Retrotransposon-Based Genetic Diversity Assessment in Wild Emmer Wheat (Triticum turgidum ssp. dicoccoides)


 , ,
, , 

Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Material and DNA Extraction
2.2. TE Sequence Sources and PCR Primer Design
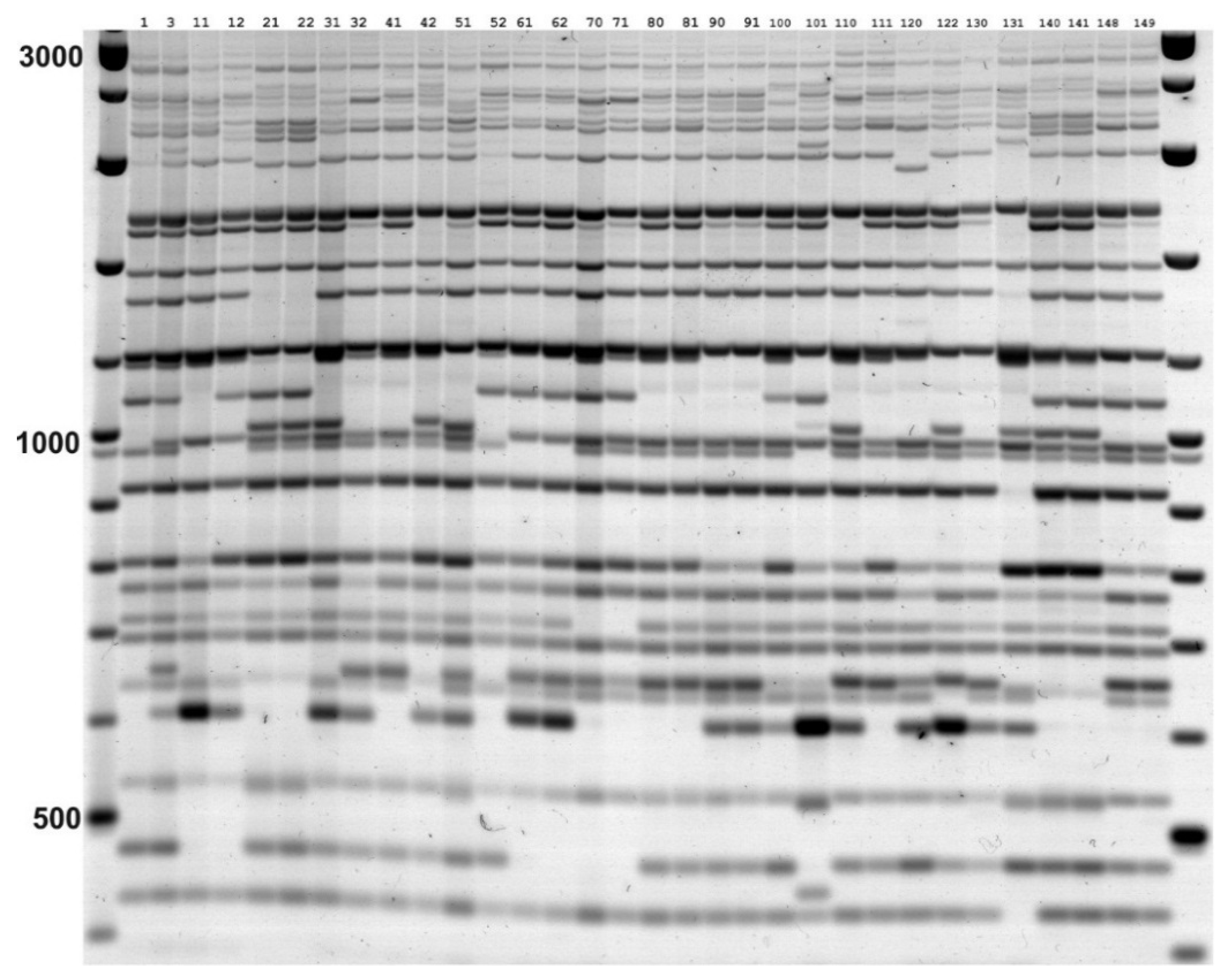
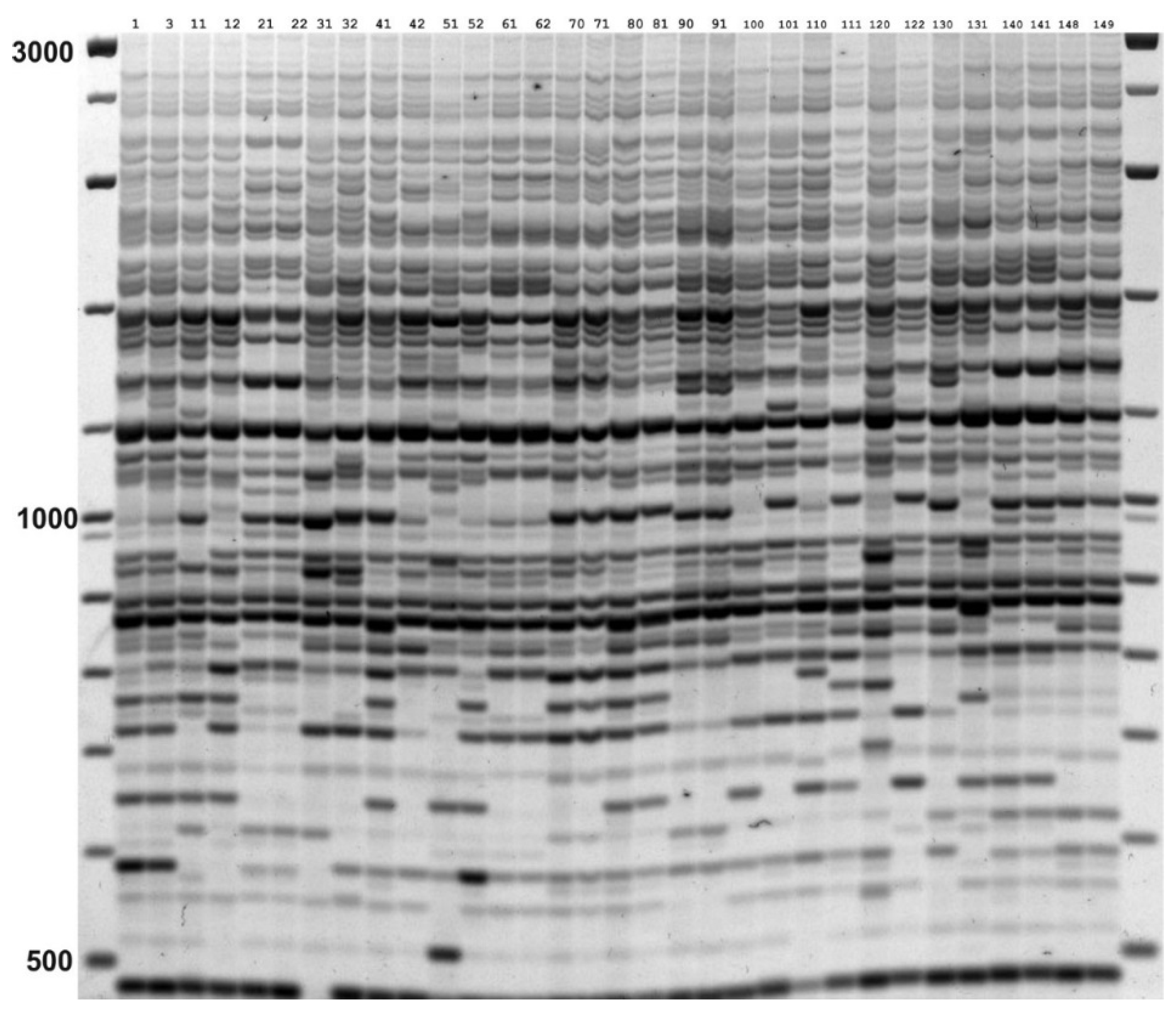
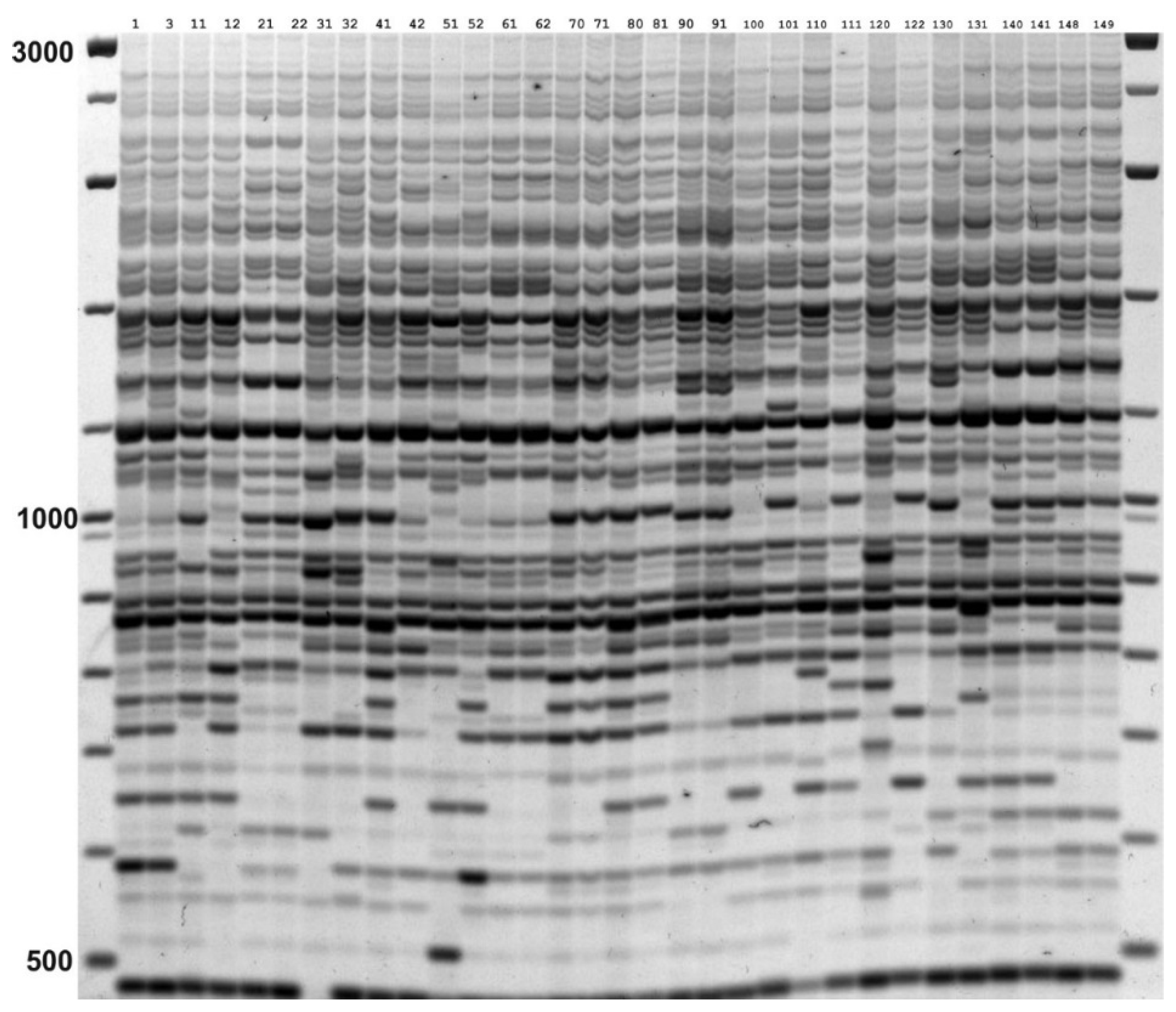
2.3. IRAP and REMAP Analysis
2.4. Data Analysis
3. Results
3.1. Genetic Diversity
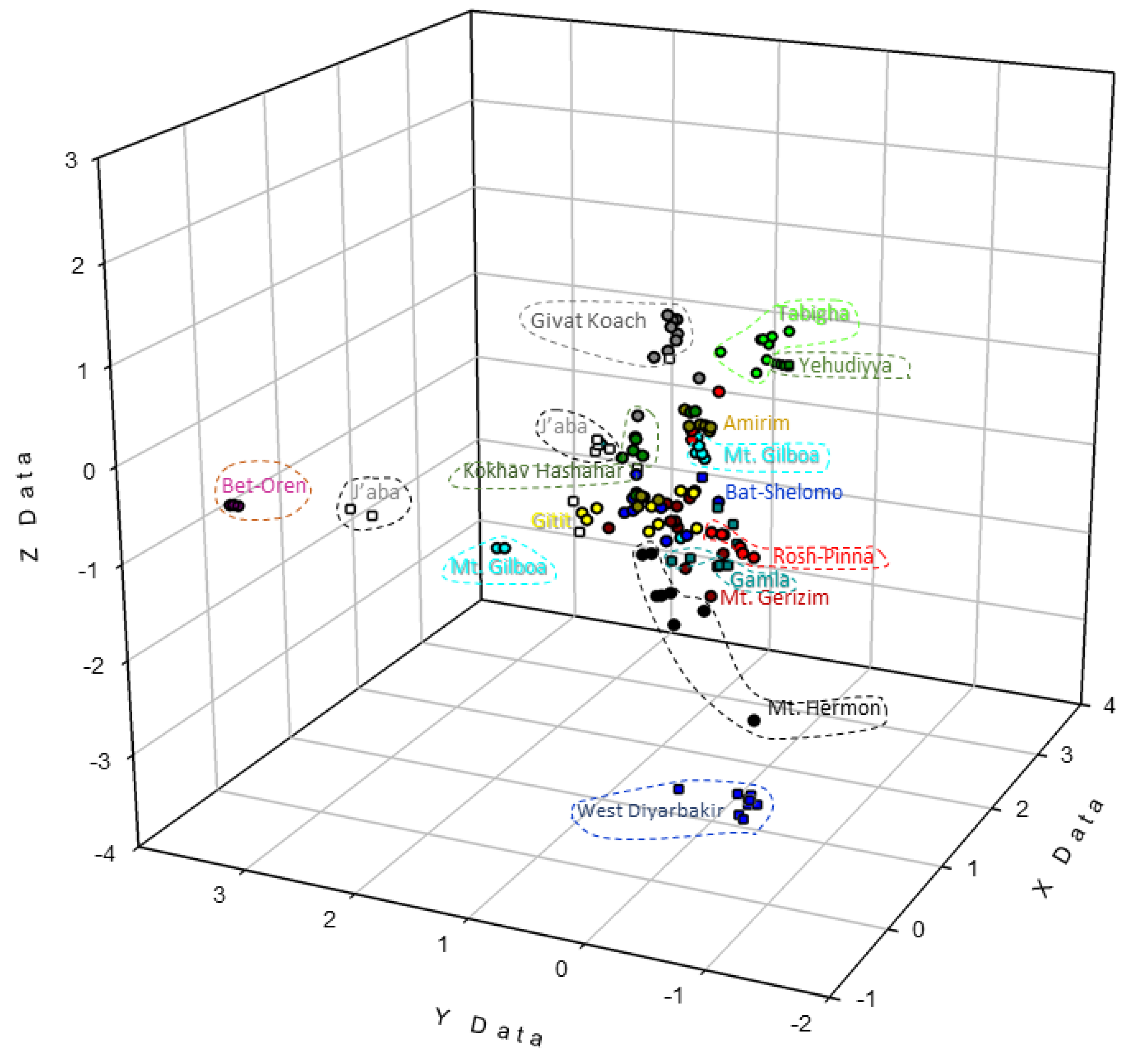
3.2. Genetic Distance
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Brunazzi, A.; Scaglione, D.; Talini, R.F.; Miculan, M.; Magni, F.; Poland, J.; Enrico Pè, M.; Brandolini, A.; Dell’Acqua, M. Molecular diversity and landscape genomics of the crop wild relative Triticum urartu across the Fertile Crescent. Plant J. 2018, 94, 670–684. [Google Scholar] [CrossRef] [PubMed]
- Merchuk-Ovnat, L.; Fahima, T.; Krugman, T.; Saranga, Y. Ancestral QTL alleles from wild emmer wheat improve grain yield, biomass and photosynthesis across environments in modern wheat. Plant Sci. 2016, 251, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Sela, H.; Loutre, C.; Keller, B.; Schulman, A.; Nevo, E.; Korol, A.; Fahima, T. Rapid linkage disequilibrium decay in the Lr10 gene in wild emmer wheat (Triticum dicoccoides) populations. Theor. Appl. Genet. 2011, 122, 175–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akhunov, E.D.; Akhunova, A.R.; Anderson, O.D.; Anderson, J.A.; Blake, N.; Clegg, M.T.; Coleman-Derr, D.; Conley, E.J.; Crossman, C.C.; Deal, K.R.; et al. Nucleotide diversity maps reveal variation in diversity among wheat genomes and chromosomes. BMC Genom. 2010, 11, 702. [Google Scholar] [CrossRef] [PubMed]
- Avni, R.; Nave, M.; Barad, O.; Baruch, K.; Twardziok, S.O.; Gundlach, H.; Hale, I.; Mascher, M.; Spannagl, M.; Wiebe, K.; et al. Wild emmer genome architecture and diversity elucidate wheat evolution and domestication. Science 2017, 357, 93–97. [Google Scholar] [CrossRef] [PubMed]
- Volis, S.; Ormanbekova, D.; Yermekbayev, K.; Song, M.; Shulgina, I. Multi-approaches analysis reveals local adaptation in the emmer wheat (Triticum dicoccoides) at macro- but not micro-geographical scale. PLoS ONE 2015, 10, e0121153. [Google Scholar] [CrossRef] [PubMed]
- Fahima, T.; Roder, M.S.; Wendehake, K.; Kirzhner, V.M.; Nevo, E. Microsatellite polymorphism in natural populations of wild emmer wheat, Triticum dicoccoides, in Israel. Theor. Appl. Genet. 2002, 104, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Wicker, T.; Sabot, F.; Hua-Van, A.; Bennetzen, J.L.; Capy, P.; Chalhoub, B.; Flavell, A.; Leroy, P.; Morgante, M.; Panaud, O.; et al. A unified classification system for eukaryotic transposable elements. Nat. Rev. Genet. 2007, 8, 973–982. [Google Scholar] [CrossRef] [PubMed]
- Schulman, A.H. Retrotransposon replication in plants. Curr. Opin. Virol. 2013, 3, 604–614. [Google Scholar] [CrossRef] [PubMed]
- Kalendar, R.; Schulman, A.H. Transposon-based tagging: IRAP, REMAP, and iPBS. Methods Mol. Biol. 2014, 1115, 233–255. [Google Scholar] [PubMed]
- Kalendar, R.; Schulman, A.H. IRAP and REMAP for retrotransposon-based genotyping and fingerprinting. Nat. Protoc. 2006, 1, 2478–2484. [Google Scholar] [CrossRef] [PubMed]
- Boyko, E.; Kalendar, R.; Korzun, V.; Fellers, J.; Korol, A.; Schulman, A.H.; Gill, B.S. A high-density cytogenetic map of the Aegilops tauschii genome incorporating retrotransposons and defense-related genes: Insights into cereal chromosome structure and function. Plant Mol. Biol. 2002, 48, 767–789. [Google Scholar] [CrossRef] [PubMed]
- Galindo-Gonzalez, L.; Mhiri, C.; Deyholos, M.K.; Grandbastien, M.A. LTR-retrotransposons in plants: Engines of evolution. Gene 2017, 626, 14–25. [Google Scholar] [CrossRef] [PubMed]
- Kalendar, R.; Flavell, A.J.; Ellis, T.H.; Sjakste, T.; Moisy, C.; Schulman, A.H. Analysis of plant diversity with retrotransposon-based molecular markers. Heredity 2011, 106, 520–530. [Google Scholar] [CrossRef] [PubMed]
- Kalendar, R.N.; Aizharkyn, K.S.; Khapilina, O.N.; Amenov, A.A.; Tagimanova, D.S. Plant diversity and transcriptional variability assessed by retrotransposon-based molecular markers. Vavilov J. Genet. Breed. 2017, 21, 128–134. [Google Scholar] [CrossRef] [Green Version]
- Mandoulakani, B.A.; Yaniv, E.; Kalendar, R.; Raats, D.; Bariana, H.S.; Bihamta, M.R.; Schulman, A.H. Development of IRAP- and REMAP-derived SCAR markers for marker-assisted selection of the stripe rust resistance gene Yr15 derived from wild emmer wheat. Theor. Appl. Genet. 2015, 128, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Grandbastien, M.A.; Audeon, C.; Bonnivard, E.; Casacuberta, J.M.; Chalhoub, B.; Costa, A.P.; Le, Q.H.; Melayah, D.; Petit, M.; Poncet, C.; et al. Stress activation and genomic impact of Tnt1 retrotransposons in Solanaceae. Cytogenet. Genome Res. 2005, 110, 229–241. [Google Scholar] [CrossRef] [PubMed]
- Jaaskelainen, M.; Chang, W.; Moisy, C.; Schulman, A.H. Retrotransposon BARE displays strong tissue-specific differences in expression. New Phytol. 2013, 200, 1000–1008. [Google Scholar] [CrossRef] [PubMed]
- Kalendar, R.; Tanskanen, J.; Immonen, S.; Nevo, E.; Schulman, A. Genome evolution of wild barley (Hordeum spontaneum) by BARE-1 retrotransposon dynamics in response to sharp microclimatic divergence. Proc. Natl. Acad. Sci. USA 2000, 97, 6603–6607. [Google Scholar] [CrossRef] [PubMed]
- Hosid, E.; Brodsky, L.; Kalendar, R.; Raskina, O.; Belyayev, A. Diversity of long terminal repeat retrotransposon genome distribution in natural populations of the wild diploid wheat Aegilops speltoides. Genetics 2012, 190, U263–U412. [Google Scholar] [CrossRef] [PubMed]
- Belyayev, A.; Kalendar, R.; Brodsky, L.; Nevo, E.; Schulman, A.H.; Raskina, O. Transposable elements in a marginal plant population: Temporal fluctuations provide new insights into genome evolution of wild diploid wheat. Mob. DNA 2010, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nevo, E.; Beiles, A. Genetic diversity of wild emmer wheat in Israel and Turkey : Structure, evolution, and application in breeding. Theor. Appl. Genet. 1989, 77, 421–455. [Google Scholar] [CrossRef] [PubMed]
- Nevo, E.; Beiles, A. Genetic Parallelism of Protein Polymorphism in Nature—Ecological test of the neutral theory of molecular evolution. Biol. J. Linn. Soc. 1988, 35, 229–245. [Google Scholar] [CrossRef]
- Corpet, F. Multiple sequence alignment with hierarchical clustering. Nucleic Acids Res. 1988, 16, 10881–10890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalendar, R.; Khassenov, B.; Ramanculov, E.; Samuilova, O.; Ivanov, K.I. FastPCR: An in silico tool for fast primer and probe design and advanced sequence analysis. Genomics 2017, 109, 312–319. [Google Scholar] [CrossRef] [PubMed]
- Kalendar, R.; Tselykh, T.V.; Khassenov, B.; Ramanculov, E.M. Introduction on using the FastPCR software and the related Java web tools for PCR and oligonucleotide assembly and analysis. Methods Mol. Biol. 2017, 1620, 33–64. [Google Scholar] [PubMed]
- Kalendar, R.; Muterko, A.; Shamekova, M.; Zhambakin, K. In Silico PCR tools for a fast primer, probe, and advanced searching. Methods Mol. Biol. 2017, 1620, 1–31. [Google Scholar] [PubMed]
- Marks, J. Molecular evolutionary genetics. Am. J. Phys. Anthropol. 1988, 75, 428–429. [Google Scholar] [CrossRef]
- Excoffier, L.; Laval, G.; Schneider, S. Arlequin (version 3.0): An integrated software package for population genetics data analysis. Evol. Bioinform. Online 2005, 1, 47–50. [Google Scholar] [CrossRef]
- Swofford, D.L. PAUP*. Phylogenetic Analysis Using Parsimony (*and Other Methods); Version 4; Sinauer Associates: Sunderland, MA, USA, 1998. [Google Scholar]
- Peakall, R.; Smouse, P.E. GenAlEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research—An update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef] [PubMed]
- Vicient, C.; Jaaskelainen, M.; Kalendar, R.; Schulman, A. Active retrotransposons are a common feature of grass genomes. Plant Physiol. 2001, 125, 1283–1292. [Google Scholar] [CrossRef] [PubMed]
- Queen, R.A.; Gribbon, B.M.; James, C.; Jack, P.; Flavell, A.J. Retrotransposon-based molecular markers for linkage and genetic diversity analysis in wheat. Mol. Genet. Genom. 2004, 271, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Fahima, T.; Sun, G.L.; Beharav, A.; Krugman, T.; Beiles, A.; Nevo, E. RAPD polymorphism of wild emmer wheat populations, Triticum dicoccoides, in Israel. Theor. Appl. Genet. 1999, 98, 434–447. [Google Scholar] [CrossRef]
- Ren, J.; Chen, L.; Sun, D.; You, F.M.; Wang, J.; Peng, Y.; Nevo, E.; Beiles, A.; Sun, D.; Luo, M.C.; et al. SNP-revealed genetic diversity in wild emmer wheat correlates with ecological factors. BMC Evol. Biol. 2013, 13, 169. [Google Scholar] [CrossRef] [PubMed]
- Anderson, O.D.; Rausch, C.; Moullet, O.; Lagudah, E.S. The wheat D-genome HMW-glutenin locus: BAC sequencing, gene distribution, and retrotransposon clusters. Funct. Integr. Genom. 2003, 3, 56–68. [Google Scholar]
- Wicker, T.; Schulman, A.H.; Tanskanen, J.; Spannagl, M.; Twardziok, S.; Mascher, M.; Springer, N.M.; Li, Q.; Waugh, R.; Li, C.; et al. The repetitive landscape of the 5100 Mbp barley genome. Mob. DNA 2017, 8, 22. [Google Scholar] [CrossRef] [PubMed]
- Sela, H.; Ezrati, S.; Ben-Yehuda, P.; Manisterski, J.; Akhunov, E.; Dvorak, J.; Breiman, A.; Korol, A. Linkage disequilibrium and association analysis of stripe rust resistance in wild emmer wheat (Triticum turgidum ssp. dicoccoides) population in Israel. Theor. Appl. Genet. 2014, 127, 2453–2463. [Google Scholar] [CrossRef] [PubMed]
- Stein, N.; Feuillet, C.; Wicker, T.; Schlagenhauf, E.; Keller, B. Subgenome chromosome walking in wheat: A 450-kb physical contig in Triticum monococcum L. spans the Lr10 resistance locus in hexaploid wheat (Triticum aestivum L.). Proc. Natl. Acad. Sci. USA 2000, 97, 13436–13441. [Google Scholar] [CrossRef] [PubMed]
- Domb, K.; Keidar, D.; Yaakov, B.; Khasdan, V.; Kashkush, K. Transposable elements generate population-specific insertional patterns and allelic variation in genes of wild emmer wheat (Triticum turgidum ssp. dicoccoides). BMC Plant Biol. 2017, 17, 175. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
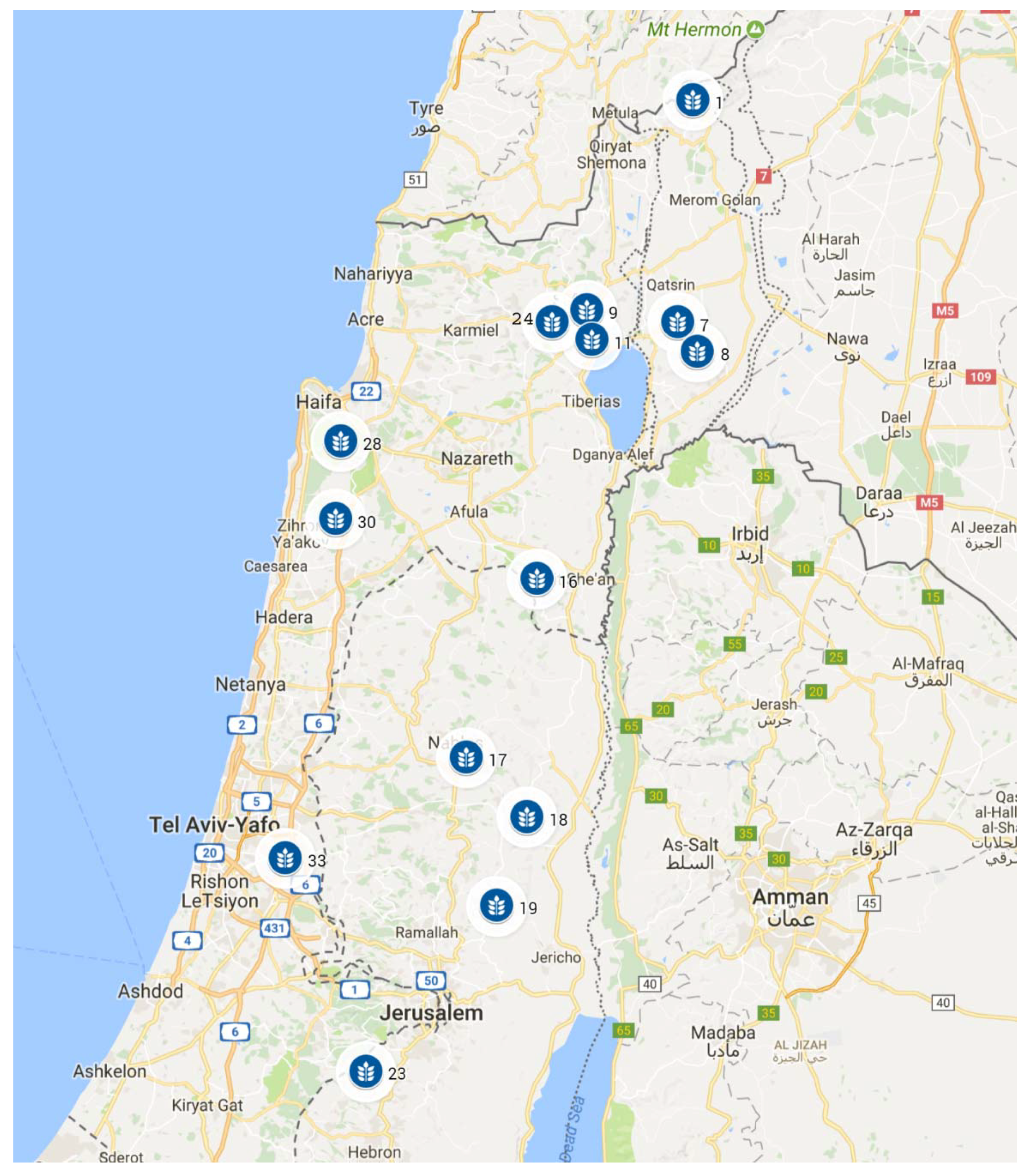
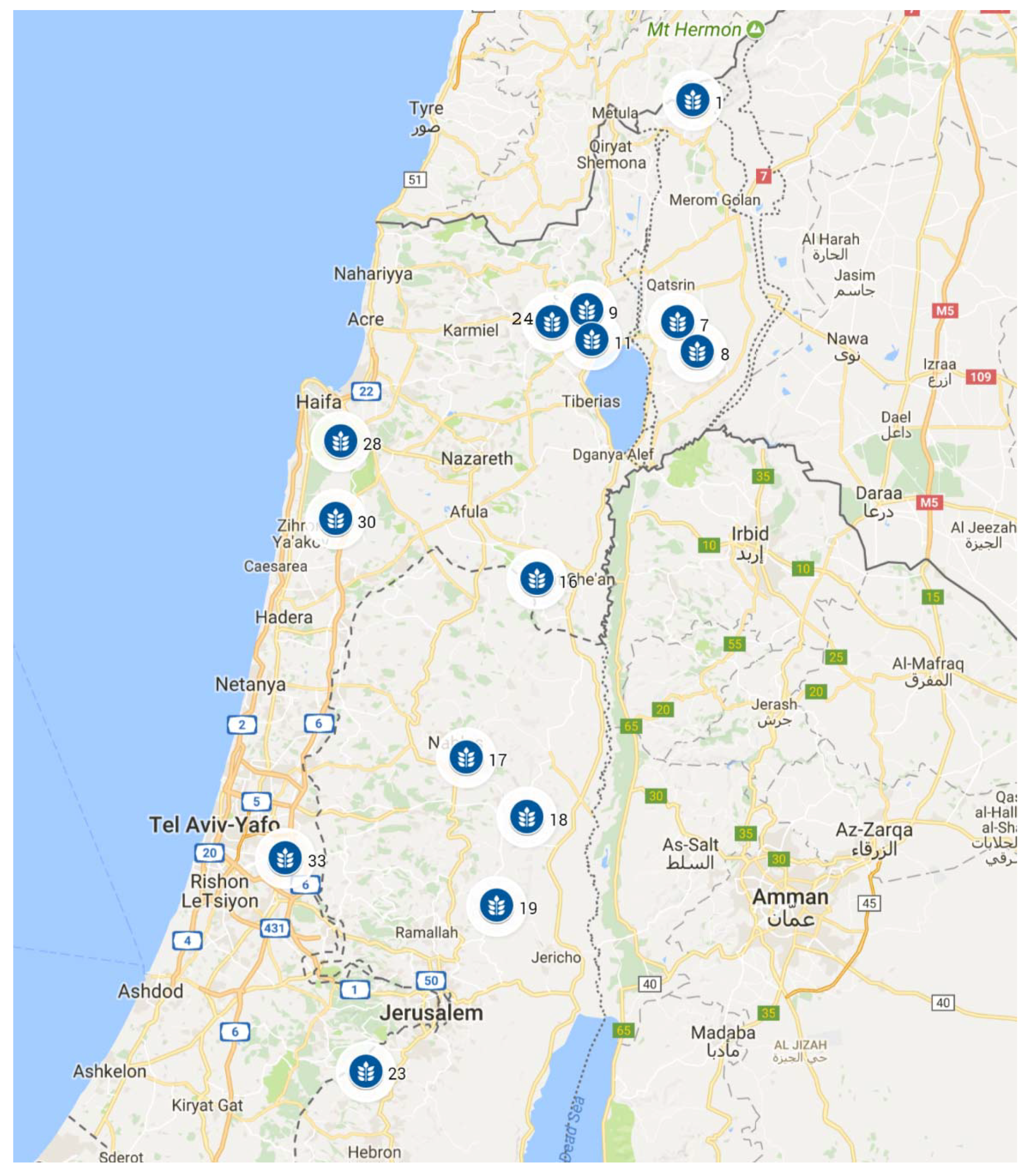
| Population a | Ln | Lt | Al | Tm | Ta | Tj | Td | Tdd | Rn | Rd | Hu14 | Huan | Dw | Trd | Ev | So | Rv | Rr | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Mt. Hermon | 35.73 | 33.30 | 1300 | 11 | 21 | 3 | 18 | 6 | 1400 | 66 | 48 | 60 | 60 | 0 | 150 | 1 | 30 | 20 |
| 7 | Yehudiyya | 35.70 | 32.93 | 200 | 19 | 27 | 11 | 16 | 12 | 550 | 47 | 42 | 58 | 58 | 100 | 160 | 5 | 38 | 25 |
| 8 | Gamla | 35.74 | 32.88 | 200 | 19 | 26 | 9 | 17 | 12 | 470 | 50 | 43 | 58 | 58 | 60 | 155 | 5 | 39 | 26 |
| 9 | Rosh Pinna | 35.52 | 32.95 | 700 | 18 | 25 | 9 | 16 | 10 | 697 | 50 | 48 | 58 | 50 | 35 | 150 | 1 | 35 | 22 |
| 11 | Tabigha | 35.53 | 32.90 | 0 | 24 | 32 | 15 | 17 | 10 | 436 | 45 | 45 | 57 | 58 | 120 | 160 | 5 | 39 | 25 |
| 16 | Mt. Gilboa | 35.42 | 32.50 | 150 | 21 | 28 | 12 | 16 | 12 | 400 | 44 | 43 | 58 | 40 | 160 | 165 | 1 | 34 | 24 |
| 17 | Mt. Gerizim | 35.28 | 32.20 | 800 | 17 | 23 | 8 | 15 | 9 | 700 | 47 | 45 | 60 | 42 | 0 | 155 | 1 | 38 | 25 |
| 18 | Gitit | 35.40 | 32.10 | 300 | 21 | 29 | 13 | 16 | 12 | 360 | 39 | 39 | 55 | 25 | 100 | 170 | 1 | 38 | 24 |
| 19 | Kokhav Hashahar | 35.34 | 31.95 | 600 | 20 | 28 | 12 | 16 | 12 | 400 | 40 | 45 | 59 | 30 | 25 | 165 | 1 | 38 | 22 |
| 23 | J’aba | 35.08 | 31.67 | 660 | 17 | 25 | 9 | 15 | 9 | 500 | 41 | 49 | 62 | 57 | 30 | 155 | 1 | 35 | 21 |
| 24 | Amirim | 35.45 | 32.93 | 600 | 15 | 24 | 8 | 16 | 8 | 850 | 61 | 48 | 60 | 53 | 13 | 153 | 1 | 35 | 23 |
| 28 | Bet-Oren | 35.03 | 32.73 | 400 | 17 | 24 | 11 | 13 | 8 | 700 | 55 | 59 | 69 | 80 | 0 | 142 | 1 | 25 | 19 |
| 30 | Bat-Shelomo | 35.02 | 32.60 | 75 | 20 | 26 | 13 | 13 | 10 | 650 | 55 | 58 | 68 | 77 | 30 | 150 | 2 | 24 | 20 |
| 33 | Givat Koach | 34.92 | 32.03 | 75 | 20 | 26 | 12 | 14 | 12 | 540 | 46 | 50 | 64 | 65 | 105 | 160 | 1 | 32 | 26 |
| 36 | W. Diyarbakir | 39.63 | 37.89 | 850 | 13 | 27 | 2 | 25 | - | 546 | 65 | - | 46 | - | - | - | 5 | - | - |
| Primer ID | Sequence (5′-3′) | Retrotransposon LTR | Tm a, °C | CG b (%) | Linguistic Complexity (%) [26] |
|---|---|---|---|---|---|
| 432 | GATAGGGTCGCATCTTGGGCGTGAC | Sukkula | 64.1 | 60.0 | 93 |
| 443 | ACACACACACACACACACT | (AC)9T microsatellite | 54.3 | 47.4 | 24 |
| 455 | TTGAATTTCTGCTACGTTCCCC | WIS2/BARE1 | 55.8 | 45.5 | 88 |
| 554 | CCAACTAGAGGCTTGCTAGGGAC | WIS2/BARE1 | 60.0 | 56.5 | 80 |
| 639 | ACACACAAAGCATTCCTCCGG | Sabrina | 58.4 | 52.4 | 79 |
| 678 | AAAGTTGTATCCGGGGCGTTAC | Sukkula | 57.8 | 50.0 | 88 |
| 679 | GGGTCGCATATTGGGCGTGAC | Sukkula | 62.0 | 61.9 | 87 |
| 692 | GCGATTGCTAAGGCGCAACG | Cereba | 61.1 | 60.0 | 89 |
| 734 | TTCCCATGCGACGTTCCCCAAC | WIS2/BARE1 | 62.6 | 59.1 | 78 |
| 738 | AATTTCTGCTACGTTCCCCTAC | WIS2/BARE1 | 55.0 | 45.5 | 80 |
| 773 | CCCTCTAGGCGACATCCACG | Nikita | 60.6 | 65.0 | 89 |
| 2105 | ACTCCATAGATGGATCTTGGTGA | WIS2/BARE1 | 54.6 | 43.5 | 88 |
| 2106 | TAATTTCTGCAACGTTCCCCAACA | WIS2/BARE1 | 57.1 | 41.7 | 83 |
| 2107 | AGCATGATGCAAAATGGACGTATCA | Wilma/Bagy2 | 56.8 | 40.0 | 84 |
| 2108 | AGAGCCTTCTGCTCCTCGTTGGGT | Wilma/Bagy2 | 63.4 | 58.3 | 83 |
| 2110 | TCGCTGCGACTGCCCGTGCACA | Daniela | 67.3 | 68.2 | 78 |
| 2113 | TACGCATCCGTGCGGCCCGAAC | Daniela | 66.6 | 68.2 | 90 |
| No. a | Population | All b | Polymorphic Loci | Fraction Loci Polymorphic | Diversity ± SD |
|---|---|---|---|---|---|
| 1 | Mt. Hermon | 197 | 93 | 0.47 | 0.204 ± 0.114 |
| 7 | Yehudiyya | 210 | 20 | 0.10 | 0.052 ± 0.030 |
| 8 | Gamla | 192 | 77 | 0.40 | 0.118 ± 0.065 |
| 9 | Rosh Pinna | 215 | 93 | 0.43 | 0.195 ± 0.105 |
| 11 | Tabigha | 221 | 48 | 0.22 | 0.109 ± 0.050 |
| 16 | Mt. Gilboa | 211 | 114 | 0.54 | 0.201 ± 0.108 |
| 17 | Mt. Gerizim | 207 | 104 | 0.50 | 0.186 ± 0.100 |
| 18 | Gitit | 214 | 102 | 0.48 | 0.189 ± 0.102 |
| 19 | Kokhav Hashahar | 201 | 78 | 0.39 | 0.157 ± 0.086 |
| 23 | J’aba | 207 | 96 | 0.46 | 0.182 ± 0.098 |
| 24 | Amirim | 217 | 58 | 0.27 | 0.112 ± 0.062 |
| 28 | Bet-Oren | 215 | 2 | 0.01 | 0.002 ± 0.002 |
| 30 | Bat-Shelomo | 175 | 82 | 0.47 | 0.186 ± 0.100 |
| 33 | Givat Koach | 204 | 48 | 0.24 | 0.063 ± 0.035 |
| 36 | W. Diyarbakir, 22 km | 202 | 77 | 0.38 | 0.118 ± 0.064 |
| Mean | 205.9 | 72.8 | 0.36 | 0.138 ± 0.063 |
| Population a | 1 | 7 | 8 | 9 | 11 | 16 | 17 | 18 | 19 | 23 | 24 | 28 | 30 | 33 | 36 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Mt. Hermon | 0.0000 | ||||||||||||||
| 7 | Yehudiyya | 0.6849 | 0.0000 | |||||||||||||
| 8 | Gamla | 0.4590 | 0.7727 | 0.0000 | ||||||||||||
| 9 | Rosh Pinna | 0.2902 | 0.6738 | 0.4605 | 0.0000 | |||||||||||
| 11 | Tabigha | 0.6284 | 0.6430 | 0.7464 | 0.6514 | 0.0000 | ||||||||||
| 16 | Mt. Gilboa | 0.2525 | 0.6391 | 0.4650 | 0.3549 | 0.6068 | 0.0000 | |||||||||
| 17 | Mt. Gerizim | 0.2593 | 0.6786 | 0.4660 | 0.3430 | 0.6443 | 0.2185 | 0.0000 | ||||||||
| 18 | Gitit | 0.3406 | 0.6641 | 0.5077 | 0.4063 | 0.6460 | 0.3132 | 0.3255 | 0.0000 | |||||||
| 19 | Kokhav H. | 0.3724 | 0.7430 | 0.5438 | 0.3786 | 0.7187 | 0.3692 | 0.3726 | 0.4259 | 0.0000 | ||||||
| 23 | J’aba | 0.3597 | 0.6724 | 0.4880 | 0.3772 | 0.6427 | 0.2721 | 0.2891 | 0.2749 | 0.3666 | 0.0000 | |||||
| 24 | Amirim | 0.4738 | 0.8041 | 0.5914 | 0.4443 | 0.7674 | 0.4472 | 0.5383 | 0.5010 | 0.4311 | 0.4948 | 0.0000 | ||||
| 28 | Bet-Oren | 0.6773 | 0.9421 | 0.8171 | 0.6909 | 0.8995 | 0.6215 | 0.6756 | 0.6961 | 0.7205 | 0.5994 | 0.8070 | 0.0000 | |||
| 30 | Bat-Shelomo | 0.2992 | 0.6730 | 0.4367 | 0.3466 | 0.6384 | 0.2305 | 0.3114 | 0.3506 | 0.3832 | 0.3180 | 0.4143 | 0.6596 | 0.0000 | ||
| 33 | Givat Koach | 0.5879 | 0.8388 | 0.7156 | 0.5573 | 0.8010 | 0.4794 | 0.5343 | 0.5499 | 0.5835 | 0.4604 | 0.6757 | 0.8894 | 0.5121 | 0.0000 | |
| 36 | W. Diyarbakir | 0.4226 | 0.7530 | 0.5860 | 0.5090 | 0.7254 | 0.4626 | 0.4720 | 0.5076 | 0.5534 | 0.4941 | 0.6163 | 0.8092 | 0.4620 | 0.7030 | 0.0000 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vuorinen, A.L.; Kalendar, R.; Fahima, T.; Korpelainen, H.; Nevo, E.; Schulman, A.H. Retrotransposon-Based Genetic Diversity Assessment in Wild Emmer Wheat (Triticum turgidum ssp. dicoccoides). Agronomy 2018, 8, 107. https://doi.org/10.3390/agronomy8070107
Vuorinen AL, Kalendar R, Fahima T, Korpelainen H, Nevo E, Schulman AH. Retrotransposon-Based Genetic Diversity Assessment in Wild Emmer Wheat (Triticum turgidum ssp. dicoccoides). Agronomy. 2018; 8(7):107. https://doi.org/10.3390/agronomy8070107
Chicago/Turabian StyleVuorinen, Anssi L., Ruslan Kalendar, Tzion Fahima, Helena Korpelainen, Eviatar Nevo, and Alan H. Schulman. 2018. "Retrotransposon-Based Genetic Diversity Assessment in Wild Emmer Wheat (Triticum turgidum ssp. dicoccoides)" Agronomy 8, no. 7: 107. https://doi.org/10.3390/agronomy8070107
APA StyleVuorinen, A. L., Kalendar, R., Fahima, T., Korpelainen, H., Nevo, E., & Schulman, A. H. (2018). Retrotransposon-Based Genetic Diversity Assessment in Wild Emmer Wheat (Triticum turgidum ssp. dicoccoides). Agronomy, 8(7), 107. https://doi.org/10.3390/agronomy8070107