4.1. Genetic Parameters
Although
C. arabica has a large and complex allotetraploid genome [
69], the 20,477 high-quality SNPs retained after filtering provided sufficient genome-wide coverage for downstream analyses, as they were derived from targeted, non-repetitive genic and intergenic regions designed to represent the entire genome. This dense and representative marker set enabled stable model fitting and the accurate estimation of genetic relationships across traits [
3,
32,
70]. Traditionally, Arabica coffee breeding programs have relied on single-trait selection processes, resulting in a loss of information regarding the interrelationship between variables, which is disadvantageous. Developing selection strategies based on multiple traits simultaneously is one way forward. However, the use of traditional MTM based on trait correlations may face challenges when modeling multiple variables simultaneously and does not incorporate cause-and-effect relationships between traits [
30,
32,
33,
35,
45,
46,
68]. To address this, latent variables (YL, LLL, VL, and NL) were constructed based on CFA (
Table 2) using observed variables, including Y, VV, CD, LL, NVN, and NRN (
Table 1). This approach was used to uncover cause-and-effect relationships among the latent variables and to reduce the complexity of the model, thereby facilitating its convergence.
Some previous studies used exploratory factor analysis, a technique that differs from CFA in that it does not pre-specify aspects of the model, including the number of factors [
71]. In the case of
Coffea canephora, Paixão et al. [
31] identified latent variables such as the vigor factor and the production factor, which captured the relationships among observed variables like plant height and canopy projection diameter for vegetative vigor and yield-related traits for production. These findings support the current results obtained from the CFA in
C. arabica, where similar dimensions of trait variation were observed. However, while Paixão et al. [
31] used exploratory factor analysis (EFA)—which does not infer causal relationships—our study advanced further by applying structural equation modeling (SEM), allowing for the identification of explicit cause-and-effect pathways among latent variables. Notably, the VL → NL → YL causal chain observed here reflects a stronger biological linkage between vegetative status, nodal development, and yield in
C. arabica, likely due to its distinct growth habit and lower branching plasticity compared to
C. canephora [
37]. These comparisons highlight that, while general vegetative–reproductive trade-offs are conserved across
Coffea species, the organization, strength, and causal connectivity among latent variables are species-specific, shaped by each species’ physiological and architectural characteristics.
After the adjustment of the latent variables, the factor scores were corrected based on the methods [
42,
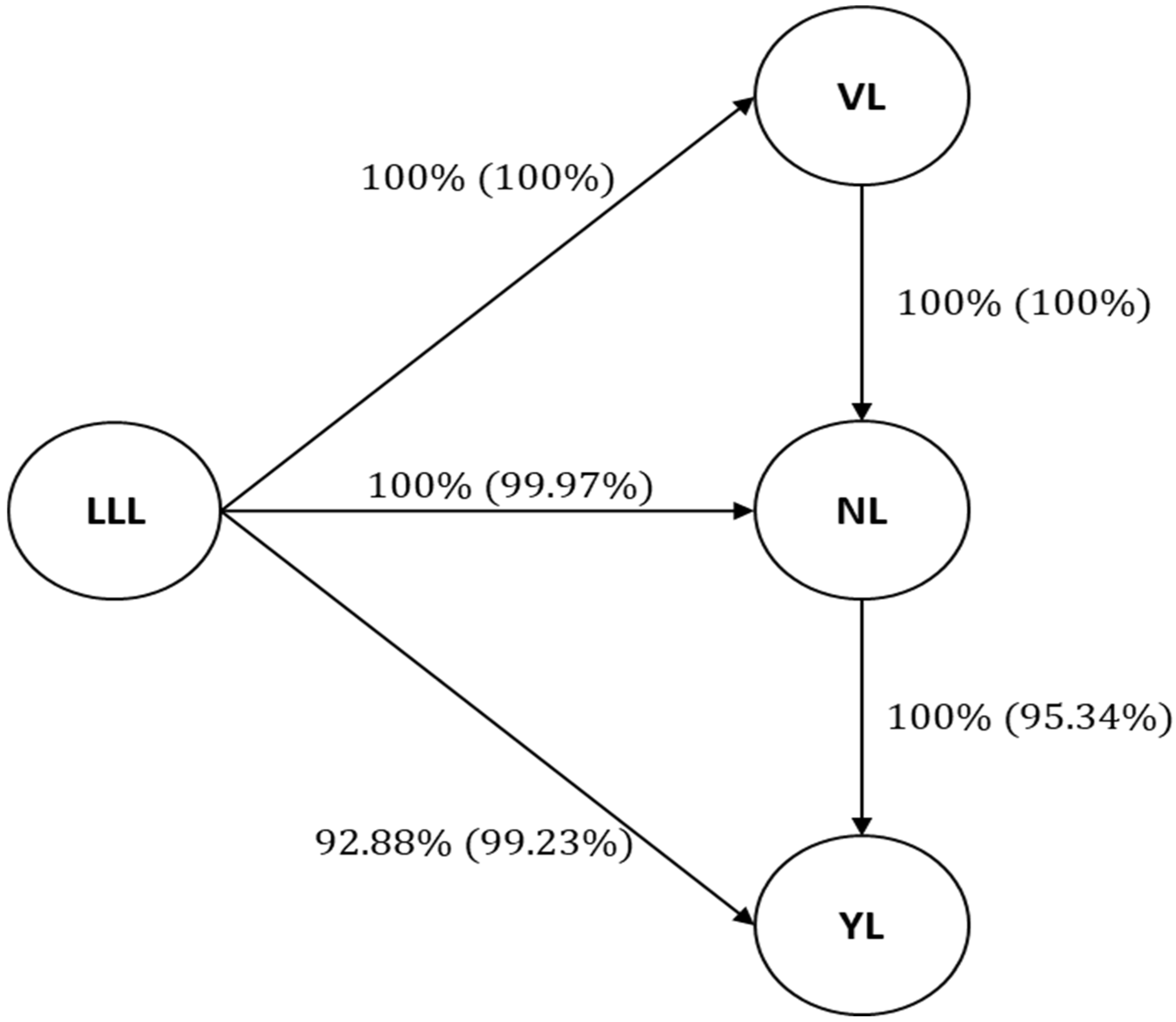
64]. After this correction, the latent variable scores were incorporated into the Bayesian network analysis. The Bayesian network revealed a structured interrelationship among the variables, with significant connections identified between LLL → VL, LLL → NL, LLL → YL, VL → NL and NL → YL (
Table 4). The results show that the most crucial connections for model fit were LLL → NL, VL → NL, and NL → YL, as their exclusion would lead to the largest reductions in BIC. Conversely, removing LLL → VL and LLL→YL had a smaller impact on model fit (
Table 4). According to Nagarajan et al. [
72], Bayesian networks are defined in terms of conditional independence and probabilistic properties; however, Pearl [
73] argues that a proper Bayesian network must represent the causal structure of the data. Thus, biologically, connections that have smaller effects on the global BIC may imply less important cause and effect, as is the case for LLL → VL and LLL → YL.
Suela et al. [
32] used the SEM approach based on observed variables in a genome-wide association study (GWAS) in the same population as in this study. They observed direct connections from vegetative vigor to number of reproductive nodes and number of reproductive nodes to yield, which supports the findings of this work. Suela et al. [
32] also highlighted the importance of the connection between vegetative vigor and number of reproductive nodes, as well as between number of reproductive nodes and yield in the general model, as indicated by BIC. Furthermore,
Figure 1 shows that 100% of the bootstrap samples exhibited directed connections, which is consistent with the earlier results of Suela et al. [
32]. They reported 100% and 81% frequencies of directed connections between vegetative vigor and number of vegetative nodes, and number of reproductive nodes and yield, respectively, with 68% and 100% of cases showing these directed connections, consistent with the previous findings.
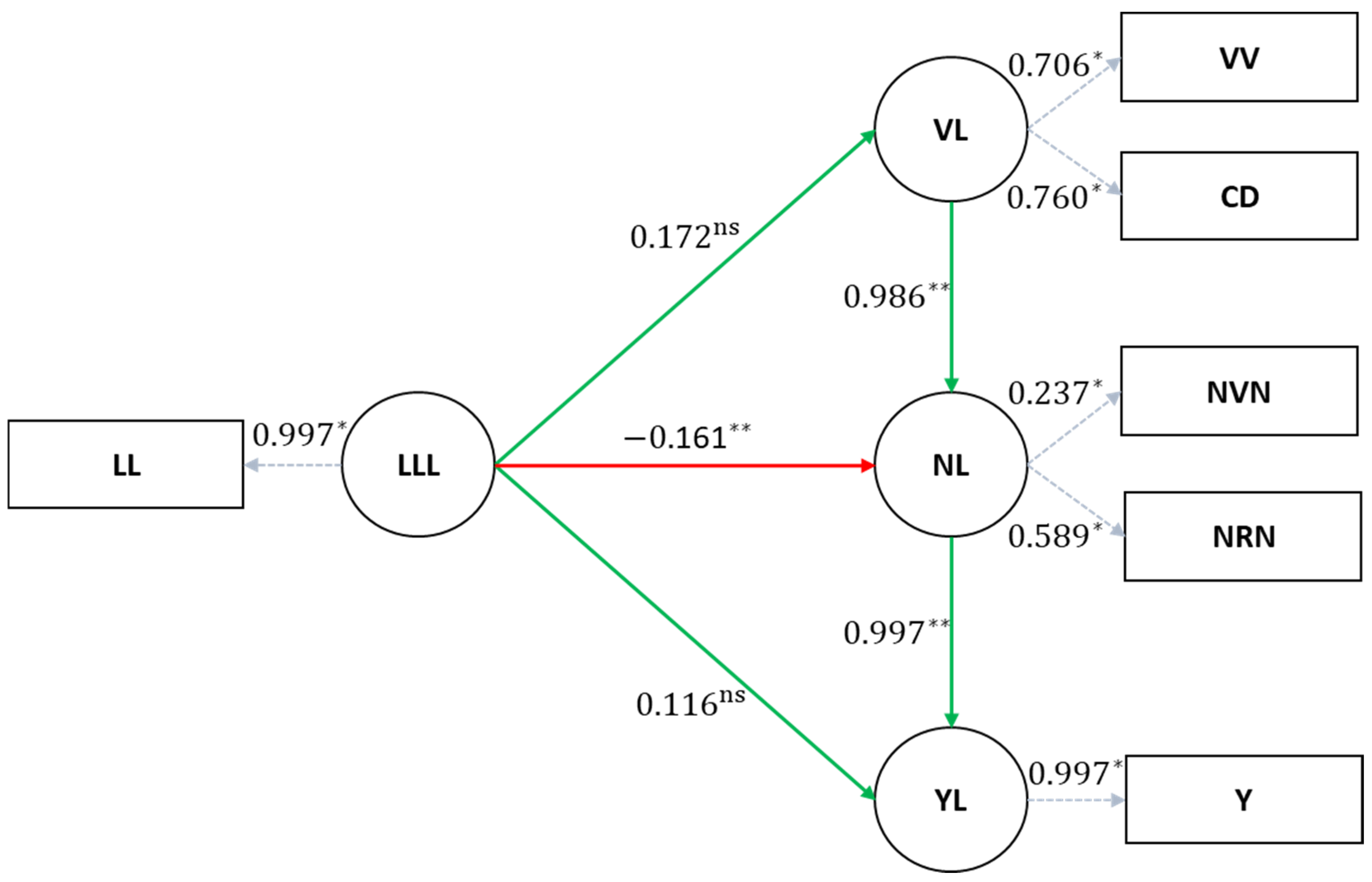
The structural coefficients, which represent cause-and-effect relationships between variables, were estimated using the SEM methodology based on the structure derived from the Bayesian networks and the previously corrected factors (
Table 5 and
Table 6). The paths LLL → NL, VL → NL, and NL → YL showed significant estimates of cause and effect between the variables. This contrasts with the findings of Suela et al. [
32], who, without using latent variables, found non-significant coefficients for all connections.
Based on
Table 3 and
Table 7, it is clear that the heritability estimates of the same latent variables were different. This difference is explained by the method of obtaining each of the results, using the MTM to calculate the estimates in
Table 3 and the univariate G-BLUP to calculate the estimates in
Table 7. In short, these models differ in their construction, with the MTM designed to exploit the information contained in the traits of correlated indicators, while univariate models do not exploit the information of correlated traits [
27].
When analyzing the results of univariate GWS, we obtained genetic parameters comparable to those reported by Sousa et al. [
3] and Suela et al. [
32], who used the same population. In both Sousa et al. [
3] and this study, low predictive abilities and selection accuracies were observed for Y and for LL. For both the observed variables and the latent variables, we found that, for Y and LL, the gains from selection using the variables themselves or their respective latent variables were identical, at 2.11% and 0.74%, respectively. This similarity is due to the fact that the CFA was conducted using only the variable itself. Studies such as Tassone et al. [
74] found yield gains for Arabica coffee ranging from 0.93 to 6.98% depending on the location and the original mean considered. However, for leaf length, the gains in the literature, as seen in the work of Chidoko et al. [
75] using Arabica coffee were higher compared to those found in this study, with a value of 16.10%; however, this magnitude can be explained by the different population used, where it presented different values of genetic parameters than those found in this study.
In contrast, when VV, CD, NVN, and NRN were selected directly, the results were different from when VL and NL were selected as latent variables. Direct selection yielded gains of 1.89% for VV, 2.39% for CD, 3.76% for NVN, and 0.46% for NRN (
Table 7). Carvalho et al. [
76] and Tassone et al. [
74] reported gains of 3% and 0.31 to 5.23% for VV, respectively. Carvalho et al. [
76] and Tassone et al. [
74] reported gains of 3% and −7.24 to −1.75% for CD, respectively. Thus, despite differences in the direction of the gains, there is consistency in the magnitude of the gains. However, the magnitude may vary due to differences in coffee species. In another study using Conilon coffee, Silva et al. [
77] found a gain of 5.23% for the number of nodes.
On the other hand, using VL for indirect selection in VV and CD resulted in selection gains of 1.40% and 1.77%, respectively (
Table 8). Similarly, selecting NL for indirect selection in NVN and NRN resulted in selection gains of 0.79% and 0.70%, respectively (
Table 8). Hence, it is evident that the use of latent variables as a selection criterion guarantees gains in all the observed variables, with only a slight reduction in the gains for VV and CD, a slight reduction in the gains for NVN, and a slight increase in NRN, compared to using the observed variables themselves.
As shown in
Table 5,
,
, and
were significant, but only MTM correlation information is required for the selection scenario [
30,
68]. By selecting VL and estimating the response in NVN and NRN, similar gains were achieved compared to selecting NL (
Table 8). This indicates that the existing cause-and-effect relationships facilitate indirect selection for Arabica coffee node number traits in our data. In the case of Y, selecting individuals for NL and estimating the indirect effect on Y resulted in a gain of 3.51%, which is 66.35% higher than what would be achieved by selection based solely on the trait Y itself. This improvement in gain with indirect selection can be attributed to the low values of
and
for the target variable (
Table 7 and
Table 8). It is also important to highlight that the predictive ability for NRN was negative (−0.1081) (
Table 7), which reflects the low heritability and low genetic correlation of this trait with other variables included in the model. Negative predictive ability values can occur when the proportion of genetic variance is low relative to residual variance, especially for traits with limited genetic control or high environmental variability [
78,
79]. From a breeding perspective, this indicates that direct selection for NRN, based on genomic predictions from this model, would be unreliable and potentially misleading. Instead, indirect selection using genetically correlated traits, such as NL, may offer a more effective strategy for improving traits associated with NRN in Arabica coffee.
Interestingly, the negative effect observed from LLL to NL suggests that genotypes with longer leaves tend to produce fewer nodes, likely reflecting a physiological trade-off between resource allocation for leaf expansion and node initiation. Longer leaves typically require the greater investment of assimilates and nutrients [
80,
81], and they are often associated with increased internode length, leading to reduced node density along the stem. Additionally, larger leaves can increase self-shading within the canopy, limiting light interception at lower nodes and reducing meristematic activity [
82]. Supporting our findings, Yirga et al. [
83] also reported a negative genotypic correlation and negative effects between leaf length and number of nodes in Ethiopian Arabica coffee germplasm.
Conversely, it is important to highlight that the paths from LLL to VL and from LLL to YL were not significant in the SEM (
Table 5), and this lack of effect is consistent with the Bayesian network analysis, which showed that removing these connections resulted in minimal changes in BIC values (
Table 4). These findings suggest that, although LLL is structurally positioned upstream in the network, its influence on downstream traits such as YL is likely weak or indirect in this population. Therefore, these pathways do not contribute substantially to the efficiency of selection and illustrate how SEM helps filter out biologically less informative relationships.
From a practical breeding standpoint, the results presented here highlight how indirect selection via latent variables—especially NL for Y—can provide substantial genetic gains while reducing reliance on traits with low heritability or those expressed late in the plant’s development. In perennial crops such as
C. arabica, where phenotyping for traits like yield is time-consuming and costly, using early measured traits (e.g., number of reproductive nodes and number of vegetative nodes) to predict target traits through SEM-based relationships can accelerate and streamline selection decisions. For instance, indirect selection using NL resulted in a 3.51% gain in yield (Y), representing a 66.35% increase compared to direct selection (2.11%), as highlighted in
Table 8. These results, emphasized in
Table 7 and
Table 8, show that SEM-based selection strategies offer a cost-effective and operationally feasible alternative for breeding programs. Once the SEM structure is defined, the computation of latent scores can be performed using standard field measurements, making this approach scalable for routine use. Therefore, incorporating SEM into multi-trait genomic selection frameworks may significantly enhance breeding efficiency, particularly when aiming for early selection, dimensionality reduction, and increased prediction accuracy in complex trait networks.
The integration of genome-wide selection (GWS) further strengthens these advantages by enabling the early and accurate identification of superior individuals based on genomic estimated breeding values. As demonstrated by Sousa et al. [
3] in Arabica coffee, GWS alone can substantially reduce breeding cycle length and operational costs by predicting complex traits before full phenotypic expression. When combined with SEM and the use of biologically meaningful latent variables such as VL and NL, this framework allows breeders to target early vegetative traits like VL to indirectly improve node-related traits (NVN and NRN) and to use NL as a key driver of yield. By prioritizing individuals with superior VL scores during early field stages, breeders can enhance genetic gains for nodal development while simultaneously shortening selection cycles by several years. Together, these strategies optimize both the speed and magnitude of genetic improvement in Arabica coffee breeding programs.
4.2. Interpretability of SEM in Biological Context
Based on the fit measures obtained from the CFA, BN, and SEM analyses, it is evident that the procedures employed were satisfactory and allowed for meaningful biological interpretation. Among these analyses, the first showed significant factor loadings in all cases, the second revealed the most robust structural relationships between the variables, and the third provided structural coefficients. However, there may be issues related to the interpretation of parameters arising from SEM in the context of a breeding program. Therefore, this section is focused on providing some explanations using the data used in this study.
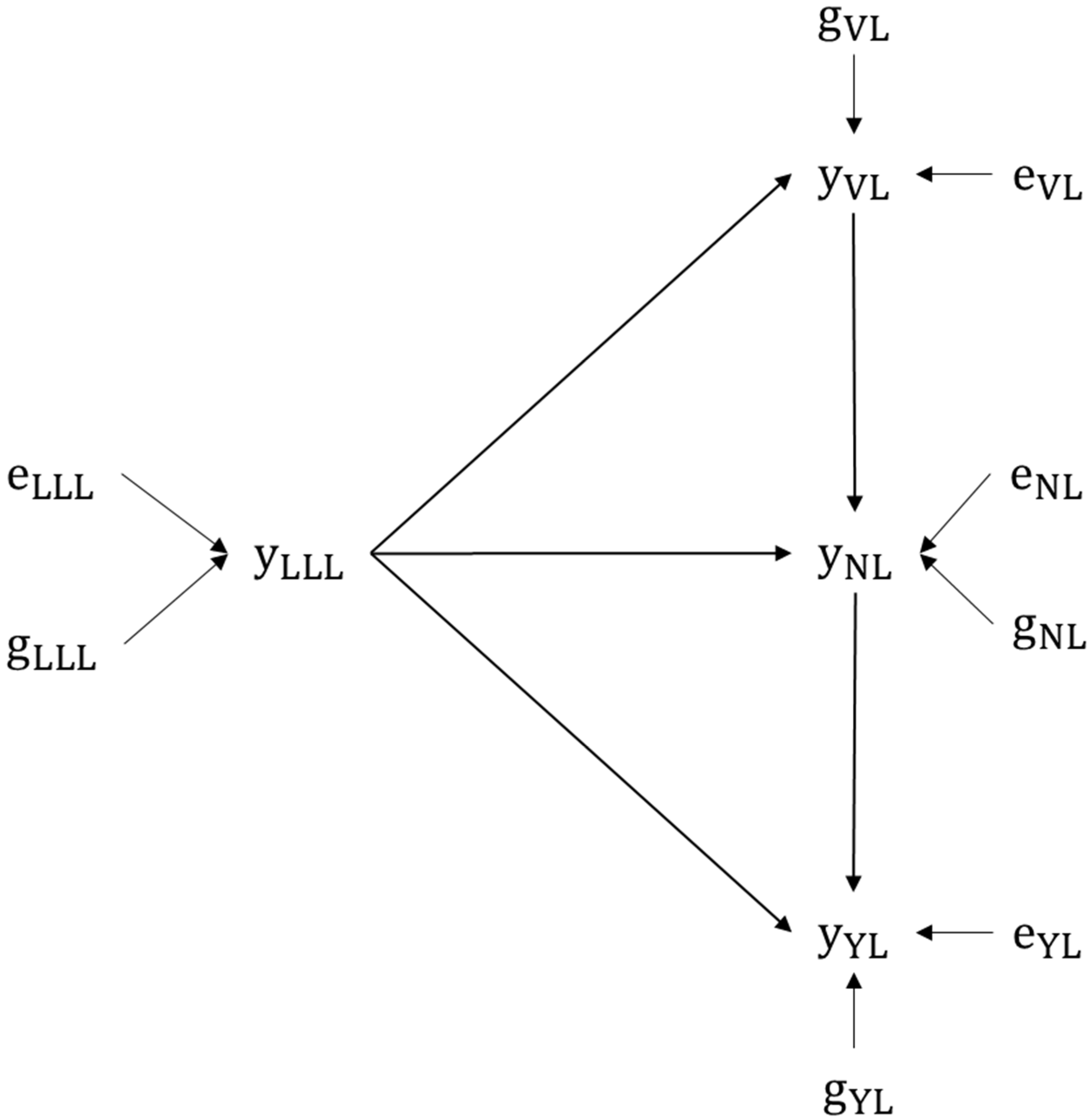
All explanations are based on the data used in this study, which are structured in the following path network (
Figure 3).
Firstly, it is important to understand that the breeding values obtained from MTM and SEM can be considered equivalent (as described in
Section 2.6), as long as the relationship
holds, where
is the vector of breeding values in MTM,
is the matrix of the structural coefficients, and
is the vector of breeding values under SEM [
33] and also due to the fact that both models produce the same joint probability distribution of phenotypes [
30]. However, genetic and environmental covariances between variables can increase or decrease depending on the sign of the structural coefficient. Any changes in covariances can result in proportional changes in other genetic parameters, such as genetic correlation, environmental correlation, and heritability [
68]. Algebraic equations of genetic parameters for the given path network can be found in the Text S1, as well as the estimated values for the MTM and SEM models (
Table S10) according to the analysis performed in
Section 2.4 and
Section 2.6, which can be validated according to the equations generated according to the path network.
According to Varona and González-Recio [
68], a noticeable pattern is that the higher the structural coefficient resulting from SEM, the greater the genetic correlation resulting from MTM, regardless of the genetic covariance between traits. This pattern can be observed in the significant genetic correlations from MTM (
Table 3). For example, the VL
NL and NL
YL connections had the highest structural coefficient and the highest genetic correlation. Additionally, when the structural coefficient is greater than 1, it indicates that the phenotypic variance of the dependent variable can also be explained by non-genetic effects, which did not occur in this data set.
While these comparisons provide insight into the relationship between the two models using genetic parameters, it is important to understand how the breeding values should be interpreted. Statistical equivalence between the models does not necessarily imply biological equivalence. Breeding values estimated by MTM are calculated based on the existence of pleiotropy or linkage disequilibrium between QTLs and common environmental effects [
68], including all additive effects of genes on the trait, even if they have a direct or indirect influence [
30]. However, SEM-based breeding values consider not only common genetic and environmental effects but also non-genetic effects that arise from the phenotypic influence of another trait. These effects represent the direct and indirect effects of genes on the trait, rather than an indirect influence through the phenotypic influence of another trait [
30]. In other words, breeding values are corrected for causality in SEM [
68].
From the perspective of a breeding program focused on the selection of superior individuals, it is recommended to use the breeding values obtained by MTM rather than SEM [
30,
68]. This is because the relevant information for selecting superior individuals is given by the overall genetic effect, which is already provided by MTM without the need for SEM [
30]. However, SEM analysis using latent variables offers advantages in terms of the model and the informativeness of the results. In terms of the model, SEM provides greater model parsimony compared to MTM [
30]. It also offers dimensionality reduction and an improved ability to converge when dealing with complex networks of interrelationships [
42]. In terms of model informativeness, SEM allows the observation of phenotypic changes resulting from causal relationships between traits and also the distinction between direct and indirect genetic effects. These changes can be understood in the context of the structural coefficient, representing a
-unit change in the predicted variables. In addition, Valente et al. [
30] pointed out that, when performing MTM, the use of different scenarios may require the construction of additional variables or the performance of new analyses, whereas SEM can handle such variations without the need for additional data processing or extended analysis time.


 ,
, 



{kind=link}
{kind=link}
{kind=link}