
Unraveling the Multifaceted Nature of CD8 T Cell Exhaustion Provides the Molecular Basis for Therapeutic T Cell Reconstitution in Chronic Hepatitis B and C

 ,
,  and
and 
Abstract
:
1. Introduction
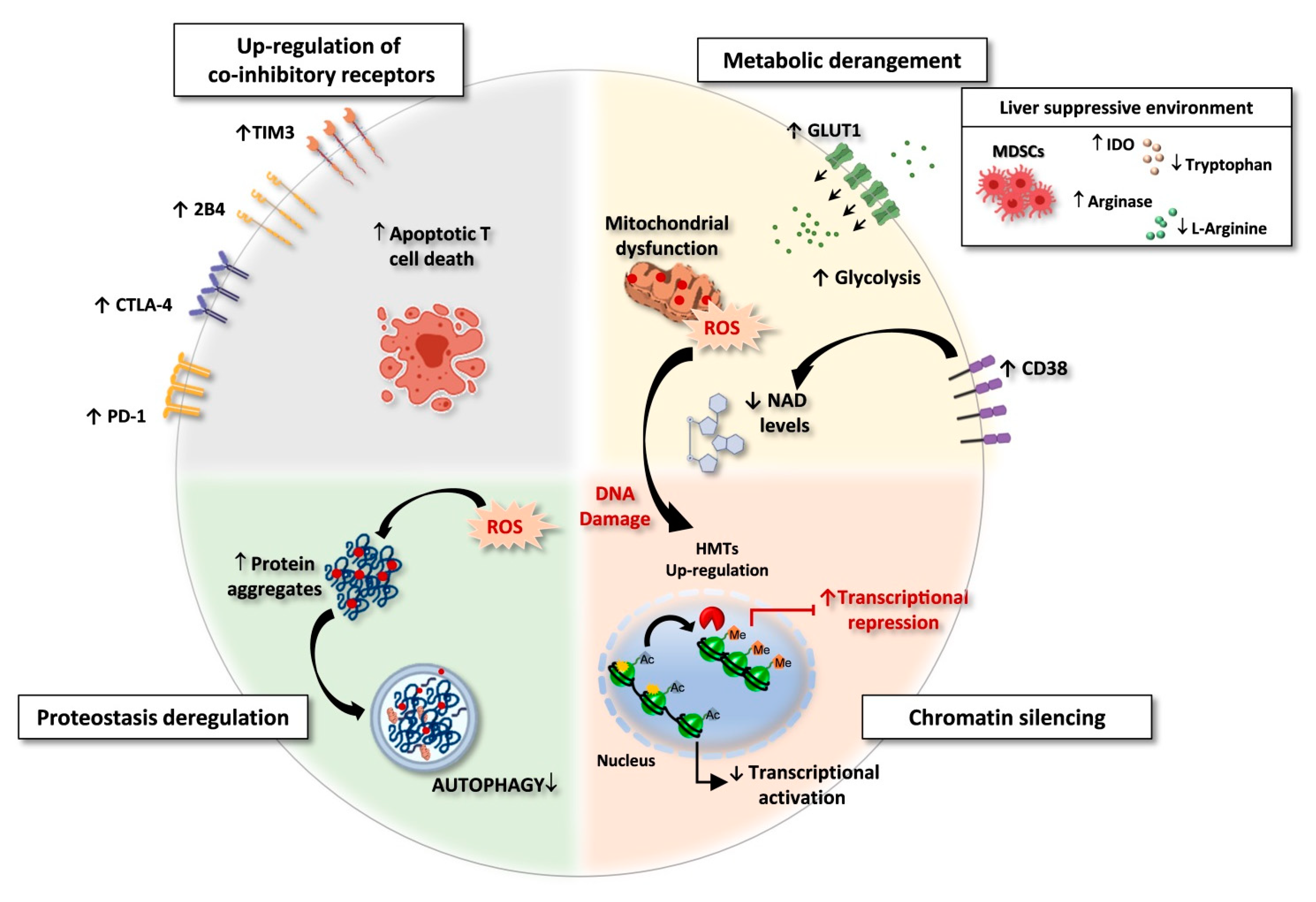
- -
- innate immunity receptors
- -
- co-inhibitory molecules
- -
- metabolic pathways
- -
- cytokine functions
- -
- epigenetic control of DNA transcription
2. Stimulation of Innate Immunity Receptors as an Immunomodulatory Strategy to Overcome HBV-Specific T Cell Exhaustion
3. Boosting Adaptive Immune Response by Blocking Co-Inhibitory Pathways
4. Targeting Metabolism to Restore T Cell Exhaustion
4.1. Early Chronic Infections
4.2. Established Chronic Infections
5. Cytokine Fueling for T Cell Restoration
6. T Cell Epigenetic Targets for Therapies during Chronic Infections
7. Future Perspectives
Funding
Conflicts of Interest
References
- Zoulim, F.; Lebossé, F.; Levrero, M. Current treatments for chronic hepatitis B virus infections. Curr. Opin. Virol. 2016, 18, 109–116. [Google Scholar] [CrossRef]
- Spearman, C.W.; Dusheiko, G.M.; Hellard, M.; Sonderup, M. Hepatitis C. Lancet 2019, 394, 1451–1466. [Google Scholar] [CrossRef]
- Rehermann, B. Pathogenesis of chronic viral hepatitis: Differential roles of T cells and NK cells. Nat. Med. 2013, 19, 859–868. [Google Scholar] [CrossRef] [Green Version]
- McLane, L.M.; Abdel-Hakeem, M.S.; Wherry, E.J. CD8 T Cell Exhaustion During Chronic Viral Infection and Cancer. Annu. Rev. Immunol. 2019, 37, 457–495. [Google Scholar] [CrossRef] [Green Version]
- Wherry, E.J.; Blattman, J.N.; Murali-Krishna, K.; van der Most, R.; Ahmed, R. Viral Persistence Alters CD8 T-Cell Immunodominance and Tissue Distribution and Results in Distinct Stages of Functional Impairment. J. Virol. 2003, 77, 4911–4927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wherry, E.J.; Kurachi, M. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 2015, 15, 486–499. [Google Scholar] [CrossRef] [PubMed]
- Ng, C.T.; Oldstone, M.B.A. IL-10: Achieving balance during persistent viral infection. Curr. Top. Microbiol. Immunol. 2014, 380, 129–144. [Google Scholar]
- Wilson, E.B.; Brooks, D.G. The Role of IL-10 in Regulating Immunity to Persistent Viral Infections. In Negative Co-Receptors and Ligands; Ahmed, R., Honjo, T., Eds.; Springer: Berlin/Heidelberg, Germany, 2011; pp. 39–65. ISBN 978-3-642-19545-7. [Google Scholar]
- Battegay, M.; Moskophidis, D.; Rahemtulla, A.; Hengartner, H.; Mak, T.W.; Zinkernagel, R.M. Enhanced establishment of a virus carrier state in adult CD4+ T-cell-deficient mice. J. Virol. 1994, 68, 4700–4704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barber, D.L.; Wherry, E.J.; Masopust, D.; Zhu, B.; Allison, J.P.; Sharpe, A.H.; Freeman, G.J.; Ahmed, R. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 2006, 439, 682–687. [Google Scholar] [CrossRef] [PubMed]
- Blackburn, S.D.; Shin, H.; Haining, W.N.; Zou, T.; Workman, C.J.; Polley, A.; Betts, M.R.; Freeman, G.J.; Vignali, D.A.A.; Wherry, E.J. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat. Immunol. 2009, 10, 29–37. [Google Scholar] [CrossRef] [Green Version]
- Chew, G.M.; Fujita, T.; Webb, G.M.; Burwitz, B.J.; Wu, H.L.; Reed, J.S.; Hammond, K.B.; Clayton, K.L.; Ishii, N.; Abdel-Mohsen, M.; et al. TIGIT Marks Exhausted T Cells, Correlates with Disease Progression, and Serves as a Target for Immune Restoration in HIV and SIV Infection. PLoS Pathog. 2016, 12, e1005349. [Google Scholar] [CrossRef] [Green Version]
- Tinoco, R.; Carrette, F.; Barraza, M.L.; Otero, D.C.; Magaña, J.; Bosenberg, M.W.; Swain, S.L.; Bradley, L.M. PSGL-1 Is an Immune Checkpoint Regulator that Promotes T Cell Exhaustion. Immunity 2016, 44, 1190–1203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, H.; Blackburn, S.D.; Intlekofer, A.M.; Kao, C.; Angelosanto, J.M.; Reiner, S.L.; Wherry, E.J. A Role for the Transcriptional Repressor Blimp-1 in CD8+ T Cell Exhaustion during Chronic Viral Infection. Immunity 2009, 31, 309–320. [Google Scholar] [CrossRef] [Green Version]
- Paley, M.A.; Kroy, D.C.; Odorizzi, P.M.; Johnnidis, J.B.; Dolfi, D.V.; Barnett, B.E.; Bikoff, E.K.; Robertson, E.J.; Lauer, G.M.; Reiner, S.L.; et al. Progenitor and terminal subsets of CD8+ T cells cooperate to contain chronic viral infection. Science 2012, 338, 1220–1225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLane, L.M.; Ngiow, S.F.; Chen, Z.; Attanasio, J.; Manne, S.; Ruthel, G.; Wu, J.E.; Staupe, R.P.; Xu, W.; Amaravadi, R.K.; et al. Role of nuclear localization in the regulation and function of T-bet and Eomes in exhausted CD8 T cells. Cell Rep. 2021, 35, 109120. [Google Scholar] [CrossRef] [PubMed]
- Blackburn, S.D.; Shin, H.; Freeman, G.J.; Wherry, E.J. Selective expansion of a subset of exhausted CD8 T cells by αPD-L1 blockade. Proc. Natl. Acad. Sci. USA 2008, 105, 15016–15021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandu, I.; Cerletti, D.; Oetiker, N.; Borsa, M.; Wagen, F.; Spadafora, I.; Welten, S.P.M.; Stolz, U.; Oxenius, A.; Claassen, M. Landscape of Exhausted Virus-Specific CD8 T Cells in Chronic LCMV Infection. Cell Rep. 2020, 32, 108078. [Google Scholar] [CrossRef] [PubMed]
- Utzschneider, D.T.; Charmoy, M.; Chennupati, V.; Pousse, L.; Ferreira, D.P.; Calderon-Copete, S.; Danilo, M.; Alfei, F.; Hofmann, M.; Wieland, D.; et al. T Cell Factor 1-Expressing Memory-like CD8+ T Cells Sustain the Immune Response to Chronic Viral Infections. Immunity 2016, 45, 415–427. [Google Scholar] [CrossRef] [Green Version]
- Im, S.J.; Hashimoto, M.; Gerner, M.Y.; Lee, J.; Kissick, H.T.; Burger, M.C.; Shan, Q.; Hale, J.S.; Lee, J.; Nasti, T.H.; et al. Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature 2016, 537, 417–421. [Google Scholar] [CrossRef]
- Li, Y.; Tang, L.; Guo, L.; Chen, C.; Gu, S.; Zhou, Y.; Ye, G.; Li, X.; Wang, W.; Liao, X.; et al. CXCL13-mediated recruitment of intrahepatic CXCR5+CD8+ T cells favors viral control in chronic HBV infection. J. Hepatol. 2020, 72, 420–430. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.; Zhu, Y.O.; Becht, E.; Aw, P.; Chen, J.; Poidinger, M.; de Sessions, P.F.; Hibberd, M.L.; Bertoletti, A.; Lim, S.G.; et al. Multifactorial heterogeneity of virus-specific T cells and association with the progression of human chronic hepatitis B infection. Sci. Immunol. 2019, 4, eaau6905. [Google Scholar] [CrossRef]
- Cheng, Y.; Gunasegaran, B.; Singh, H.D.; Dutertre, C.-A.; Loh, C.Y.; Lim, J.Q.; Crawford, J.C.; Lee, H.K.; Zhang, X.; Lee, B.; et al. Non-terminally exhausted tumor-resident memory HBV-specific T cell responses correlate with relapse-free survival in hepatocellular carcinoma. Immunity 2021, 54, 1825–1840.e7. [Google Scholar] [CrossRef]
- Schuch, A.; Salimi Alizei, E.; Heim, K.; Wieland, D.; Kiraithe, M.M.; Kemming, J.; Llewellyn-Lacey, S.; Sogukpinar, Ö.; Ni, Y.; Urban, S.; et al. Phenotypic and functional differences of HBV core-specific versus HBV polymerase-specific CD8+ T cells in chronically HBV-infected patients with low viral load. Gut 2018, 68, 905–915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wieland, D.; Kemming, J.; Schuch, A.; Emmerich, F.; Knolle, P.; Neumann-Haefelin, C.; Held, W.; Zehn, D.; Hofmann, M.; Thimme, R. TCF1+ hepatitis C virus-specific CD8+ T cells are maintained after cessation of chronic antigen stimulation. Nat. Commun. 2017, 8, 15050. [Google Scholar] [CrossRef] [Green Version]
- Hensel, N.; Gu, Z.; Sagar; Wieland, D.; Jechow, K.; Kemming, J.; Llewellyn-Lacey, S.; Gostick, E.; Sogukpinar, O.; Emmerich, F.; et al. Memory-like HCV-specific CD8+ T cells retain a molecular scar after cure of chronic HCV infection. Nat. Immunol. 2021, 22, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Heim, K.; Binder, B.; Sagar; Wieland, D.; Hensel, N.; Llewellyn-Lacey, S.; Gostick, E.; Price, D.A.; Emmerich, F.; Vingerhoet, H.; et al. TOX defines the degree of CD8+ T cell dysfunction in distinct phases of chronic HBV infection. Gut 2020, 70, 1550–1560. [Google Scholar] [CrossRef] [PubMed]
- Aregay, A.; Owusu Sekyere, S.; Deterding, K.; Port, K.; Dietz, J.; Berkowski, C.; Sarrazin, C.; Manns, M.P.; Cornberg, M.; Wedemeyer, H. Elimination of hepatitis C virus has limited impact on the functional and mitochondrial impairment of HCV-specific CD8+ T cell responses. J. Hepatol. 2019, 71, 889–899. [Google Scholar] [CrossRef]
- Tonnerre, P.; Wolski, D.; Subudhi, S.; Aljabban, J.; Hoogeveen, R.C.; Damasio, M.; Drescher, H.K.; Bartsch, L.M.; Tully, D.C.; Sen, D.R.; et al. Differentiation of exhausted CD8+ T cells after termination of chronic antigen stimulation stops short of achieving functional T cell memory. Nat. Immunol. 2021, 22, 1030–1041. [Google Scholar] [CrossRef] [PubMed]
- Yates, K.B.; Tonnerre, P.; Martin, G.E.; Gerdemann, U.; Al Abosy, R.; Comstock, D.E.; Weiss, S.A.; Wolski, D.; Tully, D.C.; Chung, R.T.; et al. Epigenetic scars of CD8+ T cell exhaustion persist after cure of chronic infection in humans. Nat. Immunol. 2021, 22, 1020–1029. [Google Scholar] [CrossRef]
- Fosdick, A.; Zheng, J.; Pflanz, S.; Frey, C.R.; Hesselgesser, J.; Halcomb, R.L.; Wolfgang, G.; Tumas, D.B. Pharmacokinetic and pharmacodynamic properties of gs-9620, a novel toll-like receptor 7 agonist, demonstrate interferon-stimulated gene induction without detectable serum interferon at low oral dosess. J. Pharmacol. Exp. Ther. 2014, 348, 96–105. [Google Scholar] [CrossRef] [Green Version]
- Menne, S.; Tumas, D.B.; Liu, K.H.; Thampi, L.; Aldeghaither, D.; Baldwin, B.H.; Bellezza, C.A.; Cote, P.J.; Zheng, J.; Halcomb, R.; et al. Sustained efficacy and seroconversion with the toll-like receptor 7 agonist GS-9620 in the woodchuck model of chronic hepatitis B. J. Hepatol. 2015, 62, 1237–1245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanford, R.E.; Guerra, B.; Chavez, D.; Giavedoni, L.; Hodara, V.L.; Brasky, K.M.; Fosdick, A.; Frey, C.R.; Zheng, J.; Wolfgang, G.; et al. GS-9620, an oral agonist of toll-like receptor-7, induces prolonged suppression of hepatitis B virus in chronically infected chimpanzees. Gastroenterology 2013, 144, 1508–1517.e10. [Google Scholar] [CrossRef] [Green Version]
- Boni, C.; Vecchi, A.; Rossi, M.; Laccabue, D.; Giuberti, T.; Alfieri, A.; Lampertico, P.; Grossi, G.; Facchetti, F.; Brunetto, M.R.; et al. TLR7 Agonist Increases Responses of Hepatitis B Virus–Specific T Cells and Natural Killer Cells in Patients with Chronic Hepatitis B Treated With Nucleos(T)Ide Analogues. Gastroenterology 2018, 154, 1764–1777.e7. [Google Scholar] [CrossRef]
- Korolowizc, K.E.; Li, B.; Huang, X.; Yon, C.; Rodrigo, E.; Corpuz, M.; Plouffe, D.M.; Kallakury, B.V.; Suresh, M.; Wu, T.Y.-H.; et al. Liver-Targeted Toll-Like Receptor 7 Agonist Combined with Entecavir Promotes a Functional Cure in the Woodchuck Model of Hepatitis B Virus. Hepatol. Commun. 2019, 3, 1296–1310. [Google Scholar] [CrossRef]
- Bertoletti, A.; Le Bert, N. Fine-Tuning TLR-7-Based Therapy for Functional HBV Cure. Hepatol. Commun. 2019, 3, 1289–1292. [Google Scholar] [CrossRef] [PubMed]
- Mackman, R.L.; Mish, M.; Chin, G.; Perry, J.K.; Appleby, T.; Aktoudianakis, V.; Metobo, S.; Pyun, P.; Niu, C.; Daffis, S.; et al. Discovery of GS-9688 (Selgantolimod) as a Potent and Selective Oral Toll-Like Receptor 8 Agonist for the Treatment of Chronic Hepatitis B. J. Med. Chem. 2020, 63, 10188–10203. [Google Scholar] [CrossRef]
- Daffis, S.; Balsitis, S.; Chamberlain, J.; Zheng, J.; Santos, R.; Rowe, W.; Ramakrishnan, D.; Pattabiraman, D.; Spurlock, S.; Chu, R.; et al. Toll-Like Receptor 8 Agonist GS-9688 Induces Sustained Efficacy in the Woodchuck Model of Chronic Hepatitis B. Hepatology 2021, 73, 53–67. [Google Scholar] [CrossRef] [Green Version]
- Amin, O.E.; Colbeck, E.J.; Daffis, S.; Khan, S.; Ramakrishnan, D.; Pattabiraman, D.; Chu, R.; Micolochick Steuer, H.; Lehar, S.; Peiser, L.; et al. Therapeutic Potential of TLR8 Agonist GS-9688 (Selgantolimod) in Chronic Hepatitis B: Remodeling of Antiviral and Regulatory Mediators. Hepatology 2021, 74, 55–71. [Google Scholar] [CrossRef]
- Gane, E.J.; Kim, H.J.; Visvanathan, K.; Kim, Y.J.; Nguyen, A.; Wallin, J.J.; Chen, D.Y.; McDonald, C.; Arora, P.; Tan, S.K.; et al. Safety, pharmacokinetics, and pharmacodynamics of the oral TLR8 agonist selgantolimod in chronic hepatitis B. Hepatology 2021, 74, 1737–1749. [Google Scholar] [CrossRef] [PubMed]
- Janovec, V.; Hodek, J.; Clarova, K.; Hofman, T.; Dostalik, P.; Fronek, J.; Chlupac, J.; Chaperot, L.; Durand, S.; Baumert, T.F.; et al. Toll-like receptor dual-acting agonists are potent inducers of PBMC-produced cytokines that inhibit hepatitis B virus production in primary human hepatocytes. Sci. Rep. 2020, 10, 12767. [Google Scholar] [CrossRef]
- Korolowicz, K.E.; Suresh, M.; Li, B.; Huang, X.; Yon, C.; Leng, X.; Kallakury, B.V.; Tucker, R.D.; Menne, S. Treatment with the immunomodulator aic649 in combination with entecavir produces antiviral efficacy in the woodchuck model of chronic hepatitis b. Viruses 2021, 13, 648. [Google Scholar] [CrossRef]
- Suresh, M.; Korolowicz, K.E.; Balarezo, M.; Iyer, R.P.; Padmanabhan, S.; Cleary, D.; Gimi, R.; Sheri, A.; Yon, C.; Kallakury, B.V.; et al. Antiviral efficacy and host immune response induction during sequential treatment with SB 9200 followed by entecavir in woodchucks. PLoS ONE 2017, 12, e0169631. [Google Scholar] [CrossRef] [Green Version]
- Jones, M.; Cunningham, M.E.; Wing, P.; DeSilva, S.; Challa, R.; Sheri, A.; Padmanabhan, S.; Iyer, R.P.; Korba, B.E.; Afdhal, N.; et al. SB 9200, a novel agonist of innate immunity, shows potent antiviral activity against resistant HCV variants. J. Med. Virol. 2017, 89, 1620–1628. [Google Scholar] [CrossRef]
- Fisicaro, P.; Valdatta, C.; Massari, M.; Loggi, E.; Ravanetti, L.; Urbani, S.; Giuberti, T.; Cavalli, A.; Vandelli, C.; Andreone, P.; et al. Combined blockade of programmed death-1 and activation of CD137 increase responses of human liver T cells against HBV, but not HCV. Gastroenterology 2012, 143, 1576–1585.e4. [Google Scholar] [CrossRef]
- Bengsch, B.; Martin, B.; Thimme, R. Restoration of HBV-specific CD8+ T cell function by PD-1 blockade in inactive carrier patients is linked to T cell differentiation. J. Hepatol. 2014, 61, 1212–1219. [Google Scholar] [CrossRef] [PubMed]
- Fisicaro, P.; Valdatta, C.; Massari, M.; Loggi, E.; Biasini, E.; Sacchelli, L.; Cavallo, M.C.; Silini, E.M.; Andreone, P.; Missale, G.; et al. Antiviral Intrahepatic T-Cell Responses Can Be Restored by Blocking Programmed Death-1 Pathway in Chronic Hepatitis B. Gastroenterology 2010, 138, 682–693.e4. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhang, E.; Ma, Z.; Wu, W.; Kosinska, A.; Zhang, X.; Möller, I.; Seiz, P.; Glebe, D.; Wang, B.; et al. Enhancing Virus-Specific Immunity In Vivo by Combining Therapeutic Vaccination and PD-L1 Blockade in Chronic Hepadnaviral Infection. PLoS Pathog. 2014, 10, e1003856. [Google Scholar] [CrossRef] [Green Version]
- Kosinska, A.D.; Liu, J.; Lu, M.; Roggendorf, M. Therapeutic vaccination and immunomodulation in the treatment of chronic hepatitis B: Preclinical studies in the woodchuck. Med. Microbiol. Immunol. 2015, 204, 103–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balsitis, S.; Gali, V.; Mason, P.J.; Chaniewski, S.; Levine, S.M.; Wichroski, M.J.; Feulner, M.; Song, Y.; Granaldi, K.; Loy, J.K.; et al. Safety and efficacy of anti-PD-L1 therapy in the woodchuck model of HBV infection. PLoS ONE 2018, 13, e0190058. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Yan, C.; Zhu, J.; Chen, X.; Fu, Q.; Zhang, H.; Tong, Z.; Liu, L.; Zheng, Y.; Zhao, P.; et al. Anti–PD-1/PD-L1 Blockade Immunotherapy Employed in Treating Hepatitis B Virus Infection–Related Advanced Hepatocellular Carcinoma: A Literature Review. Front. Immunol. 2020, 11, 11. [Google Scholar] [CrossRef]
- Gane, E.; Verdon, D.J.; Brooks, A.E.; Gaggar, A.; Nguyen, A.H.; Subramanian, G.M.; Schwabe, C.; Dunbar, P.R. Anti-PD-1 blockade with nivolumab with and without therapeutic vaccination for virally suppressed chronic hepatitis B: A pilot study. J. Hepatol. 2019, 71, 900–907. [Google Scholar] [CrossRef]
- Boni, C.; Fisicaro, P.; Valdatta, C.; Amadei, B.; Di Vincenzo, P.; Giuberti, T.; Laccabue, D.; Zerbini, A.; Cavalli, A.; Missale, G.; et al. Characterization of Hepatitis B Virus (HBV)-Specific T-Cell Dysfunction in Chronic HBV Infection. J. Virol. 2007, 81, 4215–4225. [Google Scholar] [CrossRef] [Green Version]
- Schurich, A.; Khanna, P.; Lopes, A.R.; Han, K.J.; Peppa, D.; Micco, L.; Nebbia, G.; Kennedy, P.T.F.; Geretti, A.M.; Dusheiko, G.; et al. Role of the coinhibitory receptor cytotoxic T lymphocyte antigen-4 on apoptosis-Prone CD8 T cells in persistent hepatitis B virus infection. Hepatology 2011, 53, 1494–1503. [Google Scholar] [CrossRef]
- Nebbia, G.; Peppa, D.; Schurich, A.; Khanna, P.; Singh, H.D.; Cheng, Y.; Rosenberg, W.; Dusheiko, G.; Gilson, R.; ChinAleong, J.; et al. Upregulation of the Tim-3/Galectin-9 Pathway of T Cell Exhaustion in Chronic Hepatitis B Virus Infection. PLoS ONE 2012, 7, e47648. [Google Scholar]
- Raziorrouh, B.; Schraut, W.; Gerlach, T.; Nowack, D.; Grüner, N.H.; Ulsenheimer, A.; Zachoval, R.; Wächtler, M.; Spannagl, M.; Haas, J.; et al. The immunoregulatory role of CD244 in chronic hepatitis B infection and its inhibitory potential on virus-specific CD8+ T-cell function. Hepatology 2010, 52, 1934–1947. [Google Scholar] [CrossRef] [PubMed]
- Jacobi, F.J.; Wild, K.; Smits, M.; Zoldan, K.; Csernalabics, B.; Flecken, T.; Lang, J.; Ehrenmann, P.; Emmerich, F.; Hofmann, M.; et al. OX40 stimulation and PD-L1 blockade synergistically augment HBV-specific CD4 T cells in patients with HBeAg-negative infection. J. Hepatol. 2019, 70, 1103–1113. [Google Scholar] [CrossRef] [PubMed]
- Swadling, L.; Pallett, L.J.; Diniz, M.O.; Baker, J.M.; Amin, O.E.; Stegmann, K.A.; Burton, A.R.; Schmidt, N.M.; Jeffery-Smith, A.; Zakeri, N.; et al. Human Liver Memory CD8+ T Cells Use Autophagy for Tissue Residence. Cell Rep. 2020, 30, 687–698.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schurich, A.; Pallett, L.J.; Jajbhay, D.; Wijngaarden, J.; Otano, I.; Gill, U.S.; Hansi, N.; Kennedy, P.T.; Nastouli, E.; Gilson, R.; et al. Distinct Metabolic Requirements of Exhausted and Functional Virus-Specific CD8 T Cells in the Same Host. Cell Rep. 2016, 16, 1243–1252. [Google Scholar] [CrossRef] [Green Version]
- Schurich, A.; Pallett, L.J.; Lubowiecki, M.; Singh, H.D.; Gill, U.S.; Kennedy, P.T.; Nastouli, E.; Tanwar, S.; Rosenberg, W.; Maini, M.K. The Third Signal Cytokine IL-12 Rescues the Anti-Viral Function of Exhausted HBV-Specific CD8 T Cells. PLoS Pathog. 2013, 9, e1003208. [Google Scholar] [CrossRef]
- Bénéchet, A.P.; De Simone, G.; Di Lucia, P.; Cilenti, F.; Barbiera, G.; Le Bert, N.; Fumagalli, V.; Lusito, E.; Moalli, F.; Bianchessi, V.; et al. Dynamics and genomic landscape of CD8+ T cells undergoing hepatic priming. Nature 2019, 574, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Cubero, E.; Subirá, D.; Sanz-de-Villalobos, E.; Parra-Cid, T.; Madejón, A.; Miquel, J.; Olveira, A.; González-Praetorius, A.; García-Samaniego, J.; Larrubia, J.-R. According to Hepatitis C Virus (HCV) Infection Stage, Interleukin-7 Plus 4-1BB Triggering Alone or Combined with PD-1 Blockade Increases TRAF1 low HCV-Specific CD8 + Cell Reactivity. J. Virol. 2018, 92, e01443-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.H.; Perry, C.J.; Tsui, Y.C.; Staron, M.M.; Parish, I.A.; Dominguez, C.X.; Rosenberg, D.W.; Kaech, S.M. Prostaglandin E2 and programmed cell death 1 signaling coordinately impair CTL function and survival during chronic viral infection. Nat. Med. 2015, 21, 327–334. [Google Scholar] [CrossRef] [PubMed]
- Barili, V.; Fisicaro, P.; Montanini, B.; Acerbi, G.; Filippi, A.; Forleo, G.; Romualdi, C.; Ferracin, M.; Guerrieri, F.; Pedrazzi, G.; et al. Targeting p53 and histone methyltransferases restores exhausted CD8+ T cells in HCV infection. Nat. Commun. 2020, 11, 604. [Google Scholar] [CrossRef] [Green Version]
- Fisicaro, P.; Barili, V.; Montanini, B.; Acerbi, G.; Ferracin, M.; Guerrieri, F.; Salerno, D.; Boni, C.; Massari, M.; Cavallo, M.C.; et al. Targeting mitochondrial dysfunction can restore antiviral activity of exhausted HBV-specific CD8 T cells in chronic hepatitis B. Nat. Med. 2017, 23, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Acerbi, G.; Montali, I.; Ferrigno, G.D.; Barili, V.; Schivazappa, S.; Alfieri, A.; Laccabue, D.; Loglio, A.; Borghi, M.; Massari, M.; et al. Functional reconstitution of HBV-specific CD8 T cells by in vitro polyphenol treatment in chronic hepatitis B. J. Hepatol. 2021, 74, 783–793. [Google Scholar] [CrossRef] [PubMed]
- Gane, E.J.; Weilert, F.; Orr, D.W.; Keogh, G.F.; Gibson, M.; Lockhart, M.M.; Frampton, C.M.; Taylor, K.M.; Smith, R.A.J.; Murphy, M.P. The mitochondria-targeted anti-oxidant mitoquinone decreases liver damage in a phase II study of hepatitis C patients. Liver Int. 2010, 30, 1019–1026. [Google Scholar] [CrossRef]
- Pallett, L.J.; Gill, U.S.; Quaglia, A.; Sinclair, L.V.; Jover-Cobos, M.; Schurich, A.; Singh, K.P.; Thomas, N.; Das, A.; Chen, A.; et al. Metabolic regulation of hepatitis B immunopathology by myeloid-derived suppressor cells. Nat. Med. 2015, 21, 591–600. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, N.M.; Wing, P.A.C.; Diniz, M.O.; Pallett, L.J.; Swadling, L.; Harris, J.M.; Burton, A.R.; Jeffery-Smith, A.; Zakeri, N.; Amin, O.E.; et al. Targeting human Acyl-CoA:cholesterol acyltransferase as a dual viral and T cell metabolic checkpoint. Nat. Commun. 2021, 12, 2814. [Google Scholar] [CrossRef]
- Nishida, N.; Kudo, M. Liver damage related to immune checkpoint inhibitors. Hepatol. Int. 2019, 13, 248–252. [Google Scholar] [CrossRef]
- Suslov, A.; Wieland, S.; Menne, S. Modulators of innate immunity as novel therapeutics for treatment of chronic hepatitis B. Curr. Opin. Virol. 2018, 30, 9–17. [Google Scholar] [CrossRef]
- Kumashie, K.G.; Cebula, M.; Hagedorn, C.; Kreppel, F.; Pils, M.C.; Koch-Nolte, F.; Rissiek, B.; Wirth, D. Improved Functionality of Exhausted Intrahepatic CXCR5+ CD8+ T Cells Contributes to Chronic Antigen Clearance Upon Immunomodulation. Front. Immunol. 2021, 11, 592328. [Google Scholar] [CrossRef]
- Lee, S.; Goyal, A.; Perelson, A.S.; Ishida, Y.; Saito, T.; Gale, M. Suppression of hepatitis B virus through therapeutic activation of RIG-I and IRF3 signaling in hepatocytes. iScience 2021, 24, 101969. [Google Scholar] [CrossRef] [PubMed]
- Khan, O.; Giles, J.R.; McDonald, S.; Manne, S.; Ngiow, S.F.; Patel, K.P.; Werner, M.T.; Huang, A.C.; Alexander, K.A.; Wu, J.E.; et al. TOX transcriptionally and epigenetically programs CD8+ T cell exhaustion. Nature 2019, 571, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Pauken, K.E.; Sammons, M.A.; Odorizzi, P.M.; Manne, S.; Godec, J.; Khan, O.; Drake, A.M.; Chen, Z.; Sen, D.R.; Kurachi, M.; et al. Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science 2016, 354, 1160–1165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoogeveen, R.C.; Robidoux, M.P.; Schwarz, T.; Heydmann, L.; Cheney, J.A.; Kvistad, D.; Aneja, J.; Melgaço, J.G.; Fernandes, C.A.; Chung, R.T.; et al. Phenotype and function of HBV-specific T cells is determined by the targeted epitope in addition to the stage of infection. Gut 2018, 68, 893–904. [Google Scholar] [CrossRef] [PubMed]
- Huang, A.C.; Postow, M.A.; Orlowski, R.J.; Mick, R.; Bengsch, B.; Manne, S.; Xu, W.; Harmon, S.; Giles, J.R.; Wenz, B.; et al. T-cell invigoration to tumour burden ratio associated with anti-PD-1 response. Nature 2017, 545, 60–65. [Google Scholar] [CrossRef] [Green Version]
- Miller, B.C.; Sen, D.R.; Al Abosy, R.; Bi, K.; Virkud, Y.V.; LaFleur, M.W.; Yates, K.B.; Lako, A.; Felt, K.; Naik, G.S.; et al. Subsets of exhausted CD8+ T cells differentially mediate tumor control and respond to checkpoint blockade. Nat. Immunol. 2019, 20, 326–336. [Google Scholar] [CrossRef]
- Siddiqui, I.; Schaeuble, K.; Chennupati, V.; Fuertes Marraco, S.A.; Calderon-Copete, S.; Pais Ferreira, D.; Carmona, S.J.; Scarpellino, L.; Gfeller, D.; Pradervand, S.; et al. Intratumoral Tcf1 + PD-1 + CD8 + T Cells with Stem-like Properties Promote Tumor Control in Response to Vaccination and Checkpoint Blockade Immunotherapy. Immunity 2019, 50, 195–211.e10. [Google Scholar] [CrossRef] [Green Version]
- Gide, T.N.; Quek, C.; Menzies, A.M.; Tasker, A.T.; Shang, P.; Holst, J.; Madore, J.; Lim, S.Y.; Velickovic, R.; Wongchenko, M.; et al. Distinct Immune Cell Populations Define Response to Anti-PD-1 Monotherapy and Anti-PD-1/Anti-CTLA-4 Combined Therapy. Cancer Cell 2019, 35, 238–255.e6. [Google Scholar] [CrossRef] [Green Version]
- El-Khoueiry, A.B.; Sangro, B.; Yau, T.; Crocenzi, T.S.; Kudo, M.; Hsu, C.; Kim, T.-Y.; Choo, S.-P.; Trojan, J.; Welling, T.H.; et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): An open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet 2017, 389, 2492–2502. [Google Scholar] [CrossRef]
- Klein Geltink, R.I.; Kyle, R.L.; Pearce, E.L. Unraveling the Complex Interplay Between T Cell Metabolism and Function. Annu. Rev. Immunol. 2018, 36, 461–488. [Google Scholar] [CrossRef]
- Pearce, E.L.; Poffenberger, M.C.; Chang, C.H.; Jones, R.G. Fueling immunity: Insights into metabolism and lymphocyte function. Science 2013, 342, 1242454. [Google Scholar] [CrossRef] [Green Version]
- Jacobs, S.R.; Herman, C.E.; MacIver, N.J.; Wofford, J.A.; Wieman, H.L.; Hammen, J.J.; Rathmell, J.C. Glucose Uptake Is Limiting in T Cell Activation and Requires CD28-Mediated Akt-Dependent and Independent Pathways. J. Immunol. 2008, 180, 4476–4486. [Google Scholar] [CrossRef] [Green Version]
- Chapman, N.M.; Boothby, M.R.; Chi, H. Metabolic coordination of T cell quiescence and activation. Nat. Rev. Immunol. 2020, 20, 55–70. [Google Scholar] [CrossRef] [PubMed]
- Zuniga, E.I.; Macal, M.; Lewis, G.M.; Harker, J.A. Innate and Adaptive Immune Regulation during Chronic Viral Infections. Annu. Rev. Virol. 2015, 2, 573–597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maciver, N.J.; Michalek, R.D.; Rathmell, J.C. Metabolic regulation of T lymphocytes. Annu. Rev. Immunol. 2013, 31, 259–283. [Google Scholar] [CrossRef] [Green Version]
- Bengsch, B.; Johnson, A.L.; Kurachi, M.; Odorizzi, P.M.; Pauken, K.E.; Attanasio, J.; Stelekati, E.; McLane, L.M.; Paley, M.A.; Delgoffe, G.M.; et al. Bioenergetic Insufficiencies Due to Metabolic Alterations Regulated by the Inhibitory Receptor PD-1 Are an Early Driver of CD8+ T Cell Exhaustion. Immunity 2016, 45, 358–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolski, D.; Foote, P.K.; Chen, D.Y.; Lewis-Ximenez, L.L.; Fauvelle, C.; Aneja, J.; Walker, A.; Tonnerre, P.; Torres-Cornejo, A.; Kvistad, D.; et al. Early Transcriptional Divergence Marks Virus-Specific Primary Human CD8+ T Cells in Chronic versus Acute Infection. Immunity 2017, 47, 648–663.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Staron, M.M.; Gray, S.M.; Marshall, H.D.; Parish, I.A.; Chen, J.H.; Perry, C.J.; Cui, G.; Li, M.O.; Kaech, S.M. The Transcription Factor FoxO1 Sustains Expression of the Inhibitory Receptor PD-1 and Survival of Antiviral CD8+ T Cells during Chronic Infection. Immunity 2014, 41, 802–814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kruiswijk, F.; Labuschagne, C.F.; Vousden, K.H. P53 in survival, death and metabolic health: A lifeguard with a licence to kill. Nat. Rev. Mol. Cell Biol. 2015, 16, 393–405. [Google Scholar] [CrossRef]
- Muñoz-Fontela, C.; Mandinova, A.; Aaronson, S.A.; Lee, S.W. Emerging roles of p53 and other tumour-suppressor genes in immune regulation. Nat. Rev. Immunol. 2016, 16, 741–750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canman, C.E.; Lim, D.S.; Cimprich, K.A.; Taya, Y.; Tamai, K.; Sakaguchi, K.; Appella, E.; Kastan, M.B.; Siliciano, J.D. Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science 1998, 281, 1677–1679. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [Green Version]
- Lanna, A.; Henson, S.M.; Escors, D.; Akbar, A.N. The kinase p38 activated by the metabolic regulator AMPK and scaffold TAB1 drives the senescence of human T cells. Nat. Immunol. 2014, 15, 965–972. [Google Scholar] [CrossRef] [Green Version]
- Akbar, A.N.; Henson, S.M.; Lanna, A. Senescence of T Lymphocytes: Implications for Enhancing Human Immunity. Trends Immunol. 2016, 37, 866–876. [Google Scholar] [CrossRef]
- Patsoukis, N.; Bardhan, K.; Chatterjee, P.; Sari, D.; Liu, B.; Bell, L.N.; Karoly, E.D.; Freeman, G.J.; Petkova, V.; Seth, P.; et al. PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat. Commun. 2015, 6, 6692. [Google Scholar] [CrossRef] [Green Version]
- Berger, N.A. Poly(ADP-Ribose) in the Cellular Response to DNA Damage. Radiat. Res. 1985, 101, 4. [Google Scholar] [CrossRef]
- Pascal, J.M. The comings and goings of PARP-1 in response to DNA damage. DNA Repair 2018, 71, 177–182. [Google Scholar] [CrossRef]
- Hogan, K.A.; Chini, C.C.S.; Chini, E.N. The Multi-faceted Ecto-enzyme CD38: Roles in Immunomodulation, Cancer, Aging, and Metabolic Diseases. Front. Immunol. 2019, 10, 1187. [Google Scholar] [CrossRef] [Green Version]
- Shahgaldi, S.; Kahmini, F.R. A comprehensive review of Sirtuins: With a major focus on redox homeostasis and metabolism. Life Sci. 2021, 282, 119803. [Google Scholar] [CrossRef] [PubMed]
- Brunet, A. Stress-Dependent Regulation of FOXO Transcription Factors by the SIRT1 Deacetylase. Science 2004, 303, 2011–2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bosch-Presegué, L.; Vaquero, A. Sirtuin-dependent epigenetic regulation in the maintenance of genome integrity. FEBS J. 2015, 282, 1745–1767. [Google Scholar] [CrossRef]
- Katsuyama, E.; Suarez-Fueyo, A.; Bradley, S.J.; Mizui, M.; Marin, A.V.; Mulki, L.; Krishfield, S.; Malavasi, F.; Yoon, J.; Sui, S.J.H.; et al. The CD38/NAD/SIRTUIN1/EZH2 Axis Mitigates Cytotoxic CD8 T Cell Function and Identifies Patients with SLE Prone to Infections. Cell Rep. 2020, 30, 112–123.e4. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Sauve, A.A. NAD+ metabolism: Bioenergetics, signaling and manipulation for therapy. Biochim. Biophys. Acta Proteins Proteom. 2016, 1864, 1787–1800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imai, S.; Guarente, L. NAD+ and sirtuins in aging and disease. Trends Cell Biol. 2014, 24, 464–471. [Google Scholar] [CrossRef]
- Avalos, J.L.; Bever, K.M.; Wolberger, C. Mechanism of sirtuin nhibition by nicotinamide: Altering the NAD + cosubstrate specificity of a Sir2 enzyme. Mol. Cell 2005, 17, 855–868. [Google Scholar] [CrossRef]
- Covarrubias, A.J.; Perrone, R.; Grozio, A.; Verdin, E. NAD+ metabolism and its roles in cellular processes during ageing. Nat. Rev. Mol. Cell Biol. 2021, 22, 119–141. [Google Scholar] [CrossRef]
- Fisicaro, P.; Barili, V.; Rossi, M.; Montali, I.; Vecchi, A.; Acerbi, G.; Laccabue, D.; Zecca, A.; Penna, A.; Missale, G.; et al. Pathogenetic Mechanisms of T Cell Dysfunction in Chronic HBV Infection and Related Therapeutic Approaches. Front. Immunol. 2020, 11, 849. [Google Scholar] [CrossRef] [PubMed]
- Kalia, V.; Sarkar, S.; Subramaniam, S.; Haining, W.N.; Smith, K.A.; Ahmed, R. Prolonged Interleukin-2Rα Expression on Virus-Specific CD8+ T Cells Favors Terminal-Effector Differentiation In Vivo. Immunity 2010, 32, 91–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pipkin, M.E.; Sacks, J.A.; Cruz-Guilloty, F.; Lichtenheld, M.G.; Bevan, M.J.; Rao, A. Interleukin-2 and Inflammation Induce Distinct Transcriptional Programs that Promote the Differentiation of Effector Cytolytic T Cells. Immunity 2010, 32, 79–90. [Google Scholar] [CrossRef] [Green Version]
- Spolski, R.; Li, P.; Leonard, W.J. Biology and regulation of IL-2: From molecular mechanisms to human therapy. Nat. Rev. Immunol. 2018, 18, 648–659. [Google Scholar] [CrossRef] [PubMed]
- Man, K.; Kallies, A. Synchronizing transcriptional control of T cell metabolism and function. Nat. Rev. Immunol. 2015, 15, 574–584. [Google Scholar] [CrossRef] [PubMed]
- Boni, C.; Barili, V.; Acerbi, G.; Rossi, M.; Vecchi, A.; Laccabue, D.; Penna, A.; Missale, G.; Ferrari, C.; Fisicaro, P. HBV Immune-Therapy: From Molecular Mechanisms to Clinical Applications. Int. J. Mol. Sci. 2019, 20, 2754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maini, M.K.; Burton, A.R. Restoring, releasing or replacing adaptive immunity in chronic hepatitis B. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 662–675. [Google Scholar] [CrossRef] [PubMed]
- Maini, M.K.; Pallett, L.J. Defective T-cell immunity in hepatitis B virus infection: Why therapeutic vaccination needs a helping hand. Lancet Gastroenterol. Hepatol. 2018, 3, 192–202. [Google Scholar] [CrossRef]
- Gollob, J.A.; Schnipper, C.P.; Murphy, E.A.; Ritz, J.; Frank, D.A. The functional synergy between IL-12 and IL-2 involves p38 mitogen-activated protein kinase and is associated with the augmentation of STAT serine phosphorylation. J. Immunol. 1999, 162, 4472–4481. [Google Scholar]
- Buck, M.D.; O’Sullivan, D.; Pearce, E.L. T cell metabolism drives immunity. J. Exp. Med. 2015, 212, 1345–1360. [Google Scholar] [CrossRef] [Green Version]
- Buck, M.D.; Sowell, R.T.; Kaech, S.M.; Pearce, E.L. Metabolic Instruction of Immunity. Cell 2017, 169, 570–586. [Google Scholar] [CrossRef]
- Pollizzi, K.N.; Powell, J.D. Integrating canonical and metabolic signalling programmes in the regulation of T cell responses. Nat. Rev. Immunol. 2014, 14, 435–446. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Dillon, C.P.; Shi, L.Z.; Milasta, S.; Carter, R.; Finkelstein, D.; McCormick, L.L.; Fitzgerald, P.; Chi, H.; Munger, J.; et al. The Transcription Factor Myc Controls Metabolic Reprogramming upon T Lymphocyte Activation. Immunity 2011, 35, 871–882. [Google Scholar] [CrossRef] [Green Version]
- Chou, C.; Pinto, A.K.; Curtis, J.D.; Persaud, S.P.; Cella, M.; Lin, C.-C.; Edelson, B.T.; Allen, P.M.; Colonna, M.; Pearce, E.L.; et al. c-Myc-induced transcription factor AP4 is required for host protection mediated by CD8+ T cells. Nat. Immunol. 2014, 15, 884–893. [Google Scholar] [CrossRef] [PubMed]
- Nie, Z.; Hu, G.; Wei, G.; Cui, K.; Yamane, A.; Resch, W.; Wang, R.; Green, D.R.; Tessarollo, L.; Casellas, R.; et al. c-Myc is a universal amplifier of expressed genes in lymphocytes and embryonic stem cells. Cell 2012, 151, 68–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Riggelen, J.; Yetil, A.; Felsher, D.W. MYC as a regulator of ribosome biogenesis and protein synthesis. Nat. Rev. Cancer 2010, 10, 301–309. [Google Scholar] [CrossRef]
- Lochner, M.; Berod, L.; Sparwasser, T. Fatty acid metabolism in the regulation of T cell function. Trends Immunol. 2015, 36, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Kidani, Y.; Elsaesser, H.; Hock, M.B.; Vergnes, L.; Williams, K.J.; Argus, J.P.; Marbois, B.N.; Komisopoulou, E.; Wilson, E.B.; Osborne, T.F.; et al. Sterol regulatory element–binding proteins are essential for the metabolic programming of effector T cells and adaptive immunity. Nat. Immunol. 2013, 14, 489–499. [Google Scholar] [CrossRef] [Green Version]
- Finlay, D.K.; Rosenzweig, E.; Sinclair, L.V.; Carmen, F.C.; Hukelmann, J.L.; Rolf, J.; Panteleyev, A.A.; Okkenhaug, K.; Cantrell, D.A. PDK1 regulation of mTOR and hypoxia-inducible factor 1 integrate metabolism and migration of CD8+ T cells. J. Exp. Med. 2012, 209, 2441–2453. [Google Scholar] [CrossRef] [Green Version]
- Gnanaprakasam, J.N.R.; Sherman, J.W.; Wang, R. MYC and HIF in shaping immune response and immune metabolism. Cytokine Growth Factor Rev. 2017, 35, 63–70. [Google Scholar] [CrossRef]
- Johnson, M.O.; Siska, P.J.; Contreras, D.C.; Rathmell, J.C. Nutrients and the microenvironment to feed a T cell army. Semin. Immunol. 2016, 28, 505–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, M.; Peng, M.; Yin, N.; Chhangawala, S.; Xu, K.; Leslie, C.S.; Li, M.O. Aerobic glycolysis promotes T helper 1 cell differentiation through an epigenetic mechanism. Science 2016, 6284, 481–485. [Google Scholar] [CrossRef]
- Wellen, K.E.; Hatzivassiliou, G.; Sachdeva, U.M.; Bui, T.V.; Cross, J.R.; Thompson, C.B. ATP-Citrate Lyase Links Cellular Metabolism to Histone Acetylation. Science 2009, 324, 1076–1080. [Google Scholar] [CrossRef] [Green Version]
- Gray, S.M.; Amezquita, R.A.; Guan, T.; Kleinstein, S.H.; Kaech, S.M. Polycomb Repressive Complex 2-Mediated Chromatin Repression Guides Effector CD8(+) T Cell Terminal Differentiation and Loss of Multipotency. Immunity 2017, 46, 596–608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, B.; Zhang, K.; Milner, J.J.; Toma, C.; Chen, R.; Scott-Browne, J.P.; Pereira, R.M.; Crotty, S.; Chang, J.T.; Pipkin, M.E.; et al. Epigenetic landscapes reveal transcription factors that regulate CD8(+) T cell differentiation. Nat. Immunol. 2017, 18, 573–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sen, D.R.; Kaminski, J.; Barnitz, R.A.; Kurachi, M.; Gerdemann, U.; Yates, K.B.; Tsao, H.-W.; Godec, J.; LaFleur, M.W.; Brown, F.D.; et al. The epigenetic landscape of T cell exhaustion. Science 2016, 354, 1165–1169. [Google Scholar] [CrossRef] [Green Version]
- Beltra, J.C.; Manne, S.; Abdel-Hakeem, M.S.; Kurachi, M.; Giles, J.R.; Chen, Z.; Casella, V.; Ngiow, S.F.; Khan, O.; Huang, Y.J.; et al. Developmental Relationships of Four Exhausted CD8+ T Cell Subsets Reveals Underlying Transcriptional and Epigenetic Landscape Control Mechanisms. Immunity 2020, 52, 825–841.e8. [Google Scholar] [CrossRef]
- Philip, M.; Fairchild, L.; Sun, L.; Horste, E.L.; Camara, S.; Shakiba, M.; Scott, A.C.; Viale, A.; Lauer, P.; Merghoub, T.; et al. Chromatin states define tumour-specific T cell dysfunction and reprogramming. Nature 2017, 545, 452–456. [Google Scholar] [CrossRef] [PubMed]
- Doering, T.A.; Crawford, A.; Angelosanto, J.M.; Paley, M.A.; Ziegler, C.G.; Wherry, E.J. Network analysis reveals centrally connected genes and pathways involved in CD8+ T cell exhaustion versus memory. Immunity 2012, 37, 1130–1144. [Google Scholar] [CrossRef] [Green Version]
- Lu, P.; Youngblood, B.A.; Austin, J.J.; Mohammed, A.U.R.; Butler, R.; Ahmed, R.; Boss, J.M. Blimp-1 represses CD8 T cell expression of PD-1 using a feed-forward transcriptional circuit during acute viral infection. J. Exp. Med. 2014, 211, 515–527. [Google Scholar] [CrossRef] [Green Version]
- Martinez, G.J.; Pereira, R.M.; Äijö, T.; Kim, E.Y.; Marangoni, F.; Pipkin, M.E.; Togher, S.; Heissmeyer, V.; Zhang, Y.C.; Crotty, S.; et al. The transcription factor NFAT promotes exhaustion of activated CD8+ T cells. Immunity 2015, 42, 265–278. [Google Scholar] [CrossRef] [Green Version]
- Ghoneim, H.E.; Fan, Y.; Moustaki, A.; Abdelsamed, H.A.; Dash, P.; Dogra, P.; Carter, R.; Awad, W.; Neale, G.; Thomas, P.G.; et al. De Novo Epigenetic Programs Inhibit PD-1 Blockade-Mediated T Cell Rejuvenation. Cell 2017, 170, 142–157.e19. [Google Scholar] [CrossRef] [Green Version]
- Black, J.C.; Van Rechem, C.; Whetstine, J.R. Histone Lysine Methylation Dynamics: Establishment, Regulation, and Biological Impact. Mol. Cell 2012, 48, 491–507. [Google Scholar] [CrossRef] [Green Version]
- Margueron, R.; Reinberg, D. The Polycomb complex PRC2 and its mark in life. Nature 2011, 469, 343–349. [Google Scholar] [CrossRef] [Green Version]
- Kouzarides, T. Chromatin Modifications and Their Function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [Green Version]
- Shanbhag, N.M.; Rafalska-Metcalf, I.U.; Balane-Bolivar, C.; Janicki, S.M.; Greenberg, R.A. ATM-dependent chromatin changes silence transcription in cis to DNA double-strand breaks. Cell 2010, 141, 970–981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, R.; Tsukada, Y.I.; Zhang, Y. Role of Bmi-1 and Ring1A in H2A ubiquitylation and hox gene silencing. Mol. Cell 2005, 20, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Mattiroli, F.; Vissers, J.H.A.; Van Dijk, W.J.; Ikpa, P.; Citterio, E.; Vermeulen, W.; Marteijn, J.A.; Sixma, T.K. RNF168 ubiquitinates K13-15 on H2A/H2AX to drive DNA damage signaling. Cell 2012, 150, 1182–1195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vissers, J.H.A.; Van Lohuizen, M.; Citterio, E. The emerging role of Polycomb repressors in the response to DNA damage. J. Cell Sci. 2012, 125, 3939–3948. [Google Scholar] [CrossRef] [Green Version]
- Chambers, A.L.; Brownlee, P.M.; Durley, S.C.; Beacham, T.; Kent, N.A.; Downs, J.A. The two different isoforms of the RSC chromatin remodeling complex play distinct roles in DNA damage responses. PLoS ONE 2012, 7, e32016. [Google Scholar] [CrossRef]
- Bennett, G.; Papamichos-Chronakis, M.; Peterson, C.L. DNA repair choice defines a common pathway for recruitment of chromatin regulators. Nat. Commun. 2013, 4, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Papamichos-Chronakis, M.; Peterson, C.L. Chromatin and the genome integrity network. Nat. Rev. Genet. 2013, 14, 62–75. [Google Scholar] [CrossRef] [Green Version]
- Kakaradov, B.; Arsenio, J.; Widjaja, C.E.; He, Z.; Aigner, S.; Metz, P.J.; Yu, B.; Wehrens, E.J.; Lopez, J.; Kim, S.H.; et al. Early transcriptional and epigenetic regulation of CD8(+) T cell differentiation revealed by single-cell RNA sequencing. Nat. Immunol. 2017, 18, 422–432. [Google Scholar] [CrossRef] [Green Version]
- Tumes, D.J.; Onodera, A.; Suzuki, A.; Shinoda, K.; Endo, Y.; Iwamura, C.; Hosokawa, H.; Koseki, H.; Tokoyoda, K.; Suzuki, Y.; et al. The Polycomb protein Ezh2 regulates differentiation and plasticity of CD4+ T helper Type 1 and type 2 cells. Immunity 2013, 39, 819–832. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Kinkel, S.; Maksimovic, J.; Bandala-Sanchez, E.; Tanzer, M.C.; Naselli, G.; Zhang, J.G.; Zhan, Y.; Lew, A.M.; Silke, J.; et al. The polycomb repressive complex 2 governs life and death of peripheral T cells. Blood 2014, 124, 737–749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karantanos, T.; Chistofides, A.; Barhdan, K.; Li, L.; Boussiotis, V.A. Regulation of T cell differentiation and function by EZH2. Front. Immunol. 2016, 7, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheer, S.; Zaph, C. The lysine methyltransferase G9a in immune cell differentiation and function. Front. Immunol. 2017, 8, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, S.; Aune, T.M. Dynamic changes in histone-methylation “marks” across the locus encoding interferon-γ during the differentiation of T helper type 2 cells. Nat. Immunol. 2007, 8, 723–731. [Google Scholar] [CrossRef] [PubMed]
- Goswami, S.; Apostolou, I.; Zhang, J.; Skepner, J.; Anandhan, S.; Zhang, X.; Xiong, L.; Trojer, P.; Aparicio, A.; Subudhi, S.K.; et al. Modulation of EZH2 expression in T cells improves efficacy of anti-CTLA-4 therapy. J. Clin. Investig. 2018, 128, 3813–3818. [Google Scholar] [CrossRef]
- McGoverne, I.; Dunn, J.; Batham, J.; Tu, W.J.; Chrisp, J.; Rao, S. Epitherapy and immune checkpoint blockade: Using epigenetic reinvigoration of exhausted and dysfunctional T cells to reimburse immunotherapy response. BMC Immunol. 2020, 21, 1–10. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
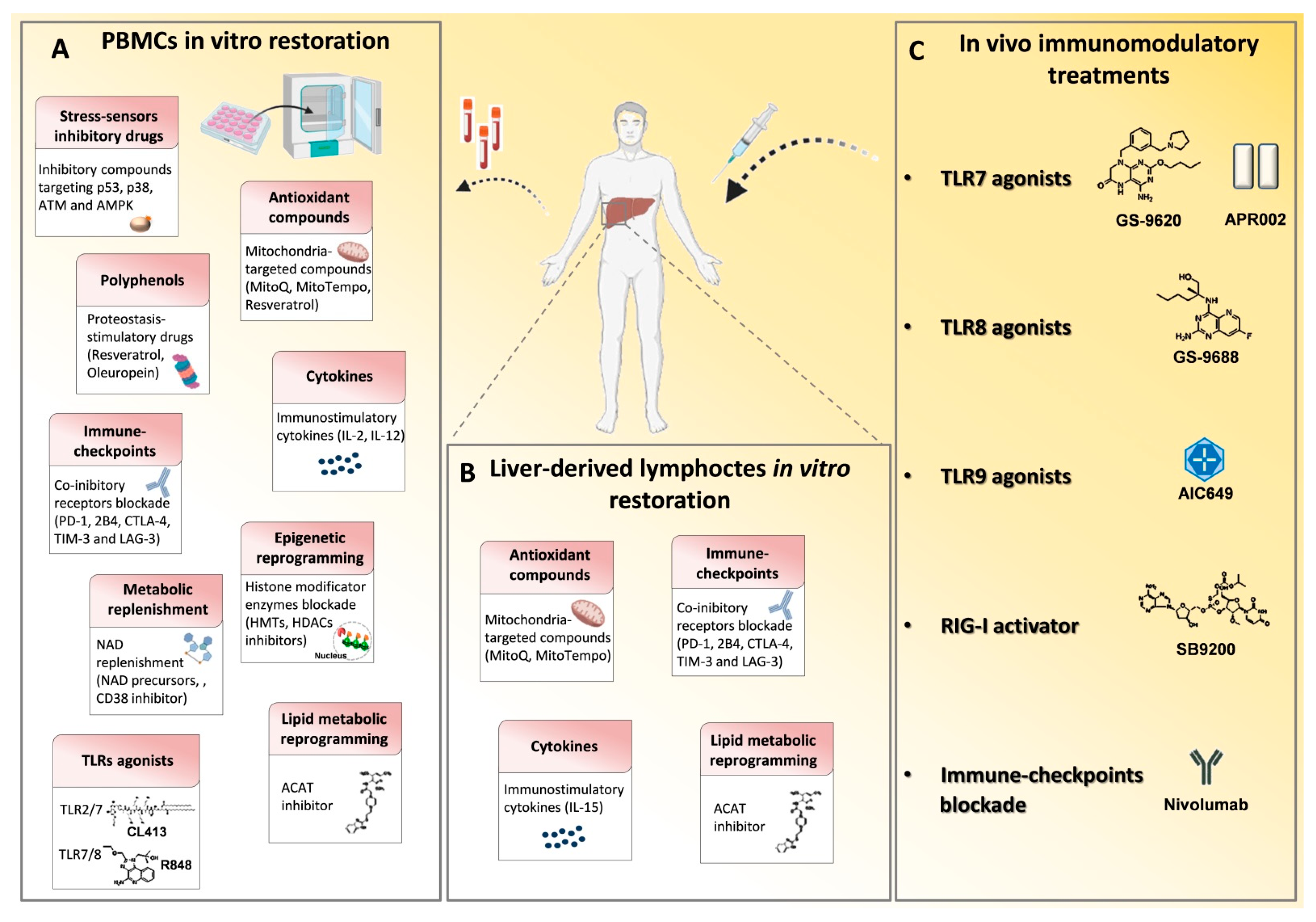
| Immune-Modulatory Interventions | Class of Agents | HBV | HCV |
|---|---|---|---|
| Stimulation of innate immunity receptors | GS-9620 | [31,32,33,34] | - |
| APR002 | [35,36] | - | |
| GS-9688/selgantolimod | [37,38,39,40] | - | |
| R848 | [41] | - | |
| CL413 | [41] | - | |
| AIC649 | [42] | - | |
| SB 9200 | [43] | [44] | |
| Co-inhibitory pathways blocking | PD-1 | [45,46,47,48,49,50,51,52,53] | [45] |
| CTLA-4 | [54] | - | |
| TIM-3 | [55] | - | |
| 2B4 | [56] | - | |
| Co-stimulatory signaling activation | CD137 | [45] | [45] |
| OX40 | [45,57] | [45] | |
| Cytokine fueling | IL-15 | [58] | - |
| IL-12 | [59,60] | - | |
| IL-2 | [61] | - | |
| IL-7 | - | [62] | |
| PGE2 inhibition | [63] | - | |
| Metabolic modulation | p53, p38, AMPK, ATM inhibitory compounds | - | [64] |
| N-acetyl-L-cysteine (NAC) | - | [64] | |
| MitoQ/MitoTempo | [65,66] | [67] | |
| Polyphenols Resveratrol and Oleuropein | [65,66] | - | |
| Arginine replenishment | [68] | - | |
| Acyl-CoA:cholesterol acyltransferase (ACAT) inhibitor | [69] | - | |
| Epigenetic intervention | Histone methyltransferses inhibitors | - | [64] |
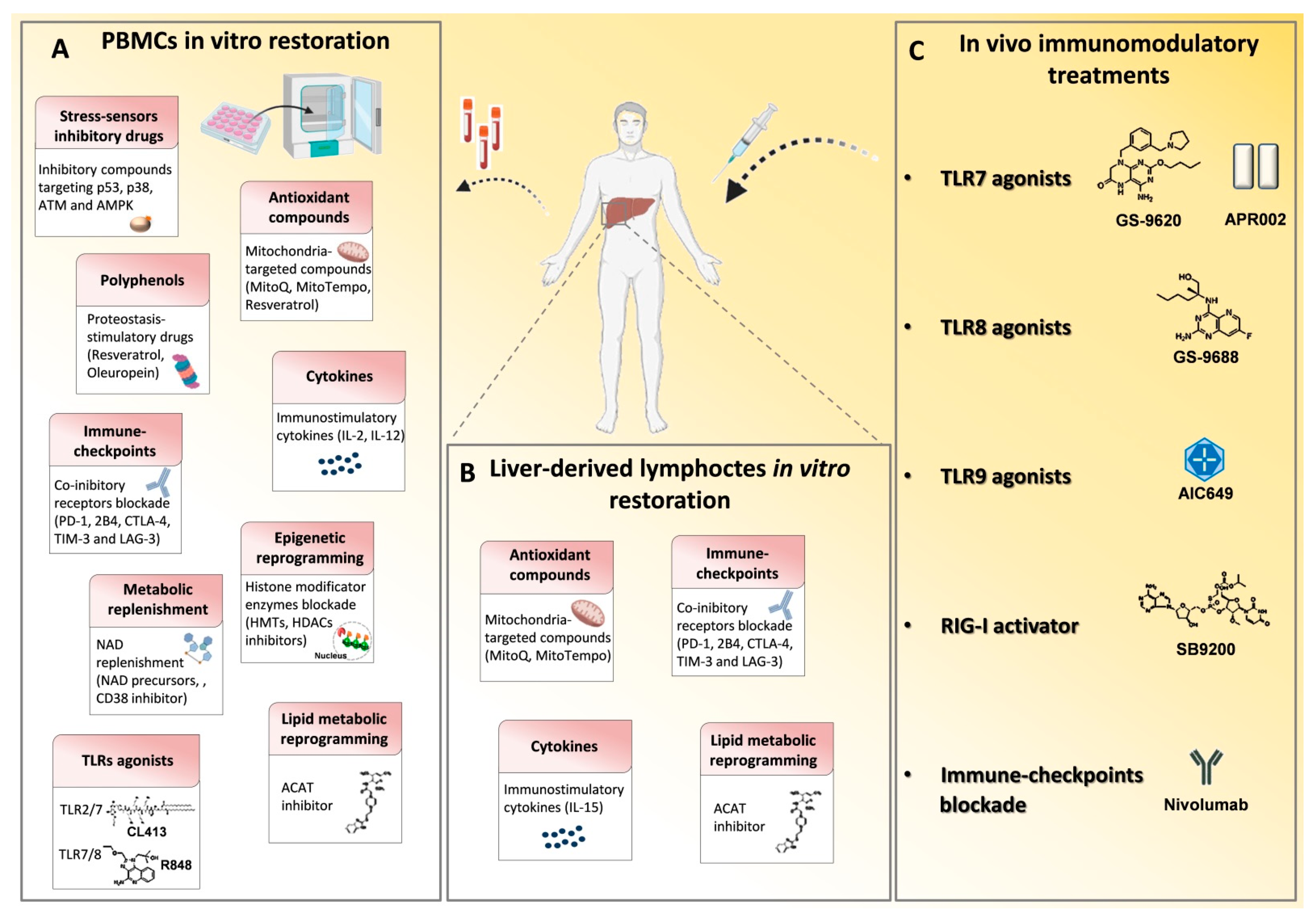
| Class of Agents | In Vitro/ In Vivo Use | Mechanism of Action | Target Cells | Disease | References |
|---|---|---|---|---|---|
| GS-9620 | In vivo | Induction of TLR7 activation | - | HBV | [31,32,33,34] |
| APR002 | In vivo | Induction of TLR7 activation | - | HBV | [35,36] |
| GS-9688/selgantolimod | In vitro/ In vivo | Agonist of endosomal TLR8 | Liver resident cells (activated DCs, mononuclear phagocytes and immune cells) | HBV | [37,38,39,40] |
| R848 | In vitro | Dual-acting TLR7/8 agonist | Hepatocytes | HBV | [41] |
| CL413 | In vitro | Dual-acting TLR2/7 agonist | Hepatocytes | HBV | [41] |
| AIC649 | In vivo | TLR9 pathway activator | - | HBV | [42] |
| SB 9200 | In vivo | RIG-I agonist | - | HBV, HCV | [43,44] |
| Checkpoint blockade | In vitro | PD-1, CTLA-4, TIM-3 and 2B4 | T cells | HBV, HCV | [45,46,47,53,54,55,56] |
| CD137 and OX40 | In vitro | Co-stimulatory CD137 or OX40 signaling activation | T cells | HBV, HCV | [45,57] |
| Nivolumab | In vivo | Anti-PD-1 for PD-1/PD-L1 blockade | T cells | HBV | [48,49,50,51,52,70] |
| p53, p38, AMPK, ATM inhibitory compounds | In vitro | Stress-sensor signaling kinase blockade | T cells | HCV | [64] |
| N-acetyl-L-cysteine (NAC) | In vitro | Anti-oxidant compounds | T cells | HCV | [64] |
| MitoQ/MitoTempo | In vitro/ In vivo | Mitochondrial anti-oxidant treatment | T cells | HBV, HCV | [65,66,67] |
| Polyphenols Resveratrol and Oleuropein | In vitro | Mitochondrial function and intracellular proteostasis restoration | T cells | HBV | [65,66] |
| Histone methyltransferses inhibitors | In vitro | EZH2 and G9a blockade | T cells | HCV | [64] |
| Arginine | In vitro | Arginine replenishment | T cells | HBV | [68] |
| acyl-CoA:cholesterol acyltransferase (ACAT) inhibitor | In vitro | Inhibition of cholesterol esterification | T cells | HBV | [69] |
| IL-15 | In vitro | Cellular proteostasis and mitochondrial function restoration | T cells | HBV | [58] |
| IL-12 | In vitro | Reverse mitochondria depolarization and glycolysis dependence | T cells | HBV | [59,60] |
| IL-2 | In vitro/ In vivo | T cell proliferate and differentiation restoration | T cells | HBV | [61] |
| IL-7 | In vitro | Exhaustion characterization and TRAF1 restoration | T cells | HCV | [62] |
| PGE2 inhibition | In vitro | PGE2 inhibitory signaling blockade | T cells | HBV | [63] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barili, V.; Vecchi, A.; Rossi, M.; Montali, I.; Tiezzi, C.; Penna, A.; Laccabue, D.; Missale, G.; Fisicaro, P.; Boni, C. Unraveling the Multifaceted Nature of CD8 T Cell Exhaustion Provides the Molecular Basis for Therapeutic T Cell Reconstitution in Chronic Hepatitis B and C. Cells 2021, 10, 2563. https://doi.org/10.3390/cells10102563
Barili V, Vecchi A, Rossi M, Montali I, Tiezzi C, Penna A, Laccabue D, Missale G, Fisicaro P, Boni C. Unraveling the Multifaceted Nature of CD8 T Cell Exhaustion Provides the Molecular Basis for Therapeutic T Cell Reconstitution in Chronic Hepatitis B and C. Cells. 2021; 10(10):2563. https://doi.org/10.3390/cells10102563
Chicago/Turabian StyleBarili, Valeria, Andrea Vecchi, Marzia Rossi, Ilaria Montali, Camilla Tiezzi, Amalia Penna, Diletta Laccabue, Gabriele Missale, Paola Fisicaro, and Carolina Boni. 2021. "Unraveling the Multifaceted Nature of CD8 T Cell Exhaustion Provides the Molecular Basis for Therapeutic T Cell Reconstitution in Chronic Hepatitis B and C" Cells 10, no. 10: 2563. https://doi.org/10.3390/cells10102563
APA StyleBarili, V., Vecchi, A., Rossi, M., Montali, I., Tiezzi, C., Penna, A., Laccabue, D., Missale, G., Fisicaro, P., & Boni, C. (2021). Unraveling the Multifaceted Nature of CD8 T Cell Exhaustion Provides the Molecular Basis for Therapeutic T Cell Reconstitution in Chronic Hepatitis B and C. Cells, 10(10), 2563. https://doi.org/10.3390/cells10102563