1. Introduction
Axonal degeneration (AxD) is a process in which axons disintegrate physiologically during nervous system development and aging, or as a pathological element of degenerative nervous system diseases [
1,
2,
3]. Apart from axonal fragments, axon swellings (also called axonal beadings, bubblings or spheroids) are a hallmark of degenerating axons [
2,
4,
5], containing disorganized cytoskeleton and organelles resulting from an interruption of axonal transport [
6,
7,
8].
It is known that axons disintegrate in different ways depending on the biological context. During development and neural circuit assembly, inappropriately grown axons can undergo axonal retraction, axonal shedding, or local AxD [
9,
10]. Axonal retraction is characterized by retraction bulb formation at the distal tip and subsequent pullback [
9]. During axonal shedding, the axon retracts, leaving behind small pieces of its distal part (axosomes) [
11]. Local AxD is characterized by axon disintegration into separated axonal fragments [
10]. Acutely and chronically injured axons may degenerate retrogradely (distal-to-proximal direction, dying-back), anterogradely (proximal-to-distal direction), or in a Wallerian degeneration pattern (distal part of the axon from injury site), ultimately resulting in the generation of axonal fragments [
6,
12,
13]. However, AxD patterns have been described primarily in extracerebral axons in models of nutrient deprivation or axotomy.
Not much is known about AxD in cortical neurons subjected to a disease-specific cytotoxic micromilieu. A distinct pathological micromilieu has recently been observed for hemorrhagic stroke, after which the lysis of erythrocytes from the hematoma leads to the release of the cytotoxic product hemin [
14,
15]. Patients suffering from hemorrhagic stroke often experience AxD that is associated with worse motor and functional outcome [
16,
17]. Importantly, AxD occurs in the subacute stages of hemorrhagic stroke. Thus, addressing AxD may not only provide a new therapeutic target, but also a much wider time window for intervention. Since not much is known about the mechanisms, morphological patterns, and the temporal progression of AxD in the context of hemorrhagic stroke, we here sought to examine the progression of AxD and its associated morphological alterations.
Several cell culture systems, such as Campenot chambers and microfluidic devices, have been developed to spatially separate axons from their somata, allowing researchers to study AxD at a molecular level and to dissect the axon-soma relationship in AxD [
18,
19]. Whereas Campenot chambers facilitate the collection of axonal material due to their open structure [
20], they are only suitable for neurons that project axons robust enough to cross the vacuum–grease barrier needed to affix the Teflon piece that separates the somata from their axons. This makes Campenot chambers unsuitable for most central nervous system neurons [
21]. In microfluidic devices, the spatial separation is enabled by a microflux established by a medium volume difference between the two opposing compartments. These opposing compartments are separated by microgrooves through which axons grow [
22]. The major limiting factor of commercially available microfluidic devices to study AxD is that they are single, individual systems. Hence, they can only be used to assess one condition, which is time-consuming and precludes high-throughput analyses [
19,
23].
As the disintegration of the axons endures from minutes to hours [
13,
24], it is necessary to continuously monitor the spatiotemporal progression of AxD and its morphological hallmarks. On the one hand, axonal fragments have been identified by binarizing microscopic images [
25,
26]. Structures that are continuously connected are interpreted as axons, while isolated, non-connected elongated structures are categorized as axonal fragments. The ratio of axonal fragments to total axons is indicated by the so-called AxD index [
25,
26]. On the other hand, the occurrence and motility of axonal swellings have been determined previously by defining a region of interest, kymograph analysis, or calculating the ratio of the number of swellings compared to the axon length [
8,
27,
28,
29].
However, conventional software solutions fail to automatically detect and quantify high axon numbers as well as axonal swellings and fragments in phase-contrast microscopic images. The reason may be two-fold: (1) binarization can lead to information loss and low sensitivity, as thin axons may not be recognized; and (2) The analysis requires subjective and time-consuming manual annotations, e.g., thresholding and defining the region of interest [
27,
30,
31]. So far, immunostained images have been used to investigate the morphological changes in AxD, as the analysis of phase-contrast images is limited by the lower target-to-background signal. Immunofluorescence images, however, entail certain disadvantages, such as photobleaching and the requirement for cell fixation, which restricts observations to a single time point. Thus, a software tool for the automatized detection and quantification of the morphological patterns of AxD in long-term live cell imaging is required to improve both sensitivity and throughput to overcome the current limitations in understanding AxD.
In this study, we demonstrate that cortical axons undergo AxD after the exposure to the hemolysis product hemin, with axonal swellings preceding axon fragmentation. Deep learning further detected the occurrence of four AxD patterns characterized as granular, retraction, swelling, and transport degeneration. This may inform downstream AxD and neurodegeneration research in health and disease. We also provide tools for the enhanced throughput analysis of AxD, including a microfluidic device containing 16 independent experimental units and the deep learning platform “EntireAxon” to analyze AxD, which will help augment our understanding of AxD and may also support the development of novel treatment approaches for neurodegenerative diseases.
2. Materials and Methods
2.1. Fabrication of an Enhanced Throughput Microfluidic Device Based on Soft Lithographic Replica Molding
In total, 32 wells were milled in a polymethyl methacrylate (PMMA) plate of the size of a conventional cell culture plate using a universal milling machine (Mikron WF21C, Mikron Holding AG) with a 1 mm triple tooth cutter (HSS-CO8 Type N, Holex, Munich, Germany) at a precision of 0.01 mm. During the milling procedure, we applied a half-synthetic cooling lubricant (Opta Cool 600 HS, Fuchs Wisura GmbH, Bremen, Germany) on a mineral base to reduce the debris. In addition, we milled screw holes in the intermediate spaces between each microfluidic unit to later detach the PMMA from the negative casting mold. To remove debris, we washed the PMMA plate by sonication (Sonicator Elmasonic S, Elma Schmidbauer GmbH, Singen, Germany) at room temperature for 30 min. Next, we lasered the microgrooves on the PMMA plate to connect both milled compartments of each individual microfluidic unit using an Excimerlaser (Excistar XS 193 nm, Coherent, Santa Clara, CA, USA). The PMMA plate was then washed again by sonication at room temperature for 30 min.
Polydimethylsiloxane (PDMS) was prepared in a 1:10 ratio and mixed properly before inducing vacuum at 0.5 Torr in a vacuum desiccator (VDC-31, Jeio Tech, Daejeon, Korea) for 30 min. After the PDMS was poured into an empty aluminum basin to cover the ground, we applied the vacuum at 0.5 Torr for 30 min to remove the air bubbles. The PDMS was cured at room temperature for 48 h. We put the PMMA plate on top of the PDMS ground with the milled and lasered structures showing upward. Half of each well of the microfluidic units was filled with PDMS before curing at room temperature for 48 h. We mixed epoxy solution in a 1:1 ratio and poured it over the microfluidic device to cover its surface by at least 1 cm. Vacuum was applied at 0.5 Torr for 10 min to remove all air bubbles located above the channel side of the microfluidic device. The epoxy was cured at room temperature for a minimum of 2 h. We subsequently detached the epoxy from the PMMA plate via a metallic block that consisted of screw holes in the intermediate spaces between the individual systems. The epoxy represented a negative casting mold to produce the microfluidic devices using PDMS.
PDMS was prepared as described above. We poured the PDMS into the negative epoxy casting mold and applied vacuum at 0.5 Torr for 30 min. The liquid PDMS was cured at 75 °C for 2 h to induce polymerization. We peeled the microfluidic devices from the casting mold and punched the wells with an 8 mm biopsy punch (DocCheck Shop GmbH, Cologne, Germany) to ensure a sufficient amount of medium for cell culture. We cleaned customized 115 × 78 × 1 mm glass slides by sonication (Sonicator Elmasonic S, Elma Schmidbauer GmbH) and subsequently cleaned them by ethanol before plasma treatment (High Power Expanded Plasma Cleaner, Harrick Plasma, Ithaca, NY, USA). Plasma was applied at 45 W and 0.5 Torr for 2 min to activate the silanol groups of the glass slides and the microfluidic devices, enabling firm attachment.
We washed the microfluidic devices with ethanol and then twice with distilled water to remove any debris. After aspirating the distilled water, except from the inside of the compartments, 0.1 mg/mL of poly-d-lysine solution in 0.02 M borate buffer (0.25% (w/v) borate acid, 0.38% (w/v) sodium tetraborate in distilled water, pH 8.5) was used for coating at 4 °C overnight. We aspirated the poly-d-lysine the next morning, not removing it from the compartments, and added 50 µg/mL of laminin as a second coating surface for incubation at 4 °C overnight. On the day of neuron isolation, the microfluidic devices were washed twice with pre-warmed medium after aspirating the laminin. Immediately prior to cell seeding, we aspirated the medium from the wells without removing it from the compartments.
2.2. Experimental Animals
Crl:CD1 (ICR) Swiss outbred mice (Charles River) were used. The animals were kept at 20–22 °C and 30–70% humidity in a 12-h/12-h light/dark cycle, and were fed a standard chow diet (Altromin Spezialfutter GmbH, Lage, Germany) ad libitum. Animal experiments were performed in accordance with the German Animal Welfare Act and the corresponding regulations and were approved by the local animal ethics committee (Ministerium für Landwirtschaft, Umwelt und ländliche Räume, Kiel, Germany, under the prospective contingent animal license number 2017-07-06 Zille).
2.3. Isolation and Culture of Primary Cortical Neurons
We isolated the primary cortical neurons from the murine E14 embryos after decapitation as previously described [
15]. We seeded the neurons at a density of 10,000 cells/mm
2 in 5 µL MEM + Glutamax medium into one compartment (soma compartment) of each microfluidic unit of the device. The cells were allowed to adhere at 37 °C for 30 min. To promote directional axon growth into the other compartment (axonal compartment) by medium microflux, 150 µL of MEM + Glutamax medium were applied to the well of the soma compartment, while 100 µL were added to the well of the axonal compartment. Neurons were cultured at 37 °C in a humidified 5% CO
2 atmosphere. The next day, we changed from MEM + Glutamax medium to Neurobasal Plus Medium containing 2% B-27 Plus Supplement, 1 mM sodium pyruvate and 1% penicillin/streptomycin. The volume differences among the wells ensured the microflux for the directional axonal growth over the following days.
2.4. Immunofluorescence
Soma and axonal compartments in the microfluidic units were fixed at room temperature for 1 h in 4% formaldehyde solution in phosphate-buffered saline (PBS). They were washed twice with PBS and permeabilized with blocking solution (2% BSA, 0.5% Triton-X-100 and 1× PBS) at room temperature for 1 h. We incubated the neurons/axons on both compartments with primary antibodies against synaptophysin (1:250) and MAP2 (1:4000) at 4 °C overnight. The next day, both compartments were washed three times with PBS and incubated with the secondary antibodies anti-mouse Alexa Fluor 546 (1:500) and anti-rabbit Alexa Fluor 488 (1:500) at room temperature for 1 h. After washing three times with PBS, both compartments were incubated with DAPI (1 µg/mL) for nuclear counterstaining at room temperature for 10 min. Both compartments were washed again three times with PBS prior to fluorescence microscopy. An Olympus IX81 time-lapse microscope (Olympus Deutschland GmbH, Hamburg, Germany) with a 10× objective (0.3 NA Ph1) and camera F-View soft Imaging system was used at room temperature. Images were acquired with Cell
TM software (Olympus Deutschland GmbH) and further processed via ImageJ (see
Section 2.10).
2.5. Selection of Microfluidic Units for Hemin Treatment and Time-Lapse Recording
After 6 or 7 days in culture, microfluidic devices were considered for recording if they met the following inclusion criteria: (i) axon growth through at least 80% of all microgrooves, and (ii) axon length of at least 150 µm from the end of the microgrooves. All included microfluidic units were randomly assigned to the experimental conditions.
2.6. Time-Lapse Recording of Axonal Degeneration
Axons were treated with 0 (vehicle), 50, 100, and 200 µM hemin. For the treatment, the medium was removed from the wells of the microfluidic units. Then, hemin was diluted in the collected media and added back to the respective wells. The media volume between the two wells was equalized during the treatment to prevent any microflux. All microfluidic units were recorded immediately after each other. We started the recordings at 1 h after treatment to allow for the adjustment of the well plates to the humidity of the incubation chamber of the microscope and the setup of the recording positions. We recorded AxD in Neurobasal Plus Medium containing 2% B-27 Plus Supplement, 1 mM sodium pyruvate, and 1% penicillin/streptomycin with a 30-min interval for 24 h using an Olympus IX81 time-lapse microscope (see
Section 2.4) at 37 °C, 5% CO
2, and 65% humidity.
2.7. Live Cell Fluorescent Staining
To evaluate axonal vitality, we washed the axonal compartment once with PBS and incubated the axonal compartment with calcein AM (4 µM) in PBS for 30 min at 37 °C at the end of the time-lapse recording or in 4 h intervals upon hemin treatment. An Olympus IX81 time-lapse microscope (see
Section 2.4) was used to record the respective images at 37 °C, 5% CO
2, and 65% humidity.
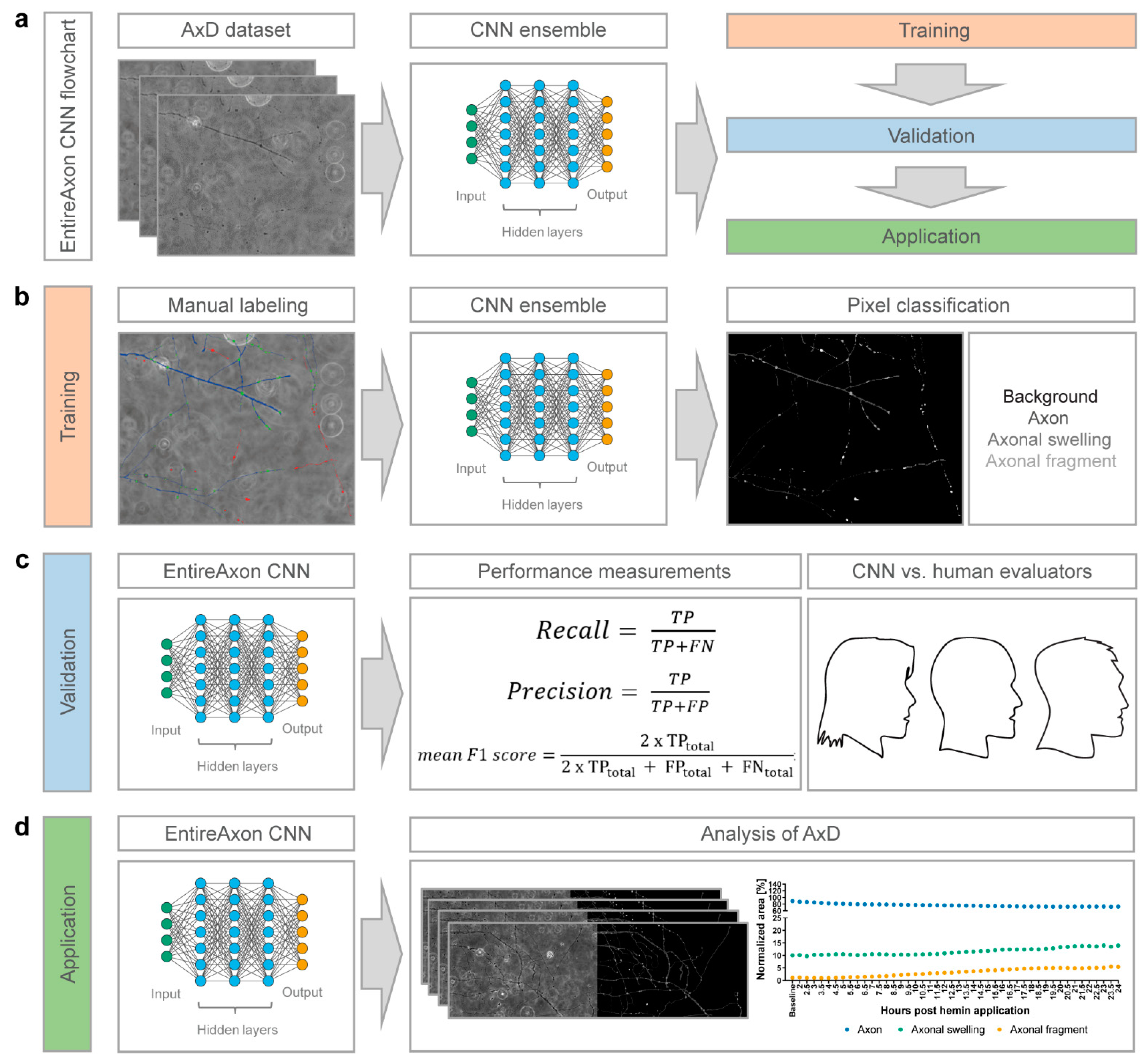
2.8. Training of the EntireAxon CNN for the Segmentation of Phase-Contrast Microscopic Images
We trained the EntireAxon CNN for the image-wise semantic segmentation of AxD features in a supervised manner. To this end, we adapted a standard u-net with ResNet-50 encoder [
32] to automatically determine the class probability for each pixel of an input image. Our segmentation aimed to classify each pixel of a microscopic image of a time-lapse recording into one of four classes: ‘background’, ‘axon’, ‘axonal swelling’, and ‘axonal fragment’.
For the training dataset, we selected 33 images and created corresponding image labels (masks) using GIMP software (v.2.10.14, RRID:SCR_003182). For each image, a label image with the same height and width was created, in which each pixel value denotes a pixel class. Specifically, the classes ‘background’, ‘axon’, ‘axonal swelling’ and ‘axonal fragment’ had the values 0, 1, 2, and 3, respectively. For each pixel of the input image, we retained 4 values that reflect the probability distribution of the pixel over the 4 classes. We assigned each pixel the most probable class to create a segmentation map. During training, the CNN observed an input image, produced an output, and compared this output to the label. The weights of the network were adapted via backpropagation so that the output better fitted the label. The weight changes were derived from a pixelwise loss function, i.e., the cross-entropy loss:
with
being the probability of class c at pixel
for the prediction and ground truth of the network, respectively.
We trained a mean ensemble consisting of 8 neural networks for 180 epochs using the Adam optimizer, a batch size of 4, and a learning rate of 0.001 that decreased by a factor of 10 after every 60 epochs. The input images were standardized by the image-net mean and standard deviation [
28]. For data augmentation, we used random cropping (size 512 × 512), image flipping along the horizontal axis, and rotation by a random angle between −90° and +90°.
2.9. Validation of the EntireAxon CNN Compared to Human Evaluatorts
To measure how well the EntireAxon CNN segments unknown images, we used a second validation set comprising eight images that were labeled by three human evaluators (A.P. (Alex Palumbo), S.K.L., L.E.H.). Importantly, the EntireAxon CNN did not update its parameters during training to fit the validation set, but only used the training set.
For each image, the EntireAxon CNN inferred a segmentation. We generated a binary mask from the prediction of the network, where 1 denotes the respective class and 0 all other classes. We computed a binary label mask in the same manner. We counted the true positive (TP), false positive (FP), and false negative (FN) pixels and computed the recall (sensitivity) and precision [
33]:
Recall and precision were calculated separately for each class on each validation image. The mean recall and precision over all eight validation images were determined subsequently.
A mean of 96.42% of pixels in the axonal images were ‘background’ pixels, while only 2.77% represented the class ‘axon’, 0.58% ‘axonal swelling’, and 0.23% ‘axonal fragment’ pixels. This reflects a challenging degree of class imbalance, where the probability of having any positives for a class in a validation image is low. Thus, we did not use the computed recall and precision of the individual images or the mean recall and precision to compute the mean F1 score, i.e., the harmonic mean of recall and precision. This has been shown to lead to bias, especially when a high degree of class imbalance is present in the dataset [
33], as it may result in undefined values for an image for recall (due to the absence of TP), precision (in case the CNN does not recognize the few positives), and F1 score (in case either recall or precision are undefined). To avoid bias, we computed the total TP, FP, and FN of all validation images from which we calculated the mean F1 score [
33]:
In addition, we computed a consensus label between human evaluator 1 and 2, between 1 and 3, and between 2 and 3, and compared the EntireAxon CNN versus the remaining evaluator (human evaluator 3, 2, and 1, respectively) to the consensus labels. Mean F1 scores for all classes were computed as described above.
2.10. Image Preprocessing
Prior to the analysis of AxD after hemin exposure, we preprocessed the time-lapse recordings in ImageJ (v1.52a, RRID: RRID:SCR_003070) using a custom-written macro. Specifically, each individual recording was converted from a 16-bit into an 8-bit recording to make it compatible with the ImageNet (8-bit) pre-trained ResNet-50. The recording was aligned automatically with the ImageJ plug-in “Linear Stack Alignment with SIFT” as described previously [
34]. The following settings were used: initial Gaussian blur of 1.6 pixels, 3 steps per scale octave, minimum image size of 64 pixels, maximum image size of 1024 pixels, feature descriptor size of 4, 8 feature descriptor orientation bins, closest/next closest ratio of 0.92, maximal alignment error of 25 pixels, inlier ratio of 0.05, expected transformation as rigid, “interpolate” and “show info” checked. Black edges that appeared on the recording after alignment were cropped.
2.11. AxD Analysis Using the EntireAxon CNN
All recordings of AxD after hemin exposure were automatically analyzed by the trained EntireAxon CNN, which classified each pixel as one of the 4 different classes: ‘background’, ‘axon’, ‘axonal swelling’, or ‘axonal fragment’. For each experimental condition (i.e., hemin concentration), the sum percentage of all pixels per class on all images of that experimental day were added at each timepoint (Axon
t1.5–24h, Axonal swelling
t1.5–24h, Axonal fragment
t1.5–24h). To determine the changes for the classes ‘axon’, ‘axonal swelling’, and ‘axonal fragment’ over time, we calculated the sum percentage of pixels for all given time points (t
i with I = 1.5 to 24 h) of the corresponding class over the sum of the pixels of all three classes at baseline:
The area under the curve was calculated as the cumulative measurement of the effect of hemin on axonal degeneration over 24 h as follows:
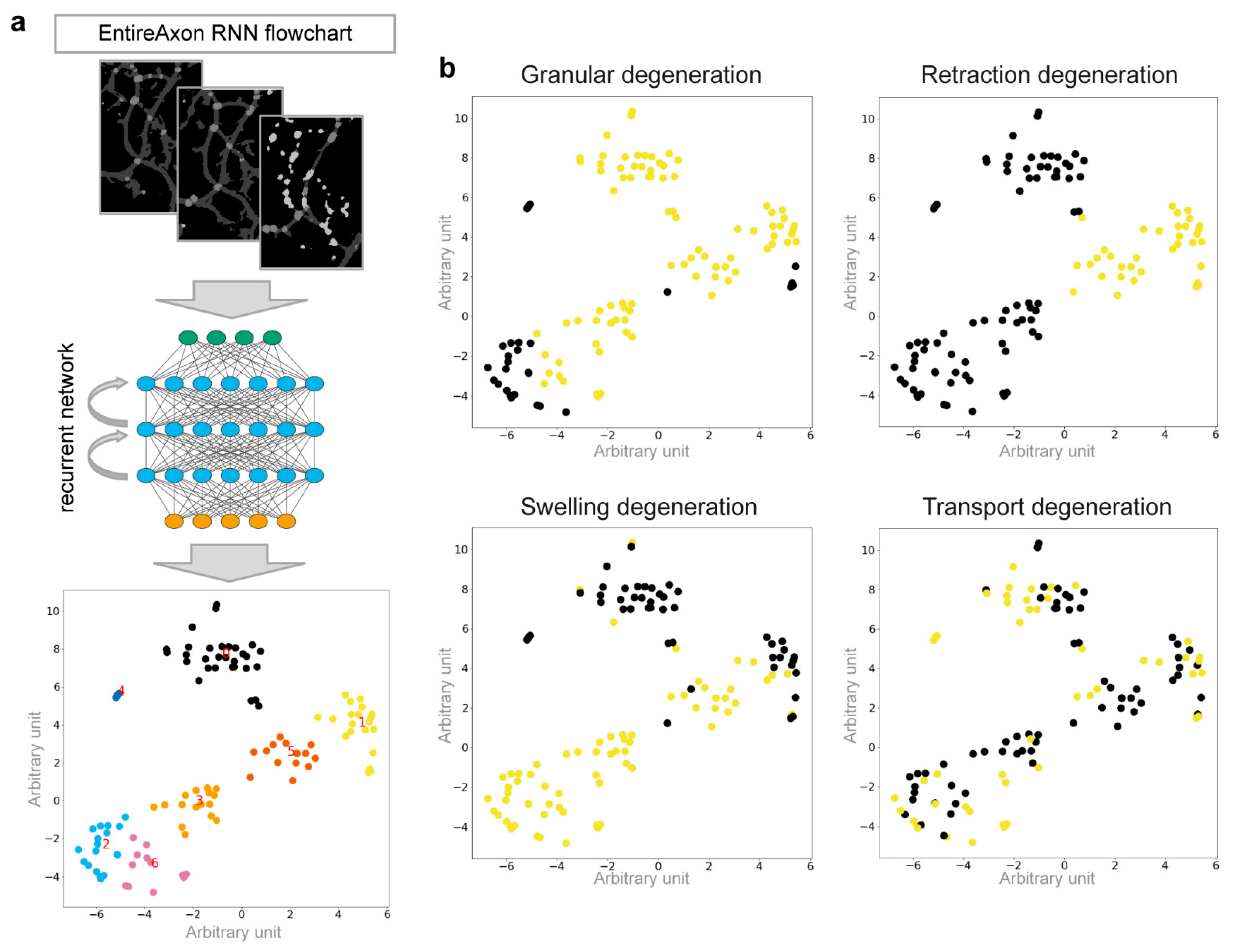
2.12. Classification of the Morphological Patterns of AxD Using an Attention-Based RNN
We used the segmentation videos derived from the original microscopic images, using the RNN to identify 4 morphological patterns of AxD: granular, retraction, swelling, and transport degeneration. We manually annotated the segmentation videos of individual, degenerating axons to classify each AxD pattern. To reduce the dimensions of the input, the segmentation video was converted into a series of normalized histograms (
H), one for each (time) frame. Thus, the RNN did not operate on the microscopic images directly, but rather on more efficient representations of the data. To compute a histogram for a frame t
i, we compared the pixels of the frames t
i and t
i+1. Each pixel was assigned to 1 of 16 classes that consisted of pairs
of the 4 segmentation classes (i.e., 4 times 4 possible configurations, 16 class pairs). For example, the class (background, axon) means that in frame t
i, the pixel was classified as background, while in frame t
i+1, it was an axon pixel. Therefore, for T time steps, we computed T-1 histograms.
is the number of pixels that belong to class
at time-frame
and that belong to
at timeframe
. In addition, we normalized each histogram to sum up to 1 (i.e., we divided by the sum over all pairs):
Of note, the histograms were computed over small patches (height and width < 90 pixels) during training and during inference on windows of size 32 × 32 pixels.
We used an encoder-decoder RNN with attention [
35]. The encoder
consisted of a gated recurrent unit (GRU) that obtained the histogram time sequence
as input. The encoder computed the hidden representation of the histograms:
For our purpose, we used an architecture that was able to base the decision for a degeneration class on the previous class predictions. To this end, the output
was computed iteratively in C + 1 steps as a sum of the previous output and the output of the decoder
C is the number of degeneration classes (4), d is the hidden dimension (we used 256), and
.
is the sigmoid function. The decoder employed a GRU that depended on the context vector
and the hidden state vector
The entries of the initial hidden vector
and output vector
were all zero. The context vector is a weighted sum of the encoder representations. At each iteration, these weights can change, enabling the network to focus on different time steps. We assumed that a specific pattern of degeneration happened only in a limited number of timeframes that was lower than the whole input video. The weights depended on the current state of the decoder and the current output:
Here,
is the concatenation of two vectors. The final output
is normalized by the sigmoid function:
Apart from the weights used by the GRUs, and are learnable weights.
The EntireAxon RNN was trained with 162 images for 60 epochs using the lamb optimizer [
36] with a batch size of 128. We used a learning rate of 0.01 that was reduced by a factor of 10 every 15 epochs and an additional weight decay of 0.0001. The two GRUs (encoder and decoder) contained three layers, and we used dropout with a
p-value of 0.9. To increase the RNN robustness against varying axon thickness, we also added eroded versions of the segmentation data using a cross-shape as kernel with the sizes three, five, and seven. Accordingly, each image existed six times in the dataset, with three eroded versions and three unchanged copies, to keep a 50% chance of having the original image for training.
2.13. RNN Cluster Analysis
The unnormalized class output was computed by the matrix-vector product where was a 256-dimensional vector representation of the input sample, computed by the model. For the classes to be linearly separable, the vector representations of each class needed to be close to each other in the 256-dimensional space. To visualize the relationships of the specific samples, we employed t-distributed stochastic neighborhood embedding (T-SNE) to compute a 2-dimensional representation of the high-dimensional data.
2.14. Ten-Fold Cross-Validation of the RNN
To validate the RNN, we used 10-fold cross-validation [
37]. The dataset
was divided into 10 subsets, ensuring that each subset included at least 1 sample of each class:
. We trained 10 models for
i = 1,...,10 on
and tested them on
. Subsequently, we combined and evaluated all test samples
. Mean recall, precision, and F1 score were determined as described above.
2.15. Analysis of the Morphological Pattern of AxD Using the EntireAxon RNN
After hemin exposure, all AxD segmentations were automatically analyzed with the trained EntireAxon RNN, which predicted the occurrence of the 4 morphological patterns of AxD in a pixel-wise manner. Of note, a pixel can be predicted to belong to 0, 1, or multiple morphological patterns. Only pixels previously identified as degenerated over time were considered by applying a ‘fragmentation mask’ that included all no-background pixels that changed to either background or fragment during the recording time.
For each experimental condition (i.e., hemin concentration), the percentage of the occurrence of each morphological pattern was calculated as the sum of all pixels per morphological pattern on all images of that experimental day divided by the ‘fragmentation mask’ as follows:
2.16. Statistical Analysis
Six biological replicates for each concentration were employed in each experiment to assess the hemin-induced AxD. We did not perform an a priori power analysis, as this was an exploratory study. Normality was evaluated with the Kolmogorov–Smirnov test, variance homogeneity using the Levené test, and sphericity by the Mauchly test. When the data were normally distributed and variance homogeneity was met, one-way ANOVA was performed, followed by the Bonferroni post-hoc test. In the case that the data were not normally distributed, the Kruskal–Wallis test was performed for multiple comparisons of independent groups, followed by the post-hoc Mann–Whitney U test with α-correction according to Bonferroni-Holm to adjust for the inflation of type I error due to multiple testing. For repeated testing with covariates, a repeated measures ANOVA was performed with Greenhouse–Geisser adjustment if the sphericity was not given. Linear regressions were performed for AxD patterns. Data are represented as mean ± 95% confidence interval (CI) except for the nonparametric data of the AUC for axonal fragments, where medians are given. A value of
p < 0.05 was considered statistically significant. The detailed statistical analyses can be found in
Tables S1–S3. All statistical analyses were performed with IBM SPSS version 23 (RRID:SCR_002865), except linear regressions that were performed with GraphPad Prism version 8 (RRID:SCR_002798).
4. Discussion
We here describe two complementary tools, a novel microfluidic device and a deep learning algorithm, that allow increasing the experimental yield, in-depth enhanced throughput analysis of AxD, and longitudinal investigation of AxD in vitro. Using these tools, we were able to demonstrate time-dependent changes of the morphological features of AxD, with axonal swellings preceding axon fragmentation in a model of hemorrhagic stroke-induced AxD as well as the occurrence of four morphological patterns of AxD under pathophysiological conditions: granular, retraction, swelling, and transport degeneration.
We here propose a novel monolithic microfluidic device consisting of 16 individual microfluidic units that enables the parallel and separated treatment and/or manipulation of axons and somata (
Figure 1). The currently available devices do not allow enhanced throughput experiments, as they comprise only single microfluidic units [
19,
23]. Although some devices can harbor multiple experimental conditions, they employ a radial design with a single soma compartment, in which one experimental condition may influence another due to the potential of retrograde signaling [
22,
43]. Another option is the parallel use of multiple individual devices, which allows handling up to 12 devices in a conventional 12-well plate [
27]. Compared to our device, this procedure is time-consuming in both the manufacturing and adjustment for recordings.
To date, the extent of AxD has mainly been investigated with a focus on axon fragmentation as the primary readout. To quantify axon fragmentation, Sasaki and colleagues introduced the AxD index as the ratio of the fragmented axon area versus the total axonal area [
26]. However, the AxD index did not include axonal swellings, which are a characteristic feature of degenerating axons [
8,
44]. Although other analyses have considered axonal swellings as a morphological feature of AxD [
7,
8], the approaches were time-consuming and required manual annotations.
We herein adapted a standard u-net with a ResNet-50 encoder [
32,
45] and used a CNN ensemble, which combines predictions from multiple CNNs to generate a final output and is superior to individual CNNs [
46,
47,
48]. The EntireAxon CNN performs an automatic segmentation and quantification of axons and morphological features relevant to AxD, including axonal swellings and fragments, on phase-contrast time-lapse microscopy images (
Figure 2). The EntireAxon CNN recognized the four classes—‘background’, ‘axon’, ‘axonal swelling’, and ‘axonal fragment’—with the highest mean F1 score for the class ‘background’ (
Table 1). The comparably lower performance of the CNN to recognize axonal fragments may be explained by the disproportional distribution of pixels in the training and validation data (‘background’ mean of 96.42% of pixels, ‘axon’ 2.77%, ‘axonal swelling’ 0.58%, ‘axonal fragment’ 0.23%). Hence, every individual segmentation error affects the false positive or false negative rate more strongly in these classes.
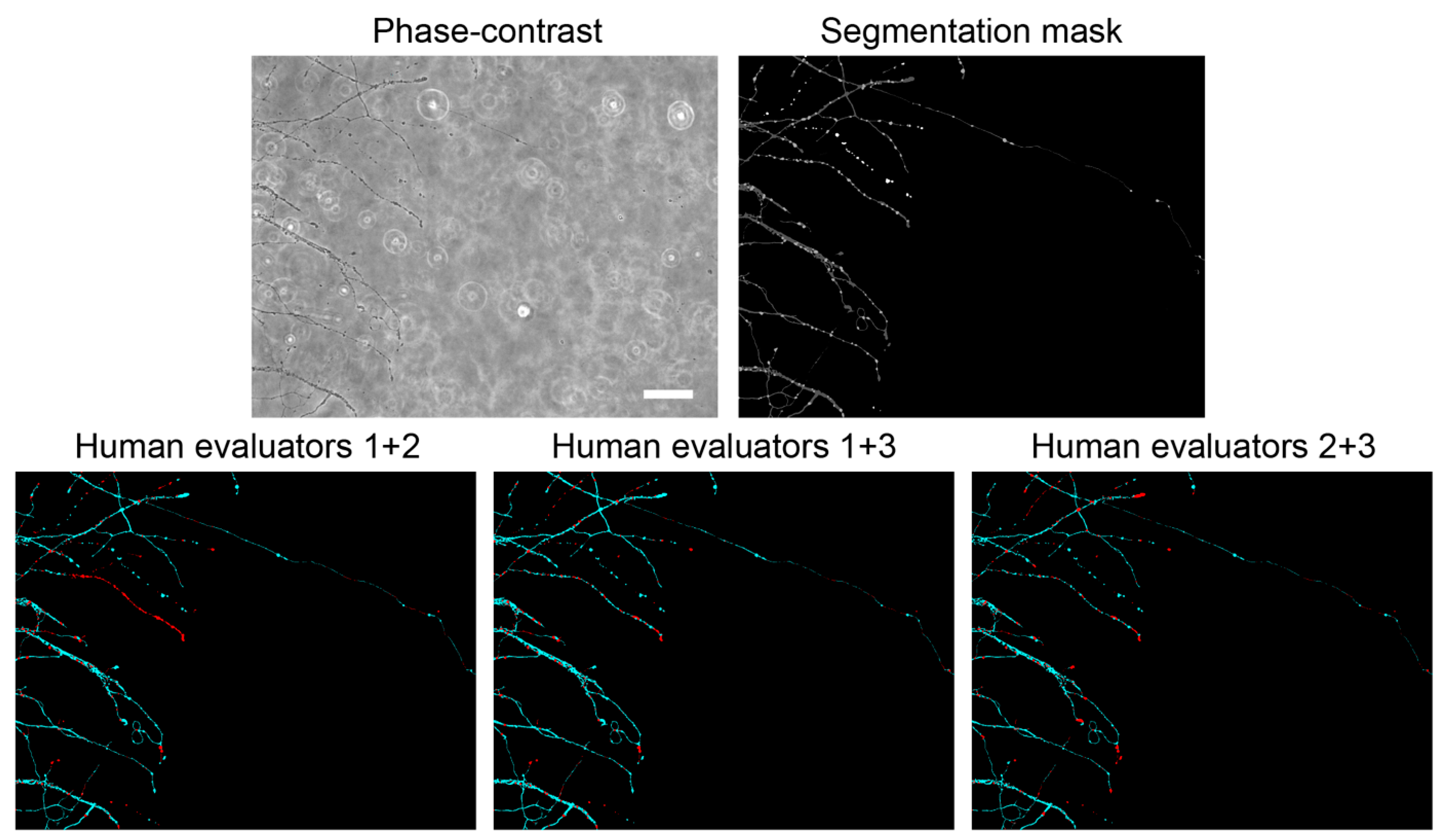
The comparison with human evaluators revealed that the EntireAxon CNN reached a similar performance level. As expected, its performance was slightly better than the human evaluators on the ground truth, as both the ground truth and training data were labeled by the same human evaluator (
Table 2). Interestingly, when comparing the EntireAxon CNN with a human evaluator on the consensus label of the other two human evaluators, not only was the EntireAxon CNN as good as or even better than the human evaluator, but the mean F1 scores were also higher than on the ground truth labels (
Figure 3 and
Table 3). This may be because pixels that were differentially assigned by the human evaluator, i.e., more difficult to classify, were excluded from the comparison. Taken together, these findings demonstrate that the EntireAxon CNN is suitable to automatically quantify AxD and its accompanying morphological changes in an enhanced throughput manner.
We then applied these novel tools to a model of hemorrhagic stroke-induced AxD by exposing axons from primary cortical neurons to the hemolysis product hemin and investigated the progression of AxD. Similar to previous results, where 100 µM hemin were sufficient to induce significant neuronal cell death in conventional cultures of somata and axons [
15], we observed that 100 µM hemin led to a significant decrease in the axon area and an increase in the axonal swelling and fragment area (
Figure 4).
It has been described previously that the progression of AxD undergoes a latent phase, during which the structural integrity of the axon is maintained, followed by a catastrophic phase with the rapid disintegration of the axon [
8]. In our model, the catastrophic phase of AxD started between 12 and 18 h after the administration of hemin (
Figure 4 and
Supplementary Figure S2). Similar durations of the latent phases of AxD have been observed in other models. For instance, under circumstances of growth factor withdrawal, the transition to the catastrophic phase occurred at 12–24 h [
8,
49,
50]. Our findings are also in line with results reported in a model of experimental autoimmune encephalomyelitis, indicating that axonal swelling anticipates fragmentation [
7]. Taken together, AxD progression depends on the severity of the insult, and axonal swellings may be reliable predictors of AxD.
Whereas we applied the EntireAxon CNN on time-lapse images, it should be noted that it is also possible to calculate the ratios between swelling or fragment area and axon area based on random images that are not necessarily in the same position over time. The respective areas are calculated for each image and class separately and independently of time or any previous images. However, we recommend including baseline recordings to allow for normalization due to differences in the neurite outgrowth in the devices.
Interestingly, axonal swellings and axonal fragments were related to different morphological patterns of AxD. Specifically, we observed axons that showed signs of axonal retraction, an enlarging of axonal swellings, and axonal transport before degeneration (
Figure 5). We therefore trained the EntireAxon RNN to quantify the occurrence of four morphological patterns of AxD, i.e., granular, retraction, swelling, and transport degeneration, based on the clusters of unique changes of classes over time (
Figure 6 and
Supplementary Figure S3). The RNN generated 7 clusters from the 16 possible class pairs. Of note, all clusters differed in the extent and timing of class changes. The RNN used a different ensemble of clusters to classify each pattern. However, some clusters overlapped between patterns, which can be explained by the fact that the eventual degeneration of the axon, which leads to fragments, is the end-product of AxD. Therefore, clusters 0, 1, 3, and 5 also occur in retraction, swelling, and transport degeneration. On the other hand, axonal swellings are a dominant feature of swelling and transport degeneration, which explains why cluster 6 was used to define both patterns.
These different AxD patterns have not yet been described to occur simultaneously in the same biological condition. However, granular degeneration was observed in retrograde, anterograde, Wallerian, and local AxD after axotomy or trophic factor deprivation [
6,
10,
12,
13]. Retraction degeneration was described in axonal retraction and shedding in developmental AxD [
9,
11]. Swelling degeneration was reported in experimental autoimmune encephalitis and growth factor deprivation [
7,
8]. Transport degeneration was shown in experimental models of amyotrophic lateral sclerosis, multiple sclerosis, oxidative stress, and genetic models [
39,
40,
41,
42].
Our data demonstrate that all four morphological degeneration patterns can occur along cortical axons (
Figure 7). Interestingly, we also observed a concentration-dependent effect in the context of hemorrhagic stroke. Granular, swelling, and transport degeneration were significantly increased with increasing hemin concentrations, with granular and swelling degeneration being more strongly correlated. The extent to which our model of hemin-induced AxD in hemorrhagic stroke is molecularly similar to developmental or pathophysiological AxD needs to be further investigated, along with the underlying molecular mechanisms of the four patterns of AxD. This could be greatly facilitated by the EntireAxon RNN, which is able to automatically detect the morphological patterns in time-lapse recording due to its capacity to relate each output to previous images in the stacks by its current units.
Limitations and Outlook
Our microfluidic device does not currently allow the investigation of AxD at more proximal axonal parts to the soma, such as the axonal initial segment. Shortening the length of the microgrooves or including a more proximal compartment are possible modifications of the current design.
Our results are based on unmyelinated axons. Co-culture with glia cells that may play a role in AxD is possible in the presented microfluidic device, and the time course and morphological changes may be different under different co-culture conditions. These studies are of high relevance to the field but are beyond the scope of the present study.
The observed effects of AxD in hemorrhagic stroke within this study were based on hemin toxicity, and we cannot exclude that other hemolysis products, such as thrombin or bilirubin, have different effects. Additional studies should investigate differences of hemolysis products to increase our understanding of the mechanisms of AxD in hemorrhagic stroke.
The overall CNN performance may be further improved with more general inputs. For example, the segmentation of fragment pixels cannot be accurately conducted based on a single image at a specific timepoint. Instead, the whole process of AxD, ultimately resulting in the disintegration of the axons (i.e., the generation of axonal fragments), needs to be considered. In principle, CNNs using 3D convolutions could perform a segmentation over an entire time-lapse recording and model the temporal dependencies. However, we decided against the 3D approach, as it severely restricts general applicability due to its greatly increased effort to label suitable time series for training. In this context, the identification of the images that will yield the best results is crucial to effectively reduce labeling costs, which we have described previously using an active learning method [
51].


 ,
, 







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}