
Lamin B1 Accumulation’s Effects on Autosomal Dominant Leukodystrophy (ADLD): Induction of Reactivity in the Astrocytes

 ,
,  , ,
, ,  and
and 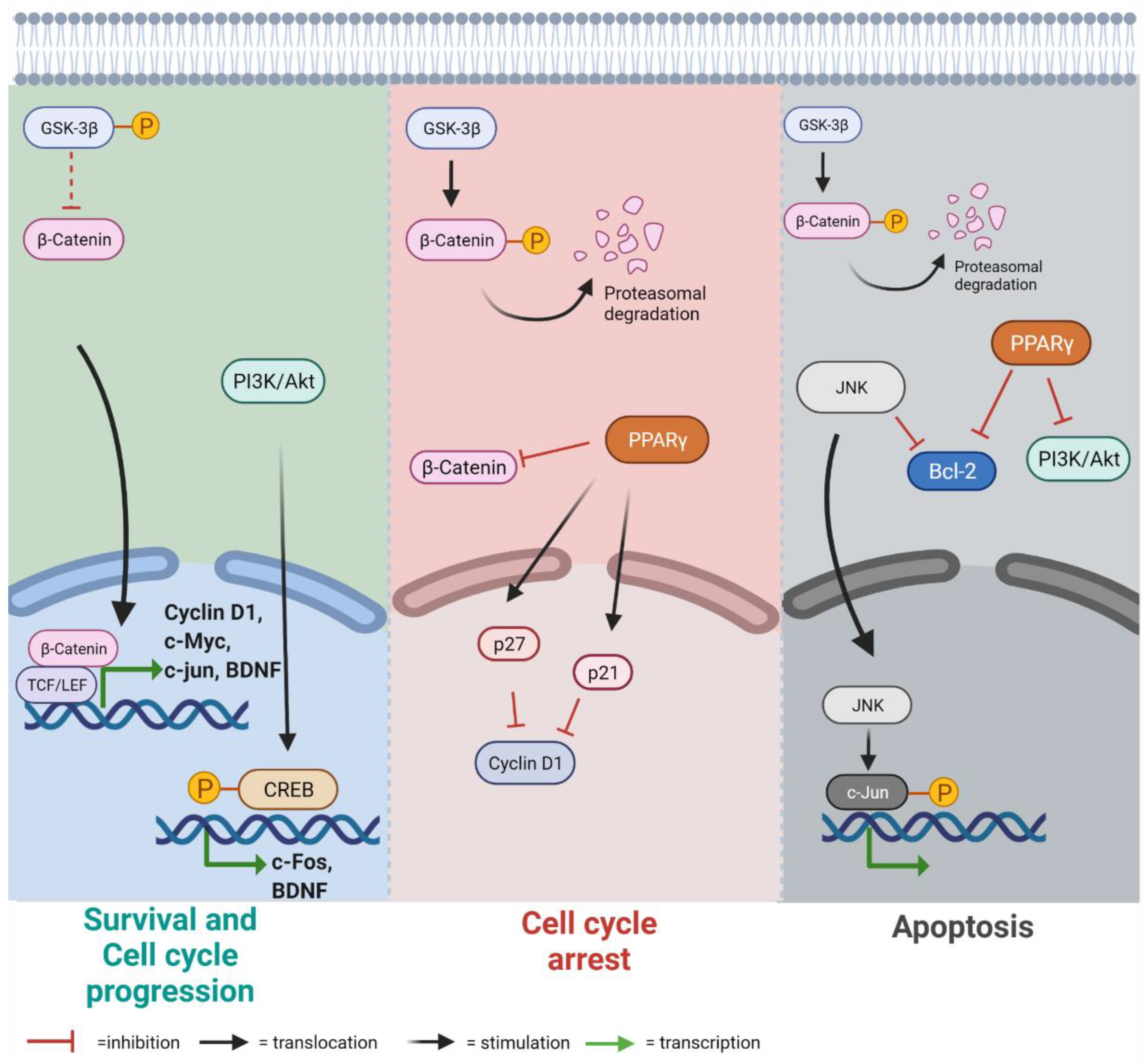{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Lentiviral Transduction
2.2. RNA Extraction, Reverse Transcription and Real-Time PCR
2.3. Protein Extraction, Nuclear Protein Extraction and Western Blot
2.4. Immunocytochemistry
2.5. Antibodies
2.6. Cell Viability, Cytotoxicity, Apoptosis Assay
2.7. Statistical Analysis
3. Results
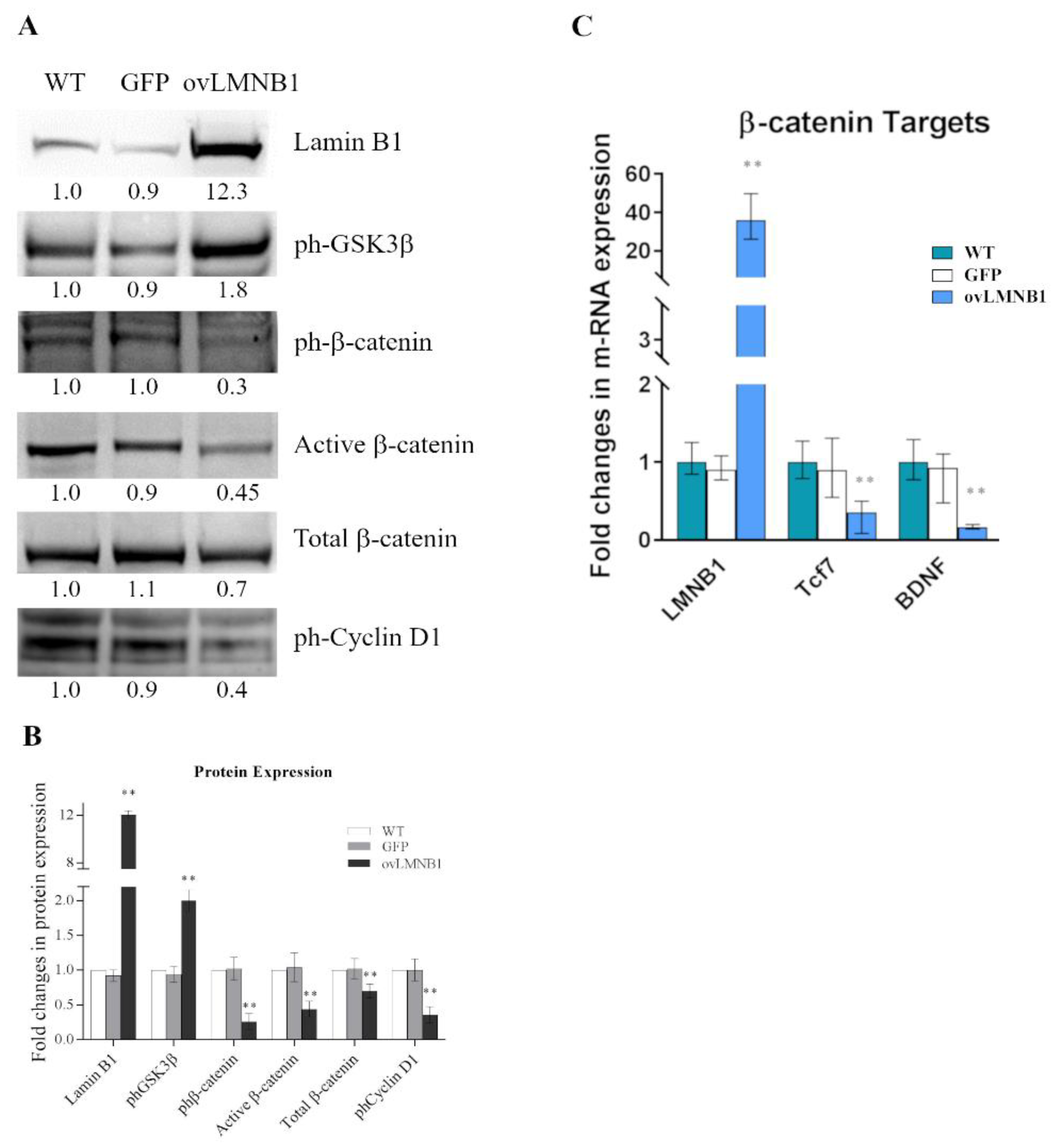
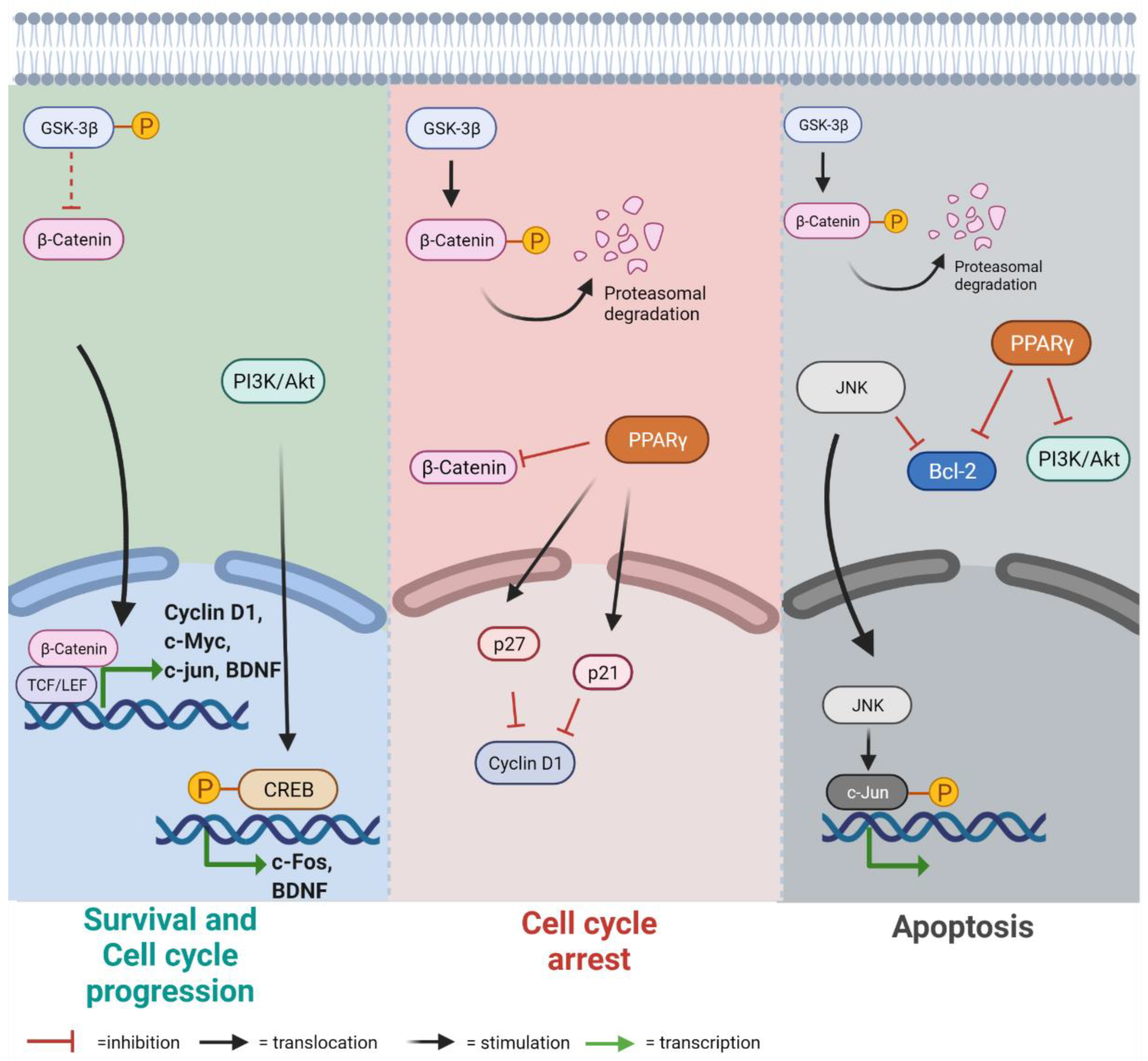
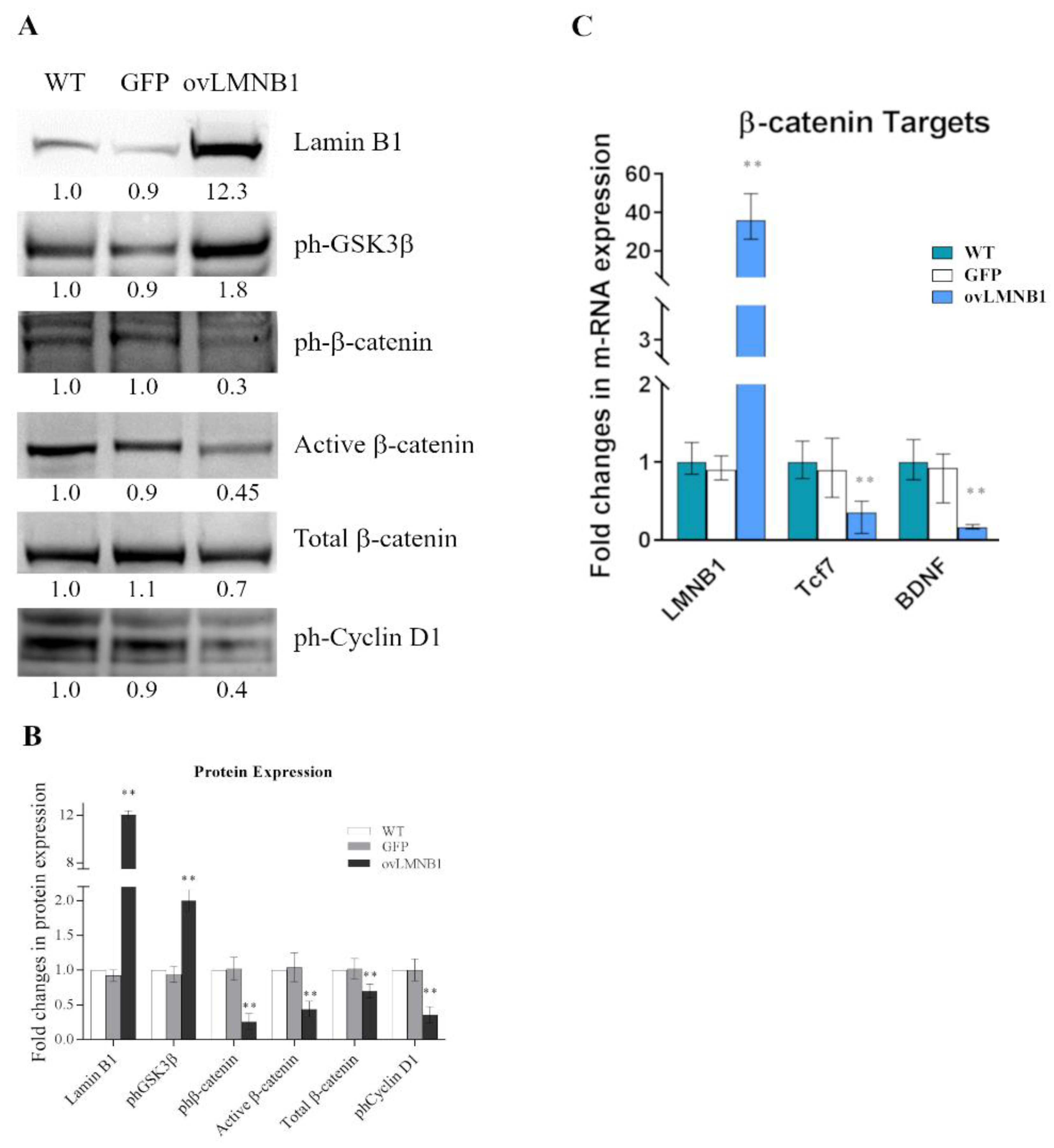
3.1. Lamin B1 Accumulation Determines GSK3β Inactivation, but Not the Upregulation of β-Catenin Targets
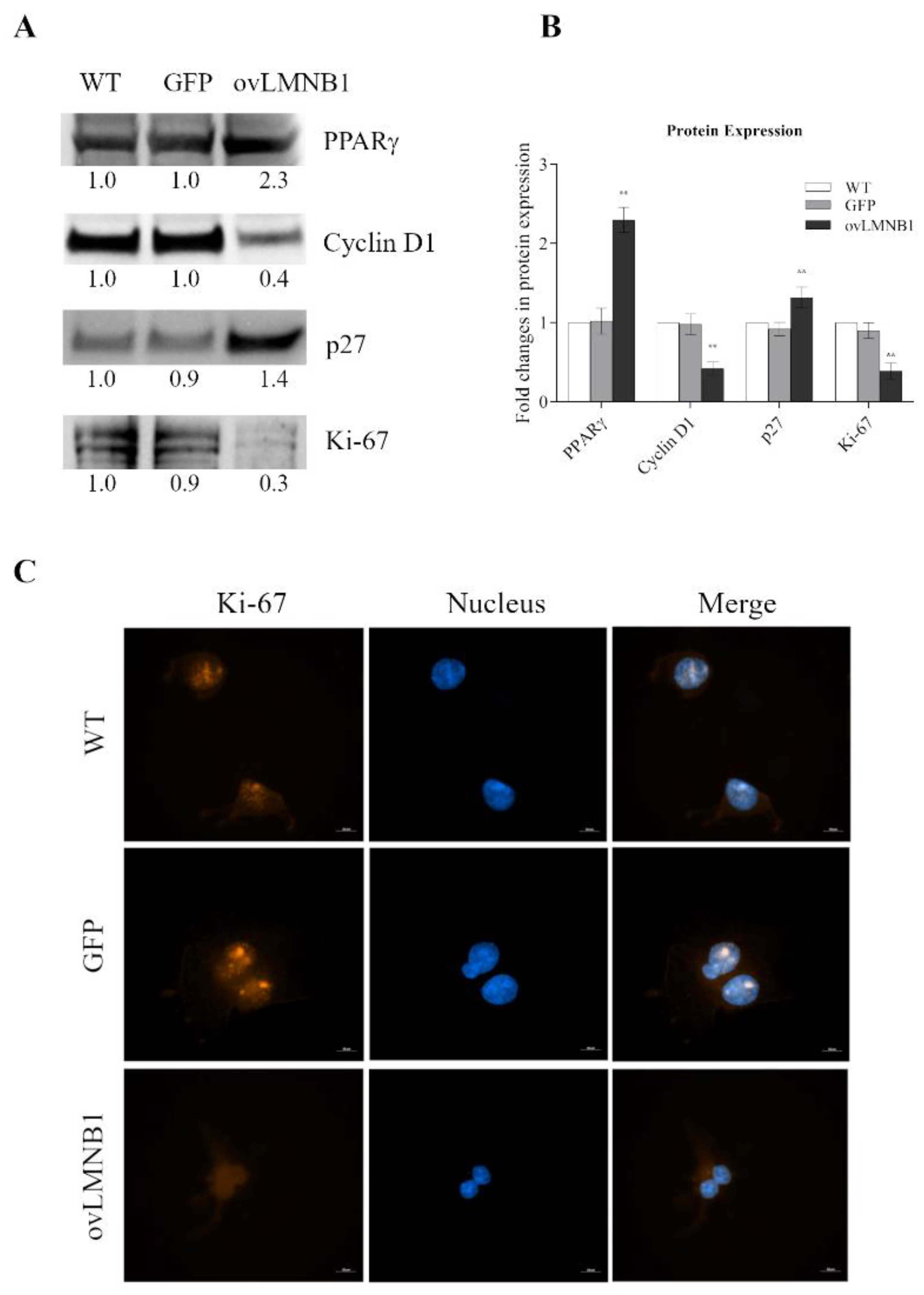
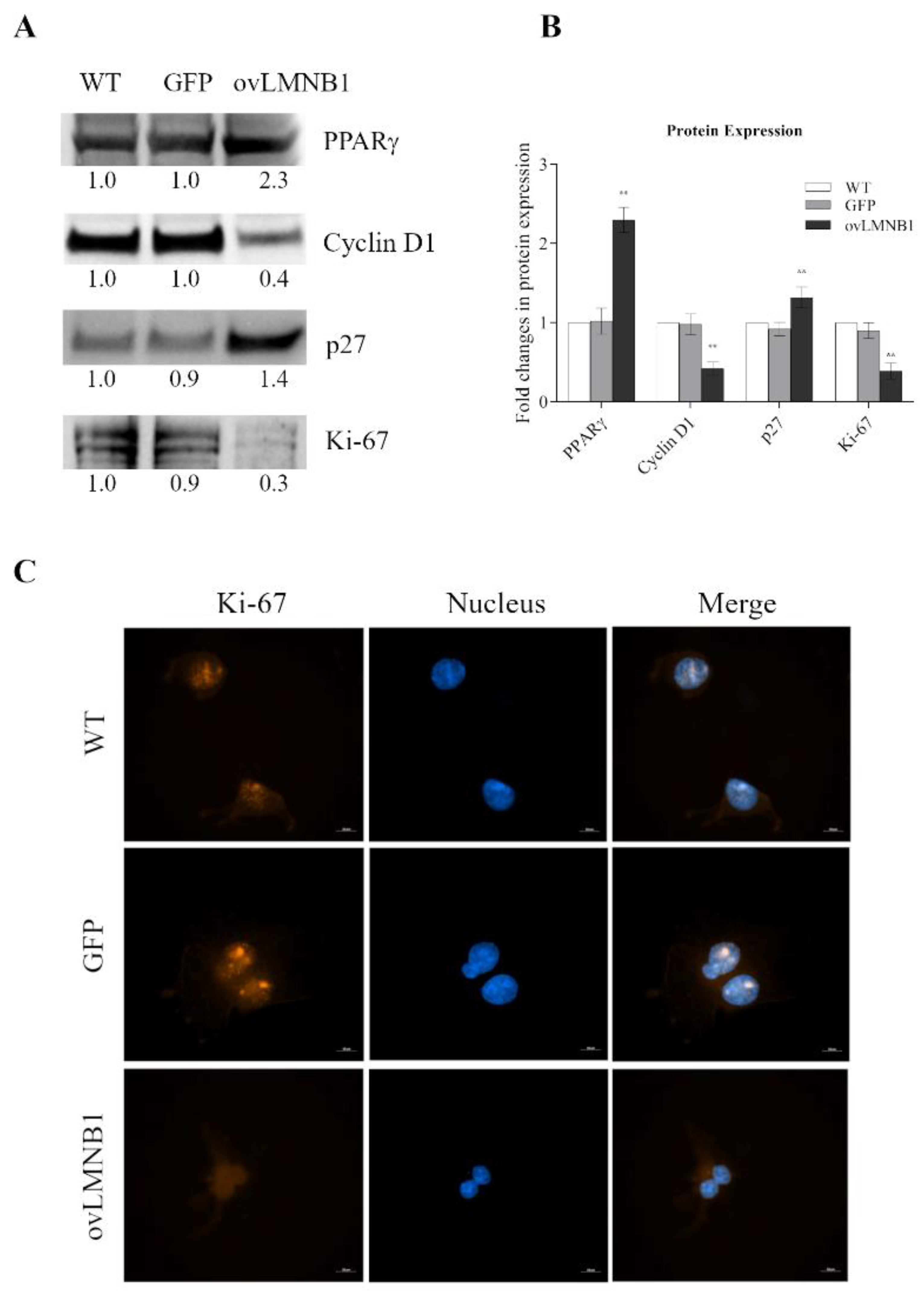
3.2. Lamin B1 Build Up Affects Proliferation and Cell Cycle Progression
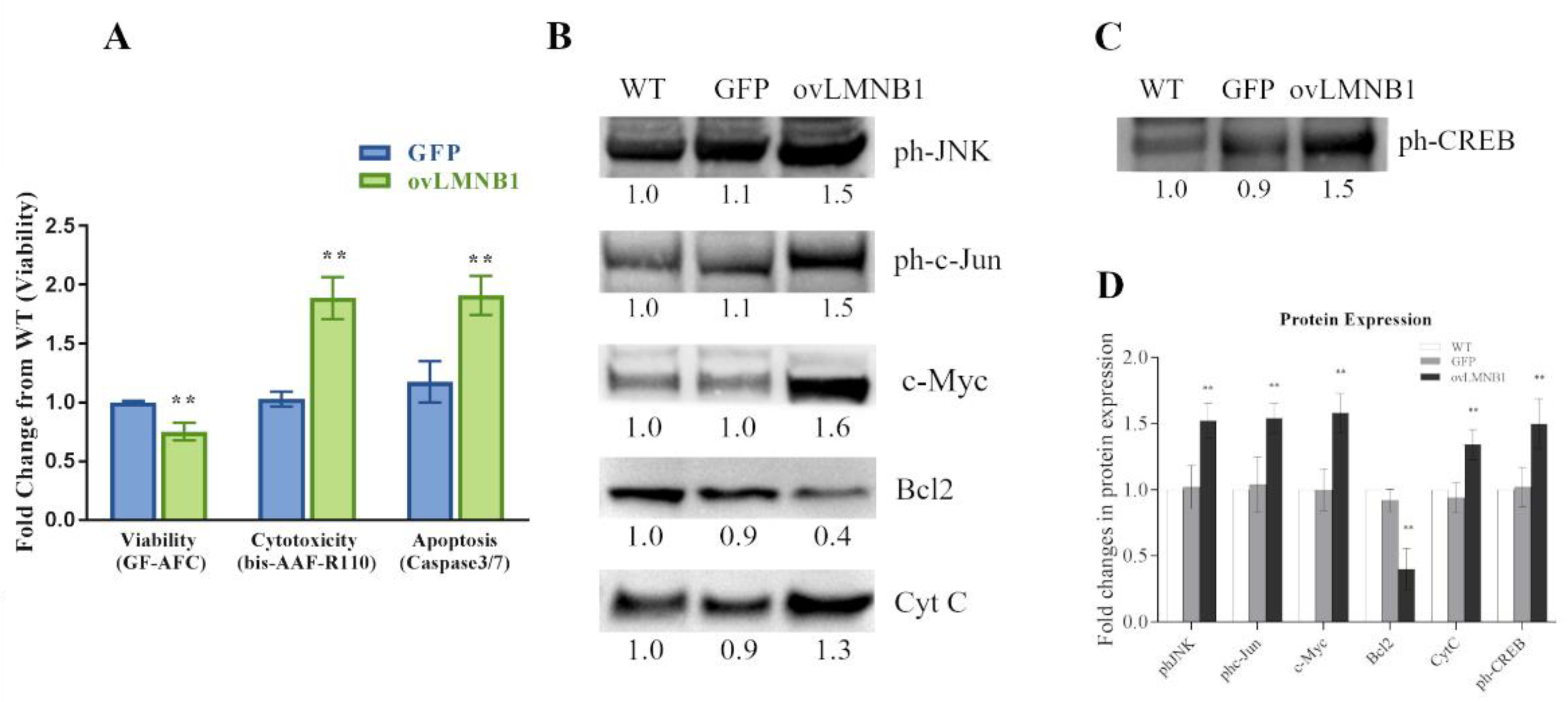
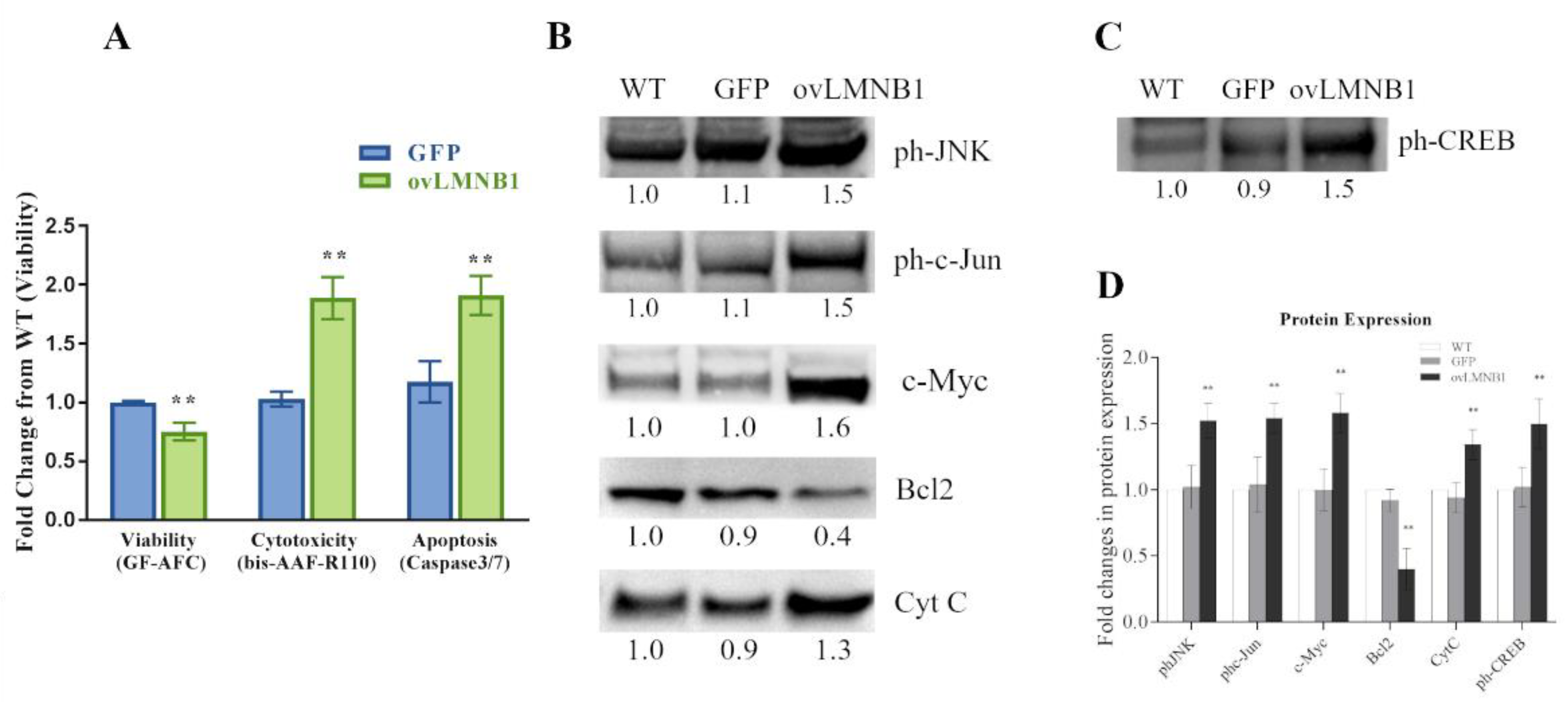
3.3. Lamin B1 Accumulation Reduces Cell Viability and Causes the Induction of Apoptosis
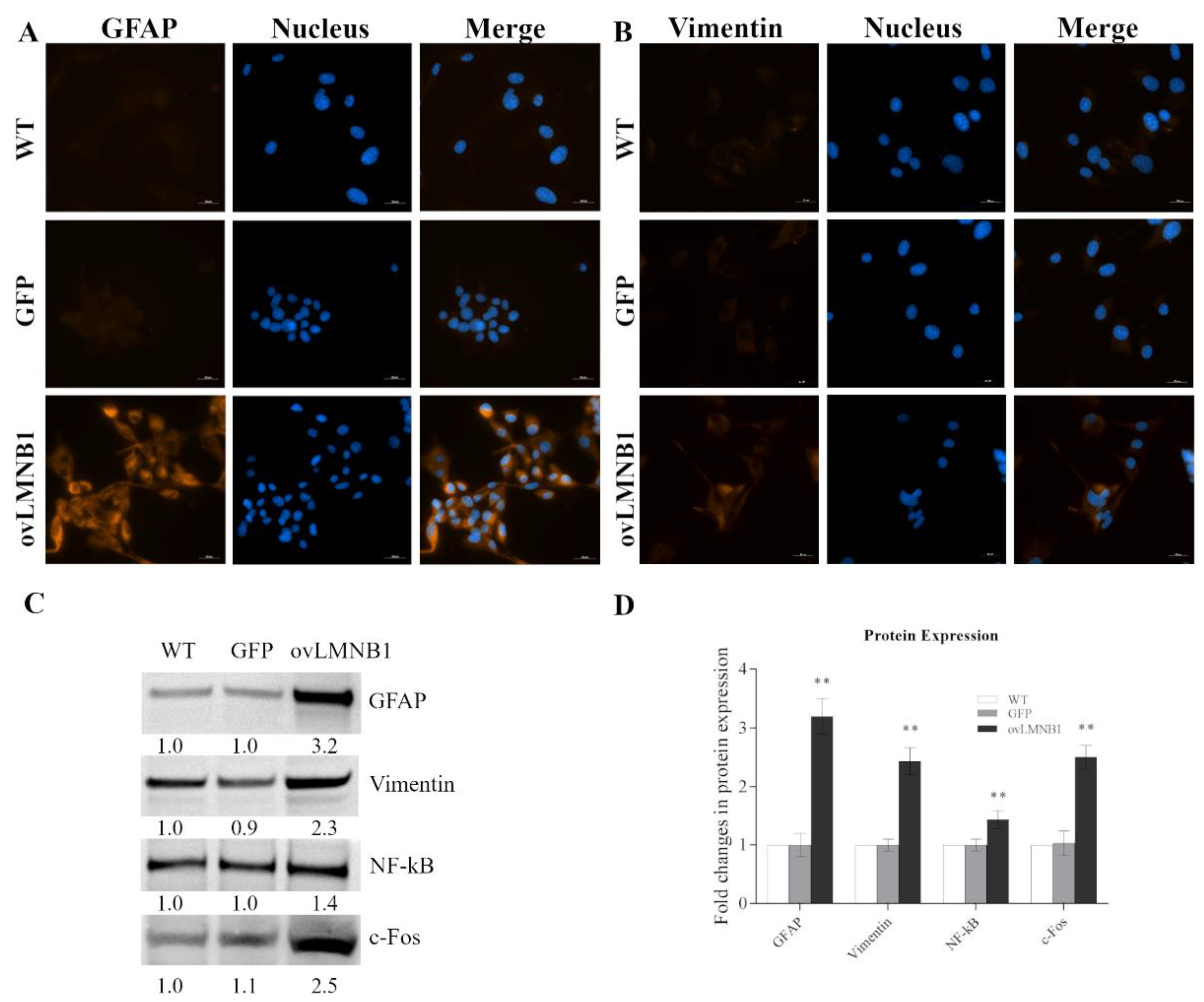
3.4. Lamin B1 Overexpression Induces Reactive Phenotype in Astrocyte-like Cells
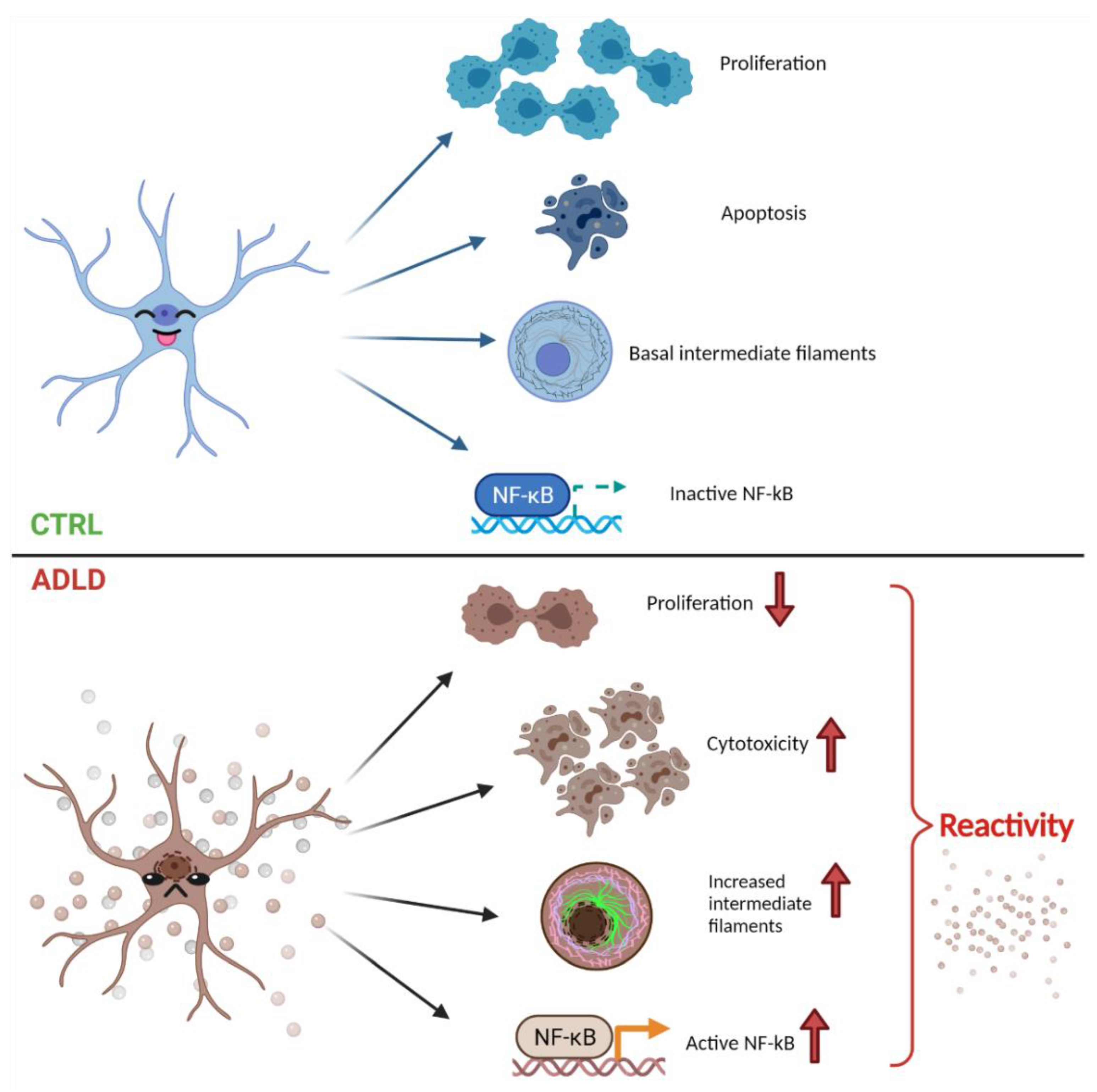
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Padiath, Q.S.; Saigoh, K.; Schiffmann, R.; Asahara, H.; Yamada, T.; Koeppen, A.; Hogan, K.; Ptáček, L.J.; Fu, Y.-H. Lamin B1 duplications cause autosomal dominant leukodystrophy. Nat. Genet. 2006, 38, 1114–1123. [Google Scholar] [CrossRef]
- Nahhas, N.; Sabet Rasekh, P.; Vanderver, A.; Padiath, Q. Autosomal Dominant Leukodystrophy with Autonomic Disease; University of Washington: Seattle, WD, USA, 1993. [Google Scholar]
- Zanigni, S.; Terlizzi, R.; Tonon, C.; Testa, C.; Manners, D.N.; Capellari, S.; Gallassi, R.; Poda, R.; Gramegna, L.L.; Calandra-Buonaura, G.; et al. Brain magnetic resonance metabolic and microstructural changes in adult-onset autosomal dominant leukodystrophy. Brain Res. Bull. 2015, 117, 24–31. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, J.; Bai, R.; Wang, J.; Peng, T.; Chen, L.; Wang, J.; Liu, Y.; Tian, T.; Lu, H. LMNB1-Related Adult-Onset Autosomal Dominant Leukodystrophy Presenting as Movement Disorder: A Case Report and Review of the Literature. Front. Neurosci. 2019, 13, 1030. [Google Scholar] [CrossRef] [Green Version]
- Dai, Y.; Ma, Y.; Li, S.; Banerjee, S.; Liang, S.; Liu, Q.; Yang, Y.; Peng, B.; Cui, L.; Jin, L. An LMNB1 duplication caused adult-onset autosomal dominant leukodystrophy in chinese family: Clinical manifestations, neuroradiology and genetic diagnosis. Front. Mol. Neurosci. 2017, 10, 215. [Google Scholar] [CrossRef]
- Sandoval-Rodríguez, V.; Cansino-Torres, M.A.; Sáenz-Farret, M.; Castañeda-Cisneros, G.; Moreno, G.; Zúñiga-Ramírez, C. Autosomal dominant leukodystrophy presenting as Alzheimer’s-type dementia. Mult. Scler. Relat. Disord. 2017, 17, 230–233. [Google Scholar] [CrossRef]
- Brussino, A.; Vaula, G.; Cagnoli, C.; Panza, E.; Seri, M.; Di Gregorio, E.; Scappaticci, S.; Camanini, S.; Daniele, D.; Bradac, G.B.; et al. A family with autosomal dominant leukodystrophy linked to 5q23.2-q23.3 without lamin B1 mutations. Eur. J. Neurol. 2010, 17, 541–549. [Google Scholar] [CrossRef]
- Giorgio, E.; Robyr, D.; Spielmann, M.; Ferrero, E.; Di Gregorio, E.; Imperiale, D.; Vaula, G.; Stamoulis, G.; Santoni, F.; Atzori, C.; et al. A large genomic deletion leads to enhancer adoption by the lamin B1 gene: A second path to autosomal dominant adult-onset demyelinating leukodystrophy (ADLD). Hum. Mol. Genet. 2015, 24, 3143–3154. [Google Scholar] [CrossRef] [Green Version]
- Nmezi, B.; Giorgio, E.; Raininko, R.; Lehman, A.; Spielmann, M.; Koenig, M.K.; Adejumo, R.; Knight, M.; Gavrilova, R.; Alturkustani, M.; et al. Genomic deletions upstream of lamin B1 lead to atypical autosomal dominant leukodystrophy. Neurol. Genet. 2019, 5, e305. [Google Scholar] [CrossRef] [Green Version]
- Giorgio, E.; Rolyan, H.; Kropp, L.; Chakka, A.B.; Yatsenko, S.; Di Gregorio, E.; Lacerenza, D.; Vaula, G.; Talarico, F.; Mandich, P.; et al. Analysis of LMNB1 duplications in autosomal dominant leukodystrophy provides insights into duplication mechanisms and allele-specific expression. Hum. Mutat. 2013, 34, 1160–1171. [Google Scholar] [CrossRef]
- Sherman, D.L.; Brophy, P.J. Mechanisms of axon ensheathment and myelin growth. Nat. Rev. Neurosci. 2005, 6, 683–690. [Google Scholar] [CrossRef]
- Barres, B.A. The Mystery and Magic of Glia: A Perspective on Their Roles in Health and Disease. Neuron 2008, 60, 430–440. [Google Scholar] [CrossRef] [Green Version]
- Kıray, H.; Lindsay, S.L.; Hosseinzadeh, S.; Barnett, S.C. The multifaceted role of astrocytes in regulating myelination. Exp. Neurol. 2016, 283, 541–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stadelmann, C.; Timmler, S.; Barrantes-Freer, A.; Simons, M. Myelin in the central nervous system: Structure, function, and pathology. Physiol. Rev. 2019, 99, 1381–1431. [Google Scholar] [CrossRef] [PubMed]
- Ratti, S.; Rusciano, I.; Mongiorgi, S.; Owusu Obeng, E.; Cappellini, A.; Teti, G.; Falconi, M.; Talozzi, L.; Capellari, S.; Bartoletti-Stella, A.; et al. Cell signaling pathways in autosomal-dominant leukodystrophy (ADLD): The intriguing role of the astrocytes. Cell. Mol. Life Sci. 2021, 78, 2781–2795. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Hlscher, C.; Liu, Y.; Li, L. GSK3: A key target for the development of novel treatments for type 2 diabetes mellitus and Alzheimer disease. Rev. Neurosci. 2012, 23, 1–11. [Google Scholar] [CrossRef] [PubMed]
- McCubrey, J.A.; Steelman, L.S.; Bertrand, F.E.; Davis, N.M.; Abrams, S.L.; Montalto, G.; D’Assoro, A.B.; Libra, M.; Nicoletti, F.; Maestro, R.; et al. Multifaceted roles of GSK-3 and Wnt/β-catenin in hematopoiesis and leukemogenesis: Opportunities for therapeutic intervention. Leukemia 2014, 28, 15–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nusse, R. Wnt signaling. Cold Spring Harb. Perspect. Biol. 2012, 4, 11. [Google Scholar] [CrossRef]
- Angers, S.; Moon, R.T. Proximal events in Wnt signal transduction. Nat. Rev. Mol. Cell Biol. 2009, 10, 468–477. [Google Scholar] [CrossRef]
- Vallée, A.; Lecarpentier, Y. Crosstalk between peroxisome proliferator-activated receptor gamma and the canonical WNT/β-catenin pathway in chronic inflammation and oxidative stress during carcinogenesis. Front. Immunol. 2018, 9, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Lecarpentier, Y.; Claes, V.; Duthoit, G.; Hébert, J.L. Circadian rhythms, Wnt/beta-catenin pathway and PPAR alpha/gamma profiles in diseases with primary or secondary cardiac dysfunction. Front. Physiol. 2014, 5, 429. [Google Scholar] [CrossRef] [Green Version]
- Shiau, C.W.; Yang, C.C.; Kulp, S.K.; Chen, K.F.; Chen, C.S.; Huang, J.W.; Chen, C.S. Thiazolidenediones mediate apoptosis in prostate cancer cells in part through inhibition of Bcl-xL/Bcl-2 functions independently of PPARγ. Cancer Res. 2005, 65, 1561–1569. [Google Scholar] [CrossRef] [Green Version]
- Yan, K.H.; Yao, C.J.; Chang, H.Y.; Lai, G.M.; Cheng, A.L.; Chuang, S.E. The synergistic anticancer effect of troglitazone combined with aspirin causes cell cycle arrest and apoptosis in human lung cancer cells. Mol. Carcinog. 2010, 49, 235–246. [Google Scholar] [CrossRef] [PubMed]
- Dhanasekaran, D.N.; Reddy, E.P. JNK signaling in apoptosis. Oncogene 2008, 27, 6245–6251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Björkblom, B.; Vainio, J.C.; Hongisto, V.; Herdegen, T.; Courtney, M.J.; Coffey, E.T. All JNKs can kill, but nuclear localization is critical for neuronal death. J. Biol. Chem. 2008, 283, 19704–19713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Xu, J.; Lazarovici, P.; Quirion, R.; Zheng, W. cAMP Response Element-Binding Protein (CREB): A Possible Signaling Molecule Link in the Pathophysiology of Schizophrenia. Front. Mol. Neurosci. 2018, 11, 1–14. [Google Scholar] [CrossRef]
- Zheng, K.B.; Xie, J.; Li, Y.T.; Yuan, Y.; Wang, Y.; Li, C.H.; Shi, Y.F. Knockdown of CERB expression inhibits proliferation and migration of glioma cells line U251. Bratisl. Med. J. 2019, 120, 309–315. [Google Scholar] [CrossRef]
- Li, X.Y.; Zhan, X.R.; Liu, X.M.; Wang, X.C. CREB is a regulatory target for the protein kinase Akt/PKB in the differentiation of pancreatic ductal cells into islet β-cells mediated by hepatocyte growth factor. Biochem. Biophys. Res. Commun. 2011, 404, 711–716. [Google Scholar] [CrossRef]
- Nieto, R.; Kukuljan, M.; Silva, H. BDNF and schizophrenia: From neurodevelopment to neuronal plasticity, learning, and memory. Front. Psychiatry 2013, 4, 45. [Google Scholar] [CrossRef] [Green Version]
- Escartin, C.; Galea, E.; Lakatos, A.; O’Callaghan, J.P.; Petzold, G.C.; Serrano-Pozo, A.; Steinhäuser, C.; Volterra, A.; Carmignoto, G.; Agarwal, A.; et al. Reactive astrocyte nomenclature, definitions, and future directions. Nat. Neurosci. 2021, 24, 312–325. [Google Scholar] [CrossRef]
- Escartin, C.; Guillemaud, O.; Carrillo-de Sauvage, M.A. Questions and (some) answers on reactive astrocytes. Glia 2019, 67, 2221–2247. [Google Scholar] [CrossRef]
- Padiath, Q.S. Lamin B1 mediated demyelination: Linking Lamins, Lipids and Leukodystrophies. Nucleus 2016, 7, 547–553. [Google Scholar] [CrossRef] [Green Version]
- Frost, B. Alzheimer’s disease: An acquired neurodegenerative laminopathy. Nucleus 2016, 7, 275–283. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.H.; Qu, J.; Suzuki, K.; Nivet, E.; Li, M.; Montserrat, N.; Yi, F.; Xu, X.; Ruiz, S.; Zhang, W.; et al. Progressive degeneration of human neural stem cells caused by pathogenic LRRK2. Nature 2012, 491, 603–607. [Google Scholar] [CrossRef] [Green Version]
- Alcalá-Vida, R.; Garcia-Forn, M.; Castany-Pladevall, C.; Creus-Muncunill, J.; Ito, Y.; Blanco, E.; Golbano, A.; Crespí-Vázquez, K.; Parry, A.; Slater, G.; et al. Neuron type-specific increase in lamin B1 contributes to nuclear dysfunction in Huntington’s disease. EMBO Mol. Med. 2021, 13, 1–25. [Google Scholar] [CrossRef]
- Hozak, P.; Sasseville, A.M.J.; Raymond, Y.; Cook, P.R. Lamin proteins form an internal nucleoskeleton as well as a peripheral lamina in human cells. J. Cell Sci. 1995, 108, 635–644. [Google Scholar] [CrossRef]
- Fracchia, A.; Asraf, T.; Salmon-Divon, M.; Gerlitz, G. Increased Lamin B1 Levels Promote Cell Migration by Altering Perinuclear Actin Organization. Cells 2020, 9, 2161. [Google Scholar] [CrossRef]
- Bedrosian, T.A.; Houtman, J.; Eguiguren, J.S.; Ghassemzadeh, S.; Rund, N.; Novaresi, N.M.; Hu, L.; Parylak, S.L.; Denli, A.M.; Randolph-Moore, L.; et al. Lamin B1 decline underlies age-related loss of adult hippocampal neurogenesis. EMBO J. 2021, 40, 1–21. [Google Scholar] [CrossRef]
- bin Imtiaz, M.K.; Jaeger, B.N.; Bottes, S.; Machado, R.A.C.; Vidmar, M.; Moore, D.L.; Jessberger, S. Declining lamin B1 expression mediates age-dependent decreases of hippocampal stem cell activity. Cell Stem Cell 2021, 28, 967–977.e8. [Google Scholar] [CrossRef]
- Zolezzi, J.M.; Santos, M.J.; Bastías-Candia, S.; Pinto, C.; Godoy, J.A.; Inestrosa, N.C. PPARs in the central nervous system: Roles in neurodegeneration and neuroinflammation. Biol. Rev. 2017, 92, 2046–2069. [Google Scholar] [CrossRef]
- Steven, A.; Friedrich, M.; Jank, P.; Heimer, N.; Budczies, J.; Denkert, C.; Seliger, B. What turns CREB on? And off? And why does it matter? Cell. Mol. Life Sci. 2020, 77, 4049–4067. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef]
- Groves, A.; Kihara, Y.; Jonnalagadda, D.; Rivera, R.; Kennedy, G.; Mayford, M.; Chun, J. A functionally defined in vivo astrocyte population identified by c-Fos activation in a mouse model of multiple sclerosis modulated by S1P signaling: Immediate-early astrocytes (ieAstrocytes). eNeuro 2018, 5, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Iglesias, J.; Morales, L.; Barreto, G.E. Metabolic and Inflammatory Adaptation of Reactive Astrocytes: Role of PPARs. Mol. Neurobiol. 2017, 54, 2518–2538. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ratti, S.; Rusciano, I.; Mongiorgi, S.; Neri, I.; Cappellini, A.; Cortelli, P.; Suh, P.-G.; McCubrey, J.A.; Manzoli, L.; Cocco, L.; et al. Lamin B1 Accumulation’s Effects on Autosomal Dominant Leukodystrophy (ADLD): Induction of Reactivity in the Astrocytes. Cells 2021, 10, 2566. https://doi.org/10.3390/cells10102566
Ratti S, Rusciano I, Mongiorgi S, Neri I, Cappellini A, Cortelli P, Suh P-G, McCubrey JA, Manzoli L, Cocco L, et al. Lamin B1 Accumulation’s Effects on Autosomal Dominant Leukodystrophy (ADLD): Induction of Reactivity in the Astrocytes. Cells. 2021; 10(10):2566. https://doi.org/10.3390/cells10102566
Chicago/Turabian StyleRatti, Stefano, Isabella Rusciano, Sara Mongiorgi, Irene Neri, Alessandra Cappellini, Pietro Cortelli, Pann-Ghill Suh, James A. McCubrey, Lucia Manzoli, Lucio Cocco, and et al. 2021. "Lamin B1 Accumulation’s Effects on Autosomal Dominant Leukodystrophy (ADLD): Induction of Reactivity in the Astrocytes" Cells 10, no. 10: 2566. https://doi.org/10.3390/cells10102566
APA StyleRatti, S., Rusciano, I., Mongiorgi, S., Neri, I., Cappellini, A., Cortelli, P., Suh, P.-G., McCubrey, J. A., Manzoli, L., Cocco, L., & Ramazzotti, G. (2021). Lamin B1 Accumulation’s Effects on Autosomal Dominant Leukodystrophy (ADLD): Induction of Reactivity in the Astrocytes. Cells, 10(10), 2566. https://doi.org/10.3390/cells10102566