
Genome-Wide Association Study and Genomic Prediction of Essential Agronomic Traits in Diversity Panel of Soybean Varieties

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Growth and Phenotyping
2.2. DNA Extraction, Genotyping by Sequencing, Variant Calling and Filtration
2.3. Phylogenetic Analysis, Linkage Disequilibrium, Principle Components Analysis and Population Structure
2.4. Genome-Wide Association Analysis and Candidate Gene Exploration
2.5. Genomic Prediction
3. Results
3.1. Phenotype Variation in the Soybean Diversity Panel
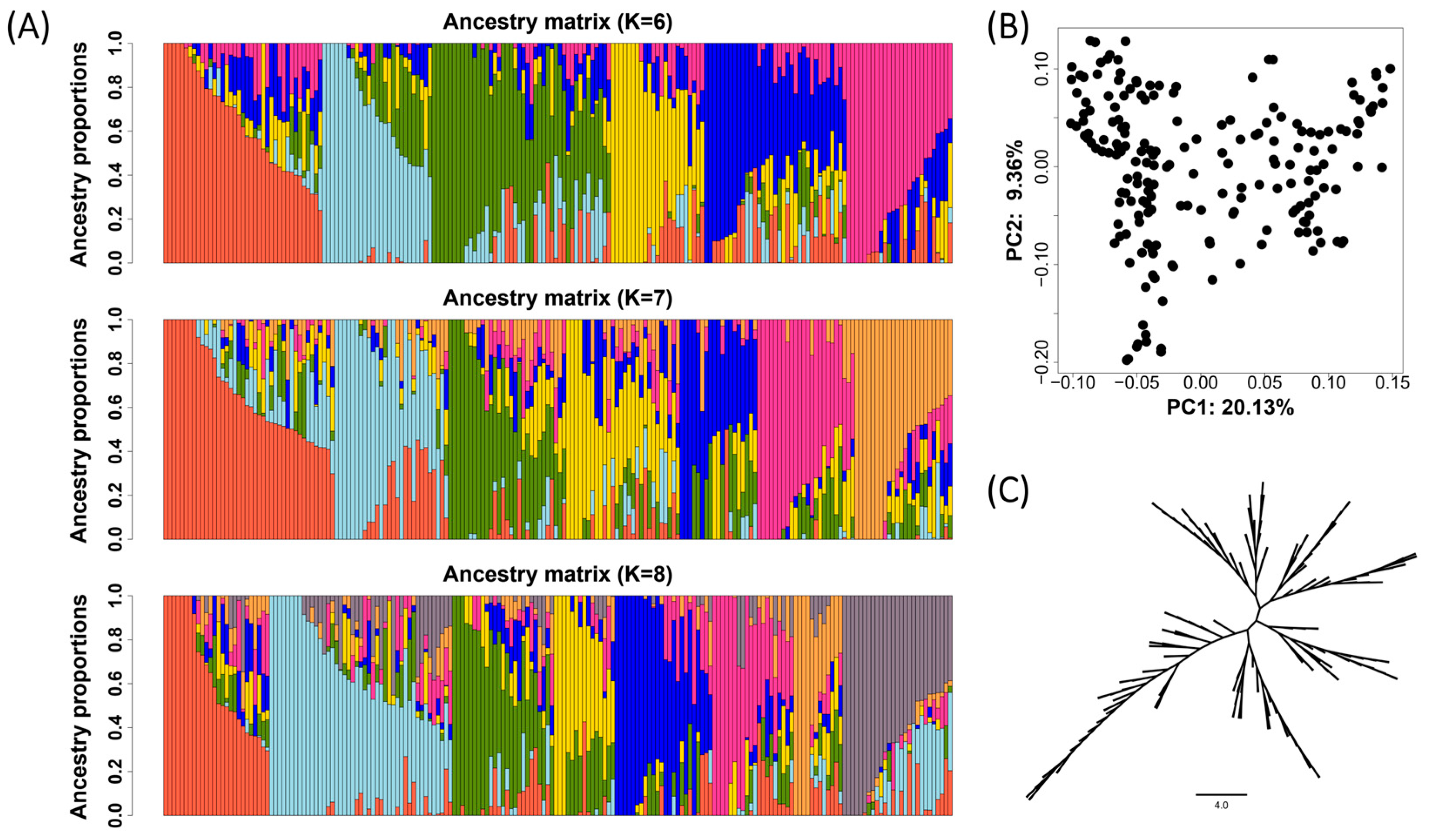
3.2. The Genetic Relationship and Population Structure of the Soybean Diversity Panel
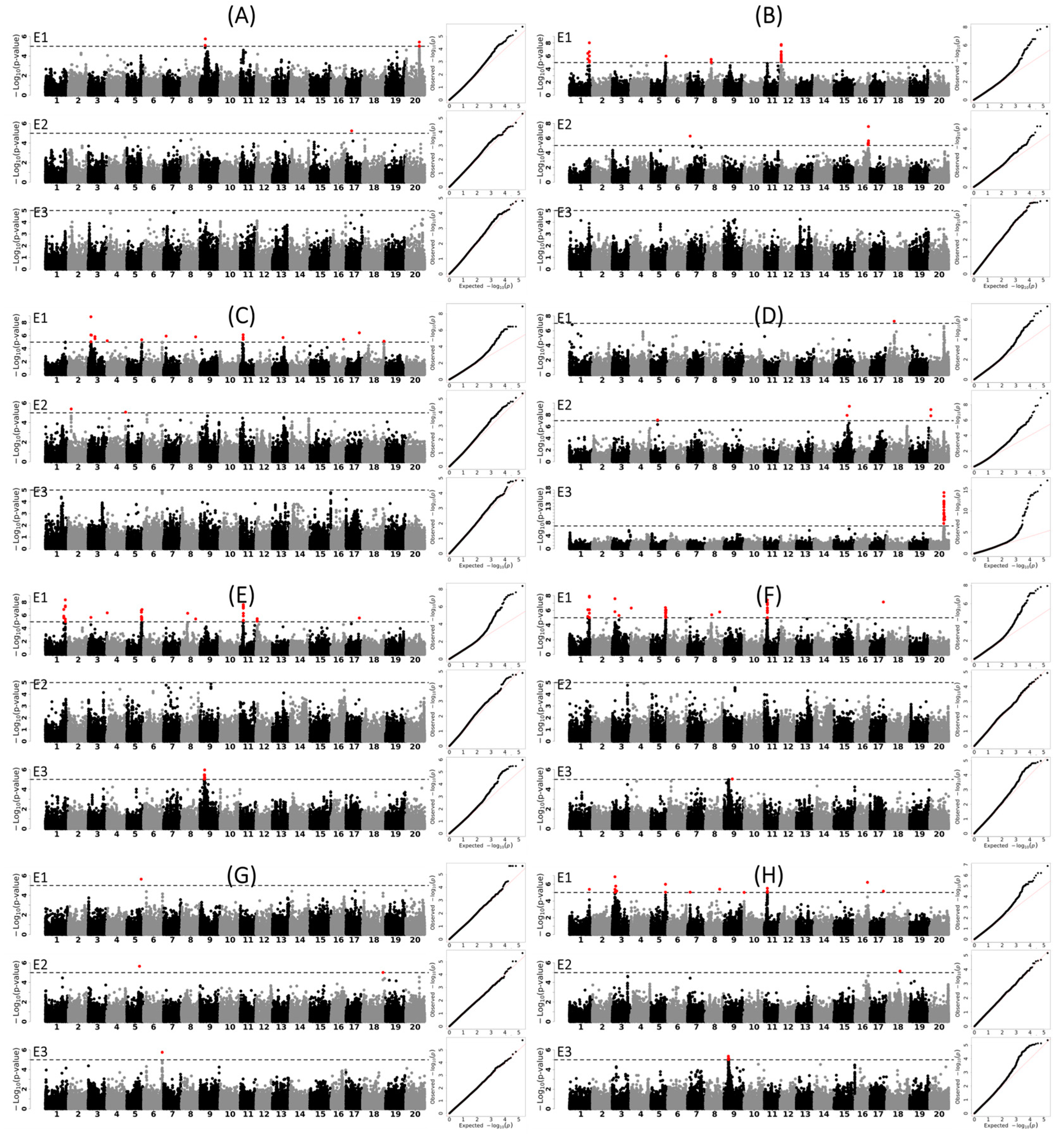
3.3. Genetic Loci and Candidate Genes Associated with Different Agronomic Traits
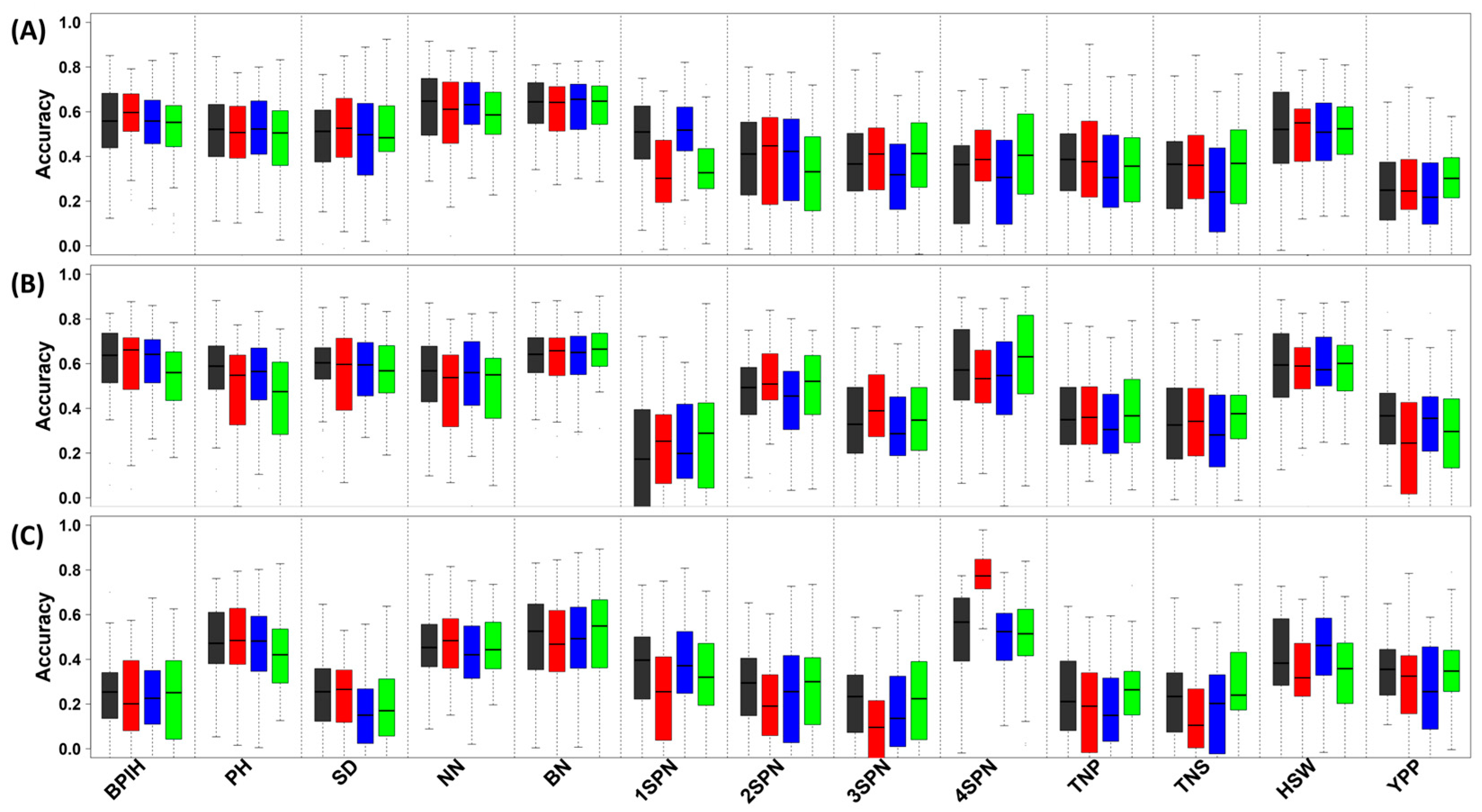
3.4. Genomic Prediction of Essential Agronomic Traits
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hartman, G.L.; West, E.D.; Herman, T.K. Crops that feed the World 2. Soybean-worldwide production, use, and constraints caused by pathogens and pests. Food Secur. 2011, 3, 5–17. [Google Scholar] [CrossRef]
- Sedivy, E.J.; Wu, F.; Hanzawa, Y. Soybean domestication: The origin, genetic architecture and molecular bases. New Phytol. 2017, 214, 539–553. [Google Scholar] [CrossRef] [PubMed]
- Tian, Z.; Nepomuceno, A.L.; Song, Q.; Stupar, R.M.; Liu, B.; Kong, F.; Ma, J.; Lee, S.H.; Jackson, S.A. Soybean2035: A decadal vision for soybean functional genomics and breeding. Mol. Plant 2025, 18, 245–271. [Google Scholar] [CrossRef] [PubMed]
- Hymowitz, T. Speciation and Cytogenetics. In Soybeans: Improvement, Production, and Uses, 3rd ed.; Boerma, H.R., Specht, J.E., Eds.; American Society of Agronomy, Inc.: Madison, WI, USA, 2004; pp. 97–136. [Google Scholar]
- Wilson, R.F. Seed Composition. In Soybeans: Improvement, Production, and Uses, 3rd ed.; Boerma, H.R., Specht, J.E., Eds.; American Society of Agronomy, Inc.: Madison, WI, USA, 2004; pp. 621–677. [Google Scholar]
- Duan, Z.; Xu, L.; Zhou, G.; Zhu, Z.; Wang, X.; Shen, Y.; Ma, X.; Tian, Z.; Fang, C. Unlocking soybean potential: Genetic resources and omics for breeding. J. Genet. Genom. 2025, in press. [Google Scholar] [CrossRef]
- Ray, D.K.; Mueller, N.D.; West, P.C.; Foley, J.A. Yield Trends Are Insufficient to Double Global Crop Production by 2050. PLoS ONE 2013, 8, e66428. [Google Scholar] [CrossRef]
- Liu, S.; Zhang, M.; Feng, F.; Tian, Z. Toward a “Green Revolution” for Soybean. Mol. Plant 2020, 13, 688–697. [Google Scholar] [CrossRef]
- Wu, T.T.; Sun, S.; Wang, C.J.; Lu, W.C.; Sun, B.C.; Song, X.Q.; Han, X.Z.; Guo, T.; Man, W.Q.; Cheng, Y.X.; et al. Characterizing Changes from a Century of Genetic Improvement of Soybean Cultivars in Northeast China. Crop Sci. 2015, 55, 2056–2067. [Google Scholar] [CrossRef]
- Guo, S.; Zhang, Z.; Zhang, F.; Yang, X. Optimizing cultivars and agricultural management practices can enhance soybean yield in Northeast China. Sci. Total Environ. 2023, 857, 159456. [Google Scholar] [CrossRef]
- Zhang, L.; Zheng, H.Y.; Li, W.J.; Olesen, J.E.; Harrison, M.T.; Bai, Z.Y.; Zou, J.; Zheng, A.X.; Bernacchi, C.; Xu, X.Y.; et al. Genetic progress battles climate variability: Drivers of soybean yield gains in China from 2006 to 2020. Agron. Sustain. Dev. 2023, 43, 50. [Google Scholar] [CrossRef]
- Liu, H.J.; Yan, J. Crop genome-wide association study: A harvest of biological relevance. Plant J. 2019, 97, 8–18. [Google Scholar] [CrossRef]
- Tibbs Cortes, L.; Zhang, Z.; Yu, J. Status and prospects of genome-wide association studies in plants. Plant Genome 2021, 14, e20077. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Zhao, X.; Han, Y.; Li, W.; Xie, F. Genome-wide association mapping for seed protein and oil contents using a large panel of soybean accessions. Genomics 2019, 111, 90–95. [Google Scholar] [CrossRef]
- Dong, L.; Fang, C.; Cheng, Q.; Su, T.; Kou, K.; Kong, L.; Zhang, C.; Li, H.; Hou, Z.; Zhang, Y.; et al. Genetic basis and adaptation trajectory of soybean from its temperate origin to tropics. Nat. Commun. 2021, 12, 5445. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.M.; Yan, J.; Jiang, S.Q.; Li, X.; Min, H.W.; Wang, X.F.; Hao, D.Y. Resequencing 250 Soybean Accessions: New Insights into Genes Associated with Agronomic Traits and Genetic Networks. Genom. Proteom. Bioinform. 2022, 20, 29–41. [Google Scholar] [CrossRef]
- Sharmin, R.A.; Karikari, B.; Chang, F.; Al Amin, G.M.; Bhuiyan, M.R.; Hina, A.; Lv, W.; Chunting, Z.; Begum, N.; Zhao, T. Genome-wide association study uncovers major genetic loci associated with seed flooding tolerance in soybean. BMC Plant Biol. 2021, 21, 497. [Google Scholar] [CrossRef]
- Bhat, J.A.; Yu, H.; Weng, L.; Yuan, Y.; Zhang, P.; Leng, J.; He, J.; Zhao, B.; Bu, M.; Wu, S.; et al. GWAS analysis revealed genomic loci and candidate genes associated with the 100-seed weight in high-latitude-adapted soybean germplasm. Theor. Appl. Genet. 2025, 138, 29. [Google Scholar] [CrossRef]
- Zhang, Q.; Sun, T.; Wang, J.; Fei, J.; Liu, Y.; Liu, L.; Wang, P. Genome-wide association study and high-quality gene mining related to soybean protein and fat. BMC Genom. 2023, 24, 596. [Google Scholar] [CrossRef] [PubMed]
- Fang, C.; Ma, Y.; Wu, S.; Liu, Z.; Wang, Z.; Yang, R.; Hu, G.; Zhou, Z.; Yu, H.; Zhang, M.; et al. Genome-wide association studies dissect the genetic networks underlying agronomical traits in soybean. Genome Biol. 2017, 18, 161. [Google Scholar] [CrossRef] [PubMed]
- Diers, B.W.; Specht, J.; Rainey, K.M.; Cregan, P.; Song, Q.J.; Ramasubramanian, V.; Graef, G.; Nelson, R.; Schapaugh, W.; Wang, D.C.; et al. Genetic Architecture of Soybean Yield and Agronomic Traits. G3 Genes Genom. Genet. 2018, 8, 3367–3375. [Google Scholar] [CrossRef]
- Bhat, J.A.; Adeboye, K.A.; Ganie, S.A.; Barmukh, R.; Hu, D.; Varshney, R.K.; Yu, D. Genome-wide association study, haplotype analysis, and genomic prediction reveal the genetic basis of yield-related traits in soybean (Glycine max L.). Front. Genet. 2022, 13, 953833. [Google Scholar] [CrossRef]
- Niu, M.; Tian, K.; Chen, Q.; Yang, C.; Zhang, M.; Sun, S.; Wang, X. A multi-trait GWAS-based genetic association network controlling soybean architecture and seed traits. Front. Plant Sci. 2023, 14, 1302359. [Google Scholar] [CrossRef] [PubMed]
- Bernardo, R. Molecular markers and selection for complex traits in plants: Learning from the last 20 years. Crop Sci. 2008, 48, 1649–1664. [Google Scholar] [CrossRef]
- Melchinger, A.E.; Utz, H.F.; Schon, C.C. Quantitative trait locus (QTL) mapping using different testers and independent population samples in maize reveals low power of QTL detection and large bias in estimates of QTL effects. Genetics 1998, 149, 383–403. [Google Scholar] [CrossRef] [PubMed]
- Meuwissen, T.H.; Hayes, B.J.; Goddard, M.E. Prediction of total genetic value using genome-wide dense marker maps. Genetics 2001, 157, 1819–1829. [Google Scholar] [CrossRef]
- Moeinizade, S.; Kusmec, A.; Hu, G.; Wang, L.; Schnable, P.S. Multi-trait Genomic Selection Methods for Crop Improvement. Genetics 2020, 215, 931–945. [Google Scholar] [CrossRef]
- Crossa, J.; Perez-Rodriguez, P.; Cuevas, J.; Montesinos-Lopez, O.; Jarquin, D.; de Los Campos, G.; Burgueno, J.; Gonzalez-Camacho, J.M.; Perez-Elizalde, S.; Beyene, Y.; et al. Genomic Selection in Plant Breeding: Methods, Models, and Perspectives. Trends Plant Sci. 2017, 22, 961–975. [Google Scholar] [CrossRef]
- Sandhu, K.; Patil, S.S.; Pumphrey, M.; Carter, A. Multitrait machine- and deep-learning models for genomic selection using spectral information in a wheat breeding program. Plant Genome 2021, 14, e20119. [Google Scholar] [CrossRef]
- Bayer, P.E.; Petereit, J.; Danilevicz, M.F.; Anderson, R.; Batley, J.; Edwards, D. The application of pangenomics and machine learning in genomic selection in plants. Plant Genome 2021, 14, e20112. [Google Scholar] [CrossRef]
- Matei, G.; Woyann, L.G.; Milioli, A.S.; Oliveira, I.D.; Zdziarski, A.D.; Zanella, R.; Coelho, A.S.G.; Finatto, T.; Benin, G. Genomic selection in soybean: Accuracy and time gain in relation to phenotypic selection. Mol. Breed. 2018, 38, 117. [Google Scholar] [CrossRef]
- Miller, M.J.; Song, Q.; Li, Z. Genomic selection of soybean (Glycine max) for genetic improvement of yield and seed composition in a breeding context. Plant Genome 2023, 16, e20384. [Google Scholar] [CrossRef]
- Wang, C.J.; Wu, T.T.; Wu, C.X.; Jiang, B.J.; Sun, S.; Hou, W.S.; Han, T.F. Changes in photo-thermal sensitivity of widely grown Chinese soybean cultivars due to a century of genetic improvement. Plant Breed. 2015, 134, 94–104. [Google Scholar] [CrossRef]
- Zhang, L.X.; Liu, W.; Tsegaw, M.; Xu, X.; Qi, Y.P.; Sepey, E.; Liu, L.P.; Wu, T.T.; Sun, S.; Han, T.F. Principles and practices of the photo-thermal adaptability improvement in soybean. J. Integr. Agric. 2020, 19, 295–310. [Google Scholar] [CrossRef]
- Bates, D.; Mächler, M.; Bolker, B.M.; Walker, S.C. Fitting Linear Mixed-Effects Models Using lme4. J. Stat. Softw. 2015, 67, 1–48. [Google Scholar] [CrossRef]
- Allen, G.C.; Flores-Vergara, M.A.; Krasynanski, S.; Kumar, S.; Thompson, W.F. A modified protocol for rapid DNA isolation from plant tissues using cetyltrimethylammonium bromide. Nat. Protoc. 2006, 1, 2320–2325. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. Gigascience 2021, 10, giab008. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef]
- Browning, B.L.; Browning, S.R. Genotype Imputation with Millions of Reference Samples. Am. J. Hum. Genet. 2016, 98, 116–126. [Google Scholar] [CrossRef]
- Felsenstein, J. Confidence Limits on Phylogenies: An Approach Using the Bootstrap. Evolution 1985, 39, 783–791. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Wang, R.; Ma, J.; Gao, H.; Deng, L.; Wang, N.; Wang, Y.; Zhang, J.; Li, K.; Zhang, W.; et al. Genome-wide association studies of yield-related traits in high-latitude japonica rice. BMC Genom. Data 2021, 22, 39. [Google Scholar] [CrossRef] [PubMed]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef]
- Zhou, X.; Stephens, M. Genome-wide efficient mixed-model analysis for association studies. Nat. Genet. 2012, 44, 821–824. [Google Scholar] [CrossRef]
- Moghaddam, S.M.; Mamidi, S.; Osorno, J.M.; Lee, R.; Brick, M.; Kelly, J.; Miklas, P.; Urrea, C.; Song, Q.; Cregan, P.; et al. Genome-Wide Association Study Identifies Candidate Loci Underlying Agronomic Traits in a Middle American Diversity Panel of Common Bean. Plant Genome 2016, 9, plantgenome2016-02. [Google Scholar] [CrossRef]
- Mamidi, S.; Chikara, S.; Goos, R.J.; Hyten, D.L.; Annam, D.; Moghaddam, S.M.; Lee, R.K.; Cregan, P.B.; McClean, P.E. Genome-Wide Association Analysis Identifies Candidate Genes Associated with Iron Deficiency Chlorosis in Soybean. Plant Genome 2011, 4, 154–164. [Google Scholar] [CrossRef]
- Crowell, S.; Korniliev, P.; Falcao, A.; Ismail, A.; Gregorio, G.; Mezey, J.; McCouch, S. Genome-wide association and high-resolution phenotyping link Oryza sativa panicle traits to numerous trait-specific QTL clusters. Nat. Commun. 2016, 7, 10527. [Google Scholar] [CrossRef]
- Endelman, J.B. Ridge Regression and Other Kernels for Genomic Selection with R Package rrBLUP. Plant Genome 2011, 4, 250–255. [Google Scholar] [CrossRef]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Bellinazzo, F.; Nadal Bigas, J.; Hogers, R.A.H.; Kodde, J.; van der Wal, F.; Kokkinopoulou, P.; Duijts, K.T.M.; Angenent, G.C.; van Dijk, A.D.J.; van Velzen, R.; et al. Evolutionary origin and functional investigation of the widely conserved plant PEBP gene stepmother of FT and TFL1 (SMFT). Plant J. 2024, 120, 1410–1420. [Google Scholar] [CrossRef]
- Tzafrir, I.; Pena-Muralla, R.; Dickerman, A.; Berg, M.; Rogers, R.; Hutchens, S.; Sweeney, T.C.; McElver, J.; Aux, G.; Patton, D.; et al. Identification of genes required for embryo development in Arabidopsis. Plant Physiol. 2004, 135, 1206–1220. [Google Scholar] [CrossRef]
- Jeong, N.; Suh, S.J.; Kim, M.H.; Lee, S.; Moon, J.K.; Kim, H.S.; Jeong, S.C. Ln is a key regulator of leaflet shape and number of seeds per pod in soybean. Plant Cell 2012, 24, 4807–4818. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Wu, J.; Romanovicz, D.; Clark, G.; Roux, S.J. Co-regulation of exine wall patterning, pollen fertility and anther dehiscence by Arabidopsis apyrases 6 and 7. Plant Physiol. Biochem. 2013, 69, 62–73. [Google Scholar] [CrossRef]
- Fitter, D.W.; Martin, D.J.; Copley, M.J.; Scotland, R.W.; Langdale, J.A. GLK gene pairs regulate chloroplast development in diverse plant species. Plant J. 2002, 31, 713–727. [Google Scholar] [CrossRef]
- Kou, K.; Yang, H.; Li, H.; Fang, C.; Chen, L.; Yue, L.; Nan, H.; Kong, L.; Li, X.; Wang, F.; et al. A functionally divergent SOC1 homolog improves soybean yield and latitudinal adaptation. Curr. Biol. 2022, 32, 1728–1742.e6. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.T.; Leiser, W.L.; Hahn, V.; Würschum, T. Identification of QTL for seed yield and agronomic traits in 944 soybean (Glycine max) RILs from a diallel cross of early-maturing varieties. Plant Breed. 2021, 140, 254–266. [Google Scholar] [CrossRef]
- Yang, K.; Jeong, N.; Moon, J.K.; Lee, Y.H.; Lee, S.H.; Kim, H.M.; Hwang, C.H.; Back, K.; Palmer, R.G.; Jeong, S.C. Genetic analysis of genes controlling natural variation of seed coat and flower colors in soybean. J. Hered. 2010, 101, 757–768. [Google Scholar] [CrossRef] [PubMed]
- Ping, J.; Liu, Y.; Sun, L.; Zhao, M.; Li, Y.; She, M.; Sui, Y.; Lin, F.; Liu, X.; Tang, Z.; et al. Dt2 is a gain-of-function MADS-domain factor gene that specifies semideterminacy in soybean. Plant Cell 2014, 26, 2831–2842. [Google Scholar] [CrossRef]
- Xun, H.; Wang, Y.; Yuan, J.; Lian, L.; Feng, W.; Liu, S.; Hong, J.; Liu, B.; Ma, J.; Wang, X. Non-CG DNA hypomethylation promotes photosynthesis and nitrogen fixation in soybean. Proc. Natl. Acad. Sci. USA 2024, 121, e2402946121. [Google Scholar] [CrossRef]
- Almeida-Silva, F.; Pedrosa-Silva, F.; Venancio, T.M. The Soybean Expression Atlas v2: A comprehensive database of over 5000 RNA-seq samples. Plant J. 2023, 116, 1041–1051. [Google Scholar] [CrossRef]
- Shen, Y.; Zhou, Z.; Wang, Z.; Li, W.; Fang, C.; Wu, M.; Ma, Y.; Liu, T.; Kong, L.A.; Peng, D.L.; et al. Global dissection of alternative splicing in paleopolyploid soybean. Plant Cell 2014, 26, 996–1008. [Google Scholar] [CrossRef]
- Varshney, R.K.; Bohra, A.; Yu, J.; Graner, A.; Zhang, Q.; Sorrells, M.E. Designing Future Crops: Genomics-Assisted Breeding Comes of Age. Trends Plant Sci. 2021, 26, 631–649. [Google Scholar] [CrossRef]
- Alemu, A.; Astrand, J.; Montesinos-Lopez, O.A.; Isidro, Y.S.J.; Fernandez-Gonzalez, J.; Tadesse, W.; Vetukuri, R.R.; Carlsson, A.S.; Ceplitis, A.; Crossa, J.; et al. Genomic selection in plant breeding: Key factors shaping two decades of progress. Mol. Plant 2024, 17, 552–578. [Google Scholar] [CrossRef] [PubMed]
- Crossa, J.; Martini, J.W.R.; Vitale, P.; Perez-Rodriguez, P.; Costa-Neto, G.; Fritsche-Neto, R.; Runcie, D.; Cuevas, J.; Toledo, F.; Li, H.; et al. Expanding genomic prediction in plant breeding: Harnessing big data, machine learning, and advanced software. Trends Plant Sci. 2025. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Xu, Y.; Hu, Z.L.; Xu, C.W. Genomic selection methods for crop improvement: Current status and prospects. Crop J. 2018, 6, 330–340. [Google Scholar] [CrossRef]
- Wang, S.; Wei, J.; Li, R.; Qu, H.; Chater, J.M.; Ma, R.; Li, Y.; Xie, W.; Jia, Z. Identification of optimal prediction models using multi-omic data for selecting hybrid rice. Heredity 2019, 123, 395–406. [Google Scholar] [CrossRef]
- Westhues, C.C.; Mahone, G.S.; da Silva, S.; Thorwarth, P.; Schmidt, M.; Richter, J.C.; Simianer, H.; Beissinger, T.M. Prediction of Maize Phenotypic Traits with Genomic and Environmental Predictors Using Gradient Boosting Frameworks. Front. Plant Sci. 2021, 12, 699589. [Google Scholar] [CrossRef]
- Montesinos-Lopez, A.; Rivera, C.; Pinto, F.; Pinera, F.; Gonzalez, D.; Reynolds, M.; Perez-Rodriguez, P.; Li, H.; Montesinos-Lopez, O.A.; Crossa, J. Multimodal deep learning methods enhance genomic prediction of wheat breeding. G3 Genes Genomes Genet. 2023, 13, jkad045. [Google Scholar] [CrossRef]
- Barreto, C.A.V.; das Gracas Dias, K.O.; de Sousa, I.C.; Azevedo, C.F.; Nascimento, A.C.C.; Guimaraes, L.J.M.; Guimaraes, C.T.; Pastina, M.M.; Nascimento, M. Genomic prediction in multi-environment trials in maize using statistical and machine learning methods. Sci. Rep. 2024, 14, 1062. [Google Scholar] [CrossRef]
- Cappa, E.P.; Chen, C.; Klutsch, J.G.; Sebastian-Azcona, J.; Ratcliffe, B.; Wei, X.; Da Ros, L.; Liu, Y.; Bhumireddy, S.R.; Benowicz, A.; et al. Revealing stable SNPs and genomic prediction insights across environments enhance breeding strategies of productivity, defense, and climate-adaptability traits in white spruce. Heredity 2025, 134, 186–199. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dong, Q.; Cheng, Y.; Li, Y.; Tong, Y.; Liu, D.; Yu, J.; Zhao, N.; Liu, B.; Ding, X.; Xu, C. Genome-Wide Association Study and Genomic Prediction of Essential Agronomic Traits in Diversity Panel of Soybean Varieties. Agronomy 2025, 15, 1181. https://doi.org/10.3390/agronomy15051181
Dong Q, Cheng Y, Li Y, Tong Y, Liu D, Yu J, Zhao N, Liu B, Ding X, Xu C. Genome-Wide Association Study and Genomic Prediction of Essential Agronomic Traits in Diversity Panel of Soybean Varieties. Agronomy. 2025; 15(5):1181. https://doi.org/10.3390/agronomy15051181
Chicago/Turabian StyleDong, Qianli, Yuting Cheng, Yiyang Li, Yan Tong, Dazhuang Liu, Jiaxin Yu, Na Zhao, Bao Liu, Xiaoyang Ding, and Chunming Xu. 2025. "Genome-Wide Association Study and Genomic Prediction of Essential Agronomic Traits in Diversity Panel of Soybean Varieties" Agronomy 15, no. 5: 1181. https://doi.org/10.3390/agronomy15051181
APA StyleDong, Q., Cheng, Y., Li, Y., Tong, Y., Liu, D., Yu, J., Zhao, N., Liu, B., Ding, X., & Xu, C. (2025). Genome-Wide Association Study and Genomic Prediction of Essential Agronomic Traits in Diversity Panel of Soybean Varieties. Agronomy, 15(5), 1181. https://doi.org/10.3390/agronomy15051181