

Genome-Resolved Metagenomics of Nitrogen Transformations in the Switchgrass Rhizosphere Microbiome on Marginal Lands

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Site Description and Management
2.2. DNA Extraction and Sequencing
2.3. Metagenomic Assembly, Annotation, Differential Abundance Statistical Analysis, and Genome Reconstruction
2.4. Read-Based mOTU Picking and Statistical Analysis
2.5. Data and Analysis Code Availability
3. Results
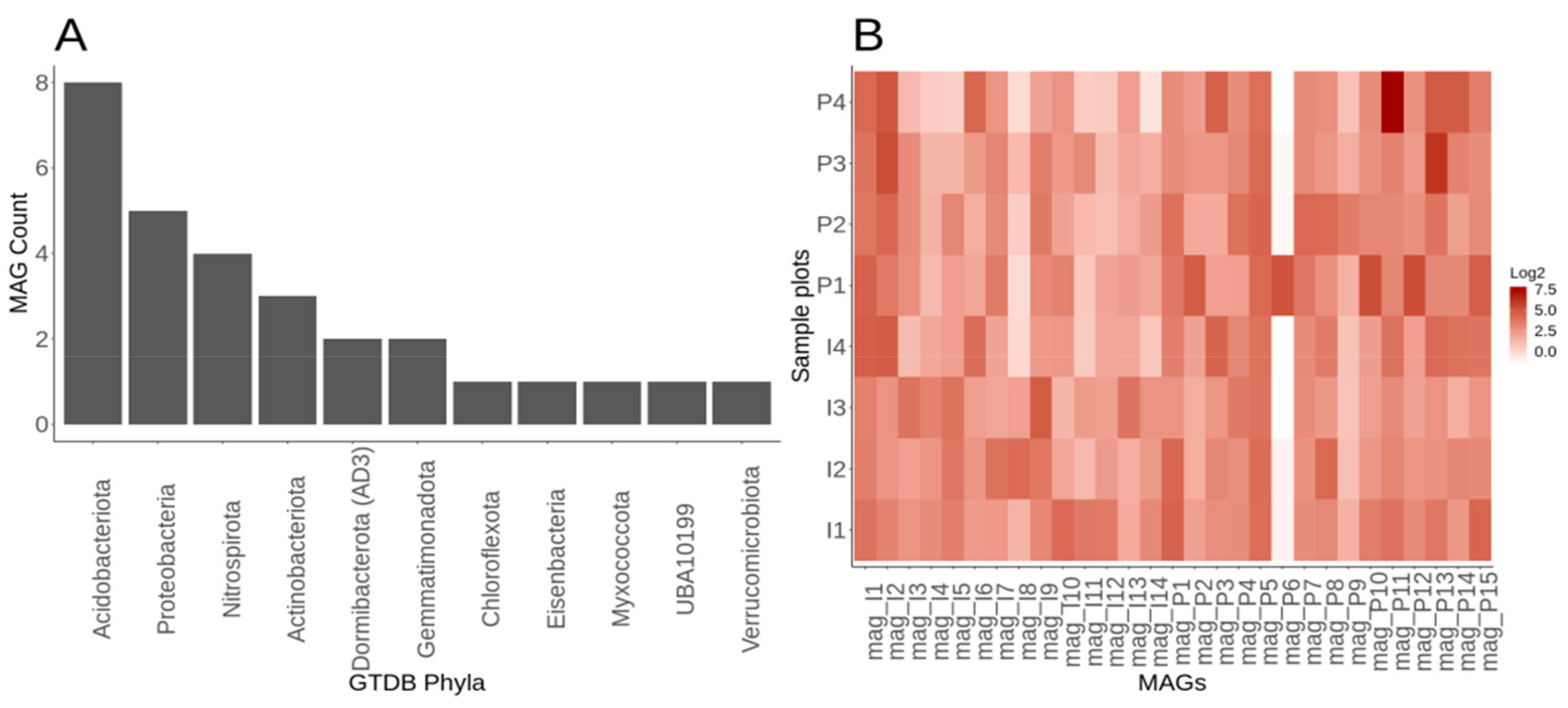
3.1. Assessment of Assembly and Metagenomic Assembled Genomes within Lux Arbor
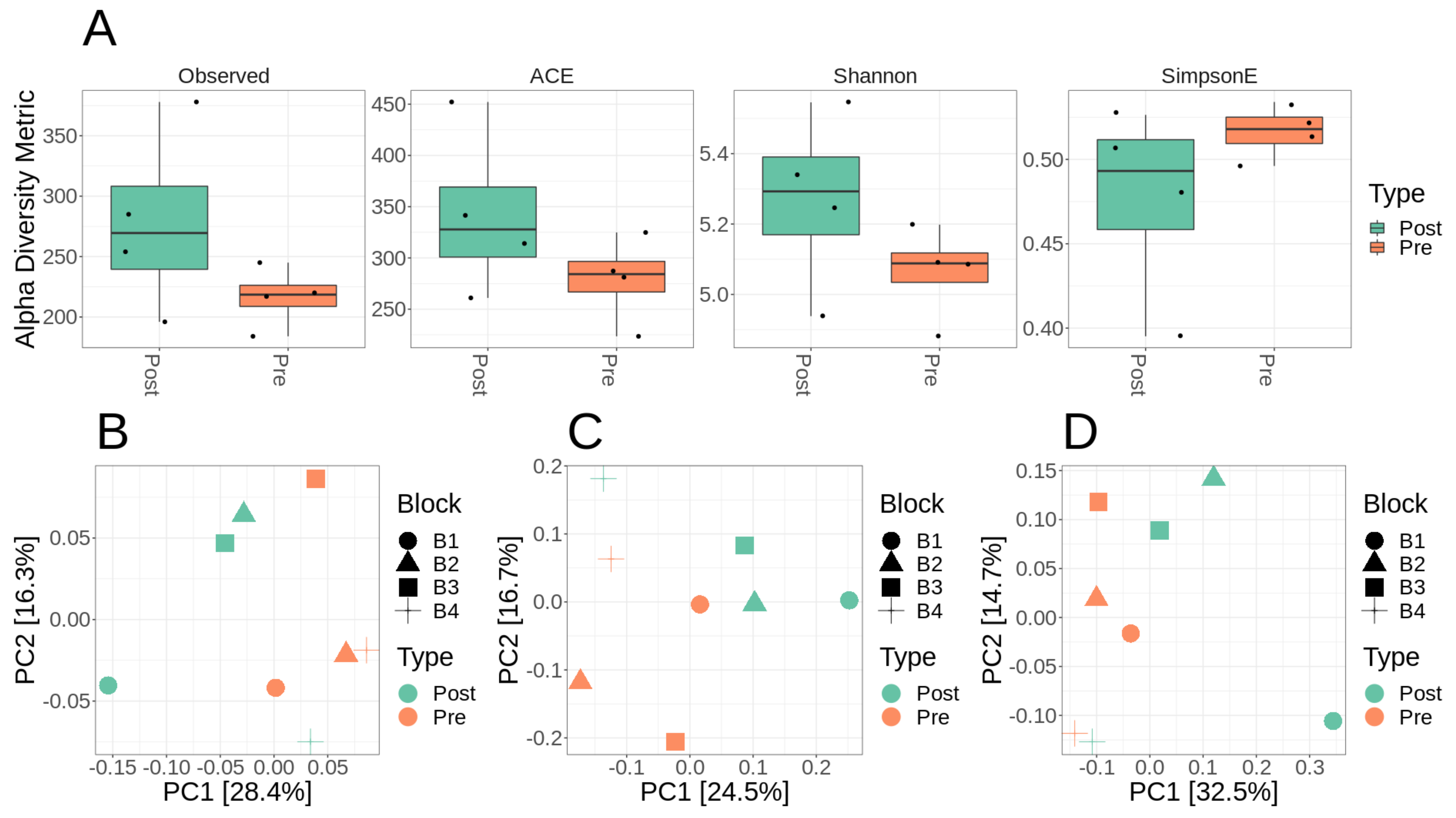
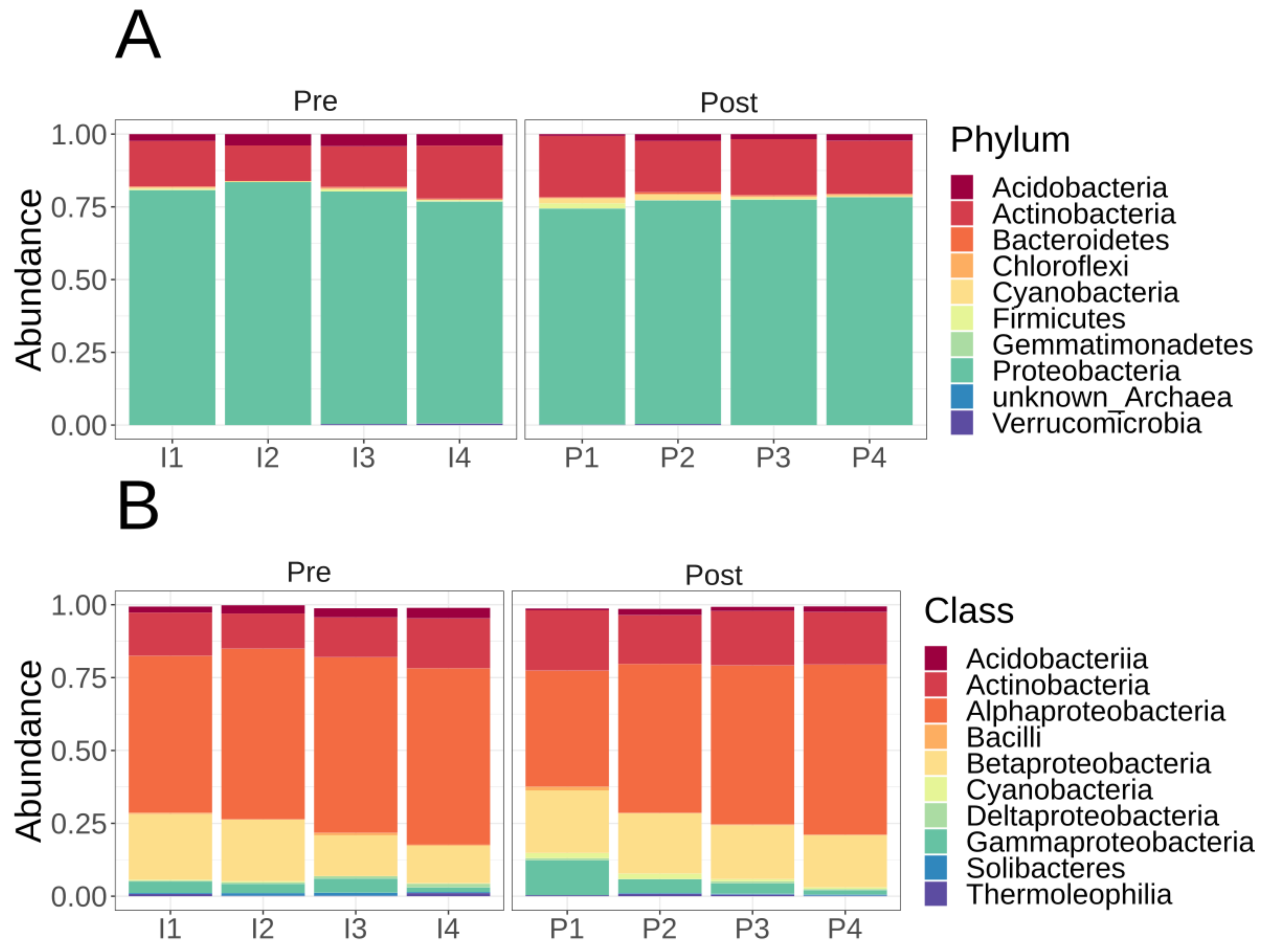
3.2. Microbiome Diversity and Composition of Lux Arbor Switchgrass Rhizosphere
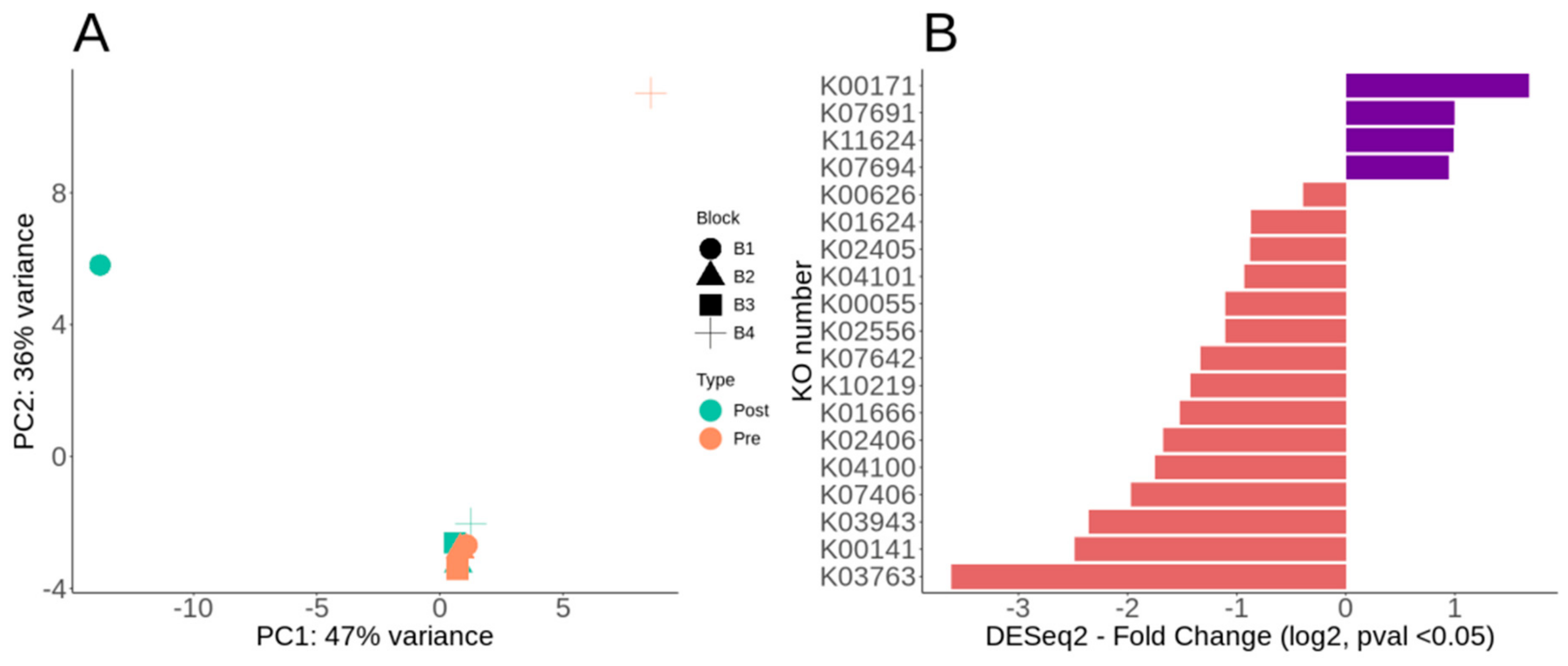
3.3. Overall Metabolic Potential and Differential Metabolic Genes of Lux Arbor Switchgrass Rhizosphere
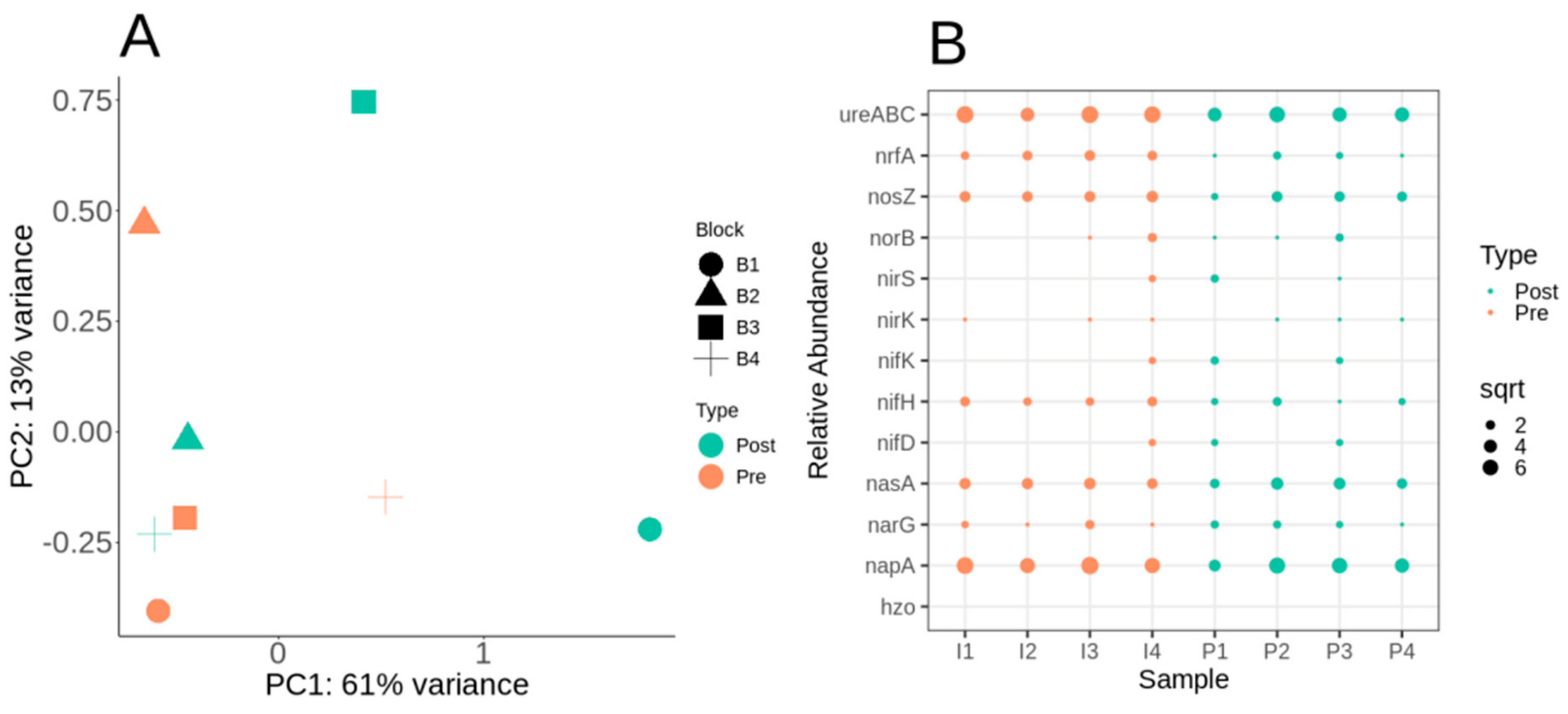
3.4. Nitrogen Cycle Metabolic Potential within the Switchgrass Rhizosphere Microbiome
3.5. Differential CAZy Potential within the Switchgrass Rhizosphere Microbiome
3.6. Genome-Resolved Metagenomics Elucidates Members of the Rare Biosphere
3.7. Betaproteobacterial MAG with Molybdenum-Based Nitrogen Fixation Gene Cluster
3.8. Acidobacteria Related to Rare Subdivision 23 with Utilization Nitrate
3.9. Nitrospira Hydrolysis of Urea, Nitrate Reduction with Limited Nitrite Reduction
3.10. Dormibacterota MAGs’ Metabolic Potential within the Switchgrass Rhizosphere
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Janzen, D.H. The natural history of mutualisms. In The Biology of Mutualism; Boucher, D.H., Ed.; Croom Helm: London, UK, 1985; Volume 3, pp. 40–99. [Google Scholar]
- Berendsen, R.L.; Pieterse, C.M.J.; Bakker, P.A.H.M. The rhizosphere microbiome and plant health. Trends Plant Sci. 2012, 17, 478–486. [Google Scholar] [CrossRef] [PubMed]
- Ramírez-Puebla, S.T.; Servín-Garcidueñas, L.E.; Jiménez-Marín, B.; Bolaños, L.M.; Rosenblueth, M.; Martínez, J.; Rogel, M.A.; Ormeño-Orrillo, E.; Martínez-Romero, E. Gut and Root Microbiota Commonalities. Appl. Environ. Microbiol. 2013, 79, 2–9. [Google Scholar] [CrossRef] [PubMed]
- White, R.A., III; Rivas-Ubach, A.; Borkum, M.I.; Köberl, M.; Bilbao, A.; Colby, S.M.; Hoyt, D.W.; Bingol, K.; Kim, Y.-M.; Wendler, J.P.; et al. The state of rhizospheric science in the era of multi-omics: A practical guide to omics technologies. Rhizosphere 2017, 3, 212–221. [Google Scholar] [CrossRef]
- White, R.A., III; Borkum, M.I.; Rivas-Ubach, A.; Bilbao, A.; Wendler, J.P.; Colby, S.M.; Köberl, M.; Jansson, C. From data to knowledge: The future of multi-omics data analysis for the rhizosphere. Rhizosphere 2017, 3, 222–229. [Google Scholar] [CrossRef]
- Ahkami, A.H.; White, R.A., III; Handakumbura, P.P.; Jansson, C. Rhizosphere engineering: Enhancing sustainable plant ecosystem productivity. Rhizosphere 2017, 3, 233–243. [Google Scholar] [CrossRef]
- Friesen, M.L.; Porter, S.S.; Stark, S.C.; von Wettberg, E.J.; Sachs, J.L.; Martinez-Romero, E. Microbially Mediated Plant Functional Traits. Annu. Rev. Ecol. Evol. Syst. 2011, 42, 23–46. [Google Scholar] [CrossRef]
- Bowers, R.M.; Kyrpides, N.C.; Stepanauskas, R.; Harmon-Smith, M.; Doud, D.; Reddy, T.B.K.; Schulz, F.; Jarett, J.; Rivers, A.R.; Eloe-Fadrosh, E.A.; et al. Minimum information about a single amplified genome (MISAG) and a metagenome-assembled genome (MIMAG) of bacteria and archaea. Nat. Biotechnol. 2017, 35, 725–731. [Google Scholar] [CrossRef]
- Howe, A.C.; Jansson, J.K.; Malfatti, S.A.; Tringe, S.G.; Tiedje, J.M.; Brown, C.T. Tackling soil diversity with the assembly of large, complex metagenomes. Proc. Natl. Acad. Sci. USA 2014, 111, 4904–4909. [Google Scholar] [CrossRef]
- Iii, R.A.W.; Bottos, E.M.; Chowdhury, T.R.; Zucker, J.D.; Brislawn, C.J.; Nicora, C.D.; Fansler, S.J.; Glaesemann, K.R.; Glass, K.; Jansson, J.K. Moleculo Long-Read Sequencing Facilitates Assembly and Genomic Binning from Complex Soil Metagenomes. Msystems 2016, 1, e00045-16. [Google Scholar] [CrossRef]
- Butterfield, C.N.; Li, Z.; Andeer, P.F.; Spaulding, S.; Thomas, B.C.; Singh, A.; Hettich, R.L.; Suttle, K.B.; Probst, A.J.; Tringe, S.G.; et al. Proteogenomic analyses indicate bacterial methylotrophy and archaeal heterotrophy are prevalent below the grass root zone. PeerJ 2016, 4, e2687. [Google Scholar] [CrossRef]
- Kroeger, M.; Delmont, T.O.; Eren, A.M.; Meyer, K.M.; Guo, J.; Khan, K.; Rodrigues, J.L.M.; Bohannan, B.J.M.; Tringe, S.G.; Borges, C.D.; et al. New Biological Insights Into How Deforestation in Amazonia Affects Soil Microbial Communities Using Metagenomics and Metagenome-Assembled Genomes. Front. Microbiol. 2018, 9, 1635. [Google Scholar] [CrossRef]
- Nelkner, J.; Henke, C.; Lin, T.W.; Pätzold, W.; Hassa, J.; Jaenicke, S.; Grosch, R.; Pühler, A.; Sczyrba, A.; Schlüter, A. Effect of Long-Term Farming Practices on Agricultural Soil Microbiome Members Represented by Metagenomically Assembled Genomes (MAGs) and Their Predicted Plant-Beneficial Genes. Genes 2019, 10, 424. [Google Scholar] [CrossRef]
- Li, Z.; Yao, Q.; Guo, X.; Crits-Christoph, A.; Mayes, M.A.; Iv, W.J.H.; Lebeis, S.L.; Banfield, J.F.; Hurst, G.B.; Hettich, R.L.; et al. Genome-Resolved Proteomic Stable Isotope Probing of Soil Microbial Communities Using 13CO2 and 13C-Methanol. Front. Microbiol. 2019, 10, 2706. [Google Scholar] [CrossRef]
- Nayfach, S.; Roux, S.; Seshadri, R.; Udwary, D.; Varghese, N.; Schulz, F.; Wu, D.; Paez-Espino, D.; Chen, I.-M.; Huntemann, M.; et al. A genomic catalog of Earth’s microbiomes. Nat. Biotechnol. 2020, 39, 499–509. [Google Scholar] [CrossRef]
- Starr, E.P.; Shi, S.; Blazewicz, S.J.; Koch, B.J.; Probst, A.J.; Hungate, B.A.; Pett-Ridge, J.; Firestone, M.K.; Banfield, J.F. Stable-Isotope-Informed, Genome-Resolved Metagenomics Uncovers Potential Cross-Kingdom Interactions in Rhizosphere Soil. Msphere 2021, 6, e00085-21. [Google Scholar] [CrossRef]
- Xu, L.; Dong, Z.; Chiniquy, D.; Pierroz, G.; Deng, S.; Gao, C.; Diamond, S.; Simmons, T.; Wipf, H.M.-L.; Caddell, D.; et al. Genome-resolved metagenomics reveals role of iron metabolism in drought-induced rhizosphere microbiome dynamics. Nat. Commun. 2021, 12, 1–17. [Google Scholar] [CrossRef]
- Mandro, J.A.; Nakamura, F.M.; Gontijo, J.B.; Tsai, S.M.; Venturini, A.M. Metagenome-Assembled Genomes from Amazonian Soil Microbial Consortia. Genome Announc. 2022, 11, e00804-22. [Google Scholar] [CrossRef]
- Wu, X.; Cui, Z.; Peng, J.; Zhang, F.; Liesack, W. Genome-resolved metagenomics identifies the particular genetic traits of phosphate-solubilizing bacteria in agricultural soil. ISME Commun. 2022, 2, 1–4. [Google Scholar] [CrossRef]
- Wrighton, K.C.; Thomas, B.C.; Sharon, I.; Miller, C.S.; Castelle, C.J.; VerBerkmoes, N.C.; Wilkins, M.J.; Hettich, R.L.; Lipton, M.S.; Williams, K.H.; et al. Fermentation, Hydrogen, and Sulfur Metabolism in Multiple Uncultivated Bacterial Phyla. Science 2012, 337, 1661–1665. [Google Scholar] [CrossRef]
- Kantor, R.S.; Wrighton, K.C.; Handley, K.M.; Sharon, I.; Hug, L.A.; Castelle, C.J.; Thomas, B.C.; Banfield, J.F. Small Genomes and Sparse Metabolisms of Sediment-Associated Bacteria from Four Candidate Phyla. mBio 2013, 4, e00708-13. [Google Scholar] [CrossRef]
- Philippot, L.; Raaijmakers, J.M.; Lemanceau, P.; van der Putten, W.H. Going back to the roots: The microbial ecology of the rhizosphere. Nat. Rev. Microbiol. 2013, 11, 789–799. [Google Scholar] [CrossRef] [PubMed]
- Zhalnina, K.; Louie, K.B.; Hao, Z.; Mansoori, N.; da Rocha, U.N.; Shi, S.; Cho, H.; Karaoz, U.; Loqué, D.; Bowen, B.P.; et al. Dynamic root exudate chemistry and microbial substrate preferences drive patterns in rhizosphere microbial community assembly. Nat. Microbiol. 2018, 3, 470–480. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, A.; Rothballer, M.; Schmid, M. Lorenz Hiltner, a pioneer in rhizosphere microbial ecology and soil bacteriology research. Plant Soil 2007, 312, 7–14. [Google Scholar] [CrossRef]
- Smercina, D.N.; Evans, S.E.; Friesen, M.L.; Tiemann, L.K. To Fix or Not To Fix: Controls on Free-Living Nitrogen Fixation in the Rhizosphere. Appl. Environ. Microbiol. 2019, 85, e02546-18. [Google Scholar] [CrossRef]
- deCatanzaro, J.B.; Beauchamp, E.G. The effect of some carbon substrates on denitrification rates and carbon utilization in soil. Biol. Fertil. Soils 1985, 1, 183–187. [Google Scholar] [CrossRef]
- Pajares, S.; Bohannan, B.J.M. Ecology of Nitrogen Fixing, Nitrifying, and Denitrifying Microorganisms in Tropical Forest Soils. Front. Microbiol. 2016, 7, 1045. [Google Scholar] [CrossRef]
- Masson-Boivin, C.; Giraud, E.; Perret, X.; Batut, J. Establishing nitrogen-fixing symbiosis with legumes: How many rhizobium recipes? Trends Microbiol. 2009, 17, 458–466. [Google Scholar] [CrossRef]
- Demanèche, S.; Philippot, L.; David, M.M.; Navarro, E.; Vogel, T.M.; Simonet, P. Characterization of Denitrification Gene Clusters of Soil Bacteria via a Metagenomic Approach. Appl. Environ. Microbiol. 2009, 75, 534–537. [Google Scholar] [CrossRef]
- Giles, M.E.; Morley, N.J.; Baggs, E.M.; Daniell, T.J. Soil nitrate reducing processes—Drivers, mechanisms for spatial variation and significance for nitrous oxide production. Front. Microbiol. 2012, 3, 407. [Google Scholar] [CrossRef]
- Koch, H.; van Kessel, M.A.H.J.; Lücker, S. Complete nitrification: Insights into the ecophysiology of comammox Nitrospira. Appl. Microbiol. Biotechnol. 2018, 103, 177–189. [Google Scholar] [CrossRef]
- McLaughlin, S.B.; Kzos, L.A. Development of switchgrass (Panicum virgatum) as a bioenergy feedstock in the United States. Biomass Bioenergy 2005, 28, 515–535. [Google Scholar] [CrossRef]
- Schmer, M.R.; Vogel, K.P.; Mitchell, R.B.; Perrin, R.K. Net energy of cellulosic ethanol from switchgrass. Proc. Natl. Acad. Sci. USA 2008, 105, 464–469. [Google Scholar] [CrossRef]
- Wright, L.L. Historical Perspective on How and Why Switchgrass Was Selected as a “Model” High-Potential Energy Crop; U.S. Department of Energy Office of Scientific and Technical Information: Washington, DC, USA, 2007. [Google Scholar] [CrossRef]
- Wright, L.; Turhollow, A. Switchgrass selection as a “model” bioenergy crop: A history of the process. Biomass Bioenergy 2010, 34, 851–868. [Google Scholar] [CrossRef]
- Ruan, L.; Bhardwaj, A.K.; Hamilton, S.K.; Robertson, G.P. Nitrogen fertilization challenges the climate benefit of cellulosic biofuels. Environ. Res. Lett. 2016, 11, 064007. [Google Scholar] [CrossRef]
- Emery, I.; Mueller, S.; Qin, Z.; Dunn, J.B. Evaluating the Potential of Marginal Land for Cellulosic Feedstock Production and Carbon Sequestration in the United States. Environ. Sci. Technol. 2016, 51, 733–741. [Google Scholar] [CrossRef]
- Duran, B.E.L.; Duncan, D.S.; Oates, L.G.; Kucharik, C.J.; Jackson, R.D. Nitrogen Fertilization Effects on Productivity and Nitrogen Loss in Three Grass-Based Perennial Bioenergy Cropping Systems. PLoS ONE 2016, 11, e0151919. [Google Scholar] [CrossRef]
- Singer, E.; Bonnette, J.; Kenaley, S.C.; Woyke, T.; Juenger, T.E. Plant compartment and genetic variation drive microbiome composition in switchgrass roots. Environ. Microbiol. Rep. 2019, 11, 185–195. [Google Scholar] [CrossRef]
- Roley, S.S.; Duncan, D.S.; Liang, D.; Garoutte, A.; Jackson, R.D.; Tiedje, J.M.; Robertson, G.P. Associative nitrogen fixation (ANF) in switchgrass (Panicum virgatum L.) across a nitrogen input gradient. PLoS ONE 2018, 13, e0197320. [Google Scholar] [CrossRef]
- Roley, S.S.; Xue, C.; Hamilton, S.K.; Tiedje, J.M.; Robertson, G.P. Isotopic evidence for episodic nitrogen fixation in switchgrass (Panicum virgatum L.). Soil Biol. Biochem. 2018, 129, 90–98. [Google Scholar] [CrossRef]
- Chen, H.; Yang, Z.K.; Yip, D.; Morris, R.H.; Lebreux, S.J.; Cregger, M.A.; Klingeman, D.M.; Hui, D.; Hettich, R.L.; Wilhelm, S.W.; et al. One-time nitrogen fertilization shifts switchgrass soil microbiomes within a context of larger spatial and temporal variation. PLoS ONE 2019, 14, e0211310. [Google Scholar] [CrossRef]
- Rinke, C.; Schwientek, P.; Sczyrba, A.; Ivanova, N.N.; Anderson, I.J.; Cheng, J.-F.; Darling, A.; Malfatti, S.; Swan, B.K.; Gies, E.A.; et al. Insights into the phylogeny and coding potential of microbial dark matter. Nature 2013, 499, 431–437. [Google Scholar] [CrossRef] [PubMed]
- Hug, L.A.; Baker, B.J.; Anantharaman, K.; Brown, C.T.; Probst, A.J.; Castelle, C.J.; Butterfield, C.N.; Hernsdorf, A.W.; Amano, Y.; Ise, K.; et al. A new view of the tree of life. Nat. Microbiol. 2016, 1, 16048. [Google Scholar] [CrossRef]
- Milanese, A.; Mende, D.R.; Paoli, L.; Salazar, G.; Ruscheweyh, H.-J.; Cuenca, M.; Hingamp, P.; Alves, R.; Costea, P.I.; Coelho, L.P.; et al. Microbial abundance, activity and population genomic profiling with mOTUs2. Nat. Commun. 2019, 10, 101. [Google Scholar] [CrossRef] [PubMed]
- White, R.A., III; Brown, J.; Colby, S.; Overall, C.C.; Lee, J.; Zucker, J.D.; Glaesemann, K.R.; Jansson, C.; Jansson, J.K. ATLAS (Automatic Tool for Local Assembly Structures)—A comprehensive infrastructure for assembly, annotation, and genomic binning of metagenomic and metatranscriptomic data. PeerJ 2017, 5, e2843v1. [Google Scholar]
- Li, D.; Liu, C.-M.; Luo, R.; Sadakane, K.; Lam, T.-W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef]
- Seemann, T. Prokka: Rapid Prokaryotic Genome Annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Huerta-Cepas, J.; Forslund, K.; Coelho, L.P.; Szklarczyk, D.; Jensen, L.J.; von Mering, C.; Bork, P. Fast genome-wide functional annotation through orthology assignment by eggNOG-Mapper. Mol. Biol. Evol. 2017, 34, 2115–2122. [Google Scholar] [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2014, 12, 59–60. [Google Scholar] [CrossRef]
- Zhang, H.; Yohe, T.; Huang, L.; Entwistle, S.; Wu, P.; Yang, Z.; Busk, P.K.; Xu, Y.; Yin, Y. dbCAN2: A meta server for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 2018, 46, W95–W101. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Alneberg, J.; Bjarnason, B.S.; de Bruijn, I.; Schirmer, M.; Quick, J.; Ijaz, U.Z.; Lahti, L.; Loman, N.; Andersson, A.; Quince, C. Binning metagenomic contigs by coverage and composition. Nat. Methods 2014, 11, 1144–1146. [Google Scholar] [CrossRef]
- Wu, Y.-W.; Simmons, B.A.; Singer, S.W. MaxBin 2.0: An automated binning algorithm to recover genomes from multiple metagenomic datasets. Bioinformatics 2015, 32, 605–607. [Google Scholar] [CrossRef]
- Kang, D.; Li, F.; Kirton, E.S.; Thomas, A.; Egan, R.S.; An, H.; Wang, Z. MetaBAT2: An adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ 2019, 7, e27522v1. [Google Scholar] [CrossRef]
- Uritskiy, G.V.; DiRuggiero, J.; Taylor, J. MetaWRAP—A flexible pipeline for genome-resolved metagenomic data analysis. Microbiome 2018, 6, 158. [Google Scholar] [CrossRef]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef]
- Richter, M.; Rosselló-Móra, R.; Oliver Glöckner, F.; Peplies, J. JSpeciesWS: A web server for prokaryotic species circumscription based on pairwise genome comparison. Bioinformatics 2016, 32, 929–931. [Google Scholar] [CrossRef]
- Chaumeil, P.-A.; Mussig, A.J.; Hugenholtz, P.; Parks, D.H. GTDB-Tk: A toolkit to classify genomes with the Genome Taxonomy Database. Bioinformatics 2020, 36, 1925–1927. [Google Scholar] [CrossRef]
- McMurdie, P.J.; Holmes, S. phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef]
- Lan, T.; Han, Y.; Roelcke, M.; Nieder, R.; Cai, Z. Effects of the nitrification inhibitor dicyandiamide (DCD) on gross N transformation rates and mitigating N2O emission in paddy soils. Soil Biol. Biochem. 2013, 67, 174–182. [Google Scholar] [CrossRef]
- Dvortsov, I.A.; Lunina, N.A.; Chekanovskaya, L.A.; Schwarz, W.H.; Zverlov, V.V.; Velikodvorskaya, G.A. Carbohydrate-binding properties of a separately folding protein module from beta-1,3-glucanase Lic16A of Clostridium thermocellum. Microbiology 2009, 155, 2442–2449. [Google Scholar] [CrossRef]
- Liu, S.Y.; Tang, Y.X.; Wang, D.C.; Lin, N.Q.; Zhou, J.N. Identification and characterization of a new Enterobacter onion bulb decay caused by Lelliottia amnigena in China. App. Micro. 2016, 2, 114. [Google Scholar] [CrossRef]
- Woodcroft, B.J.; Singleton, C.M.; Boyd, J.A.; Evans, P.N.; Emerson, J.B.; Zayed, A.A.F.; Hoelzle, R.D.; Lamberton, T.O.; McCalley, C.K.; Hodgkins, S.B.; et al. Genome-centric view of carbon processing in thawing permafrost. Nature 2018, 560, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Diamond, S.; Andeer, P.F.; Li, Z.; Crits-Christoph, A.; Burstein, D.; Anantharaman, K.; Lane, K.R.; Thomas, B.C.; Pan, C.; Northen, T.R.; et al. Mediterranean grassland soil C-N compound turnover is dependent on rainfall and depth, and is mediated by genomically divergent microorganisms. Nat. Microbiol. 2019, 4, 1356–1367. [Google Scholar] [CrossRef] [PubMed]
- Naether, A.; Foesel, B.U.; Naegele, V.; Wüst, P.K.; Weinert, J.; Bonkowski, M.; Alt, F.; Oelmann, Y.; Polle, A.; Lohaus, G.; et al. Environmental Factors Affect Acidobacterial Communities below the Subgroup Level in Grassland and Forest Soils. Appl. Environ. Microbiol. 2012, 78, 7398–7406. [Google Scholar] [CrossRef] [PubMed]
- Losey, N.A.; Stevenson, B.S.; Busse, H.-J.; Damsté, J.S.S.; Rijpstra, W.I.C.; Rudd, S.; Lawson, P.A. Thermoanaerobaculum aquaticum gen. nov., sp. nov., the first cultivated member of Acidobacteria subdivision 23, isolated from a hot spring. Int. J. Syst. Evol. Microbiol. 2013, 63, 4149–4157. [Google Scholar] [CrossRef]
- Parks, D.H.; Rinke, C.; Chuvochina, M.; Chaumeil, P.-A.; Woodcroft, B.J.; Evans, P.N.; Hugenholtz, P.; Tyson, G.W. Recovery of nearly 8,000 metagenome-assembled genomes substantially expands the tree of life. Nat. Microbiol. 2017, 2, 1533–1542. [Google Scholar] [CrossRef]
- Carini, P. A “cultural” renaissance: Genomics breathes new life into an old craft. Msystems 2019, 4, e00092-19. [Google Scholar] [CrossRef]
- Choi, J.; Yang, F.; Stepanauskas, R.; Cardenas, E.; Garoutte, A.; Williams, R.; Flater, J.; Tiedje, J.M.; Hofmockel, K.S.; Gelder, B.; et al. Strategies to improve reference databases for soil microbiomes. ISME J. 2016, 11, 829–834. [Google Scholar] [CrossRef]
- Ji, M.; Greening, C.; Vanwonterghem, I.; Carere, C.R.; Bay, S.K.; Steen, J.A.; Montgomery, K.; Lines, T.; Beardall, J.; Van Dorst, J.; et al. Atmospheric trace gases support primary production in Antarctic desert surface soil. Nature 2017, 552, 400–403. [Google Scholar] [CrossRef]
- Brewer, T.E.; Aronson, E.L.; Arogyaswamy, K.; Billings, S.A.; Botthoff, J.K.; Campbell, A.N.; Dove, N.C.; Fairbanks, D.; Rachel, E.G.; Hart, S.C.; et al. Ecological and genomic attributes of novel bacterial taxa that thrive in subsurface soil horizons. mbio 2019, 10, e01318-19. [Google Scholar] [CrossRef]
- Anantharaman, K.; Brown, C.T.; Burstein, D.; Castelle, C.J.; Probst, A.J.; Thomas, B.C.; Williams, K.H.; Banfield, J.F. Analysis of five complete genome sequences for members of the class Peribacteria in the recently recognized Peregrinibacteria bacterial phylum. PeerJ 2016, 4, e1607. [Google Scholar] [CrossRef]
- Hara, S.; Desyatkin, R.; Hashidoko, Y. Investigation of the mechanisms underlying the high acetylene-reducing activity exhibited by the soil bacterial community from BC2 horizon in the permafrost zone of the East Siberian larch forest bed. J. Appl. Microbiol. 2014, 116, 865–876. [Google Scholar] [CrossRef]
- VanInsberghe, D.; Maas, K.R.; Cardenas, E.; Strachan, C.R.; Hallam, S.J.; Mohn, W.W. Non-symbiotic Bradyrhizobium ecotypes dominate North American forest soils. ISME J. 2015, 9, 2435–2441. [Google Scholar] [CrossRef]
- Jones, F.P.; Clark, I.M.; King, R.; Shaw, L.J.; Woodward, M.J.; Hirsch, P.R. Novel european free-living, non-diazotrophic Bradyrhizobium isolates from contrasting soils that lack nodulation and nitrogen fixation genes—A genome comparison. Sci. Rep. 2016, 6, 25858. [Google Scholar] [CrossRef]
- Ning, J.; Ai, S.; Cui, L. Dicyandiamide has more inhibitory activities on nitrification than thiosulfate. PLoS ONE 2018, 13, e0200598. [Google Scholar] [CrossRef]
- Yang, W.; Wang, Y.; Tago, K.; Tokuda, S.; Hayatsu, M. Comparison of the Effects of Phenylhydrazine Hydrochloride and Dicyandiamide on Ammonia-Oxidizing Bacteria and Archaea in Andosols. Front. Microbiol. 2017, 8, 2226. [Google Scholar] [CrossRef]
- Di, H.J.; Cameron, K.C.; Podolyan, A.; Robinson, A. Effect of soil moisture status and a nitrification inhibitor, dicyandiamide, on ammonia oxidizer and denitrifier growth and nitrous oxide emissions in a grassland soil. Soil Biol. Biochem. 2014, 73, 59–68. [Google Scholar] [CrossRef]
- Orellana, L.H.; Chee-Sanford, J.C.; Sanford, R.; Löffler, F.E.; Konstantinidis, K.T. Year-Round Shotgun Metagenomes Reveal Stable Microbial Communities in Agricultural Soils and Novel Ammonia Oxidizers Responding to Fertilization. Appl. Environ. Microbiol. 2018, 84, e01646-17. [Google Scholar] [CrossRef]
- Cantarella, H.; Otto, R.; Soares, J.R.; de Brito Silva, A.G. Agronomic efficiency of NBPT as a urease inhibitor: A review. J. Adv. Res. 2018, 13, 19–27. [Google Scholar] [CrossRef]
- Zhang, L.; Chen, W.; Burger, M.; Yang, L.; Gong, P.; Wu, Z. Changes in Soil Carbon and Enzyme Activity As a Result of Different Long-Term Fertilization Regimes in a Greenhouse Field. PLoS ONE 2015, 10, e0118371. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample_ID | Sample_Dis | #Reads | #Raw Bases | Trim-Decon Reads | #Contigs | Size | N50 |
|---|---|---|---|---|---|---|---|
| P1 | Post-fertilization | 6.78 × 108 | 1.02 × 1011 | 4.60 × 108 | 4.36 × 106 | 2.62 × 109 | 607 |
| P2 | Post-fertilization | 7.49 × 108 | 1.12 × 1011 | 4.98 × 108 | 5.54 × 109 | 3.88 × 109 | 743 |
| P3 | Post-fertilization | 6.64 × 108 | 9.95 × 1010 | 4.47 × 108 | 4.95 × 109 | 3.31 × 109 | 701 |
| P4 | Post-fertilization | 5.48 × 108 | 8.22 × 1010 | 3.41 × 108 | 3.84 × 109 | 2.66 × 109 | 736 |
| I1 | Pre-fertilization | 6.77 × 108 | 1.02 × 1011 | 4.48 × 108 | 4.83 × 109 | 3.42 × 109 | 754 |
| I2 | Pre-fertilization | 6.45 × 108 | 9.67 × 1010 | 4.43 × 108 | 4.89 × 109 | 3.35 × 109 | 722 |
| I3 | Pre-fertilization | 7.17 × 108 | 1.08 × 1011 | 4.59 × 108 | 4.77 × 109 | 3.54 × 109 | 825 |
| I4 | Pre-fertilization | 6.93 × 108 | 1.04 × 1011 | 4.22 × 108 | 4.10 × 109 | 3.01 × 109 | 810 |
| Average | 5.37 × 109 | 8.06 × 1011 | 3.52 × 109 | 4.66 × 109 | 3.22 × 109 | 737 | |
| sample_ID | #contigs_1K | size_1K | N50_1K | #contigs_5K | size_5k | N50_5k | GC% |
| P1 | 387,207 | 9.98 × 107 | 1741 | 9864 | 7.01 × 108 | 10,169 | 62.30 |
| P2 | 721,633 | 2.93 × 108 | 2034 | 31,195 | 1.45 × 109 | 9402 | 62.76 |
| P3 | 595,079 | 1.89 × 108 | 1914 | 20,404 | 1.14 × 109 | 9058 | 62.22 |
| P4 | 504,757 | 1.69 × 108 | 1951 | 18,173 | 9.84 × 108 | 9139 | 62.55 |
| I1 | 649,392 | 2.57 × 108 | 2027 | 28,056 | 1.30 × 109 | 9123 | 61.98 |
| I2 | 618,357 | 2.14 × 108 | 1969 | 23,648 | 1.21 × 109 | 8891 | 60.92 |
| I3 | 726,107 | 3.05 × 108 | 2140 | 32,660 | 1.50 × 109 | 9013 | 61.35 |
| I4 | 613,447 | 2.46 × 108 | 2081 | 26,172 | 1.25 × 109 | 9243 | 61.37 |
| Average | 601,997 | 2.21 × 108 | 1982 | 23,771 | 1.19 × 109 | 9254 | 61.93 |
| Accession | Funtaxa | Phyla | Class | Habitat | log2 Fold Change | p-Value |
|---|---|---|---|---|---|---|
| ANY66681.1 | Paenibacillus sp. BIHB4019 (CBM54) | Firmicutes | Bacilli | Rhizosphere | −3.38 | 0.02 |
| AKB38096.1 | Methanosarcina siciliae C2J (GT4) | Euryarchaeota | Methanomicrobia | Unknown | −3.03 | 0.04 |
| ALJ82902.1 | Irpex lacteus (AA3_1|AA8) | Basidiomycota | Agaricomycetes | Wood | −3.02 | 0.04 |
| AUD02463.1 | Spirosoma pollinicola (CBM6) | Bacteroidetes | Cytophagia | Pollen | −2.72 | 0.02 |
| ANS78621.1 | Serinicoccus sp. JLT9 (CBM48|GH13_9) | Actinobacteria | Actinobacteria | Thermal | −2.22 | 0.05 |
| AFY81829.1 | Oscillatoria acuminata PCC6304 (GH65) | Cyanobacteria | Cyanophyceae | Soil | −2.11 | 0.02 |
| AAR38497.1 | Uncultured marine bacterium 583 (GT41) | Uncultured | Uncultured | Ocean | −1.97 | 0.02 |
| BAZ44095.1 | Chondrocystis sp. NIES-4102 (GT2) | Cyanobacteria | Cyanophyceae | Unknown | −1.82 | 0.04 |
| BAL56682.1 | Uncultured Gammaproteobacteria (PL0) | Proteobacteria | Gammaproteobacteria | Microbial mat | −1.4 | 0.05 |
| ARX88346.1 | Streptomyces alboflavus (GT2|CE4) | Actinobacteria | Actinomycetes | Rhizosphere | −1.23 | 0.02 |
| ATF41409.1 | Weissella paramesenteroides (CBM50) | Firmicutes | Bacilli | Unknown | −1.07 | 0.02 |
| ALG08540.1 | Kibdelosporangium phytohabitans (GH16) | Actinobacteria | Actinobacteria | Phyllosphere | −0.84 | 0.04 |
| ATU64527.1 | Rhizobacter gummiphilus (CBM41) | Proteobacteria | Gammaproteobacteria | Soil | −0.52 | 0.05 |
| AMG83817.1 | Microbacterium sp. PAMC 28,756 (CE14) | Actinobacteria | Actinobacteria | Lichen | 0.26 | 0.04 |
| ACL17090.1 | Methanosphaerula palustris E1–9c (CBM6) | Euryarchaeota | Methanomicrobia | Peatland Soil | 0.69 | 0.03 |
| AKP50194.1 | Cyclobacterium amurskyense (GH33) | Bacteroidetes | Flavobacteria | Ocean | 0.88 | 0.05 |
| AMT93207.1 | Brevibacterium linens (GT51) | Actinobacteria | Actinomycetes | Sediment | 1.95 | 0.02 |
| ACO33523.1 | Acidobacterium capsulatum (GT2) | Acidobacteria | Acidobacteria | Soil | 2.16 | 0.02 |
| ACN58963.1 | Uncultured bacterium BLR10 (GH9) | Uncultured | Uncultured | Soil | 2.37 | 0.05 |
| AGA24658.1 | Singulisphaera acidiphila DSM18658 (GT28) | Planctomycetes | Planctomycetacia | Peat bog wetland | 2.41 | 0.04 |
| BBA71022.1 | Geobacter sulfurreducens (GT41) | Proteobacteria | Deltaproteobacteria | Sediment | 2.43 | 0.04 |
| GTDB-Tk Taxonomy | Size | Contigs | N50 | GC | Completeness | Contamination | MIMAG Quality | |
|---|---|---|---|---|---|---|---|---|
| magI1 | Actinobacteriota; Thermoleophilia; 20CM-4-69-9; 20CM-4-69-9 | 3,377,677 | 169 | 24,250 | 69.9% | 81 | 1.293 | Medium |
| magI2 | Acidobacteriota; Thermoanaerobaculia | 5,321,068 | 131 | 66,773 | 66.0% | 95.96 | 2.849 | Medium |
| magI3 | Nitrospirota; Nitrospiria; Nitrospirales; Nitrospiraceae; Nitrospira_C | 3,293,157 | 304 | 11,734 | 58.7% | 87.87 | 7.929 | Medium |
| magI4 | Eisenbacteria; RBG-16-71-46 | 2,561,625 | 211 | 13,768 | 67.9% | 84.84 | 1.098 | Medium |
| magI5 | Gemmatimonadota; Gemmatimonadetes; Gemmatimonadales; GWC2-71-9 | 3,461,988 | 62 | 148,829 | 67.7% | 94.18 | 2.197 | Medium |
| magI6 | Acidobacteriota; Acidobacteriae; Acidobacteriales; Koribacteraceae | 4,052,648 | 57 | 120,172 | 56.0% | 91.05 | 0.854 | Medium |
| magI7 | Acidobacteriota; Thermoanaerobaculia | 7,820,903 | 527 | 18,142 | 67.6% | 80.65 | 3.703 | Medium |
| magI8 | Nitrospirota; Nitrospiria; Nitrospirales; Nitrospiraceae; GCA-2737345 | 4,109,394 | 341 | 12,463 | 56.4% | 84.66 | 4.545 | Medium |
| magI9 | Myxococcota; Polyangia; Kofleriales; Kofleriaceae | 11,007,611 | 231 | 77,779 | 69.0% | 81.93 | 1.474 | Medium |
| magI10 | Dormibacterota; Dormibacteria | 3,788,213 | 181 | 27,454 | 71.7% | 88 | 1.851 | Medium |
| magI11 | Nitrospirota; Nitrospiria; Nitrospirales; Nitrospiraceae; Nitrospira_C | 4,541,993 | 243 | 24,855 | 55.3% | 96.36 | 3.989 | Medium |
| magI12 | Gemmatimonadota; Gemmatimonadetes; Gemmatimonadales; GWC2-71-9; 40CM-2-70-7 | 2,649,947 | 193 | 16,824 | 67.7% | 82 | 1.098 | Medium |
| magI13 | Dormibacterota; Dormibacteria | 4,029,220 | 85 | 69,215 | 70.7% | 98.61 | 0.925 | Medium |
| magI14 | Nitrospirota; Nitrospiria; Nitrospirales; Nitrospiraceae; Nitrospira_C | 3,065,303 | 170 | 26,094 | 56.7% | 82.01 | 6.363 | Medium |
| magP1 | Verrucomicrobiota; Verrucomicrobiae; Pedosphaerales; Pedosphaeraceae | 8,909,748 | 261 | 55,946 | 57.6% | 98.64 | 8.108 | Medium |
| magP2 | Proteobacteria; Alphaproteobacteria; Sphingomonadales; Sphingomonadaceae | 2,721,540 | 119 | 37,362 | 62.5% | 90.75 | 3.921 | Medium |
| magP3 | Acidobacteriota; Acidobacteriae; Acidobacteriales; Koribacteraceae | 4,391,229 | 117 | 63,672 | 55.8% | 96.58 | 6.41 | Medium |
| magP4 | Acidobacteriota; Acidobacteriae; Acidobacteriales | 5,394,372 | 259 | 26,773 | 54.9% | 92.02 | 1.994 | Medium |
| magP5 | Actinobacteriota; Acidimicrobiia; IMCC26256 | 4,337,493 | 355 | 14,277 | 69.3% | 83.52 | 0.925 | Medium |
| magP6 | Proteobacteria; Gammaproteobacteria; Enterobacterales; Enterobacteriaceae; Lelliottia | 5,354,450 | 93 | 88,703 | 55.2% | 99.06 | 0.715 | Medium |
| magP7 | Actinobacteriota; Thermoleophilia; Solirubrobacterales; 70-9; 70-9 | 2,620,611 | 92 | 38,941 | 67.4% | 95.13 | 0.948 | Medium |
| magP8 | Proteobacteria; Gammaproteobacteria; Betaproteobacteriales; UKL13-2 | 3,375,344 | 364 | 14,636 | 65.7% | 84.82 | 4.31 | Medium |
| magP9 | UBA10199; UBA10199; UBA10199 | 3,108,333 | 222 | 17,369 | 57.6% | 83.87 | 1.29 | Medium |
| magP10 | Proteobacteria; Gammaproteobacteria; Betaproteobacteriales; Burkholderiaceae; Janthinobacterium | 4,309,612 | 337 | 15,502 | 66.0% | 87.65 | 2.613 | Medium |
| magP11 | Chloroflexota; Ellin6529; CSP1-4; CSP1-4; UBA5189 | 2,799,987 | 10 | 520,935 | 70.5% | 95.83 | 1.157 | Medium |
| magP12 | Acidobacteriota; Thermoanaerobaculia | 5,401,737 | 147 | 56,396 | 63.2% | 97.53 | 5.47 | Medium |
| magP13 | Acidobacteriota; Thermoanaerobaculia | 5,296,372 | 38 | 193,920 | 66.0% | 99.14 | 3.703 | Medium |
| magP14 | Proteobacteria; Alphaproteobacteria; Sphingomonadales; Sphingomonadaceae; Sphingomonas_A | 2,439,972 | 109 | 32,058 | 62.8% | 96.5 | 2.507 | Medium |
| magP15 | Acidobacteriota; Acidobacteriae; Acidobacteriales; Koribacteraceae | 3,923,518 | 235 | 22,362 | 57.5% | 92.46 | 3.019 | Medium |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
White, R.A., III; Garoutte, A.; Mclachlan, E.E.; Tiemann, L.K.; Evans, S.; Friesen, M.L. Genome-Resolved Metagenomics of Nitrogen Transformations in the Switchgrass Rhizosphere Microbiome on Marginal Lands. Agronomy 2023, 13, 1294. https://doi.org/10.3390/agronomy13051294
White RA III, Garoutte A, Mclachlan EE, Tiemann LK, Evans S, Friesen ML. Genome-Resolved Metagenomics of Nitrogen Transformations in the Switchgrass Rhizosphere Microbiome on Marginal Lands. Agronomy. 2023; 13(5):1294. https://doi.org/10.3390/agronomy13051294
Chicago/Turabian StyleWhite, Richard Allen, III, Aaron Garoutte, Emily E. Mclachlan, Lisa K. Tiemann, Sarah Evans, and Maren L. Friesen. 2023. "Genome-Resolved Metagenomics of Nitrogen Transformations in the Switchgrass Rhizosphere Microbiome on Marginal Lands" Agronomy 13, no. 5: 1294. https://doi.org/10.3390/agronomy13051294
APA StyleWhite, R. A., III, Garoutte, A., Mclachlan, E. E., Tiemann, L. K., Evans, S., & Friesen, M. L. (2023). Genome-Resolved Metagenomics of Nitrogen Transformations in the Switchgrass Rhizosphere Microbiome on Marginal Lands. Agronomy, 13(5), 1294. https://doi.org/10.3390/agronomy13051294