; Popel, A.S. Lymphatic endothelial cells support tumor growth in breast cancer. Sci. Rep. 2014, 4, 5853. [Google Scholar] [CrossRef]
- Liu, X.; De la Cruz, E.; Gu, X.; Balint, L.; Oxendine-Burns, M.; Terrones, T.; Ma, W.; Kuo, H.H.; Lantz, C.; Bansal, T.; et al. Lymphoangiocrine signals promote cardiac growth and repair. Nature 2020, 588, 705–711. [Google Scholar] [CrossRef]
- Song, L.; Chen, X.; Swanson, T.A.; LaViolette, B.; Pang, J.; Cunio, T.; Nagle, M.W.; Asano, S.; Hales, K.; Shipstone, A.; et al. Lymphangiogenic therapy prevents cardiac dysfunction by ameliorating inflammation and hypertension. Elife 2020, 9. [Google Scholar] [CrossRef]
- Yeo, K.P.; Lim, H.Y.; Thiam, C.H.; Azhar, S.H.; Tan, C.; Tang, Y.; See, W.Q.; Koh, X.H.; Zhao, M.H.; Phua, M.L.; et al. Efficient aortic lymphatic drainage is necessary for atherosclerosis regression induced by ezetimibe. Sci. Adv. 2020, 6. [Google Scholar] [CrossRef] [PubMed]
- Itkin, M.; Rockson, S.G.; Burkhoff, D. Pathophysiology of the Lymphatic System in Patients with Heart Failure: JACC State-of-the-Art Review. J. Am. Coll Cardiol. 2021, 78, 278–290. [Google Scholar] [CrossRef] [PubMed]
- Sinha, S.; Lee, E.W.; Dori, Y.; Katsuhide, M. Advances in lymphatic imaging and interventions in patients with congenital heart disease. Prog. Pediatr. Cardiol. 2021, 61, 101376. [Google Scholar] [CrossRef]
- Yamakawa, M.; Doh, S.J.; Santosa, S.M.; Montana, M.; Qin, E.C.; Kong, H.; Han, K.Y.; Yu, C.; Rosenblatt, M.I.; Kazlauskas, A.; et al. Potential lymphangiogenesis therapies: Learning from current antiangiogenesis therapies—A review. Med. Res. Rev. 2018, 38, 1769–1798. [Google Scholar] [CrossRef] [PubMed]
- Quyyumi, A.A.; Vasquez, A.; Kereiakes, D.J.; Klapholz, M.; Schaer, G.L.; Abdel-Latif, A.; Frohwein, S.; Henry, T.D.; Schatz, R.A.; Dib, N.; et al. PreSERVE-AMI: A Randomized, Double-Blind, Placebo-Controlled Clinical Trial of Intracoronary Administration of Autologous CD34+ Cells in Patients With Left Ventricular Dysfunction Post STEMI. Circ. Res. 2017, 120, 324–331. [Google Scholar] [CrossRef]
- Tompkins, B.A.; Balkan, W.; Winkler, J.; Gyongyosi, M.; Goliasch, G.; Fernandez-Aviles, F.; Hare, J.M. Preclinical Studies of Stem Cell Therapy for Heart Disease. Circ. Res. 2018, 122, 1006–1020. [Google Scholar] [CrossRef] [PubMed]
- Rurik, J.G.; Aghajanian, H.; Epstein, J.A. Immune Cells and Immunotherapy for Cardiac Injury and Repair. Circ. Res. 2021, 128, 1766–1779. [Google Scholar] [CrossRef]


{kind=link}
| Cell Type | Subtypes | Markers | Timeline [4,5] | Function | KO Effect | References |
|---|---|---|---|---|---|---|
| Neutrophils | N1 | CD45+CD11b+Ly6C+ Ly6G+CD206− | From Day 1 to day 5 | Clear debris and dead cells. Pro-inflammatory | Neutrophil deletion worsens cardiac function and increases fibrosis * | [6] |
| N2 | CD45+CD11b+Ly6C+ Ly6G+CD206+ | From Day 5 | Anti-inflammatory | [7,8] | ||
| Monocytes | Inflammatory | CD14+Ly-6Chigh CCR2highCX3CR1low | From the 1st day and peak at day 3–day 5 | Phagocytose and proteolytic activities. Inflammatory | Cardiac Fibrosis Heart Failure | [9,10,11] |
| Non-Classical | CD14+Ly-6Clow CCR2lowCX3CR1high | From day 5 onwards | Reparative process: Angiogenesis & ECM deposition | Acute inflammatory reaction 7 days post MI. No long-term differences in scar formation | [10] | |
| Macrophages | Residents | CD45+CD11b+ Ly6G–F4/80+ | Present before MI. Quickly replaced by other immune cells | Maintain homeostasis. Recruit monocytes and promote neutrophil extravasation post-MI | Adverse remodelling | [12,13,14,15] |
| M1-Like | CD45+ Ly6G–CD11bhigh F4/80+CD206− Ly-6Chigh | Peaks at day 3 | Phagocytose and proinflammatory | High mortality rate. Increase remodelling | [16,17] | |
| M2-Like | CD45+ Ly6G–CD11blow F4/80+CD206+ Ly-6Clow | Appear 3 to 5 days post-MI and onwards | Anti-inflammatory. Promote cell proliferation, angiogenesis, and ECM production | Cardiac rupture | [18,19] | |
| Dendritic Cells | cDC | CD45+MHCII+ CD11c+Zbtb46+ | Infiltrate at day 1 and peak at day 5 * | [20] | ||
| pDC | CD45+MHCII+ CD11c+BDCA2+ | Antigen Presentation to T-Lymphocytes * | Improves cardiac function & attenuation of inflammation | [21,22] * | ||
| toDC | CD45+MHCIIlow CD11c+ | Activate Tregs | Total DC depletion promotes inflammation and increases cardiac rupture * | [23,24] | ||
| T- Lymphocytes | CD4+ | CD45+CD11b− CD3+CD4+ | Infiltrate at day 1 and peak at day 7 | Produce pro-inflammatory cytokines | Increase pro-inflammatory monocytes. Impair collagen matrix formation | [25,26] |
| Tregs | CD45+CD11b− CD3+ CD4+CD25+FoxP3+ | Infiltrate at day 1 and peak at day 7 | Promote inflammatory-to-reparative macrophage | Cardiac inflammation | [27,28,29] | |
| CD8+ | CD45+CD11b− CD3+CD8+ | Gradually increase post-MI | Removal of necrotic tissue. Cytotoxic effect | Infarct size and fibrosis decreased. Heart function improved but cardiac rupture | [30,31] | |
| B- Lymphocytes | N/A | CD45+CD11b− CD3−CD19+ | Peaks at Day 5–7 | Monocyte mobilization Sustain myocardial inflammation | Reduce infarct size and improve cardiac function | [32,33,34] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ravaud, C.; Ved, N.; Jackson, D.G.; Vieira, J.M.; Riley, P.R. Lymphatic Clearance of Immune Cells in Cardiovascular Disease. Cells 2021, 10, 2594. https://doi.org/10.3390/cells10102594
Ravaud C, Ved N, Jackson DG, Vieira JM, Riley PR. Lymphatic Clearance of Immune Cells in Cardiovascular Disease. Cells. 2021; 10(10):2594. https://doi.org/10.3390/cells10102594
Chicago/Turabian StyleRavaud, Christophe, Nikita Ved, David G. Jackson, Joaquim Miguel Vieira, and Paul R. Riley. 2021. "Lymphatic Clearance of Immune Cells in Cardiovascular Disease" Cells 10, no. 10: 2594. https://doi.org/10.3390/cells10102594
APA StyleRavaud, C., Ved, N., Jackson, D. G., Vieira, J. M., & Riley, P. R. (2021). Lymphatic Clearance of Immune Cells in Cardiovascular Disease. Cells, 10(10), 2594. https://doi.org/10.3390/cells10102594