
Transcriptomic and Metabolic Analyses Elucidate the Metabolomic Variation in Leaf Development of a Calcium-Rich Vegetable (Primulina eburnea)


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. Determination of Physiological Traits
2.3. RNA-seq and Expression Analysis
2.4. Metabolite Analysis
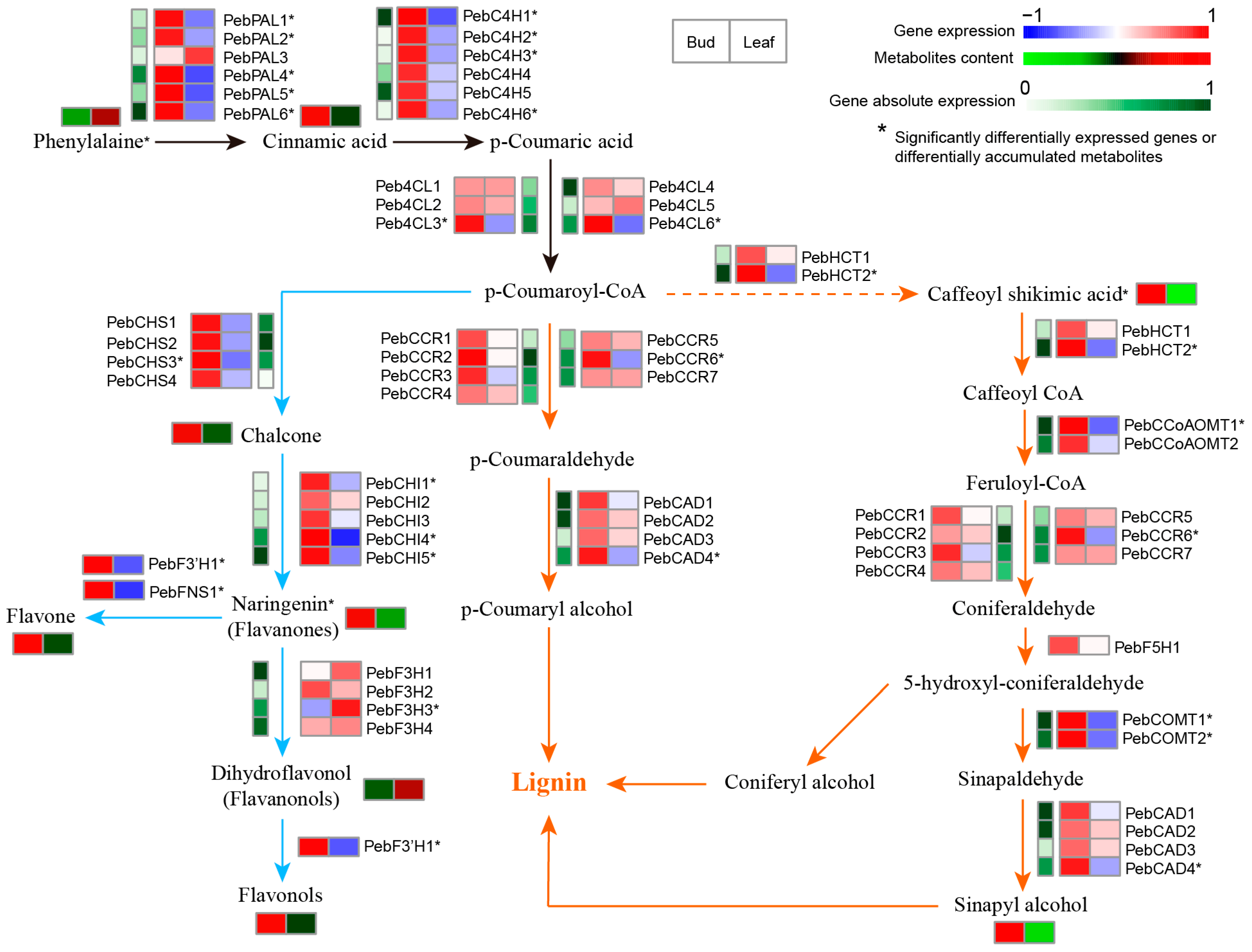
2.5. Metabolite Biosynthesis Pathway Analysis
2.6. Validation by Quantitative Real-Time PCR (qRT-PCR)
2.7. Statistical Analysis Network Construction
3. Results
3.1. Physiological Characteristics of Primulina eburnea Leaf during Development
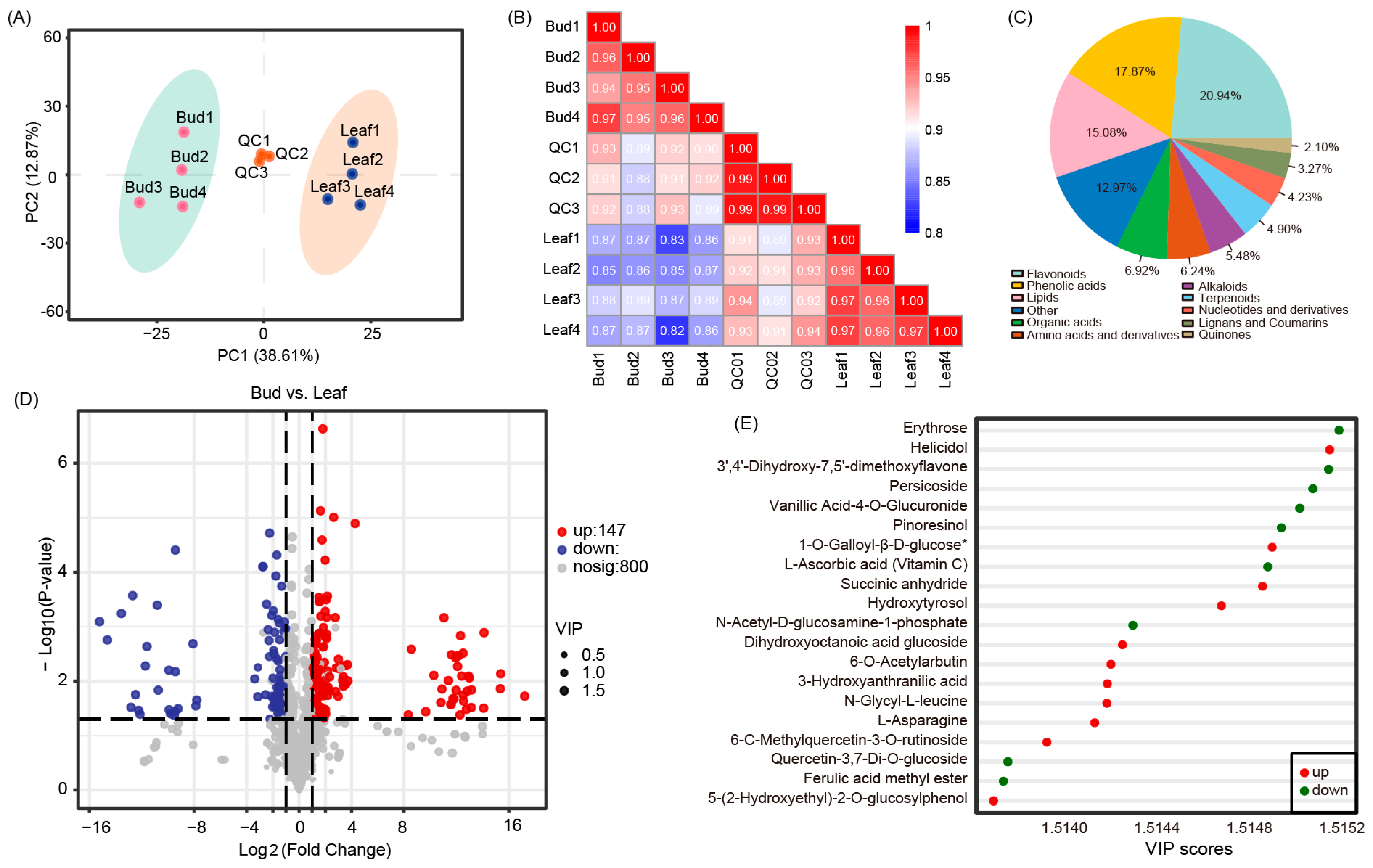
3.2. Metabolome Profiling
3.3. Differentially Accumulated Metabolites (DAMs) in Bud and Mature Leaves
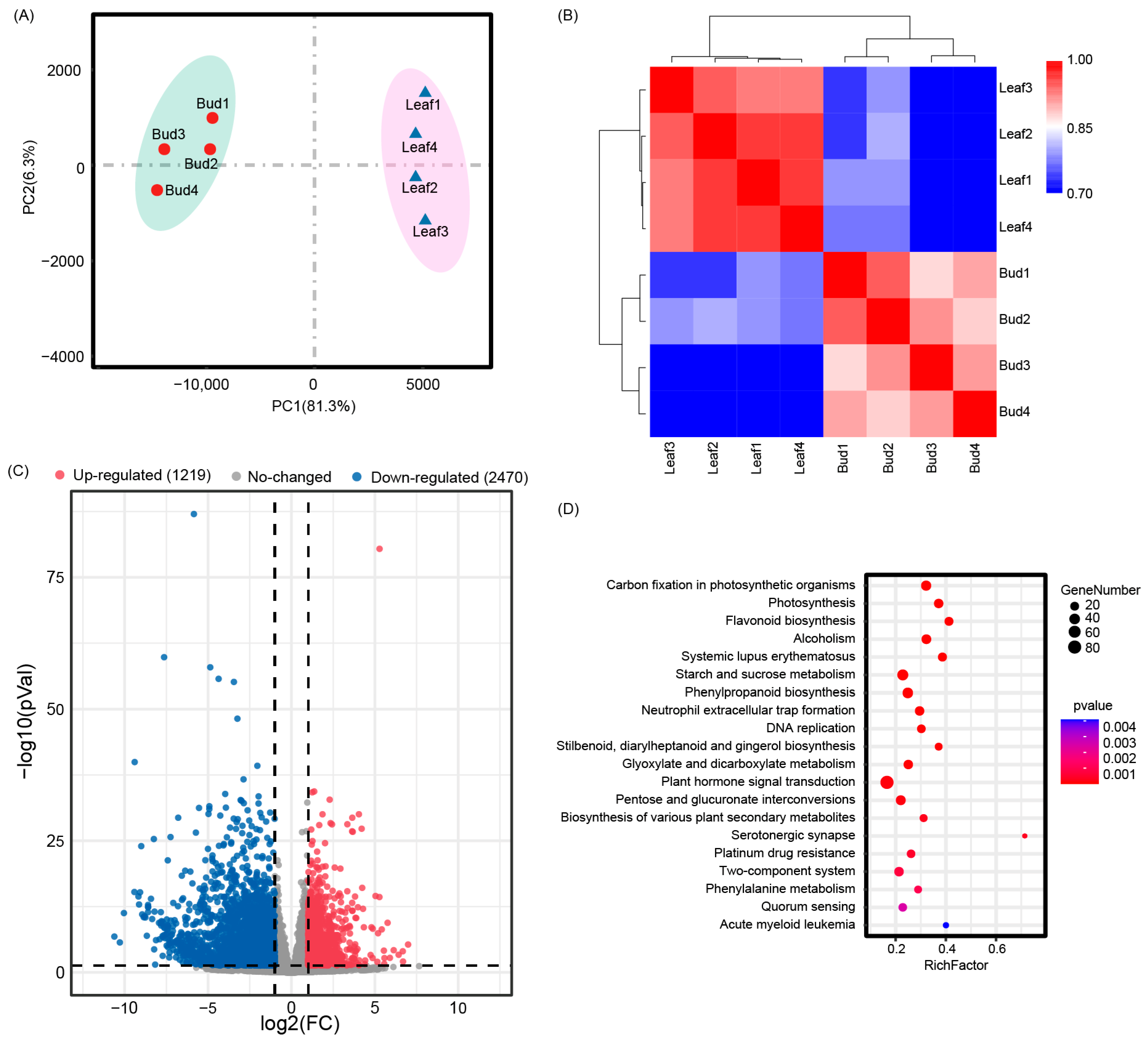
3.4. Transcriptome Profiles of Bud and Mature Leaves
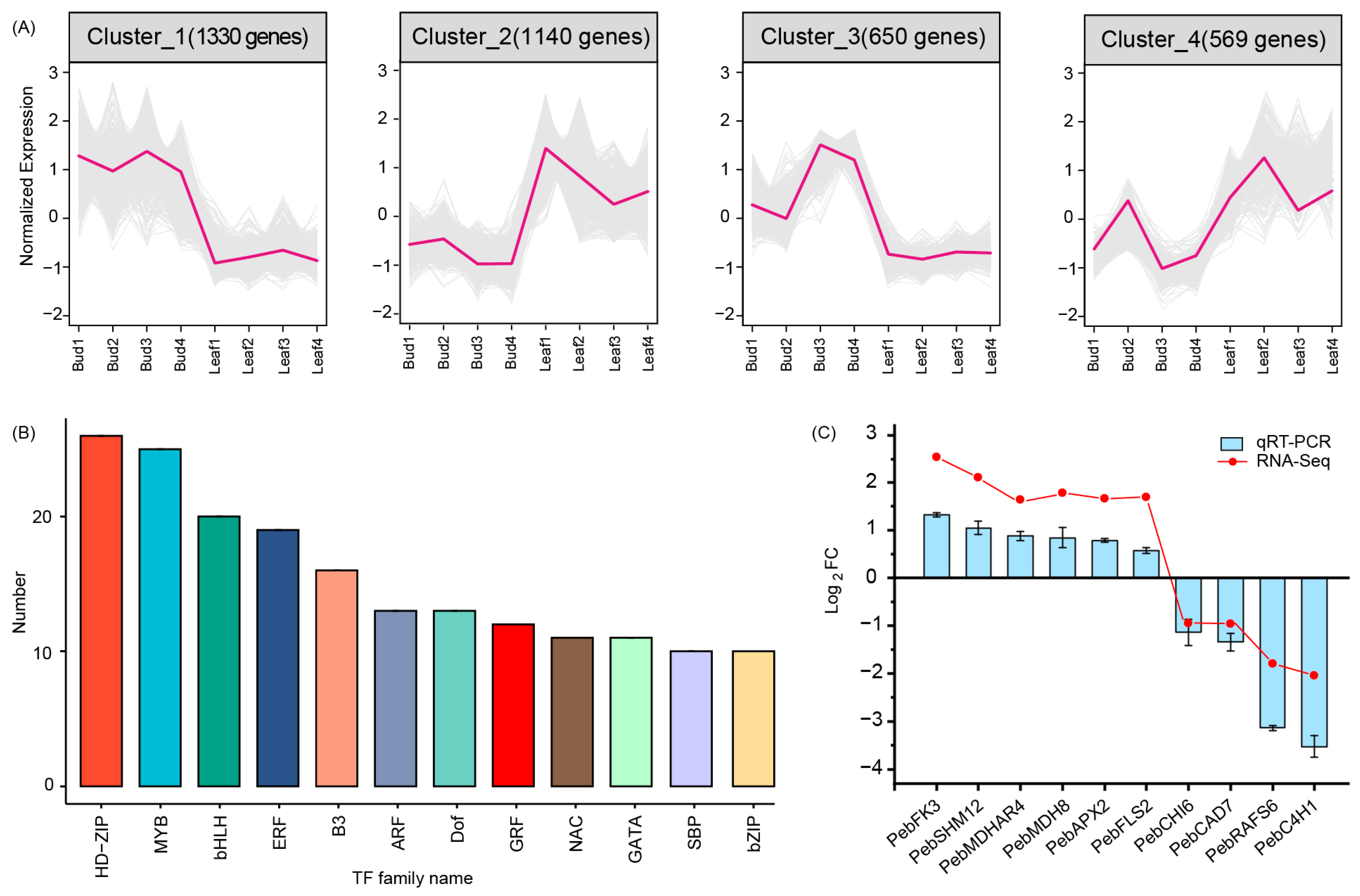
3.5. Differentially Expressed Genes between Bud and Mature Leaves
3.6. qRT-PCR Validation
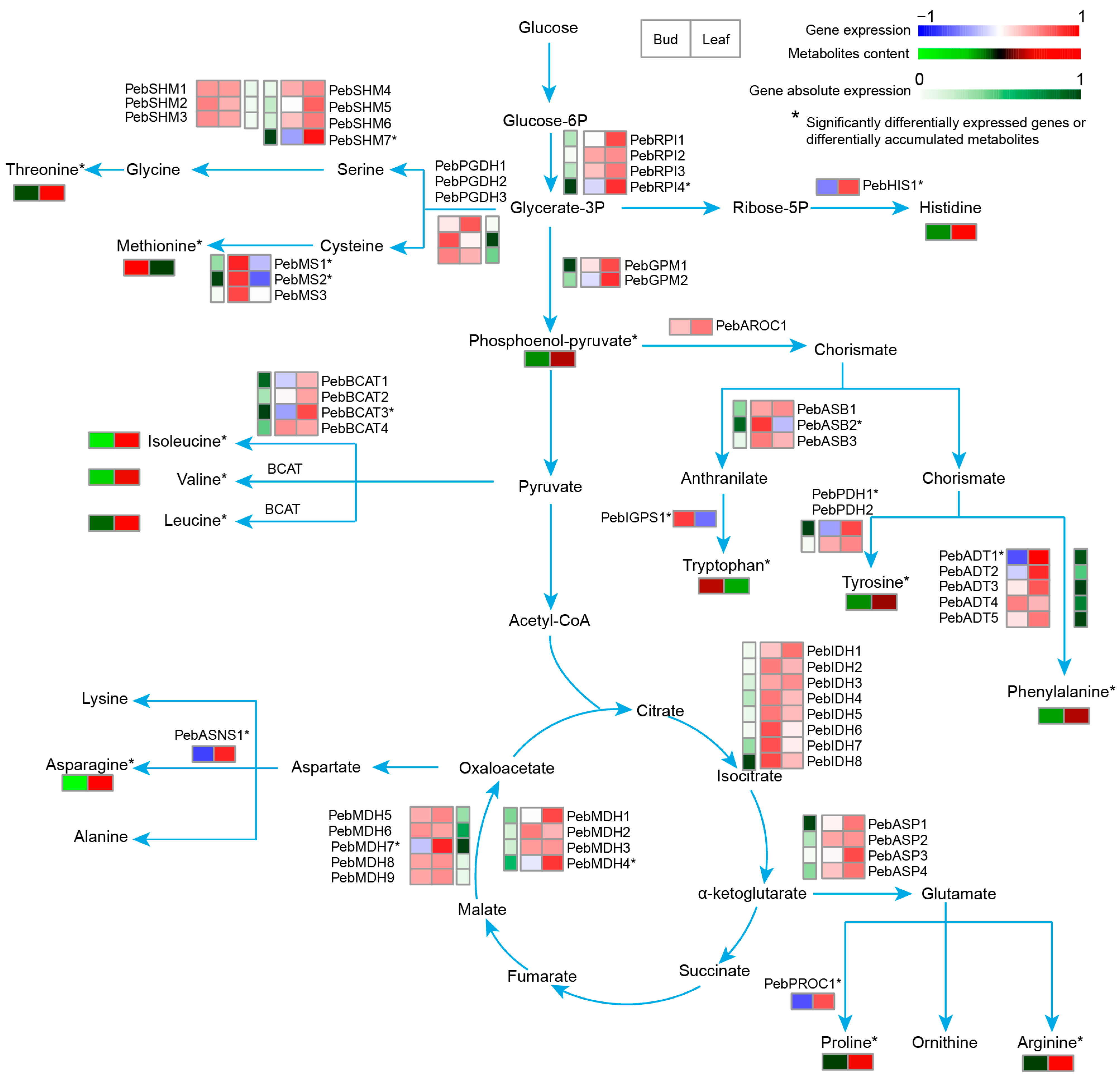
3.7. Related Genes in P. eburnea Flavor and Nutrition
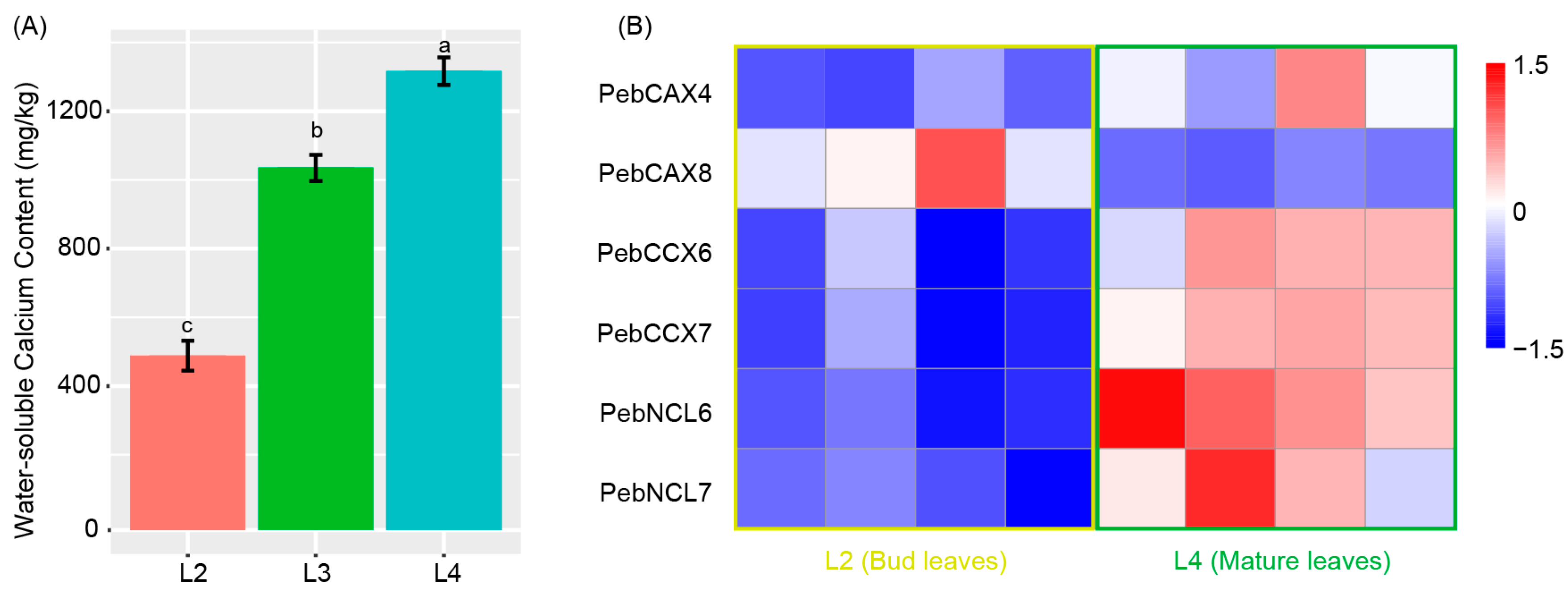
3.8. Metabolism of Water-Soluble Calcium in P. eburnea
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Glade, M.J.; Altman, R.D.; Bezman, R.J.; Davis, R.M.; Genel, M.; Howe, J.P.; Karlan, M.S.; Numan, P.J.; Riggs, J.A.; Skelton, W.D.; et al. Intake of dietary calcium to reduce the incidence of osteoporosis. Arch. Fam. Med. 1997, 6, 495–499. [Google Scholar]
- Balk, E.M.; Adam, G.P.; Langberg, V.N.; Earley, A.; Clark, P.; Ebeling, P.R.; Mithal, A.; Rizzoli, R.; Zerbini, C.A.F.; Pierroz, D.D.; et al. Global dietary calcium intake among adults: A systematic review. Osteoporos. Int. 2017, 28, 3315–3324. [Google Scholar] [CrossRef]
- Yao, X.C.; Hu, J.T.; Kong, X.H.; Zhu, Z.X. Association between Dietary Calcium Intake and Bone Mineral Density in Older Adults. Ecol. Food Nutr. 2021, 60, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Albertson, A.M.; Tobelmann, R.C.; Marquart, L. Estimated dietary calcium intake and food sources for adolescent females: 1980–1992. J. Adolesc. Health 1997, 20, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Weaver, C.M.; Alexander, D.D.; Boushey, C.J.; Dawson-Hughes, B.; Lappe, J.M.; LeBoff, M.S.; Liu, S.; Looker, A.C.; Wallace, T.C.; Wang, D.D. Calcium plus vitamin D supplementation and risk of fractures: An updated meta-analysis from the National Osteoporosis Foundation. Osteoporos. Int. 2016, 27, 367–376. [Google Scholar] [CrossRef]
- Haas, E. Staying Healthy with Nutrition; Celestial Arts Publisher: Toronto, ON, Canada, 2015. [Google Scholar]
- Wang, Y.-Z.; Mao, R.-B.; Liu, Y.; Li, J.-M.; Dong, Y.; Li, Z.-Y.; Smith, J.F. Phylogenetic reconstruction of Chirita and allies (Gesneriaceae) with taxonomic treatments. J. Syst. Evol. 2011, 49, 50–64. [Google Scholar] [CrossRef]
- Qi, Q.; Hao, Z.; Tao, J.; Kang, M. Diversity of calcium speciation in leaves of Primulina species (Gesneriaceae). Biodivers. Sci. 2014, 21, 715–722. [Google Scholar] [CrossRef]
- Zhang, Y.; Fu, C.; Zhou, R.; Liu, Z.; Feng, C. Effect of prechilling and exogenous gibberellin on seed germination of Primulina eburnea: A calcium-rich vegetable. Seed Sci. Technol. 2023, 51, 1–6. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, J.; Zou, S.; Liu, Z.; Huang, H.; Feng, C. Genome-wide analysis of the cellulose toolbox of Primulina eburnea, a calcium-rich vegetable. BMC Plant Biol. 2023, 23, 259. [Google Scholar] [CrossRef]
- Cai, X.H.; Luo, X.D.; Zhou, J.; Hao, X.J. A new naphthaquinone derivative from Chirita eburnea. J. Asian Nat. Prod. Res. 2006, 8, 351–353. [Google Scholar] [CrossRef]
- Wang, X.; Peng, Y.; Bai, Z.; Liu, Y. Antibacterial activity of plants of Gesneriaceae. J. Med. Pharm. Chin. Minor. 2011, 1, 37–38. [Google Scholar]
- Liu, H.; Li, B. A review of research developments in Primulina eburnea. J. Guilin Norm. Coll. 2018, 122, 110–113. [Google Scholar]
- Yang, L.; Wang, C.; Chen, J.; Qiu, J.; Du, C.; Wei, Y.; Hao, X.; Gu, W. Chemical constituents and bioactivitie of whole plant of Primulina eburnea from Guizhou. Chin. Tradit. Herb. Drugs 2023, 54, 3430–3437. [Google Scholar]
- Li, J.J.; Yu, X.J.; Shan, Q.R.; Shi, Z.B.; Li, J.H.; Zhao, X.T.; Chang, C.F.; Yu, J.J. Integrated volatile metabolomic and transcriptomic analysis provides insights into the regulation of floral scents between two contrasting varieties of Lonicera japonica. Front. Plant Sci. 2022, 13, 989036. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.L.; Zhang, S.B.; Li, X.; Wang, X.Q.; Li, Z.D.; Zhu, X.Y.; Liu, X.X.; Li, H.X.; Zhang, J.; Chen, X.L. Integrative analysis of high temperature-induced transcriptome and metabolome alterations in the leaves of five raspberry (Rubus ideaus L.) cultivars. Environ. Exp. Bot. 2022, 203, 105038. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, W.; Huang, W.; Biao, A.; Lin, S.; Wang, Y.; Yan, S.; Zeng, S. Salt stress affects the fruit quality of Lycium ruthenicum Murr. Ind. Crops Prod. 2023, 193, 116240. [Google Scholar] [CrossRef]
- Feng, C.; Feng, C.; Lin, X.; Liu, S.; Li, Y.; Kang, M. A chromosome-level genome assembly provides insights into ascorbic acid accumulation and fruit softening in guava (Psidium guajava). Plant Biotechnol. J. 2020, 19, 717–730. [Google Scholar] [CrossRef]
- Yi, H.; Wang, J.; Wang, J.; Rausher, M.; Kang, M. Genomic insights into inter- and intraspecific mating system shifts in Primulina. Mol. Ecol. 2022, 31, 5699–5713. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.-C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef]
- Saeed, A.I.; Bhagabati, N.K.; Braisted, J.C.; Liang, W.; Sharov, V.; Howe, E.A.; Li, J.; Thiagarajan, M.; White, J.A.; Quackenbush, J. TM4 microarray software suite. Methods Enzymol. 2006, 411, 134–193. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Shi, Q.; Wang, B.; Ma, A.; Wang, Y.; Xue, Q.; Shen, B.; Hamaila, H.; Tang, T.; Qi, X.; et al. Jujube metabolome selection determined the edible properties acquired during domestication. Plant J. 2022, 109, 1116–1133. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Gao, W.; Song, M.; Li, M.; He, D.; Wang, Z. Integrated Metabolome and Transcriptome Analysis of Fruit Flavor and Carotenoids Biosynthesis Differences Between Mature-Green and Tree-Ripe of cv. “Golden Phoenix” Mangoes (Mangifera indica L.). Front. Plant Sci. 2022, 13, 816492. [Google Scholar] [CrossRef]
- Chen, W.; Gong, L.; Guo, Z.; Wang, W.; Zhang, H.; Liu, X.; Yu, S.; Xiong, L.; Luo, J. A Novel Integrated Method for Large-Scale Detection, Identification, and Quantification of Widely Targeted Metabolites: Application in the Study of Rice Metabolomics. Mol. Plant 2013, 6, 1769–1780. [Google Scholar] [CrossRef] [PubMed]
- Kohl, M.; Wiese, S.; Warscheid, B. Cytoscape: Software for Visualization and Analysis of Biological Networks. In Data Mining in Proteomics: From Standards to Applications; Hamacher, M., Eisenacher, M., Stephan, C., Eds.; Humana Press: Totowa, NJ, USA, 2011; pp. 291–303. [Google Scholar]
- Allan, A.C.; Espley, R.V. MYBs Drive Novel Consumer Traits in Fruits and Vegetables. Trends Plant Sci. 2018, 23, 693–705. [Google Scholar] [CrossRef] [PubMed]
- Pireyre, M.; Burow, M. Regulation of MYB and bHLH transcription factors: A glance at the protein level. Mol. Plant 2015, 8, 378–388. [Google Scholar] [CrossRef]
- Riechmann, J.L.; Meyerowitz, E.M. The AP2/EREBP family of plant transcription factors. Biol. Chem. 1998, 379, 633–646. [Google Scholar] [CrossRef]
- Chinnusamy, V.; Schumaker, K.; Zhu, J.K. Molecular genetic perspectives on cross-talk and specificity in abiotic stress signalling in plants. J. Exp. Bot. 2004, 55, 225–236. [Google Scholar] [CrossRef]
- Falcone Ferreyra, M.L.; Rius, S.P.; Casati, P. Flavonoids: Biosynthesis, biological functions, and biotechnological applications. Front. Plant Sci. 2012, 3, 222. [Google Scholar] [CrossRef]
- Haug-Collet, K.; Pearson, B.; Webel, R.; Szerencsei, R.T.; Winkfein, R.J.; Schnetkamp, P.P.M.; Colley, N.J. Cloning and characterization of a potassium-dependent sodium/calcium exchanger in Drosophila. J. Cell Biol. 1999, 147, 659–669. [Google Scholar] [CrossRef]
- Sanders, D.; Brownlee, C.; Harper, J.F. Communicating with calcium. Plant Cell 1999, 11, 691–706. [Google Scholar] [CrossRef] [PubMed]
- Axelsen, K.B.; Palmgren, M.G. Inventory of the superfamily of P-type ion pumps in Arabidopsis. Plant Physiol. 2001, 126, 696–706. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Capanoglu, E.; Jassbi, A.R.; Miron, A. Advance on the Flavonoid C-glycosides and Health Benefits. Crit. Rev. Food Sci. Nutr. 2016, 56 (Suppl. S1), S29–S45. [Google Scholar] [CrossRef]
- Umer, M.J.; Bin Safdar, L.; Gebremeskel, H.; Zhao, S.; Yuan, P.; Zhu, H.; Kaseb, M.O.; Anees, M.; Lu, X.; He, N.; et al. Identification of key gene networks controlling organic acid and sugar metabolism during watermelon fruit development by integrating metabolic phenotypes and gene expression profiles. Hortic. Res. 2020, 7, 193. [Google Scholar] [CrossRef] [PubMed]
- Pattanagul, W.; Thitisaksakul, M. Effect of salinity stress on growth and carbohydrate metabolism in three rice (Oryza sativa L.) cultivars differing in salinity tolerance. Indian J. Exp. Biol. 2008, 46, 736–742. [Google Scholar]
- Du, Y.; Zhao, Q.; Chen, L.; Yao, X.; Zhang, W.; Zhang, B.; Xie, F. Effect of drought stress on sugar metabolism in leaves and roots of soybean seedlings. Plant Physiol. Biochem. 2020, 146, 1–12. [Google Scholar] [CrossRef]
- Boriboonkaset, T.; Theerawitaya, C.; Yamada, N.; Pichakum, A.; Supaibulwatana, K.; Cha-um, S.; Takabe, T.; Kirdmanee, C. Regulation of some carbohydrate metabolism-related genes, starch and soluble sugar contents, photosynthetic activities and yield attributes of two contrasting rice genotypes subjected to salt stress. Protoplasma 2013, 250, 1157–1167. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Liang, Y.S.; Zhang, B.Q.; Song, X.P.; Li, Y.R.; Li, C.N.; Qin, Z.Q.; Li, D.W.; Wei, J.G.; Wu, J.M. Comparative Transcriptome Analysis Reveals Potential Gene Modules Associated with Cold Tolerance in Sugarcane (Saccharum officinarum L.). J. Plant Growth Regul. 2022, 41, 2614–2628. [Google Scholar] [CrossRef]
- Krasensky, J.; Jonak, C. Drought, salt, and temperature stress-induced metabolic rearrangements and regulatory networks. J. Exp. Bot. 2012, 63, 1593–1608. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.Y.; Song, C.C.; Brummell, D.A.; Qi, S.N.; Lin, Q.; Bi, J.F.; Duan, Y.Q. Salicylic acid treatment mitigates chilling injury in peach fruit by regulation of sucrose metabolism and soluble sugar content. Food Chem. 2021, 358, 129867. [Google Scholar] [CrossRef]
- Liu, X.; Chen, L.; Shi, W.; Xu, X.; Li, Z.; Liu, T.; He, Q.; Xie, C.; Nie, B.; Song, B. Comparative transcriptome reveals distinct starch-sugar interconversion patterns in potato genotypes contrasting for cold-induced sweetening capacity. Food Chem. 2021, 334, 127550. [Google Scholar] [CrossRef] [PubMed]
- Ding, P.; Ding, Y. Stories of Salicylic Acid: A Plant Defense Hormone. Trends Plant Sci. 2020, 25, 549–565. [Google Scholar] [CrossRef] [PubMed]
- Dempsey, D.A.; Vlot, A.C.; Wildermuth, M.C.; Klessig, D.F. Salicylic Acid biosynthesis and metabolism. Arab. Book 2011, 9, e0156. [Google Scholar] [CrossRef] [PubMed]
- Bhuiyan, N.H.; Selvaraj, G.; Wei, Y.; King, J. Role of lignification in plant defense. Plant Signal. Behav. 2009, 4, 158–159. [Google Scholar] [CrossRef] [PubMed]
- Soares, A.R.; de Cássia Siqueira-Soares, R.; Salvador, V.H.; de Lourdes Lucio Ferrarese, M.; Ferrarese-Filho, O. The effects of l-DOPA on root growth, lignification and enzyme activity in soybean seedlings. Acta Physiol. Plant. 2012, 34, 1811–1817. [Google Scholar] [CrossRef]
- Soares, A.R.; Marchiosi, R.; Siqueira-Soares Rde, C.; Barbosa de Lima, R.; Dantas dos Santos, W.; Ferrarese-Filho, O. The role of L-DOPA in plants. Plant Signal. Behav. 2014, 9, e28275. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Yang, E.; Liu, Q.; Feng, C. Transcriptomic and Metabolic Analyses Elucidate the Metabolomic Variation in Leaf Development of a Calcium-Rich Vegetable (Primulina eburnea). Agronomy 2023, 13, 2157. https://doi.org/10.3390/agronomy13082157
Zhang Y, Yang E, Liu Q, Feng C. Transcriptomic and Metabolic Analyses Elucidate the Metabolomic Variation in Leaf Development of a Calcium-Rich Vegetable (Primulina eburnea). Agronomy. 2023; 13(8):2157. https://doi.org/10.3390/agronomy13082157
Chicago/Turabian StyleZhang, Yi, Endian Yang, Qin Liu, and Chen Feng. 2023. "Transcriptomic and Metabolic Analyses Elucidate the Metabolomic Variation in Leaf Development of a Calcium-Rich Vegetable (Primulina eburnea)" Agronomy 13, no. 8: 2157. https://doi.org/10.3390/agronomy13082157
APA StyleZhang, Y., Yang, E., Liu, Q., & Feng, C. (2023). Transcriptomic and Metabolic Analyses Elucidate the Metabolomic Variation in Leaf Development of a Calcium-Rich Vegetable (Primulina eburnea). Agronomy, 13(8), 2157. https://doi.org/10.3390/agronomy13082157