Abstract
The extracellular matrix (ECM) has been identified as a critical factor affecting synaptic function. It forms a functional scaffold that provides both the structural support and the reservoir of signaling molecules necessary for communication between cellular constituents of the central nervous system (CNS). Among numerous ECM components and modifiers that play a role in the physiological and pathological synaptic plasticity, matrix metalloproteinase 9 (MMP-9) has recently emerged as a key molecule. MMP-9 may contribute to the dynamic remodeling of structural and functional plasticity by cleaving ECM components and cell adhesion molecules. Notably, MMP-9 signaling was shown to be indispensable for long-term memory formation that requires synaptic remodeling. The core regulators of the dynamic reorganization of the actin cytoskeleton and cell adhesion are the Rho family of GTPases. These proteins have been implicated in the control of a wide range of cellular processes occurring in brain physiology and pathology. Here, we discuss the contribution of Rho GTPases to MMP-9-dependent signaling pathways in the brain. We also describe how the regulation of Rho GTPases by post-translational modifications (PTMs) can influence these processes.
1. Introduction
Accumulating data support the importance of interactions between extracellular matrix molecules with pre- and postsynaptic neuronal elements for the formation and remodeling of central synapses. One of the key components of the extracellular matrix (ECM) that plays a role in synaptic plasticity, learning and memory formation is matrix metalloproteinase 9 (MMP-9) [1,2]. It is implicated in ECM remodeling via cleavage of ECM components such as laminin and aggrecan, and cell adhesion molecules [3,4]. Several cell adhesion proteins have been proposed as substrates of MMP-9 in neurons [5,6] but their role in the structural and functional plasticity has not yet been established. At the same time, MMP-9 signaling has been shown to contribute to long-term memory, its underlying synaptic plasticity [1,2,7,8,9,10], formation and maintenance of dendritic spines [11,12,13,14], and dendritic protrusions associated with neuropsychiatric disorders (rev. in [15]). Thus, it appears to be an ideal candidate molecule responsible for synaptic remodeling, although the key substrates and downstream mechanisms of MMP-9′s enzymatic activity in synaptic plasticity and learning remain unclear [2].
It is known, however, that dynamic changes in synaptic organization require remodeling of the actin cytoskeleton, which is regulated by Rho GTPases. They constitute a family of molecular switches that regulate signal transduction pathways by interconverting between inactive GDP-bound and active GTP-bound conformational states. They can be activated by Rho-specific guanine nucleotide exchange factors (GEFs) and inhibited by GTPase-activating proteins (GAPs). Guanine nucleotide dissociation inhibitors (GDIs) sequester the GDP-bound form of some GTPases in the cytosol and prevent them from localizing to membranes or being activated by GEFs. The action of Rho GTPases can also be regulated by PTMs [16]. Rho GTPases are activated by signals originating from various cell-surface receptors and, in the GTP-bound active conformation, they interact with a range of effector proteins, including kinases, actin regulators, and adaptor proteins, leading to changes in cell behavior [17]. Thus, Rho GTPases regulate cytoskeletal rearrangements, cell motility, cell polarity, vesicle trafficking, and the cell cycle (rev. in [18]). In neurons, GTPases of the Rho family regulate spinogenesis and synaptogenesis [16], axon guidance [19], dendritogenesis, and synaptic plasticity [20]. Increasing evidence argues for the role of PTMs of Rho GTPases in controlling the structure and function of synapses [21]. Although GTPases play an important role in regulating cell function and are often at the center of many signaling pathways, their activity, specificity, and therefore cellular response depend on upstream proteins.
Our previous data indicate that the proteolytic action of MMP-9 in primary hippocampal neurons reduced the complexity of dendritic arbors, and this effect was associated with enhanced Cdc42 activity [22]. Furthermore, short-term stimulation of cortical neurons in vitro with autoactivating MMP-9 (aaMMP-9) induced rapid activation of Cdc42 and Rac1, as revealed by the pull-down assay (Figure 1). At the same time, we did not observe any changes in RhoA activity (data not shown). These results suggest that small GTPases are downstream effectors of MMP-9 signaling in the brain.
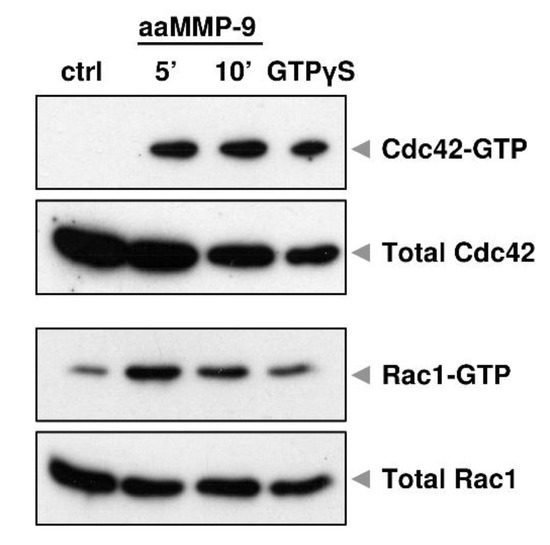
Figure 1.
MMP-9 proteolytic activity causes Cdc42 and Rac1 activation in cultured cortical neurons. Cells were treated with autoactivaiting matrix metalloproteinase 9 (aaMMP-9) for 5 and 10 min and the activity of Rho-GTPases was analyzed using a pull-down assay. Western blots show levels of the activated form (coupled with GTP) and total levels of Cdc42 and Rac1. GTPγS served as a positive control for pull-down assay.
Since MMP-9 is a known modifier of the ECM [1,3,4], here we review the intracellular pathways and MMP-9 substrates that are involved in the activation of small Rho GTPases, thought to underlie ECM remodeling and synaptic plasticity. We focus on several well-described MMP-9 substrates in the central nervous system (CNS): DG (dystroglycan), CD44, ICAM-5 (intercellular adhesion molecule-5, also known as telencephalin/TLN), NLGN1 (neuroligin 1), BDNF (brain-derived neurotrophic factor), NCAM (neural cell adhesion molecule), EphB2 (ephrin type-B receptor 2), and CRMP2 (collapsin response mediator protein 2). We also discuss post-translational modifications of RhoGTPases that may play a key role in these processes.
2. Regulation of MMP-9 Substrates by Small Rho GTPases
2.1. DG
DG is a cell adhesion molecule and a central component of the dystrophin-glycoprotein complex [23]. It was originally found in skeletal muscle but is also highly expressed in other developing and adult tissues. DG is composed of two non-covalently linked α and β subunits derived from post-translational processing of a single protein encoded by the Dag1 gene [24]. Extracellular α-DG is highly glycosylated and therefore interacts with several ECM proteins, such as laminin, agrin, perlecan, and neurexin [25,26,27]. The transmembrane β-DG anchors α-DG to the cell membrane via the N-terminal domain and interacts with the cytoskeletal proteins dystrophin and utrophin via the C-terminal cytoplasmic domain [28]. Thus, DG forms a structural linkage between ECM and the intracellular actin cytoskeleton. However, this linkage must be dynamically modulated in contexts where cellular plasticity is required, such as cell migration, wound healing, or the function of neuronal synapses. One of the mechanisms regulating the interaction between DG and ECM is post-translational cleavage by matrix metalloproteinases.
The role of DG in neurons and glia has been investigated using mice carrying a floxed Dag1 allele because constitutive deletion of Dag1 causes early embryonic lethality [29]. These studies indicate a crucial role for extracellular α-DG interactions since the cerebral cortex developed normally in transgenic mice that lacked the DG intracellular domain [30]. On the other hand, β-DG serves as a scaffold for various proteins involved in signal transduction pathways, such as the adapter protein Grb2 (growth factor receptor-bound protein 2), the kinases ERK (extracellular-signal-related kinase), and MAPK (mitogen-activated protein kinase) [31,32], and therefore deletion of β-DG may lead to serious disturbances in cell signaling processes.
In the adult mouse brain, DG was found at postsynaptic specializations of the cerebral cortex, hippocampus, olfactory bulb, and cerebellum [33]. Immunocytochemical studies in dissociated neuronal cultures revealed that DG is selectively associated with inhibitory GABAergic synapses and can be used as a marker of these synapses [34,35]. It has also been shown that prolonged elevation of neuronal activity leads to increased expression of glycosylated DG and the aggregation of α-DG and GABAA receptors at the postsynaptic surface [36]. Interestingly, this inhibitory upscaling is partly reproduced by the addition of agrin to the culture medium, indicating that ligand-induced signaling through DG is responsible for the modulation of GABAergic synaptic strength. In the light of these data, it is not surprising that breaking the link between DG subunits may lead to a disruption of DG interactions with other proteins, which is of great importance for physiological processes taking place in the CNS.
It has been established that β-DG is a native substrate of MMP-9 in the brain that is cleaved in response to enhanced neuronal activity [5]. In this study, stimulation of neuronal cultures with either glutamate or bicuculline leads to the formation of a 30 kDa product, readily detectable by Western blot. Therefore, the β-DG cleavage assay is commonly used to estimate the endogenous MMP-9 activity. Since then, MMP-9-dependent proteolytic cleavage of β-DG has been shown to contribute to physiological synaptic plasticity, learning and memory formation. Enhanced MMP-9 activation and subsequent β-DG proteolysis in the mouse amygdala, hippocampus, and prefrontal cortex have been linked to fear conditioning [37]. Increased cleavage of β-DG has also been found in protein lysates from primary cortical neurons exposed to a mixture of forskolin, rolipram, and picrotoxin, i.e., chemically induced long-term potentiation (cLTP) that is known to upregulate endogenous MMP-9 activity [13]. Importantly, this enhanced proteolytic activity was accompanied by the structural and functional remodeling of dendritic spines. Live imaging of hippocampal neurons revealed that cLTP stimulation led to the formation of spine head protrusions (SHPs). Notably, spines with SHPs contained more AMPA receptors, providing evidence for the involvement of MMP-9 in alterations of synaptic connectivity.
On the other hand, enhanced proteolysis of β-DG has been associated with several neuronal pathologies ranging from a stroke to neurodegeneration and epileptogenesis. The appearance of a 30 kDa β-DG degradation product has been detected in the hippocampus of mice injected intraperitoneally with PTZ (pentylenetetrazole), a proconvulsant and GABAA receptor antagonist [5,38]. Increased proteolytic degradation of β-DG has also been observed in organotypic hippocampal cultures treated with kainic acid, known to induce neurotoxicity [38]. Importantly, the application of DP-b99, a membrane-activated zinc chelator, effectively reduced β-DG cleavage and had a neuroprotective effect.
There is still little direct experimental evidence linking MMP-9-dependent cleavage of β-DG in the CNS with activation of Rho GTPases. A study on the involvement of DG in dendritic morphogenesis showed that MMP-9-mediated β-DG proteolysis inhibits dendritic tree growth and arborization of hippocampal primary neurons, and these structural changes are closely related to increased Cdc42 activation [22]. It is well established that synaptic formation, modulation, and stabilization depend on the dynamics of actin filaments, which (as mentioned above) is strictly regulated by Rho-family GTPases. Since β-DG has been shown to interact directly with a RhoGEF (Dbl) that is responsible for Cdc42 activation and thus leads to filopodia formation in fibroblasts [39], it is reasonable to speculate that DG may also play an important role in synaptic remodeling. However, further studies are required to confirm this statement.
2.2. CD44
CD44 is a transmembrane receptor for hyaluronan (HA), which represents the main component of the ECM influencing both structural and functional aspects of neuronal plasticity [40,41]. CD44 is a highly glycosylated protein that consists of several functional domains. While the extracellular N-terminal domain is responsible for the binding of HA and other ligands, e.g., matrix metalloproteinases, the C-terminal cytoplasmatic region is involved in the interaction with many different effectors including small Rho GTPases [42].
CD44 plays an important role in physiological and pathological processes in the nervous system. It was shown to be involved in the regulation of axon growth, dendritic tree arborization, calcium signaling, synaptogenesis and synaptic plasticity, astrocyte morphology, neurogenesis, epileptogenesis, ischemia, multiple sclerosis, Alzheimer’s disease, and brain tumors (rev. in [43]).
Proteolytic cleavage of the CD44 ectodomain by MMP-9 was initially described in glioblastoma cells [44]. Recently, we identified CD44 as an MMP-9 substrate in neurons [45]. We showed that 5-HT7 receptor stimulation increases local MMP-9 activity, triggering extracellular cleavage of CD44 by MMP-9, followed by Cdc42 activation to regulate synaptogenesis, long-term potentiation (LTP), and the structural plasticity of dendritic spines.
Moreover, CD44 modulates the functional and structural plasticity of dendritic spines via the Rho GTPases-dependent mechanism [46]. Specific knockdown of CD44 in neurons cultured in vitro resulted in changes in dendritic spine morphology, reduced number of functional synapses, and disturbed activity-dependent structural plasticity of dendritic spines. Using FRET-based biosensors of Rho GTPases, we have shown that CD44 regulates the activity of RhoA, Rac1, and Cdc42 in dendritic spines. Higher levels of activated Cdc42 and Rac1 and lower activation of RhoA were observed in CD44-depleted neurons. Defects in dendritic spine morphology that were induced by CD44 knockdown were rescued by inhibition of Cdc42 activity.
Astrocytic processes that are associated with synapses, enwrapping, and interacting with dendritic spines and synaptic terminals actively contribute to structural plasticity of dendritic spines, synapse formation, and stabilization [47,48,49,50,51,52,53,54,55]. Interestingly, CD44 regulates astrocyte morphology via Rac1 signaling [56]. Its depletion in astrocytes leads to stellate-like morphology of the cells and elevated level of Rac1 activity, whereas the overexpression results in the increased cell area, flattened morphology of astrocytes, and reduced activity of Rac1. Moreover, CD44 knockdown-induced stellation was inhibited by blocking Rac1 activity.
2.3. ICAM-5
The neuron-specific ICAM-5 belongs to the immunoglobulin (Ig) superfamily of adhesion proteins and is a neuronal cell surface protein. The external part of ICAM-5 is formed by 9 Ig domains, where binding sites for integrins and vitronectin are localized. The cytoplasmic domain of ICAM-5 interacts with α-actinin and ERM (ezrin, radixin, moesin) family of cytoplasmic proteins [57].
ICAM-5 shows low expression during embryonic development but increases rapidly after birth [58]. At the same time, dendritic elongation and branching as well as synapse formation, occur. ICAM-5 stimulates neurite outgrowth and plays an important role in spine morphogenesis [59,60]. Interestingly, unlike other adhesion molecules, ICAM-5 inhibits the maturation and stabilization of synapses. This adhesion molecule is present at the soma and dendrites and is particularly abundant in dendritic filopodia to support their formation and maintenance. ICAM-5 deficiency increases spine maturation, while its overexpression favors filopodia formation, thereby slowing spine morphogenesis.
Abnormally high expression levels of ICAM-5 were found in the brains of MMP-2- and MMP-9–deficient mice [61]. This suggested that the MMPs are responsible for the proteolytic processing of ICAM-5. Indeed, long-term treatment (16 h) of hippocampal cultures with N-methyl-D-aspartic acid (NMDA) or α-amino-3-hydroxy-5-methylisoxazole-propionic acid (AMPA) led to the MMP-2- and MMP-9-dependent shedding of ICAM-5 accompanied by spine enlargement. Likewise, short-term exposure (15-30 min) of primary cortical neurons to NMDA, as well as high-frequency tetanic stimulation of hippocampal slices, also promote MMPs-dependent cleavage of ICAM-5. [62]. These findings indicate that enhanced neuronal activity induces MMPs-mediated shedding of membrane-bound ICAM-5 that may facilitate the maturation of dendritic spines.
MMP-9 has been also implicated in ECM degradation, synaptic dynamics, and experience-dependent plasticity in the mouse visual cortex [63,64,65,66]. The subcellular localization of ICAM-5 in the visual cortex is developmentally regulated and depends on MMP-9 activity [67]. Ultrastructural studies of ICAM-5 distribution before and during the critical period for ocular dominance plasticity showed that during early visual development, ICAM-5 occurs mainly in immature dendritic protrusions, while in later developmental stages it is present in dendritic shafts. However, this developmental shift in ICAM-5 localization does not occur in MMP-9 knockout mice. Additionally, in the absence of MMP-9, the ICAM-5 immuno-reactive elements make significantly more contacts with axonal terminals [68]. Together this may indicate that MMP-9 cleaves ICAM-5 to determine its subcellular localization and in turn induces spine maturation during the development of the visual cortex.
Since the spines are largely devoid of ICAM-5, its exclusion from the filopodia surface appears to be required in the process of the dendritic spine maturation. Using HeLa cells and primary hippocampal neurons, it was demonstrated that internalization of ICAM-5 from the cell membrane is mediated by the small GTPase ADP-ribosylation factor 6 (ARF6), recruited by ICAM-5 and activated by GEF, EFA6A [69]. Endocytosis of this adhesion molecule affects filopodia-to-spine transition and requires Rac1-mediated dephosphorylation and release of actin-binding ERM proteins from ICAM-5 [70]. In summary, ICAM-5 is cleaved by MMPs upon activation of glutamate receptors or degraded by RhoGTPase-dependent endocytosis, and both processes promote the maturation of dendritic spines.
2.4. NLGN1
NLGN1 is a transmembrane cell adhesion protein that belongs to the highly conserved neuroligin family. Five isoforms of NLGN have been identified in mammals, each exhibiting a specific pattern of expression and subcellular distribution [71,72]. NLGN1 is located mainly on the postsynaptic side of excitatory synapses [73,74]. The extracellular domain of NLGN1 interacts with presynaptic neurexin1β (NRX1β), forming a trans-synaptic complex [75,76], whereas its intracellular domain binds to postsynaptic density protein-95 (PSD-95; [77]). PSD-95 is required for efficient NLGN1 surface expression in cultured neurons and importantly, PKA-dependent phosphorylation of NLGN1 controls its interaction with PSD-95 [78].
Many studies indicate that NLGN1 plays a crucial role in synapse maturation and brain function [72,79,80,81]. Live imaging of primary cortical neurons from NLGN1-deficient mice has shown destabilization of excitatory synaptic organization and impaired tenacity of these synapses [82]. NLGN1-deficient mice also exhibit impaired LTP [83,84], and NLGN1 knockdown in epileptic rats reduced seizure severity. Whole-cell patch-clamp recordings of pyramidal hippocampal neurons from epileptic rats revealed a decrease in the amplitude of NMDA receptor-dependent excitatory postsynaptic currents [85]. Moreover, silencing of NLGN1 in mice blocks the storage of fear memory by reducing NMDAR-mediated currents [83]. Importantly, a genomic microarray test detected partial loss of NLGN1 in a patient with seizures and severe intellectual disability [86]. Mutations in the NLGN1 gene have been proposed to be associated with autism and other neuropsychiatric disorders [87,88].
Neuronal activity-driven cleavage of NLGN1 by MMP-9 was reported by Peixoto et al. [10]. Notably, the authors showed that MMP-9-dependent shedding of NLGN1 leads to the destabilization of presynaptic NRX1β and downregulation of synaptic transmission. Recent data suggest that the soluble extracellular domain of NLGN1 binds to metabotropic glutamate receptor 2 (mGluR2), and thereby decreases synaptic strength [89].
There have been several reports describing mechanisms by which NLGN1 affects actin reorganization and spine stability. An elegant study by Liu et al. demonstrated that the cytoplasmic C-terminal domain of NLGN1, remaining after proteolytic cleavage, may interact with the spine-associated Rap GTPase-activating protein (SPAR) leading to activation of Rap1/Rac1/LIMK1/cofilin pathway and subsequent spine growth and synapse development [90]. Furthermore, NLGN1 appears to promote excitatory synaptogenesis by cooperation with the postsynaptic adhesion-G protein-coupled receptor (A-GPCR) BAI1, which also results in local activation of Rac1 within spines [91]. Recently, quantitative proteomic analysis of rat brain lysate revealed that NLGN1 interacts with Kalirin-7, an essential RhoGEF of the postsynaptic density. NLGN1-mediated spine formation and glutamatergic synapse function require RhoGEF signaling, indicating that Kalirin-7 is a crucial intracellular effector of NLGN1 function [92].
2.5. BDNF
BDNF is a pleiotropic protein, a member of the neurotrophic factor family, playing an important role in neuronal growth and differentiation [93,94,95]. BDNF exerts its biological function mainly upon binding to tyrosine kinase B (TrkB) receptors [96,97]. The existence of multiple alternative splicing variants of BDNF [98,99,100] and the regulation of its expression by several promoters [101,102] make this neurotrophic factor a variable regulator orchestrating neurite development, learning, and memory formation in the young and adult brain. BDNF is expressed in neuronal and non-neuronal cells, and its tissue-specific localization is developmentally regulated [103]. Neuronal BDNF immunoreactivity was found in several regions of the CNS, as well as in the peripheral and enteric nervous system [104,105]. However, BDNF occurs prominently in the brain and can be found in the hippocampus, cortex, amygdala, striatum, and hypothalamus (rev. in [106]), and localized mostly at presynaptic terminals [107,108] and postsynapses [109,110]. In addition to neurons, astrocytes and microglia are also important sources of BDNF [111,112]. Altered levels of BDNF in the CNS are involved in the pathogenesis of neurodegenerative diseases (e.g., Alzheimer’s and Parkinson’s) [113] and mental disorders (e.g., depression and schizophrenia) [114]. Abnormalities in the regulation of BDNF secretion also affect the proper development of neonates and probably contribute to autism spectrum disorders [115,116].
BDNF is synthesized as a precursor proBDNF, about 32 kDa protein [117], that is responsible for correct folding of the mature form (mBDNF). Importantly, proBDNF can target the secretory pathway and through interaction with the p75 neurotrophin receptor (p75NTR) may act as an active signaling molecule. The conversion of proBDNF to mBDNF occurs through many different proteolytic mechanisms, leading to the release of a polypeptide of approximately 14 kDa. ProBDNF can be cleaved intracellularly by serine proteases, such as convertases PC1/3 and PC7 [118,119], and/or furin [120]. However, at synapses, this neurotrophic factor is predominantly secreted as proBDNF and then digested by extracellular proteases. Extracellular conversion of proBDNF to mBDNF is shown to be essential for hippocampal late-phase LTP (L-LTP), and this is mediated by various proteases including tPA (tissue plasminogen activator)/plasmin [121], and/or metalloproteinases MMP-3, MMP-7, and MMP-9 [120]. Importantly, proBDNF and mBDNF can have opposing effects on neuronal structure and synaptic plasticity, and therefore the extracellular levels of both proteins are tightly controlled by neuronal activity [122,123,124,125,126,127,128].
In the PTZ kindling mouse model of epilepsy, enhanced hippocampal expression of MMP-9 was found to be associated with increased levels of mBDNF, while downregulation of this neurotrophic factor was observed in kindled MMP-9-deficient mice. In addition, decreasing mBDNF by injecting the mice with the BDNF scavenger TrkB-Fc, significantly suppressed the kindling development in wild-type mice, but not in MMP-deficient mice [129].
A growing body of evidence indicates that GTPases are potent mediators of BDNF signaling implicated in synapse formation and plasticity. The conversion of nonfunctional synaptic contacts between hippocampal neurons in vitro into functional synapses occurs through activation of the BDNF/TrkB signaling pathway and can be abolished by the transfection with a plasmid encoding dominant-negative Cdc42. Moreover, treatment of the cultures with Cdc42 activator, bradykinin, also promotes presynaptic maturation. Thus, rapid induction of functional synapses requires the activation of Cdc42, a downstream effector of TrkB [130]. In neonatal sympathetic neurons that do not express the TrkB receptor, binding of BDNF to p75NTR activates the RhoA/ROCK pathway, promoting axonal degeneration [131]. This mechanism may be of general importance as p75NTR is highly upregulated in most post-traumatic CNS neurons [132,133].
Postsynaptic BDNF/TrkB signaling pathway and downstream activation of small GTPases are crucial for structural and functional plasticity. In CA1 hippocampal neurons, glutamate uncaging evokes BDNF release from stimulated dendritic spines and subsequent TrkB activation on these same spines, assessed using a FRET-based biosensor for TrkB [110]. The expression of structural LTP (sLTP) in stimulated spines is associated with the distinct spatial activation of Cdc42 and RhoA [134]. Additionally, imaging the activity of GTPases in organotypic hippocampal slices from BDNF or TrkB conditional knockout mice revealed the spatiotemporal activation patterns of these GTPases during sLTP. Notably, activation of Cdc42 is preferentially enriched in stimulated spines and overlaps the TrkB signaling pattern, while Rac1 signaling seems to spread wider than local activation, affecting nearby spines. Moreover, inhibition of Rac1 by either pharmacological treatment, or single-cell knockout, blocks sLTP in the neighboring spines, but do not affect sLTP of the stimulated spine. Finally, activation of both Cdc42 and Rac1 depends on postsynaptic, autocrine BDNF and corresponds to the local sLTP over spine-specific (Cdc42) and heterosynaptic (Rac1) domains. Interestingly, the spreading of RhoA activation is independent on BDNF/TrkB but is also necessary for synaptic crosstalk [135].
2.6. NCAM
NCAM is a glycoprotein from the Ig immunoglobulin superfamily with three main MW-named alternative splicing variants: NCAM-180 and NCAM-140 expressed in neurons, and NCAM-120 identified in glia. They share an identical extracellular domain, differing on membrane anchoring and cytoplasmic region. The extracellular domain of NCAM consists of 5 Ig-like domains (Ig1-Ig5) followed by 2 fibronectin III domains [136,137] and is polysialylated in the Ig5 domain. This polysialic acid modification is necessary for synaptic plasticity, formation, and consolidation of memory [138,139,140,141].
NCAM-140 is involved in the maturation of olfactory granule cell precursors [142], whereas NCAM-180 is found in postsynaptic densities of mature neurons [143,144]. NCAM plays a role in axonal and dendritic growth, synaptogenesis, and plasticity. It was shown to be required for the development and stabilization of LTP [145].
NCAM is associated with neurodegenerative and psychiatric disorders in humans such as Alzheimer’s disease, bipolar disorder, schizophrenia, and cerebral ischemic stress [146,147,148]. Elevated levels of its 105–115 kDa isoform, possibly the N-terminal cleavage product of NCAM-180, were found in the hippocampus, prefrontal cortex, and cerebrospinal fluid of patients with schizophrenia [149].
All three main isoforms of NCAM undergo MMP-dependent shedding. In cultured cortical neurons exposed to oxidative stress upregulated MMP-9 induces cleavage of NCAM-180 producing extracellular (110–115 kDa) and intracellular (65–70 kDa) ectodomains (EC-NCAM) [150]. The involvement of MMP-9-mediated proteolysis of NCAM-180 in the development of neuronal ischemic damage in vivo was confirmed in middle cerebral artery occlusion (MCAO) model mice [151]. In this study, downregulation of MMPs activity using a broad-spectrum inhibitor (GM6001) or a knockdown of MMP-9 gene expression increases the levels of NCAM-180 accompanied by a decrease in EC-NCAM. MMP-dependent shedding of NCAM occurs in primary hippocampal neurons and this effect is enhanced by ATP added to the culture medium [152]. Application of the MMP inhibitor evokes neuronal aggregation, indicating that MMP-mediated proteolytic cleavage of NCAM is involved in the regulation of neurite outgrowth. Soluble NCAM fragments were detected in the dentate gyrus of rats after the induction of LTP in vivo [153], as well as in rat hippocampal cell culture after exposure to 17β-estradiol, which mediates enhancement of LTP in vitro [154].
Ligand affinity chromatography and subsequent peptide mass fingerprinting of rat brain extracts enabled identifying several intracellular NCAM180-interacting proteins such as α- and β-tubulin, α-actinin-1, and RhoA-binding kinase-α [155]. Moreover, NCAM associates with the receptor tyrosine kinase EphA3 in murine cortical GABAergic interneurons, promoting ephrin-A5-dependent receptor clustering. This triggers EphA3 kinase signaling and downstream RhoA-GTPase activation, leading to growth cone collapse of interneurons and hence regulation of the perisomatic synapse density [156].
2.7. EphB2
EphB2 transmembrane receptor is a member of the largest subfamily of receptor tyrosine kinases (RTKs). It consists of a highly conserved extracellular domain necessary for ligand recognition and binding, followed by a cysteine-rich region, two fibronectin repeats, an intracellular highly conserve kinase domain that catalyzes tyrosine phosphorylation of protein substrates, a C-terminal sterile alpha motif (SAM), and a PDZ-binding motif [157].
EphB receptors bind to their corresponding ephrin-B membrane-bound ligands, thus requiring a direct cell-cell contact. Their interaction upon the activation can be bi-directional—into Eph-expressing cells (forward signaling) or ephrin-expressing cells (reverse signaling) [158]. In the nervous system, the EphB2 receptor interacts directly with NMDA receptors and its absence may cause an impairment in the long-term potentiation and depression [159]. Additionally, EphB2 plays a crucial role in axon guidance and growth cone migration [160].
It has been demonstrated that MMP-9 protein can cleave the EphB2 receptor. MMP-induced cleavage causes a prolonged EphB2 activation by sustaining its phosphorylation. That leads to strong cytoskeletal responses, recruitment of focal adhesion kinase (FAK), and RhoA activation. It is suggested that those changes may also further impact other small Rho GTPases’ signaling pathways [161]. Studies performed on the hippocampal primary cultures have shown that ephrin-B1-induced activation of the EphB2 receptor can increase RhoA activity as a result of the FAK stimulation. That suggests the shortening of dendritic filopodia and their transformation into spine-like protrusions is mediated by EphB2 [162]. The EphB2/FAK downstream signaling through RhoA/ROCK/LIMK-1 pathway partly controls the stability of mature dendritic spines by inhibiting their cofilin-mediated remodeling [163]. Furthermore, EphB2/ephrin reverse signaling through the RhoA/ROCK pathway can negatively regulate branching and axonal outgrowth [158].
One of the exceptional characteristics of the Eph/ephrin signaling is the ability to form higher-order receptor-ligand clusters that, in comparison to monomers or dimers, invoke stronger cell repulsion response through their trans-endocytosis [164,165]. This type of reverse endocytosis is highly important for axon withdrawal during growth cone collapse [166]. In vitro studies on the primary mouse cortical neurons indicate that EphB2 trans-endocytosis is positively regulated by the activity of Rac GTPase and its specific GEF—Tiam2 [167]. The downstream signaling pathway, after the EphB2/ephrin-B clustering, can also induce the synaptic concentration of the Rho-GEF Kalirin, resulting in the local activation of Rac1 and spine morphogenesis [168].
Although most studies emphasize the importance of RhoGTPases in the regulation of actin cytoskeleton, it has been confirmed that Arg kinase activated by the EphB2 receptor can trigger actin polymerization in the dendritic spine independently of RhoGTPases. It suggests that Rac1 and Cdc42 are required particularly in the reorganization of the synapse structure but not always of the dendritic region [169]. Still, the interaction of EphB2 and Zizimin1 (Cdc42-GEF), and its downstream signaling through p21-activated kinase-3 (PAK3) was shown to regulate cell surface targeting of AMPA receptors, therefore impacting synaptic plasticity [170]. Moreover, the synergistic effect of EphB2 and neural Wiskott-Aldrich syndrome protein (WASP) highly upregulates Cdc42-GEF intersectin and, by Cdc42 activation, triggers actin polymerization and spine morphogenesis [171].
EphB2/ephrin signaling pathway also includes another small GTPase—Ras, that is considered to be engaged in the long-lasting synaptic changes, learning and memory formation, and also promotes protective mechanisms in the nervous system [172,173]. It has been shown that the activation of EphB2 in the NG108 neuronal cell line leads to a reduction of the GTP-bound Ras level in the Ras/MAPK pathway, a retraction of filopodial extensions, and a collapse of the neurite structures [160,174]. Ras/MAPK signaling was also observed to be inhibited by ephrin-B3 (EphB2 ligand) through direct binding to ERK1/2, suggesting the role of EphB2/ephrin-B3 interaction in the formation of dendritic spines and the control of synapse density [175]. Apart from that, Eph2B receptor signaling may also inactivate R-Ras by the joint effects of tyrosine phosphorylation and heightened GTP hydrolysis, causing growth cone collapse in hippocampal neurons [176].
2.8. CRMP2
CRMP2 is a cytosolic phosphoprotein whose monomeric structure consists of a triosephosphate isomerase (TIM) barrel, a small β-sheet domain formed by the N-terminal residues, C-terminus close segments, and a C-terminal helix that is the only region on the CRMP2 surface with a positive charge potential [177]. In the brain, it is expressed in the oligodendrocytes and developing neurons, showing its abundance especially in the hippocampus, cerebellum, and olfactory system [178,179].
CRMP2 plays a crucial role in the regulation of several signaling pathways impacting axon formation, as well as growth cone guidance and collapse, thereby participating in the establishment and maintenance of neuronal polarity [180,181,182]. Its N-terminal globular domain, particularly the helix H19, binds to the soluble GTP-tubulin dimers to promote the growth of GTP-state microtubules; while the C-terminal flexible tail further binds to those polymerized microtubules to stabilize them [183].
With the use of the two-dimensional fluorescence difference gel electrophoresis (2D-DIGE) technique and tandem mass spectrometry, CRMP2 was proposed as one of the MMP-9 extracellular targets in the synapse [6]. Results derived from Western blot analysis confirmed that MMP-9 can cleave CRMP protein. Those changes may then impact the latter’s binding ability to tubulin, thereby affecting axonal elongation.
Studies on numerous Rho GTPase effectors indicate that CRMP2 is directly dependent on the activity of those small G proteins. Activated Rho/Rho-kinase signal induces phosphorylation of CRMP2 at threonine 555, hence negatively regulating its ability to bind tubulin heterodimers, which may lead to a decrease in neurite length [184]. Although most studies portray CRMP2 as a Rho substrate, it has been suggested that it can have a negative effect on both upstream and downstream of Rho signaling. In combination with active Rho or Rac GTPases, CRMP2 may act as a cyclical switch reversing their usual morphological effects, therefore stimulating dynamic neuronal shape changes [185]. Additionally, Ras protein also can play an essential role in the regulation of neuronal polarity by inhibiting phosphorylation of CRMP2 in the PI3-kinase/Akt/GSK3α/CRMP2 signaling pathway [186]. Most probably, the erroneous signaling in the Rho/ROCK/CRMP2 pathway could have lineal implications in the development of conditions like cancer, Alzheimer’s disease, Guillain Barrè Syndrome, ischemic stroke, or peripheral nerve and spinal cord injuries [187,188,189,190,191,192].
3. Post-Translational Modifications of Rho GTPases Involved in Neuronal Plasticity
Rho GTPases can be modified by a variety of post-translational modifications that cause differences in localization, activity, and directly affect their function [16,193]. PTMs known to regulate the role of Rho GTPases in synaptic plasticity include lipidation, phosphorylation, and ubiquitination [16,193,194].
Lipidations such as prenylation or palmitoylation are the first and most frequently described PTMs of small GTPases. This modification consists of covalent binding of a lipid group to a peptide chain, which may affect the activity of the protein and alter its subcellular localization. The location of Rho GTPases plays a critical role in determining their spatially unique functions.
Many Rho proteins cycle from the cytosol to various cellular membranes, guided by a prenylation [195,196]. Prenylation of small GTPases directs them to a plasma membrane (PM) where they are activated by interaction with GEFs. In the absence of prenyl modification, proteins remain in the cytosol and are unable to function properly [197,198]. Prenylation plays a critical role in regulating Rac1 activity in neurons by targeting Rac1 to the neuronal PM [199]. Moreover, newly synthesized Rho GTPases such as Rac1, Cdc42, or RhoA were shown to be rapidly prenylated in the cytosol and then transferred to the endoplasmic reticulum (ER) for further modifications [195,200,201,202]. Once attached to the membrane, specific GTPases carry out a variety of functions, from neurite outgrowth and guidance (Rac1) to inhibiting neuronal extensions and promoting growth cone collapse (RhoA) [203,204,205,206,207,208,209,210]. Importantly, transport of these proteins from the endoplasmic reticulum to the PM requires another modification which is palmitoylation.
Palmitoylation is a post-translational covalent binding of the palmitate to the cysteine residue of proteins by thioester bond. In contrast to prenylation, palmitoylation is a dynamic and reversible cysteine modification [211]. A recent study showed that Rac1 undergoes palmitoylation, which enhances its recruitment to the PM and induces translocation to lipid rafts [212]. This PTM targets Rac1 for stabilization at actin cytoskeleton-linked ordered membrane regions. Inhibition of Rac1 palmitoylation significantly reduces the PM localization and GTP loading of Rac1, leading to downregulation of its effector protein, p21-activated kinase (PAK). Palmitoylation might also be a regulatory mechanism for other small GTPases such as Cdc42 [213,214]. Interestingly, the canonical Cdc42 form associates with membranes through prenylation whereas the brain-specific form of Cdc42 is palmitoylated [213,214]. Although prenylated and palmitoylated isoforms coexist in the developing neurons, only palmitoylated Cdc42 is critically involved in the extension of dendritic filopodia, which develop into dendritic spines [214,215]. Importantly, in response to glutamate, Cdc42 undergoes rapid depalmitoylation and dislocates from dendritic spines. Therefore, the level of Cdc42 in dendritic spines is rapidly modified by neuronal activity and may be responsible for dynamic changes in spine morphology [214,216]. It is also worth to mention, that there are interesting differences in palmitoylation between Ras isoforms. H-Ras is stabilized by two cysteines that can bind palmitate, while N-Ras and K-Ras4A have only one modified cysteine residue [217,218,219,220,221]. On the other hand, K-Ras4B lacks a palmitoylation site [222]. The level of Ras palmitoylation could vary between membrane compartments and might even depend on the activation status of the Ras isoforms [220]. A recent study showed that activation of H-Ras and N-Ras leads to depalmitoylation and redistribution to the ER/Golgi, where they can be repalmitoylated [217,219]. This cycle is crucial for preventing the nonspecific accumulation of Ras isoforms on all membranes. Furthermore, a detailed analysis of H-Ras trafficking indicated that specific palmitoylated cysteines play different roles in the protein localization. Palmitoylation of the first cysteine stabilizes the localization of H-Ras in the PM, whereas modification of the second cysteine facilitates redistribution between cell membranes [219,223]. The different distribution of N-Ras and H-Ras proteins in the membrane compartments is crucial for the ability of the Ras proteins to control so many distinct cellular processes.
It is not surprising that Rho GTPases can also undergo phosphorylation. Several kinases are involved in this process, namely protein kinase A (PKA), protein kinase C (PKC), and Src family kinases [224]. Phosphorylation of RhoA and Cdc42 significantly increases their interaction with RhoGDI and thus plays a key role in the activation-inactivation cycle of these GTPases, controlling the actin cytoskeletal organization in the brain under physiological conditions [225]. Rac1 phosphorylation is carried out by Src and FAK [199]. Recent data suggest that Rac1 phosphorylation negatively regulates cell spreading, focal adhesion localization, and interaction with GTP, RhoGDI, GEFs, and an effector protein PAK [226]. It was also demonstrated that PKC regulates the activation of Rac1 during sLTP of dendritic spines without affecting Cdc42 [227]. Additionally, PKC-mediated phosphorylation of K-Ras4B on serine residue initiates its dissociation from the PM [228].
It is known that Rho-family GTPases can be modified by ubiquitination, which affects their cellular localization and activity state [229]. Recent studies have shown that PM-tethered H-Ras and N-Ras undergo ubiquitination, regardless of their activation state, and such modification leads to their internalization into endosomes [230]. Ubiquitination of K-Ras enhances GDP to GTP conversion and downstream signaling. Importantly, ubiquitin-mediated degradation of RhoA protein promotes neurite outgrowth [231]. Although a growing body of evidence underlines the importance of PTMs for the signaling roles of GTPases, the consequences of these modifications for the regulation of individual proteins have not yet been fully elucidated.
4. Conclusions
Evidence has been accumulated that MMP-9-dependent proteolysis that occurs at the synapse is crucial for neuronal function and remodeling of synapses during physiological and aberrant plasticity. However, questions remain about the downstream cellular signaling pathways involved in these processes. In this review, we highlight the Rho family of GTPases as potentially important players in MMP-9-controlled signaling in the brain (Figure 2). We also discuss the role of PTMs of Rho GTPases, as such modifications have a direct impact on the localization and function of these proteins.
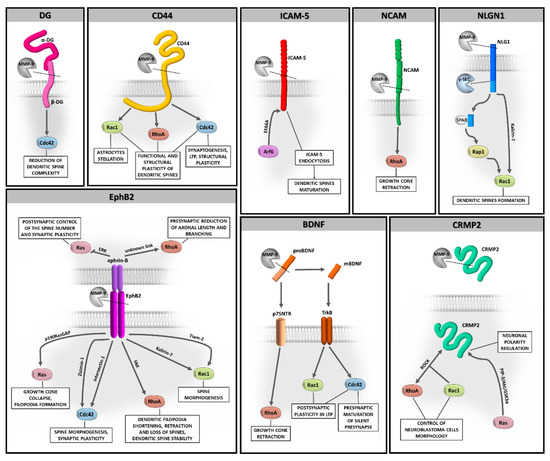
Figure 2.
The potential relationship between MMP-9-driven substrates shedding and Rho GTPases signaling in brain plasticity.
While the proteolytic cleavage of the described MMP-9 substrates and its possible regulation by activated Rho GTPases have been studied, the relationship between these two processes remains speculative. So far, only the MMP-9/CD44/Cdc42 pathway has been well proven [45]. However, the continuous development of the live-cell single-molecule tracking methods should provide an understanding of the role of Rho GTPases in the MMP-9 downstream signaling at the synapse.
Author Contributions
I.F. and P.K.K. contributed equally to this work (co-first authors). All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Science Centre, grant number UMO-2015/17/B/NZ4/02540 and UMO-2015/19/B/NZ3/01376.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Acknowledgments
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| AMPA | α-amino-3-hydroxy-5-methylisoxazole-propionic acid |
| BDNF | brain-derived neurotrophic factor |
| cLTP | chemically induced long-term potentiation |
| CNS | central nervous system |
| CRMP2 | collapsin response mediator protein 2 |
| DG | dystroglycan |
| ECM | extracellular matrix |
| EphB2 | ephrin type-B receptor 2 |
| ER | endoplasmic reticulum |
| ERK | extracellular-signal-regulated kinase |
| ERM | ezrin, radixin, moesin |
| FAK | focal adhesion kinase |
| GABAA | γ-aminobutyric acid type A |
| GAP | GTPase activating protein |
| GDI | guanine nucleotide dissociation inhibitor |
| GEF | guanine nucleotide exchange factor |
| ICAM-5 | intracellular adhesion molecule-5 |
| L-LTP | late-phase long-term potentiation |
| LTP | long-term potentiation |
| MAPK | mitogen-activated protein kinase |
| MMP | matrix metalloproteinase |
| NCAM | neural cell adhesion molecule |
| NLGN1 | neuroligin 1 |
| NMDA | N-methyl-D-aspartic acid |
| PAK | p21-activated kinase |
| PKA | protein kinase A |
| PM | plasma membrane |
| PTM | post-translational modifications |
| PTZ | pentylenetetrazole |
| ROCK | Rho-associated protein kinase |
| sLTP | structural long-term potentiation |
| TrkB | tyrosine kinase B |
References
- Huntley, G.W. Synaptic circuit remodelling by matrix metalloproteinases in health and disease. Nat. Rev. Neurosci. 2012, 13, 743–757. [Google Scholar] [CrossRef] [PubMed]
- Tsien, R.Y. Very long-term memories may be stored in the pattern of holes in the perineuronal net. Proc. Natl. Acad. Sci. USA 2013, 110, 12456–12461. [Google Scholar] [CrossRef] [PubMed]
- Dityatev, A. Remodeling of extracellular matrix and epileptogenesis. Epilepsia 2010, 51, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Ethell, I.M.; Ethell, D.W. Matrix metalloproteinases in brain development and remodeling: Synaptic functions and targets. J. Neurosci. Res. 2007, 85, 2813–2823. [Google Scholar] [CrossRef] [PubMed]
- Michaluk, P.; Kolodziej, L.R.; Mioduszewska, B.; Wilczynski, G.M.; Dzwonek, J.; Jaworski, J.; Gorecki, D.C.; Ottersen, O.P.; Kaczmarek, L. β-Dystroglycan as a Target for MMP-9, in Response to Enhanced Neuronal Activity. J. Biol. Chem. 2007, 282, 16036–16041. [Google Scholar] [CrossRef] [PubMed]
- Bajor, M.; Michaluk, P.; Gulyássy, P.; Kékesi, K.A.; Juhász, G.; Kaczmarek, L. Synaptic cell adhesion molecule-2 and collapsin response mediator protein-2 are novel members of the matrix metalloproteinase-9 degradome. J. Neurochem. 2012, 122, 775–788. [Google Scholar] [CrossRef]
- Meighan, S.E.; Meighan, P.C.; Choudhury, P.; Davis, C.J.; Olson, M.L.; Zornes, P.A.; Wright, J.W.; Harding, J.W. Effects of extracellular matrix-degrading proteases matrix metalloproteinases 3 and 9 on spatial learning and synaptic plasticity. J. Neurochem. 2006, 96, 1227–1241. [Google Scholar] [CrossRef] [PubMed]
- Nagy, V.; Bozdagi, O.; Matynia, A.; Balcerzyk, M.; Okulski, P.; Dzwonek, J.; Costa, R.M.; Silva, A.J.; Kaczmarek, L.; Huntley, G.W. Matrix Metalloproteinase-9 Is Required for Hippocampal Late-Phase Long-Term Potentiation and Memory. J. Neurosci. 2006, 26, 1923–1934. [Google Scholar] [CrossRef]
- Okulski, P.; Jay, T.M.; Jaworski, J.; Duniec, K.; Dzwonek, J.; Konopacki, F.A.; Wilczynski, G.M.; Sánchez-Capelo, A.; Mallet, J.; Kaczmarek, L. TIMP-1 Abolishes MMP-9-Dependent Long-lasting Long-term Potentiation in the Prefrontal Cortex. Biol. Psychiatry 2007, 62, 359–362. [Google Scholar] [CrossRef]
- Peixoto, R.T.; Kunz, P.A.; Kwon, H.; Mabb, A.M.; Sabatini, B.L.; Philpot, B.D.; Ehlers, M.D. Transsynaptic Signaling by Activity-Dependent Cleavage of Neuroligin-1. Neuron 2012, 76, 396–409. [Google Scholar] [CrossRef]
- Wang, X.B.; Bozdagi, O.; Nikitczuk, J.S.; Zhai, Z.W.; Zhou, Q.; Huntley, G.W. Extracellular Proteolysis by Matrix Metallopro-teinase-9 Drives Dendritic Spine Enlargement and Long-Term Potentiation Coordinately. Proc. Natl. Acad. Sci. USA 2008, 105, 19520–19525. [Google Scholar] [CrossRef] [PubMed]
- Michaluk, P.; Wawrzyniak, M.; Alot, P.; Szczot, M.; Wyrembek, P.; Mercik, K.; Medvedev, N.; Wilczek, E.; De Roo, M.; Zuschratter, W.; et al. Influence of matrix metalloproteinase MMP-9 on dendritic spine morphology. J. Cell Sci. 2011, 124, 3369–3380. [Google Scholar] [CrossRef] [PubMed]
- Szepesi, Z.; Bijata, M.; Ruszczycki, B.; Kaczmarek, L.; Włodarczyk, J. Matrix Metalloproteinases Regulate the Formation of Dendritic Spine Head Protrusions during Chemically Induced Long-Term Potentiation. PLoS ONE 2013, 8, e63314. [Google Scholar] [CrossRef] [PubMed]
- Szepesi, Z.; Hosy, E.; Ruszczycki, B.; Bijata, M.; Pyskaty, M.; Bikbaev, A.; Heine, M.; Choquet, D.; Kaczmarek, L.; Włodarczyk, J. Synaptically Released Matrix Metalloproteinase Activity in Control of Structural Plasticity and the Cell Surface Distribution of GluA1-AMPA Receptors. PLoS ONE 2014, 9, e98274. [Google Scholar] [CrossRef] [PubMed]
- Stawarski, M.; Stefaniuk, M.; Wlodarczyk, J. Matrix Metalloproteinase-9 Involvement in the Structural Plasticity of Dendritic Spines. Front. Neuroanat. 2014, 8, 68. [Google Scholar] [CrossRef] [PubMed]
- Hodge, R.G.; Ridley, A.J. Regulating Rho GTPases and Their Regulators. Nat. Rev. Mol. Cell Biol. 2016, 17, 496–510. [Google Scholar] [CrossRef]
- Kjøller, L.; Hall, A. Signaling to Rho GTPases. Exp. Cell Res. 1999, 253, 166–179. [Google Scholar] [CrossRef]
- Heasman, S.J.; Ridley, A.J. Mammalian Rho GTPases: New insights into their functions from in vivo studies. Nat. Rev. Mol. Cell Biol. 2008, 9, 690–701. [Google Scholar] [CrossRef]
- Niftullayev, S.; Lamarche-Vane, N. Regulators of Rho GTPases in the Nervous System: Molecular Implication in Axon Guid-ance and Neurological Disorders. Int. J. Mol. Sci. 2019, 20, 1497. [Google Scholar] [CrossRef]
- Martino, A.; Ettorre, M.; Musilli, M.; Lorenzetto, E.; Buffelli, M.; Diana, G. Rho GTPase-dependent plasticity of dendritic spines in the adult brain. Front. Cell. Neurosci. 2013, 7, 62. [Google Scholar] [CrossRef]
- Shinde, S.R.; Maddika, S. Post Translational Modifications of Rab GTPases. Small GTPases 2018, 9, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Bijata, M.; Włodarczyk, J.; Figiel, I. Dystroglycan controls dendritic morphogenesis of hippocampal neurons in vitro. Front. Cell. Neurosci. 2015, 9, 199. [Google Scholar] [CrossRef] [PubMed]
- Ervasti, J.M.; Campbell, K.P. Membrane organization of the dystrophin-glycoprotein complex. Cell 1991, 66, 1121–1131. [Google Scholar] [CrossRef]
- Ibraghimov-Beskrovnaya, O.; Ervasti, J.M.; Leveille, C.J.; Slaughter, C.A.; Sernett, S.W.; Campbell, K.P. Primary structure of dystrophin-associated glycoproteins linking dystrophin to the extracellular matrix. Nat. Cell Biol. 1992, 355, 696–702. [Google Scholar] [CrossRef] [PubMed]
- Gee, S.H.; Montanaro, F.; Lindenbaum, M.H.; Carbonetto, S. Dystroglycan-α, a dystrophin-associated glycoprotein, is a functional agrin receptor. Cell 1994, 77, 675–686. [Google Scholar] [CrossRef]
- Talts, J.F.; Andac, Z.; Gohring, W.; Brancaccio, A.; Timpl, R. Binding of the G Domains of Laminin Alpha1 and Alpha2 Chains and Perlecan to Heparin, Sulfatides, Alpha-Dystroglycan and Several Extracellular Matrix Proteins. EMBO J. 1999, 18, 863–870. [Google Scholar] [CrossRef]
- Sugita, S.; Saito, F.; Tang, J.; Satz, J.; Campbell, K.; Südhof, T.C. A stoichiometric complex of neurexins and dystroglycan in brain. J. Cell Biol. 2001, 154, 435–446. [Google Scholar] [CrossRef]
- Barresi, R.; Campbell, K.P. Dystroglycan: From Biosynthesis to Pathogenesis of Human Disease. J. Cell Sci. 2006, 119, 199–207. [Google Scholar] [CrossRef]
- Williamson, R.A.; Henry, M.D.; Daniels, K.J.; Hrstka, R.F.; Lee, J.C.; Sunada, Y.; Ibraghimov-Beskrovnaya, O.; Campbell, K.P. Dystroglycan Is Essential for Early Embryonic Development: Disruption of Reichert’s Membrane in Dag1-Null Mice. Hum. Mol. Genet. 1997, 6, 831–841. [Google Scholar] [CrossRef]
- Satz, J.S.; Ostendorf, A.P.; Hou, S.; Turner, A.; Kusano, H.; Lee, J.C.; Turk, R.; Nguyen, H.; Ross-Barta, S.E.; Westra, S.; et al. Distinct Functions of Glial and Neuronal Dystroglycan in the Developing and Adult Mouse Brain. J. Neurosci. 2010, 30, 14560–14572. [Google Scholar] [CrossRef]
- Russo, K.; Di Stasio, E.; Macchia, G.; Rosa, G.; Brancaccio, A.; Petrucci, T.C. Characterization of the Beta-Dystroglycan-Growth Factor Receptor 2 (Grb2) Interaction. Biochem. Biophys. Res. Commun. 2000, 274, 93–98. [Google Scholar] [CrossRef]
- Spence, H.J.; Dhillon, A.S.; James, M.; Winder, S.J. Dystroglycan, a scaffold for the ERK–MAP kinase cascade. EMBO Rep. 2004, 5, 484–489. [Google Scholar] [CrossRef]
- Zaccaria, M.; Di Tommaso, F.; Brancaccio, A.; Paggi, P.; Petrucci, T. Dystroglycan distribution in adult mouse brain: A light and electron microscopy study. Neuroscience 2001, 104, 311–324. [Google Scholar] [CrossRef]
- Levi, S.; Grady, R.M.; Henry, M.D.; Campbell, K.P.; Sanes, J.R.; Craig, A.M. Dystroglycan Is Selectively Associated with In-hibitory GABAergic Synapses but Is Dispensable for Their Differentiation. J. Neurosci. 2002, 22, 4274–4285. [Google Scholar] [CrossRef]
- Briatore, F.; Patrizi, A.; Viltono, L.; Sassoè-Pognetto, M.; Wulff, P. Quantitative Organization of GABAergic Synapses in the Molecular Layer of the Mouse Cerebellar Cortex. PLoS ONE 2010, 5, e12119. [Google Scholar] [CrossRef]
- Pribiag, H.; Peng, H.; Shah, W.A.; Stellwagen, D.; Carbonetto, S. Dystroglycan mediates homeostatic synaptic plasticity at GABAergic synapses. Proc. Natl. Acad. Sci. USA 2014, 111, 6810–6815. [Google Scholar] [CrossRef]
- Ganguly, K.; Rejmak, E.; Mikosz, M.; Nikolaev, E.; Knapska, E.; Kaczmarek, L. Matrix Metalloproteinase (MMP) 9 Transcription in Mouse Brain Induced by Fear Learning. J. Biol. Chem. 2013, 288, 20978–20991. [Google Scholar] [CrossRef]
- Yeghiazaryan, M.; Rutkowska-Wlodarczyk, I.; Konopka, A.; Wilczyński, G.M.; Melikyan, A.; Korkotian, E.; Kaczmarek, L.; Figiel, I. DP-b99 Modulates Matrix Metalloproteinase Activity and Neuronal Plasticity. PLoS ONE 2014, 9, e99789. [Google Scholar] [CrossRef]
- Batchelor, C.L.; Higginson, J.R.; Chen, Y.-J.; Vanni, C.; Eva, A.; Winder, S.J. Recruitment of Dbl by Ezrin and Dystroglycan Drives Membrane Proximal Cdc42 Activation and Filopodia Formation. Cell Cycle 2007, 6, 353–363. [Google Scholar] [CrossRef]
- Kochlamazashvili, G.; Henneberger, C.; Bukalo, O.; Dvoretskova, E.; Senkov, O.; Lievens, P.M.-J.; Westenbroek, R.; Engel, A.K.; Catterall, W.A.; Rusakov, D.A.; et al. The Extracellular Matrix Molecule Hyaluronic Acid Regulates Hippocampal Synaptic Plasticity by Modulating Postsynaptic L-Type Ca2+ Channels. Neuron 2010, 67, 116–128. [Google Scholar] [CrossRef]
- Wlodarczyk, J.; Mukhina, I.; Kaczmarek, L.; Dityatev, A. Extracellular Matrix Molecules, Their Receptors, and Secreted Pro-teases in Synaptic Plasticity. Dev. Neurobiol. 2011, 71, 1040–1053. [Google Scholar] [CrossRef] [PubMed]
- Bourguignon, L.Y.W.; Gilad, E.; Rothman, K.; Peyrollier, K. Hyaluronan-CD44 Interaction with IQGAP1 Promotes Cdc42 and ERK Signaling, Leading to Actin Binding, Elk-1/Estrogen Receptor Transcriptional Activation, and Ovarian Cancer Progression. J. Biol. Chem. 2005, 280, 11961–11972. [Google Scholar] [CrossRef] [PubMed]
- Dzwonek, J.; Wilczyński, G.M. CD44: Molecular interactions, signaling and functions in the nervous system. Front. Cell. Neurosci. 2015, 9, 175. [Google Scholar] [CrossRef] [PubMed]
- Chetty, C.; Vanamala, S.K.; Gondi, C.S.; Dinh, D.H.; Gujrati, M.; Rao, J.S. MMP-9 induces CD44 cleavage and CD44 mediated cell migration in glioblastoma xenograft cells. Cell. Signal. 2012, 24, 549–559. [Google Scholar] [CrossRef]
- Bijata, M.; Labus, J.; Guseva, D.; Stawarski, M.; Butzlaff, M.; Dzwonek, J.; Schneeberg, J.; Bohm, K.; Michaluk, P.; Rusakov, D.A.; et al. Synaptic Remodeling Depends on Signaling between Ser-otonin Receptors and the Extracellular Matrix. Cell Rep. 2017, 19, 1767–1782. [Google Scholar] [CrossRef]
- Roszkowska, M.; Skupien, A.; Wójtowicz, T.; Konopka, A.; Gorlewicz, A.; Kisiel, M.; Bekisz, M.; Ruszczycki, B.; Dolezyczek, H.; Rejmak, E.; et al. CD44: A novel synaptic cell adhesion molecule regulating structural and functional plasticity of dendritic spines. Mol. Biol. Cell 2016, 27, 4055–4066. [Google Scholar] [CrossRef]
- Haber, M.; Zhou, L.; Murai, K.K. Cooperative Astrocyte and Dendritic Spine Dynamics at Hippocampal Excitatory Synapses. J. Neurosci. 2006, 26, 8881–8891. [Google Scholar] [CrossRef]
- Theodosis, D.T.; Poulain, D.A.; Oliet, S.H. Activity-Dependent Structural and Functional Plasticity of Astrocyte-Neuron In-teractions. Physiol. Rev. 2008, 88, 983–1008. [Google Scholar] [CrossRef]
- Volterra, A.; Meldolesi, J. Astrocytes, from brain glue to communication elements: The revolution continues. Nat. Rev. Neurosci. 2005, 6, 626–640. [Google Scholar] [CrossRef]
- Perea, G.; Navarrete, M.; Araque, A. Tripartite synapses: Astrocytes process and control synaptic information. Trends Neurosci. 2009, 32, 421–431. [Google Scholar] [CrossRef]
- Halassa, M.M.; Haydon, P.G. Integrated Brain Circuits: Astrocytic Networks Modulate Neuronal Activity and Behavior. Annu. Rev. Physiol. 2010, 72, 335–355. [Google Scholar] [CrossRef]
- Araque, A.; Parpura, V.; Sanzgiri, R.P.; Haydon, P.G. Tripartite synapses: Glia, the unacknowledged partner. Trends Neurosci. 1999, 22, 208–215. [Google Scholar] [CrossRef]
- Araque, A.; Carmignoto, G.; Haydon, P.G.; Oliet, S.H.R.; Robitaille, R.; Volterra, A. Gliotransmitters Travel in Time and Space. Neuron 2014, 81, 728–739. [Google Scholar] [CrossRef]
- Ullian, E.M.; Sapperstein, S.K.; Christopherson, K.S.; Barres, B.A. Control of Synapse Number by Glia. Science 2001, 291, 657–661. [Google Scholar] [CrossRef]
- Bernardinelli, Y.; Randall, J.; Janett, E.; Nikonenko, I.; Konig, S.; Jones, E.V.; Flores, C.E.; Murai, K.K.; Bochet, C.G.; Holtmaat, A.; et al. Activity-Dependent Structural Plasticity of Perisynaptic Astrocytic Domains Promotes Excitatory Synapse Stability. Curr. Biol. 2014, 24, 1679–1688. [Google Scholar] [CrossRef]
- Konopka, A.; Zeug, A.; Skupien, A.; Kaza, B.; Mueller, F.; Chwedorowicz, A.; Ponimaskin, E.; Wilczynski, G.M.; Dzwonek, J. Cleavage of Hyaluronan and CD44 Adhesion Molecule Regulate Astrocyte Morphology via Rac1 Signalling. PLoS ONE 2016, 11, e0155053. [Google Scholar] [CrossRef]
- Gahmberg, C.G.; Ning, L.; Paetau, S. ICAM-5: A neuronal dendritic adhesion molecule involved in immune and neuronal functions. Adv. Neurobiol. 2014, 8, 117–132. [Google Scholar] [CrossRef]
- Yoshihara, Y.; Oka, S.; Nemoto, Y.; Watanabe, Y.; Nagata, S.; Kagamiyama, H.; Mori, K. An ICAM-Related Neuronal Glycoprotein, Telencephalin, with Brain Segment-Specific Expression. Neuron 1994, 12, 541–553. [Google Scholar] [CrossRef]
- Matsuno, H.; Okabe, S.; Mishina, M.; Yanagida, T.; Mori, K.; Yoshihara, Y. Telencephalin Slows Spine Maturation. J. Neurosci. 2006, 26, 1776–1786. [Google Scholar] [CrossRef]
- Furutani, Y.; Matsuno, H.; Kawasaki, M.; Sasaki, T.; Mori, K.; Yoshihara, Y. Interaction between Telencephalin and ERM Family Proteins Mediates Dendritic Filopodia Formation. J. Neurosci. 2007, 27, 8866–8876. [Google Scholar] [CrossRef]
- Tian, L.; Stefanidakis, M.; Ning, L.; Van Lint, P.; Nyman-Huttunen, H.; Libert, C.; Itohara, S.; Mishina, M.; Rauvala, H.; Gahmberg, C.G. Activation of NMDA receptors promotes dendritic spine development through MMP-mediated ICAM-5 cleavage. J. Cell Biol. 2007, 178, 687–700. [Google Scholar] [CrossRef]
- Conant, K.; Wang, Y.; Szklarczyk, A.; Dudak, A.; Mattson, M.P.; Lim, S.T. Matrix metalloproteinase-dependent shedding of intercellular adhesion molecule-5 occurs with long-term potentiation. Neuroscience 2010, 166, 508–521. [Google Scholar] [CrossRef]
- Kelly, E.A.; Russo, A.S.; Jackson, C.D.; Lamantia, C.E.; Majewska, A. Proteolytic regulation of synaptic plasticity in the mouse primary visual cortex: Analysis of matrix metalloproteinase 9 deficient mice. Front. Cell. Neurosci. 2015, 9, 368. [Google Scholar] [CrossRef]
- Reinhard, S.M.; Razak, K.; Ethell, I.M. A delicate balance: Role of MMP-9 in brain development and pathophysiology of neurodevelopmental disorders. Front. Cell. Neurosci. 2015, 9, 280. [Google Scholar] [CrossRef]
- Murase, S.; Lantz, C.L.; Quinlan, E.M. Light Reintroduction after Dark Exposure Reactivates Plasticity in Adults via Peri-synaptic Activation of MMP-9. eLife 2017, 6, e27345. [Google Scholar] [CrossRef]
- Murase, S.; Winkowski, D.; Liu, J.; Kanold, P.O.; Quinlan, E.M. Homeostatic Regulation of Perisynaptic Matrix Metallopro-teinase 9 (MMP9) Activity in the Amblyopic Visual Cortex. eLife 2019, 8, e52503. [Google Scholar] [CrossRef]
- Kelly, E.A.; Tremblay, M.-È.; Gahmberg, C.G.; Tian, L.; Majewska, A. Subcellular localization of intercellular adhesion molecule-5 (telencephalin) in the visual cortex is not developmentally regulated in the absence of matrix metalloproteinase-9. J. Comp. Neurol. 2013, 522, 676–688. [Google Scholar] [CrossRef]
- Kelly, E.A.; Tremblay, M.-È.; Gahmberg, C.G.; Tian, L.; Majewska, A. Interactions between intercellular adhesion molecule-5 positive elements and their surroundings in the rodent visual cortex. Commun. Integr. Biol. 2013, 6, e27315. [Google Scholar] [CrossRef]
- Raemaekers, T.; Peric, A.; Baatsen, P.; Sannerud, R.; Declerck, I.; Baert, V.; Michiels, C.; Annaert, W. ARF6-Mediated Endo-somal Transport of Telencephalin Affects Dendritic Filopodia-to-Spine Maturation. EMBO J. 2012, 31, 3252–3269. [Google Scholar] [CrossRef]
- Choi, S.; Ko, J.; Lee, J.-R.; Lee, H.W.; Kim, K.; Chung, H.S.; Kim, H.; Kim, E. ARF6 and EFA6A Regulate the Development and Maintenance of Dendritic Spines. J. Neurosci. 2006, 26, 4811–4819. [Google Scholar] [CrossRef]
- Dean, C.; Dresbach, T. Neuroligins and neurexins: Linking cell adhesion, synapse formation and cognitive function. Trends Neurosci. 2006, 29, 21–29. [Google Scholar] [CrossRef]
- Südhof, T.C. Neuroligins and neurexins link synaptic function to cognitive disease. Nat. Cell Biol. 2008, 455, 903–911. [Google Scholar] [CrossRef] [PubMed]
- Song, J.-Y.; Ichtchenko, K.; Südhof, T.C.; Brose, N. Neuroligin 1 is a postsynaptic cell-adhesion molecule of excitatory synapses. Proc. Natl. Acad. Sci. USA 1999, 96, 1100–1105. [Google Scholar] [CrossRef]
- Chubykin, A.A.; Atasoy, D.; Etherton, M.R.; Brose, N.; Kavalali, E.T.; Gibson, J.R.; Sudhof, T.C. Activity-Dependent Validation of Excitatory versus Inhibitory Synapses by Neuroligin-1 versus Neuroligin-2. Neuron 2007, 54, 919–931. [Google Scholar] [CrossRef]
- Ichtchenko, K.; Hata, Y.; Nguyen, T.; Ullrich, B.; Missler, M.; Moomaw, C.; Sudhof, T.C. Neuroligin 1: A Splice Site-Specific Ligand for Beta-Neurexins. Cell 1995, 81, 435–443. [Google Scholar] [CrossRef]
- Nguyen, T.; Sudhof, T.C. Binding Properties of Neuroligin 1 and Neurexin 1beta Reveal Function as Heterophilic Cell Adhesion Molecules. J. Biol. Chem. 1997, 272, 26032–26039. [Google Scholar] [CrossRef]
- Irie, M.; Hata, Y.; Takeuchi, M.; Ichtchenko, K.; Toyoda, A.; Hirao, K.; Takai, Y.; Rosahl, T.W.; Sudhof, T.C. Binding of Neu-roligins to PSD-95. Science 1997, 277, 1511–1515. [Google Scholar] [CrossRef]
- Jeong, J.; Pandey, S.; Li, Y.; Badger, J.D.; Lu, W.; Roche, K.W. PSD-95 Binding Dynamically Regulates NLGN1 Trafficking and Function. Proc. Natl. Acad. Sci. USA 2019, 116, 12035–12044. [Google Scholar] [CrossRef]
- Varoqueaux, F.; Aramuni, G.; Rawson, R.L.; Mohrmann, R.; Missler, M.; Gottmann, K.; Zhang, W.; Südhof, T.C.; Brose, N. Neuroligins Determine Synapse Maturation and Function. Neuron 2006, 51, 741–754. [Google Scholar] [CrossRef]
- Krueger-Burg, D.; Tuffy, L.P.; Papadopoulos, T.; Brose, N. The role of neurexins and neuroligins in the formation, maturation, and function of vertebrate synapses. Curr. Opin. Neurobiol. 2012, 22, 412–422. [Google Scholar] [CrossRef]
- Jedlicka, P.; Muellerleile, J.; Schwarzacher, S.W. Synaptic Plasticity and Excitation-Inhibition Balance in the Dentate Gyrus: Insights fromIn VivoRecordings in Neuroligin-1, Neuroligin-2, and Collybistin Knockouts. Neural Plast. 2018, 2018, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Zeidan, A.; Ziv, N.E. Neuroligin-1 Loss Is Associated with Reduced Tenacity of Excitatory Synapses. PLoS ONE 2012, 7, e42314. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Jung, S.-Y.; Lee, Y.K.; Park, S.; Choi, J.-S.; Lee, C.J.; Kim, H.-S.; Choi, Y.-B.; Scheiffele, P.; Bailey, C.H.; et al. Neuroligin-1 is required for normal expression of LTP and associative fear memory in the amygdala of adult animals. Proc. Natl. Acad. Sci. USA 2008, 105, 9087–9092. [Google Scholar] [CrossRef] [PubMed]
- Jedlicka, P.; Vnencak, M.; Krueger, D.D.; Jungenitz, T.; Brose, N.; Schwarzacher, S.W. Neuroligin-1 regulates excitatory synaptic transmission, LTP and EPSP-spike coupling in the dentate gyrus in vivo. Brain Struct. Funct. 2015, 220, 47–58. [Google Scholar] [CrossRef]
- Fang, M.; Wei, J.-L.; Tang, B.; Liu, J.; Chen, L.; Tang, Z.-H.; Luo, J.; Chen, G.-J.; Wang, X.-F. Neuroligin-1 Knockdown Suppresses Seizure Activity by Regulating Neuronal Hyperexcitability. Mol. Neurobiol. 2016, 53, 270–284. [Google Scholar] [CrossRef]
- Millson, A.; Lagrave, D.; Willis, M.J.; Rowe, L.R.; Lyon, E.; South, S.T. Chromosomal loss of 3q26.3-3q26.32, involving a partial neuroligin 1 deletion, identified by genomic microarray in a child with microcephaly, seizure disorder, and severe intellectual disability. Am. J. Med. Genet. 2011, 158, 159–165. [Google Scholar] [CrossRef]
- Nakanishi, M.; Nomura, J.; Ji, X.; Tamada, K.; Arai, T.; Takahashi, E.; Bucan, M.; Takumi, T. Functional Significance of Rare Neuroligin 1 Variants Found in Autism. PLoS Genet. 2017, 13, e1006940. [Google Scholar] [CrossRef]
- Trobiani, L.; Meringolo, M.; Diamanti, T.; Bourne, Y.; Marchot, P.; Martella, G.; Dini, L.; Pisani, A.; De Jaco, A.; Bonsi, P. The neuroligins and the synaptic pathway in Autism Spectrum Disorder. Neurosci. Biobehav. Rev. 2020, 119, 37–51. [Google Scholar] [CrossRef]
- Gjorlund, M.D.; Carlsen, E.M.M.; Konig, A.B.; Dmytrieva, O.; Petersen, A.V.; Jacobsen, J.; Berezin, V.; Perrier, J.F.; Owczarek, S. Soluble Ectodomain of Neuroligin 1 Decreases Synaptic Activity by Activating Metabotropic Glutamate Receptor 2. Front. Mol. Neurosci. 2017, 10, 116. [Google Scholar] [CrossRef]
- Liu, A.; Zhou, Z.; Dang, R.; Zhu, Y.; Qi, J.; He, G.; Leung, C.; Pak, D.; Jia, Z.; Xie, W. Neuroligin 1 regulates spines and synaptic plasticity via LIMK1/cofilin-mediated actin reorganization. J. Cell Biol. 2016, 212, 449–463. [Google Scholar] [CrossRef]
- Tu, Y.K.; Duman, J.G.; Tolias, K.F. The Adhesion-GPCR BAI1 Promotes Excitatory Synaptogenesis by Coordinating Bidirec-tional Trans-Synaptic Signaling. J. Neurosci. 2018, 38, 8388–8406. [Google Scholar] [CrossRef] [PubMed]
- Paskus, J.D.; Tian, C.; Fingleton, E.; Shen, C.; Chen, X.; Li, Y.; Myers, S.A.; Badger, J.D.; Bemben, M.A.; Herring, B.E.; et al. Synaptic Kalirin-7 and Trio Interactomes Reveal a GEF Protein-Dependent Neuroligin-1 Mechanism of Action. Cell Rep. 2019, 29, 2944–2952. [Google Scholar] [CrossRef] [PubMed]
- Cabelli, R.J.; Hohn, A.; Shatz, C.J. Inhibition of Ocular Dominance Column Formation by Infusion of NT-4/5 or BDNF. Science 1995, 267, 1662–1666. [Google Scholar] [CrossRef] [PubMed]
- Lu, B. BDNF and Activity-Dependent Synaptic Modulation. Learn. Mem. 2003, 10, 86–98. [Google Scholar] [CrossRef]
- Poo, M.-M. Neurotrophins as synaptic modulators. Nat. Rev. Neurosci. 2001, 2, 24–32. [Google Scholar] [CrossRef]
- Kang, H.; Welcher, A.A.; Shelton, D.; Schuman, E.M. Neurotrophins and Time: Different Roles for TrkB Signaling in Hippo-campal Long-Term Potentiation. Neuron 1997, 19, 653–664. [Google Scholar] [CrossRef]
- Lohof, A.M.; Ip, N.Y.; Poo, M.-M. Potentiation of developing neuromuscular synapses by the neurotrophins NT-3 and BDNF. Nat. Cell Biol. 1993, 363, 350–353. [Google Scholar] [CrossRef]
- Fang, H.; Chartier, J.; Sodja, C.; Desbois, A.; Ribecco-Lutkiewicz, M.; Walker, P.R.; Sikorska, M. Transcriptional Activation of the Human Brain-derived Neurotrophic Factor Gene Promoter III by Dopamine Signaling in NT2/N Neurons. J. Biol. Chem. 2003, 278, 26401–26409. [Google Scholar] [CrossRef]
- Liu, Q.-R.; Walther, D.; Drgon, T.; Polesskaya, O.; Lesnick, T.G.; Strain, K.J.; De Andrade, M.; Bower, J.H.; Maraganore, D.M.; Uhl, G.R. Human brain derived neurotrophic factor (BDNF) genes, splicing patterns, and assessments of associations with substance abuse and Parkinson’s Disease. Am. J. Med. Genet. Neuropsychiatr. Genet. 2005, 134 B, 93–103. [Google Scholar] [CrossRef]
- Pruunsild, P.; Kazantseva, A.; Aid, T.; Palm, K.; Timmusk, T. Dissecting the human BDNF locus: Bidirectional transcription, complex splicing, and multiple promoters. Genomics 2007, 90, 397–406. [Google Scholar] [CrossRef]
- Aid, T.; Kazantseva, A.; Piirsoo, M.; Palm, K.; Timmusk, T. Mouse and ratBDNF gene structure and expression revisited. J. Neurosci. Res. 2007, 85, 525–535. [Google Scholar] [CrossRef]
- Timmusk, T.; Palm, K.; Metsis, M.; Reintam, T.; Paalme, V.; Saarma, M.; Persson, H. Multiple promoters direct tissue-specific expression of the rat BDNF gene. Neuron 1993, 10, 475–489. [Google Scholar] [CrossRef]
- Yang, J.; Siao, C.-J.; Nagappan, G.; Marinic, T.; Jing, D.; McGrath, K.; Chen, Z.-Y.; Mark, W.; Tessarollo, L.; Lee, F.S.; et al. Neuronal release of proBDNF. Nat. Neurosci. 2009, 12, 113–115. [Google Scholar] [CrossRef]
- Ernfors, P.; Ibanez, C.F.; Ebendal, T.; Olson, L.; Persson, H. Molecular cloning and neurotrophic activities of a protein with structural similarities to nerve growth factor: Developmental and topographical expression in the brain. Proc. Natl. Acad. Sci. USA 1990, 87, 5454–5458. [Google Scholar] [CrossRef]
- Katoh-Semba, R.; Takeuchi, I.K.; Semba, R.; Kato, K. Distribution of Brain-Derived Neurotrophic Factor in Rats and Its Changes with Development in the Brain. J. Neurochem. 2002, 69, 34–42. [Google Scholar] [CrossRef]
- Edelmann, E.; Lessmann, V.; Brigadski, T. Pre- and postsynaptic twists in BDNF secretion and action in synaptic plasticity. Neuropharmacology 2014, 76, 610–627. [Google Scholar] [CrossRef]
- Andreska, T.; Aufmkolk, S.; Sauer, M.; Blum, R. High abundance of BDNF within glutamatergic presynapses of cultured hippocampal neurons. Front. Cell. Neurosci. 2014, 8, 107. [Google Scholar] [CrossRef]
- Dieni, S.; Matsumoto, T.; Dekkers, M.; Rauskolb, S.; Ionescu, M.S.; Deogracias, R.; Gundelfinger, E.D.; Kojima, M.; Nestel, S.; Frotscher, M.; et al. BDNF and its pro-peptide are stored in presynaptic dense core vesicles in brain neurons. J. Cell Biol. 2012, 196, 775–788. [Google Scholar] [CrossRef]
- Brigadski, T.; Hartmann, M.; Lessmann, V. Differential Vesicular Targeting and Time Course of Synaptic Secretion of the Mammalian Neurotrophins. J. Neurosci. 2005, 25, 7601–7614. [Google Scholar] [CrossRef]
- Harward, S.C.; Hedrick, N.G.; Hall, C.E.; Parra-Bueno, P.; Milner, T.A.; Pan, E.; Laviv, T.; Hempstead, B.L.; Yasuda, R.; McNamara, J.O. Autocrine BDNF-TrkB Signalling within a Single Dendritic Spine. Nature 2016, 538, 99–103. [Google Scholar] [CrossRef]
- Coull, J.A.M.; Beggs, S.; Boudreau, D.; Boivin, D.; Tsuda, M.; Inoue, K.; Gravel, C.; Salter, M.W.; De Koninck, Y. BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nat. Cell Biol. 2005, 438, 1017–1021. [Google Scholar] [CrossRef] [PubMed]
- Su, C.; Cunningham, R.L.; Rybalchenko, N.; Singh, M. Progesterone Increases the Release of Brain-Derived Neurotrophic Factor from Glia via Progesterone Receptor Membrane Component 1 (Pgrmc1)-Dependent ERK5 Signaling. Endocrinology 2012, 153, 4389–4400. [Google Scholar] [CrossRef] [PubMed]
- Amooeian, V.G.; Rashidi, E. Dysfunction in Brain-Derived Neurotrophic Factor Signaling Pathway and Susceptibility to Schizophrenia, Parkinson’s and Alzheimer’s Diseases. Curr. Gene Ther. 2018, 18, 45–63. [Google Scholar] [CrossRef]
- Castrén, E.; Hen, R. Neuronal plasticity and antidepressant actions. Trends Neurosci. 2013, 36, 259–267. [Google Scholar] [CrossRef]
- Qin, X.Y.; Feng, J.C.; Cao, C.; Wu, H.T.; Loh, Y.P.; Cheng, Y. Association of Peripheral Blood Levels of Brain-Derived Neu-rotrophic Factor with Autism Spectrum Disorder in Children: A Systematic Review and Meta-Analysis. JAMA Pediatr. 2016, 170, 1079–1086. [Google Scholar] [CrossRef]
- Zheng, Z.; Zhang, L.; Zhu, T.; Huang, J.; Qu, Y.; Mu, D. Peripheral brain-derived neurotrophic factor in autism spectrum disorder: A systematic review and meta-analysis. Sci. Rep. 2016, 6, 31241. [Google Scholar] [CrossRef]
- Mowla, S.J.; Farhadi, H.F.; Pareek, S.; Atwal, J.K.; Morris, S.J.; Seidah, N.G.; Murphy, R.A. Biosynthesis and Post-translational Processing of the Precursor to Brain-derived Neurotrophic Factor. J. Biol. Chem. 2001, 276, 12660–12666. [Google Scholar] [CrossRef]
- Zhang, X.-Y.; Liu, F.; Chen, Y.; Guo, W.-C.; Zhang, Z.-H. Proprotein Convertase 1/3-Mediated down-Regulation of Brain-Derived Neurotrophic Factor in Cortical Neurons Induced by Oxygen-Glucose Deprivation. Neural Regen. Res. 2020, 15, 1066. [Google Scholar]
- Wetsel, W.C.; Rodriguiz, R.M.; Guillemot, J.; Rousselet, E.; Essalmani, R.; Kim, I.H.; Bryant, J.C.; Marcinkiewicz, J.; Desjardins, R.; Day, R.; et al. Disruption of the expression of the proprotein convertase PC7 reduces BDNF production and affects learning and memory in mice. Proc. Natl. Acad. Sci. USA 2013, 110, 17362–17367. [Google Scholar] [CrossRef]
- Lee, R.; Kermani, P.; Teng, K.K.; Hempstead, B.L. Regulation of Cell Survival by Secreted Proneurotrophins. Science 2001, 294, 1945–1948. [Google Scholar] [CrossRef]
- Pang, P.T.; Teng, H.K.; Zaitsev, E.; Woo, N.T.; Sakata, K.; Zhen, S.; Teng, K.K.; Yung, W.H.; Hempstead, B.L.; Lu, B. Cleavage of ProBDNF by TPA/Plasmin Is Essential for Long-Term Hippocampal Plasticity. Science 2004, 306, 487–491. [Google Scholar] [CrossRef] [PubMed]
- Nagappan, G.; Zaitsev, E.; Senatorov, V.V.; Yang, J.; Hempstead, B.L.; Lu, B. Control of extracellular cleavage of ProBDNF by high frequency neuronal activity. Proc. Natl. Acad. Sci. USA 2009, 106, 1267–1272. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Harte-Hargrove, L.C.; Siao, C.-J.; Marinic, T.; Clarke, R.; Ma, Q.; Jing, D.; Lafrancois, J.J.; Bath, K.G.; Mark, W.; et al. proBDNF Negatively Regulates Neuronal Remodeling, Synaptic Transmission, and Synaptic Plasticity in Hippocampus. Cell Rep. 2014, 7, 796–806. [Google Scholar] [CrossRef] [PubMed]
- Fleitas, C.; Piñol-Ripoll, G.; Marfull, P.; Rocandio, D.; Ferrer, I.; Rampon, C.; Egea, J.; Espinet, C. ProBDNF Is Modified by Advanced Glycation End Products in Alzheimer’s Disease and Causes Neuronal Apoptosis by Inducing P75 Neurotrophin Receptor Processing 11 Medical and Health Sciences 1109 Neurosciences. Mol. Brain 2018, 11, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Koshimizu, H.; Kiyosue, K.; Hara, T.; Hazama, S.; Suzuki, S.; Uegaki, K.; Nagappan, G.; Zaitsev, E.; Hirokawa, T.; Tatsu, Y.; et al. Multiple functions of precursor BDNF to CNS neurons: Negative regulation of neurite growth, spine formation and cell survival. Mol. Brain 2009, 2, 27. [Google Scholar] [CrossRef]
- Rösch, H.; Schweigreiter, R.; Bonhoeffer, T.; Barde, Y.-A.; Korte, M. The neurotrophin receptor p75NTR modulates long-term depression and regulates the expression of AMPA receptor subunits in the hippocampus. Proc. Natl. Acad. Sci. USA 2005, 102, 7362–7367. [Google Scholar] [CrossRef]
- Woo, N.H.; Teng, H.K.; Siao, C.-J.; Chiaruttini, C.; Pang, P.T.; Milner, T.A.; Hempstead, B.L.; Lu, B. Activation of p75NTR by proBDNF facilitates hippocampal long-term depression. Nat. Neurosci. 2005, 8, 1069–1077. [Google Scholar] [CrossRef]
- Korte, M.; Carroll, P.; Wolf, E.; Brem, G.; Thoenen, H.; Bonhoeffer, T. Hippocampal long-term potentiation is impaired in mice lacking brain-derived neurotrophic factor. Proc. Natl. Acad. Sci. USA 1995, 92, 8856–8860. [Google Scholar] [CrossRef]
- Mizoguchi, H.; Nakade, J.; Tachibana, M.; Ibi, D.; Someya, E.; Koike, H.; Kamei, H.; Nabeshima, T.; Itohara, S.; Takuma, K.; et al. Matrix Metalloproteinase-9 Contributes to Kindled Seizure Development in Pentylene-tetrazole-Treated Mice by Converting pro-BDNF to Mature BDNF in the Hippocampus. J. Neurosci. 2011, 31, 12963–12971. [Google Scholar] [CrossRef]
- Shen, W.; Wu, B.; Zhang, Z.; Dou, Y.; Rao, Z.-R.; Chen, Y.-R.; Duan, S. Activity-Induced Rapid Synaptic Maturation Mediated by Presynaptic Cdc42 Signaling. Neuron 2006, 50, 401–414. [Google Scholar] [CrossRef]
- Park, K.J.; Grosso, C.A.; Aubert, I.; Kaplan, D.R.; Miller, F.D. p75NTR-dependent, myelin-mediated axonal degeneration regulates neural connectivity in the adult brain. Nat. Neurosci. 2010, 13, 559–566. [Google Scholar] [CrossRef] [PubMed]
- Roux, P.P.; Colicos, M.A.; Barker, P.A.; Kennedy, T.E. p75 Neurotrophin Receptor Expression Is Induced in Apoptotic Neurons After Seizure. J. Neurosci. 1999, 19, 6887–6896. [Google Scholar] [CrossRef] [PubMed]
- Kokaia, Z.; Andsberg, G.; Martinez-Serrano, A.; Lindvall, O. Focal cerebral ischemia in rats induces expression of p75 neurotrophin receptor in resistant striatal cholinergic neurons. Neuroscience 1998, 84, 1113–1125. [Google Scholar] [CrossRef]
- Murakoshi, H.; Wang, H.; Yasuda, R. Local, persistent activation of Rho GTPases during plasticity of single dendritic spines. Nat. Cell Biol. 2011, 472, 100–104. [Google Scholar] [CrossRef] [PubMed]
- Hedrick, N.G.; Harward, S.C.; Hall, C.E.; Murakoshi, H.; McNamara, J.O.; Yasuda, R. Rho GTPase Complementation Underlies BDNF-Dependent Homo- and Heterosynaptic Plasticity. Nature 2016, 538, 104–108. [Google Scholar] [CrossRef]
- Finne, J.; Finne, U.; Deagostini-Bazin, H.; Goridis, C. Occurrence of α2–8 linked polysialosyl units in a neural cell adhesion molecule. Biochem. Biophys. Res. Commun. 1983, 112, 482–487. [Google Scholar] [CrossRef]
- Panicker, A.K.; Buhusi, M.; Thelen, K.; Maness, P.F. Cellular Signalling Mechanisms of Neural Cell Adhesion Molecules. Front. Biosci. 2003, 8, d900–d911. [Google Scholar]
- Becker, C.G.; Artola, A.; Gerardy-Schahn, R.; Becker, T.; Welzl, H.; Schachner, M. The Polysialic Acid Modification of the Neural Cell Adhesion Molecule Is Involved in Spatial Learning and Hippocampal Long-Term Potentiation. J. Neurosci. Res. 1996, 45, 143–152. [Google Scholar] [CrossRef]
- Senkov, O.; Sun, M.; Weinhold, B.; Gerardy-Schahn, R.; Schachner, M.; Dityatev, A. Polysialylated Neural Cell Adhesion Molecule Is Involved in Induction of Long-Term Potentiation and Memory Acquisition and Consolidation in a Fear-Conditioning Paradigm. J. Neurosci. 2006, 26, 10888–109898. [Google Scholar] [CrossRef]
- Stoenica, L.; Senkov, O.; Gerardy-Schahn, R.; Weinhold, B.; Schachner, M.; Dityatev, A. In Vivo Synaptic Plasticity in the Dentate Gyrus of Mice Deficient in the Neural Cell Adhesion Molecule NCAM or Its Polysialic Acid. Eur. J. Neurosci. 2006, 23, 2255–2264. [Google Scholar] [CrossRef]
- Suzuki, M.; Angata, K.; Nakayama, J.; Fukuda, M. Polysialic Acid and Mucin TypeO-Glycans on the Neural Cell Adhesion Molecule Differentially Regulate Myoblast Fusion. J. Biol. Chem. 2003, 278, 49459–49468. [Google Scholar] [CrossRef] [PubMed]
- Stork, O.; Welzl, H.; Wolfer, D.; Schuster, T.; Mantei, N.; Stork, S.; Hoyer, D.; Lipp, H.-P.; Obata, K.; Schachner, M. Recovery of emotional behaviour in neural cell adhesion molecule (NCAM) null mutant mice through transgenic expression of NCAM180. Eur. J. Neurosci. 2000, 12, 3291–3306. [Google Scholar] [CrossRef] [PubMed]
- Dityatev, A.; Dityateva, G.; Schachner, M. Synaptic Strength as a Function of Post- versus Presynaptic Expression of the Neural Cell Adhesion Molecule NCAM. Neuron 2000, 26, 207–217. [Google Scholar] [CrossRef]
- Persohn, E.; Schachner, M. Immunohistological Localization of the Neural Adhesion Molecules L1 and N-CAM in the Developing Hippocampus of the Mouse. J. Neurocytol. 1990, 19, 807–819. [Google Scholar] [CrossRef] [PubMed]
- Lüthi, A.; Laurent, J.-P.; Figurovt, A.; Mullert, D.; Schachnert, M. Hippocampal long-term potentiation and neural cell adhesion molecules L1 and NCAM. Nat. Cell Biol. 1994, 372, 777–779. [Google Scholar] [CrossRef]
- Todaro, L.; Puricelli, L.; Gioseffi, H.; Pallotta, M.G.; Lastiri, J.; Joffé, E.B.D.K.; Varela, M.; De Lustig, E.S. Neural cell adhesion molecule in human serum. Increased levels in dementia of the Alzheimer type. Neurobiol. Dis. 2004, 15, 387–393. [Google Scholar] [CrossRef]
- Santuccione, A.; Sytnyk, V.; Leshchyns’Ka, I.; Schachner, M. Prion protein recruits its neuronal receptor NCAM to lipid rafts to activate p59fyn and to enhance neurite outgrowth. J. Cell Biol. 2005, 169, 341–354. [Google Scholar] [CrossRef]
- Vawter, M.P. Dysregulation of the neural cell adhesion molecule and neuropsychiatric disorders. Eur. J. Pharmacol. 2000, 405, 385–395. [Google Scholar] [CrossRef]
- Vawter, M.P.; Usen, N.; Thatcher, L.; Ladenheim, B.; Zhang, P.; Vanderputten, D.M.; Conant, K.; Herman, M.M.; Van Kammen, D.P.; Sedvall, G.; et al. Characterization of Human Cleaved N-CAM and Association with Schizophrenia. Exp. Neurol. 2001, 172, 29–46. [Google Scholar] [CrossRef]
- Fujita-Hamabe, W.; Tokuyama, S. The involvement of cleavage of neural cell adhesion molecule in neuronal death under oxidative stress conditions in cultured cortical neurons. Biol. Pharm. Bull. 2012, 35, 624–628. [Google Scholar] [CrossRef]
- Shichi, K.; Fujita-Hamabe, W.; Harada, S.; Mizoguchi, H.; Yamada, K.; Nabeshima, T.; Tokuyama, S. Involvement of Matrix Metalloproteinase-Mediated Proteolysis of Neural Cell Adhesion Molecule in the Development of Cerebral Ischemic Neuronal Damage. J. Pharmacol. Exp. Ther. 2011, 338, 701–710. [Google Scholar] [CrossRef] [PubMed]
- Hübschmann, M.V.; Skladchikova, G.; Bock, E.; Berezin, V. Neural Cell Adhesion Molecule Function Is Regulated by Metal-loproteinase- Mediated Ectodomain Release. J. Neurosci. Res. 2005, 80, 826–837. [Google Scholar] [CrossRef] [PubMed]
- Fazeli, M.; Breen, K.C.; Errington, M.; Bliss, T. Increase in extracellular NCAM and amyloid precursor protein following induction of long-term potentiation in the dentate gyrus of anaesthetized rats. Neurosci. Lett. 1994, 169, 77–80. [Google Scholar] [CrossRef]
- Kim, J.S.; Kim, H.Y.; Kim, J.H.; Shin, H.K.; Lee, S.H.; Lee, Y.S.; Son, H. Enhancement of Rat Hippocampal Long-Term Poten-tiation by 17β-Estradiol Involves Mitogen-Activated Protein Kinase-Dependent and -Independent Components. Neurosci. Lett. 2002, 332, 65–69. [Google Scholar] [CrossRef]
- Büttner, B.; Kannicht, C.; Reutter, W.; Horstkorte, R. The neural cell adhesion molecule is associated with major components of the cytoskeleton. Biochem. Biophys. Res. Commun. 2003, 310, 967–971. [Google Scholar] [CrossRef]
- Sullivan, C.S.; Kümper, M.; Temple, B.S.; Maness, P.F. The Neural Cell Adhesion Molecule (NCAM) Promotes Clustering and Activation of EphA3 Receptors in GABAergic Interneurons to Induce Ras Homolog Gene Family, Member A (RhoA)/Rho-associated protein kinase (ROCK)-mediated Growth Cone Collapse. J. Biol. Chem. 2016, 291, 26262–26272. [Google Scholar] [CrossRef]
- Himanen, J.-P.; Nikolov, D.B. Eph receptors and ephrins. Int. J. Biochem. Cell Biol. 2003, 35, 130–134. [Google Scholar] [CrossRef]
- Takeuchi, S.; Katoh, H.; Negishi, M. Eph/ephrin reverse signalling induces axonal retraction through RhoA/ROCK pathway. J. Biochem. 2015, 158, 245–252. [Google Scholar] [CrossRef]
- Klein, R. Excitatory Eph receptors and adhesive ephrin ligands. Curr. Opin. Cell Biol. 2001, 13, 196–203. [Google Scholar] [CrossRef]
- Elowe, S.; Holland, S.J.; Kulkarni, S.; Pawson, T. Downregulation of the Ras–Mitogen-Activated Protein Kinase Pathway by the EphB2 Receptor Tyrosine Kinase Is Required for Ephrin-Induced Neurite Retraction. Mol. Cell. Biol. 2001, 21, 7429–7441. [Google Scholar] [CrossRef]
- Lin, K.-T.; Sloniowski, S.; Ethell, D.W.; Ethell, I.M. Ephrin-B2-induced Cleavage of EphB2 Receptor Is Mediated by Matrix Metalloproteinases to Trigger Cell Repulsion. J. Biol. Chem. 2008, 283, 28969–28979. [Google Scholar] [CrossRef] [PubMed]
- Moeller, M.L.; Shi, Y.; Reichardt, L.F.; Ethell, I.M. EphB Receptors Regulate Dendritic Spine Morphogenesis through the Re-cruitment/Phosphorylation of Focal Adhesion Kinase and RhoA Activation. J. Biol. Chem. 2006, 281, 1587–1598. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Pontrello, C.G.; DeFea, K.A.; Reichardt, L.F.; Ethell, I.M. Focal Adhesion Kinase Acts Downstream of EphB Receptors to Maintain Mature Dendritic Spines by Regulating Cofilin Activity. J. Neurosci. 2009, 29, 8129–8142. [Google Scholar] [CrossRef] [PubMed]
- Lauterbach, J.; Klein, R. Release of Full-Length EphB2 Receptors from Hippocampal Neurons to Cocultured Glial Cells. J. Neurosci. 2006, 26, 11575–11581. [Google Scholar] [CrossRef] [PubMed]
- Schaupp, A.; Sabet, O.; Dudanova, I.; Ponserre, M.; Bastiaens, P.; Klein, R. The composition of EphB2 clusters determines the strength in the cellular repulsion response. J. Cell Biol. 2014, 204, 409–422. [Google Scholar] [CrossRef]
- Zimmer, M.; Palmer, A.; Köhler, J.; Klein, R. EphB-EphrinB Bi-Directional Endocytosis Terminates Adhesion Allowing Contact Mediated Repulsion. Nat. Cell Biol. 2003, 5, 869–878. [Google Scholar] [CrossRef]
- Gaitanos, T.N.; Koerner, J.; Klein, R. Tiam-Rac Signaling Mediates Trans-Endocytosis of Ephrin Receptor EphB2 and Is Im-portant for Cell Repulsion. J. Cell Biol. 2016, 214, 735. [Google Scholar] [CrossRef]
- Penzes, P.; Beeser, A.; Chernoff, J.; Schiller, M.R.; Eipper, B.A.; Mains, R.E.; Huganir, R.L. Rapid Induction of Dendritic Spine Morphogenesis by Trans-Synaptic EphrinB-EphB Receptor Activation of the Rho-GEF Kalirin. Neuron 2003, 37, 263–274. [Google Scholar] [CrossRef]
- Locke, C.; Machida, K.; Tucker, C.L.; Wu, Y.; Yu, J. Optogenetic activation of EphB2 receptor in dendrites induced actin polymerization by activating Arg kinase. Biol. Open 2017, 6, 1820–1830. [Google Scholar] [CrossRef]
- Hussain, N.K.; Thomas, G.M.; Luo, J.; Huganir, R.L. Regulation of AMPA receptor subunit GluA1 surface expression by PAK3 phosphorylation. Proc. Natl. Acad. Sci. USA 2015, 112, E5883–E5890. [Google Scholar] [CrossRef]
- Irie, F.; Yamaguchi, Y. EphB receptors regulate dendritic spine development via intersectin, Cdc42 and N-WASP. Nat. Neurosci. 2002, 5, 1117–1118. [Google Scholar] [CrossRef] [PubMed]
- Brambilla, R.; Gnesutta, N.; Minichiello, L.; White, G.; Roylance, A.J.; Herron, C.E.; Ramsey, M.; Wolfer, D.P.; Cestari, V.; Rossi-Arnaud, C.; et al. A role for the Ras signalling pathway in synaptic transmission and long-term memory. Nat. Cell Biol. 1997, 390, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Heumann, R.; Goemans, C.; Bartsch, D.; Lingenhöhl, K.; Waldmeier, P.C.; Hengerer, B.; Allegrini, P.R.; Schellander, K.; Wagner, E.F.; Arendt, T.; et al. Transgenic Activation of Ras in Neurons Promotes Hypertrophy and Protects from Lesion-Induced Degeneration. J. Cell Biol. 2000, 151, 1537–1548. [Google Scholar] [CrossRef] [PubMed]
- Tong, J.; Elowe, S.; Nash, P.; Pawson, T. Manipulation of EphB2 Regulatory Motifs and SH2 Binding Sites Switches MAPK Signaling and Biological Activity. J. Biol. Chem. 2003, 278, 6111–6119. [Google Scholar] [CrossRef] [PubMed]
- McClelland, A.C.; Hruska, M.; Coenen, A.J.; Henkemeyer, M.; Dalva, M.B. Trans-synaptic EphB2-ephrin-B3 interaction regulates excitatory synapse density by inhibition of postsynaptic MAPK signaling. Proc. Natl. Acad. Sci. USA 2010, 107, 8830–8835. [Google Scholar] [CrossRef]
- Dail, M.; Richter, M.; Godement, P.; Pasquale, E.B. Eph receptors inactivate R-Ras through different mechanisms to achieve cell repulsion. J. Cell Sci. 2006, 119, 1244–1254. [Google Scholar] [CrossRef][Green Version]
- Stenmark, P.; Ogg, D.; Flodin, S.; Flores, A.; Kotenyova, T.; Nyman, T.; Nordlund, P.; Kursula, P. The structure of human collapsin response mediator protein 2, a regulator of axonal growth. J. Neurochem. 2006, 101, 906–917. [Google Scholar] [CrossRef]
- Ricard, D.; Stankoff, B.; Bagnard, D.; Aguera, M.; Rogemond, V.; Antoine, J.; Spassky, N.; Zalc, B.; Lubetzki, C.; Belin, M.; et al. Differential Expression of Collapsin Response Mediator Proteins (CRMP/ULIP) in Subsets of Oligodendrocytes in the Postnatal Rodent Brain. Mol. Cell. Neurosci. 2000, 16, 324–337. [Google Scholar] [CrossRef]
- Wang, L.-H.; Strittmatter, S.M. A Family of Rat CRMP Genes Is Differentially Expressed in the Nervous System. J. Neurosci. 1996, 16, 6197–6207. [Google Scholar] [CrossRef]
- Arimura, N.; Ménager, C.; Fukata, Y.; Kaibuchi, K. Role of CRMP-2 in neuronal polarity. J. Neurobiol. 2003, 58, 34–47. [Google Scholar] [CrossRef]
- Inagaki, N.; Chihara, K.; Arimura, N.; Ménager, C.; Kawano, Y.; Matsuo, N.; Nishimura, T.; Amano, M.; Kaibuchi, K. CRMP-2 induces axons in cultured hippocampal neurons. Nat. Neurosci. 2001, 4, 781–782. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.P.; Strittmatter, S.M. Semaphorin-mediated axonal guidance via Rho-related G proteins. Curr. Opin. Cell Biol. 2001, 13, 619–626. [Google Scholar] [CrossRef]
- Niwa, S.; Nakamura, F.; Tomabechi, Y.; Aoki, M.; Shigematsu, H.; Matsumoto, T.; Yamagata, A.; Fukai, S.; Hirokawa, N.; Goshima, Y.; et al. Structural basis for CRMP2-induced axonal microtubule formation. Sci. Rep. 2017, 7, 10681. [Google Scholar] [CrossRef] [PubMed]
- Arimura, N.; Inagaki, N.; Chihara, K.; Ménager, C.; Nakamura, N.; Amano, M.; Iwamatsu, A.; Goshima, Y.; Kaibuchi, K. Phosphorylation of Collapsin Response Mediator Protein-2 by Rho-Kinase: Evidence for Two Separate Signaling Pathways for Growth Cone Collapse. J. Biol. Chem. 2000, 275, 23973–23980. [Google Scholar] [CrossRef] [PubMed]
- Hall, C.; Brown, M.; Jacobs, T.; Ferrari, G.; Cann, N.; Teo, M.; Monfries, C.; Lim, L. Collapsin Response Mediator Protein Switches RhoA and Rac1 Morphology in N1E-115 Neuroblastoma Cells and Is Regulated by Rho Kinase. J. Biol. Chem. 2001, 276, 43482–43486. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, T.; Arimura, N.; Kawano, Y.; Kawabata, S.; Wang, S.; Kaibuchi, K. Ras regulates neuronal polarity via the PI3-kinase/Akt/GSK-3β/CRMP-2 pathway. Biochem. Biophys. Res. Commun. 2006, 340, 62–68. [Google Scholar] [CrossRef]
- Mimura, F.; Yamagishi, S.; Arimura, N.; Fujitani, M.; Kubo, T.; Kaibuchi, K.; Yamashita, T. Myelin-associated Glycoprotein Inhibits Microtubule Assembly by a Rho-kinase-dependent Mechanism. J. Biol. Chem. 2006, 281, 15970–15979. [Google Scholar] [CrossRef]
- Nan, L.; Yang, L.; Zheng, Y.; He, Y.; Xie, Q.; Chen, Z.; Li, H.; Huang, M. Effects of Gualou Guizhi Decoction Aqueous Extract on Axonal Regeneration in Organotypic Cortical Slice Culture after Oxygen-Glucose Deprivation. Evid. Based Complement. Altern. Med. 2017, 2017, 1–11. [Google Scholar] [CrossRef]
- Petratos, S.; Li, Q.-X.; George, A.J.; Hou, X.; Kerr, M.L.; Unabia, S.E.; Hatzinisiriou, I.; Maksel, D.; Aguilar, M.-I.; Small, D.H. The β-amyloid protein of Alzheimer’s disease increases neuronal CRMP-2 phosphorylation by a Rho-GTP mechanism. Brain 2007, 131, 90–108. [Google Scholar] [CrossRef]
- Pilotte, J.; Kiosses, W.; Chan, S.W.; Makarenkova, H.P.; Dupont-Versteegden, E.; Vanderklish, P.W. Morphoregulatory functions of the RNA-binding motif protein 3 in cell spreading, polarity and migration. Sci. Rep. 2018, 8, 1–19. [Google Scholar] [CrossRef]
- Quarta, S.; Camprubí-Robles, M.; Schweigreiter, R.; Matusica, D.; Haberberger, R.V.; Proia, R.L.; Bandtlow, C.E.; Ferrer-Montiel, A.; Kress, M. Sphingosine-1-Phosphate and the S1P3 Receptor Initiate Neuronal Retraction via RhoA/ROCK Associated with CRMP2 Phosphorylation. Front. Mol. Neurosci. 2017, 10, 317. [Google Scholar] [CrossRef] [PubMed]
- Rozés Salvador, V.; Heredia, F.; Berardo, A.; Palandri, A.; Wojnacki, J.; Vivinetto, A.L.; Sheikh, K.A.; Caceres, A.; Lopez, P.H.H. Anti-Glycan Antibodies Halt Axon Regeneration in a Model of Guillain Barrè Syndrome Axonal Neuropathy by Inducing Microtubule Disorganization via RhoA-ROCK-Dependent Inactivation of CRMP-2. Exp. Neurol. 2016, 278, 42–53. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.-H.; Sun, Z.-L.; Li, H.-J.; Feng, D.-F. Rho GTPase-activating proteins: Regulators of Rho GTPase activity in neuronal development and CNS diseases. Mol. Cell. Neurosci. 2017, 80, 18–31. [Google Scholar] [CrossRef] [PubMed]
- Ahearn, I.M.; Haigis, K.M.; Bar-Sagi, D.; Philips, M.R. Regulating the regulator: Post-translational modification of RAS. Nat. Rev. Mol. Cell Biol. 2011, 13, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Roberts, P.J.; Mitin, N.; Keller, P.J.; Chenette, E.J.; Madigan, J.P.; Currin, R.O.; Cox, A.D.; Wilson, O.; Kirschmeier, P.; Der, C.J. Rho Family GTPase Modification and Dependence on CAAX Motif-Signaled Posttranslational Modification. J. Biol. Chem. 2008, 283, 25150–25163. [Google Scholar] [CrossRef] [PubMed]
- Mitin, N.; Roberts, P.J.; Chenette, E.J.; Der, C.J. Posttranslational Lipid Modification of Rho Family Small GTPases. Adv. Struct. Saf. Stud. 2011, 827, 87–95. [Google Scholar] [CrossRef]
- Lane, K.T.; Beese, L.S. Thematic Review Series: Lipid Posttranslational Modifications. Structural Biology of Protein Farnesyl-transferase and Geranylgeranyltransferase Type I. J. Lipid Res. 2006, 47, 681–699. [Google Scholar] [CrossRef]
- Wang, M.; Casey, P.J. Protein prenylation: Unique fats make their mark on biology. Nat. Rev. Mol. Cell Biol. 2016, 17, 110–122. [Google Scholar] [CrossRef]
- Abdrabou, A.; Wang, Z. Post-Translational Modification and Subcellular Distribution of Rac1: An Update. Cells 2018, 7, 263. [Google Scholar] [CrossRef]
- Samuel, F.; Hynds, D.L. RHO GTPase Signaling for Axon Extension: Is Prenylation Important? Mol. Neurobiol. 2010, 42, 133–142. [Google Scholar] [CrossRef]
- Spillane, M.; Gallo, G. Involvement of Rho-family GTPases in axon branching. Small GTPases 2014, 5, e27974. [Google Scholar] [CrossRef] [PubMed]
- Reddy, J.M.; Samuel, F.G.; McConnell, J.A.; Reddy, C.P.; Beck, B.W.; Hynds, D.L. Non-prenylatable, cytosolic Rac1 alters neurite outgrowth while retaining the ability to be activated. Cell. Signal. 2015, 27, 630–637. [Google Scholar] [CrossRef] [PubMed]
- Berzat, A.C.; Brady, N.C.; Fiordalisi, J.J.; Cox, A.D. Using Inhibitors of Prenylation to Block Localization and Transforming Activity. Methods Enzymol. 2006, 407, 575–597. [Google Scholar] [CrossRef] [PubMed]
- Tricarico, P.M.; Romeo, A.; Gratton, R.; Crovella, S.; Celsi, F. Lack of Prenylated Proteins, Autophagy Impairment and Apoptosis in SH-SY5Y Neuronal Cell Model of Mevalonate Kinase Deficiency. Cell. Physiol. Biochem. 2017, 41, 1649–1660. [Google Scholar] [CrossRef]
- Shimizu, H.; Toma-Fukai, S.; Kontani, K.; Katada, T.; Shimizu, T. GEF mechanism revealed by the structure of SmgGDS-558 and farnesylated RhoA complex and its implication for a chaperone mechanism. Proc. Natl. Acad. Sci. USA 2018, 115, 9563–9568. [Google Scholar] [CrossRef]
- Akula, M.K.; Ibrahim, M.X.; Ivarsson, E.G.; Khan, O.M.; Kumar, I.T.; Erlandsson, M.; Karlsson, C.; Xu, X.; Brisslert, M.; Brakebusch, C.; et al. Protein prenylation restrains innate immunity by inhibiting Rac1 effector interactions. Nat. Commun. 2019, 10, 1–13. [Google Scholar] [CrossRef]
- Aslam, M.; Troidl, C.; Tanislav, C.; Rohrbach, S.; Gündüz, D.; Hamm, C.W. Inhibition of Protein Prenylation of GTPases Alters Endothelial Barrier Function. Int. J. Mol. Sci. 2019, 21, 2. [Google Scholar] [CrossRef]
- Kalpachidou, T.; Spiecker, L.; Kress, M.; Quarta, S. Rho GTPases in the Physiology and Pathophysiology of Peripheral Sensory Neurons. Cells 2019, 8, 591. [Google Scholar] [CrossRef]
- Xu, Z.; Chen, Y. Spatiotemporal Regulation of Rho GTPases in Neuronal Migration. Cells 2019, 8, 568. [Google Scholar] [CrossRef]
- Zmurchok, C.; Holmes, W.R. Simple Rho GTPase Dynamics Generate a Complex Regulatory Landscape Associated with Cell Shape. Biophys. J. 2020, 118, 1438–1454. [Google Scholar] [CrossRef]
- Zaręba-Kozioł, M.; Figiel, I.; Bartkowiak-Kaczmarek, A.; Włodarczyk, J. Insights Into Protein. Front. Mol. Neurosci. 2018, 11, 175. [Google Scholar] [CrossRef] [PubMed]
- Navarro-Lérida, I.; Sánchez-Perales, S.; Calvo, M.; Rentero, C.; Zheng, Y.; Enrich, C.; Del Pozo, M.A. A palmitoylation switch mechanism regulates Rac1 function and membrane organization. EMBO J. 2011, 31, 534–551. [Google Scholar] [CrossRef] [PubMed]
- Kang, R.; Wan, J.; Arstikaitis, P.; Takahashi, H.; Huang, K.; Bailey, A.O.; Thompson, J.X.; Roth, A.F.; Drisdel, R.C.; Mastro, R.; et al. Neural palmitoyl-proteomics reveals dynamic synaptic palmitoylation. Nat. Cell Biol. 2008, 456, 904–909. [Google Scholar] [CrossRef] [PubMed]
- Wirth, A.; Chen-Wacker, C.; Wu, Y.-W.; Gorinski, N.; Filippov, M.A.; Pandey, G.; Ponimaskin, E. Dual lipidation of the brain-specific Cdc42 isoform regulates its functional properties. Biochem. J. 2013, 456, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Albanesi, J.P.; Barylko, B.; DeMartino, G.N.; Jameson, D.M. Palmitoylated Proteins in Dendritic Spine Remodeling. Front. Synaptic Neurosci. 2020, 12, 22. [Google Scholar] [CrossRef] [PubMed]
- Moutin, E.; Nikonenko, I.; Stefanelli, T.; Wirth, A.; Ponimaskin, E.; De Roo, M.; Muller, D. Palmitoylation of cdc42 Promotes Spine Stabilization and Rescues Spine Density Deficit in a Mouse Model of 22q11.2 Deletion Syndrome. Cereb. Cortex 2016, 27, 3618–3629. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, J.S.; Drake, K.R.; Rogers, C.; Wright, L.; Lippincott-Schwartz, J.; Philips, M.R.; Kenworthy, A.K. Depalmitoylated Ras traffics to and from the Golgi complex via a nonvesicular pathway. J. Cell Biol. 2005, 170, 261–272. [Google Scholar] [CrossRef]
- Rocks, O.; Peyker, A.; Kahms, M.; Verveer, P.J.; Koerner, C.; Lumbierres, M.; Kuhlmann, J.; Waldmann, H.; Wittinghofer, A.; Bastiaens, P.I.H. An Acylation Cycle Regulates Localization and Activity of Palmitoylated Ras Isoforms. Science 2005, 307, 1746–1752. [Google Scholar] [CrossRef]
- Roy, S.; Plowman, S.; Rotblat, B.; Prior, I.A.; Muncke, C.; Grainger, S.; Parton, R.G.; Henis, Y.I.; Kloog, Y.; Hancock, J.F. Indi-vidual Palmitoyl Residues Serve Distinct Roles in H-Ras Trafficking, Microlocalization, and Signaling. Mol. Cell Biol. 2005, 25, 6722–6733. [Google Scholar] [CrossRef]
- Laude, A.J.; Prior, I.A. Palmitoylation and localisation of RAS isoforms are modulated by the hypervariable linker domain. J. Cell Sci. 2008, 121, 421–427. [Google Scholar] [CrossRef]
- Rocks, O.; Gerauer, M.; Vartak, N.; Koch, S.; Huang, Z.-P.; Pechlivanis, M.; Kuhlmann, J.; Brunsveld, L.; Chandra, A.; Ellinger, B.; et al. The Palmitoylation Machinery Is a Spatially Organizing System for Peripheral Membrane Proteins. Cell 2010, 141, 458–471. [Google Scholar] [CrossRef] [PubMed]
- Silvius, J.R.; Bhagatji, P.; Leventis, R.; Terrone, D. K-Ras4B and Prenylated Proteins Lacking “Second Signals” Associate Dy-namically with Cellular Membranes. Mol. Biol Cell. 2006, 17, 192–202. [Google Scholar] [CrossRef] [PubMed]
- Prior, I.A.; Harding, A.; Yan, J.; Sluimer, J.C.; Parton, R.G.; Hancock, J.F. GTP-dependent segregation of H-ras from lipid rafts is required for biological activity. Nat. Cell Biol. 2001, 3, 368–375. [Google Scholar] [CrossRef] [PubMed]
- Woolfrey, K.M.; Srivastava, D.P. Control of Dendritic Spine Morphological and Functional Plasticity by Small GTPases. Neural Plast. 2016, 2016, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Forget, M.A.; Desrosiers, R.R.; Gingras, D.; Béliveau, R. Phosphorylation States of Cdc42 and RhoA Regulate Their Interactions with Rho GDP Dissociation Inhibitor and Their Extraction from Biological Membranes. Biochem. J. 2002, 361, 243–254. [Google Scholar] [CrossRef]
- Chang, F.; Lemmon, C.A.; Lietha, D.; Eck, M.; Romer, L.H. Tyrosine Phosphorylation of Rac1: A Role in Regulation of Cell Spreading. PLoS ONE 2011, 6, e28587. [Google Scholar] [CrossRef]
- Tu, X.; Yasuda, R.; Colgan, L.A. Rac1 is a downstream effector of PKCα in structural synaptic plasticity. Sci. Rep. 2020, 10, 1–9. [Google Scholar] [CrossRef]
- Álvarez-Moya, B.; López-Alcalá, C.; Drosten, M.; Bachs, O.; Agell, N. K-Ras4B phosphorylation at Ser181 is inhibited by calmodulin and modulates K-Ras activity and function. Oncogene 2010, 29, 5911–5922. [Google Scholar] [CrossRef]
- Mabb, A.M.; Ehlers, M.D. Ubiquitination in Postsynaptic Function and Plasticity. Annu. Rev. Cell Dev. Biol. 2010, 26, 179–210. [Google Scholar] [CrossRef]
- Jura, N.; Scotto-Lavino, E.; Sobczyk, A.; Bar-Sagi, D. Differential Modification of Ras Proteins by Ubiquitination. Mol. Cell 2006, 21, 679–687. [Google Scholar] [CrossRef]
- Bryan, B.; Cai, Y.; Wrighton, K.; Wu, G.; Feng, X.-H.; Liu, M. Ubiquitination of RhoA by Smurf1 promotes neurite outgrowth. FEBS Lett. 2005, 579, 1015–1019. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).