


Tissue-Specific Transcriptomes Outline Halophyte Adaptive Strategies in the Gray Mangrove (Avicennia marina)

 , , , ,
, , , ,  , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Sequence Sources
2.2. Sequence Processing and Alignment
2.3. Uniform Manifold Approximation Projection
2.4. Hierarchical Clustering
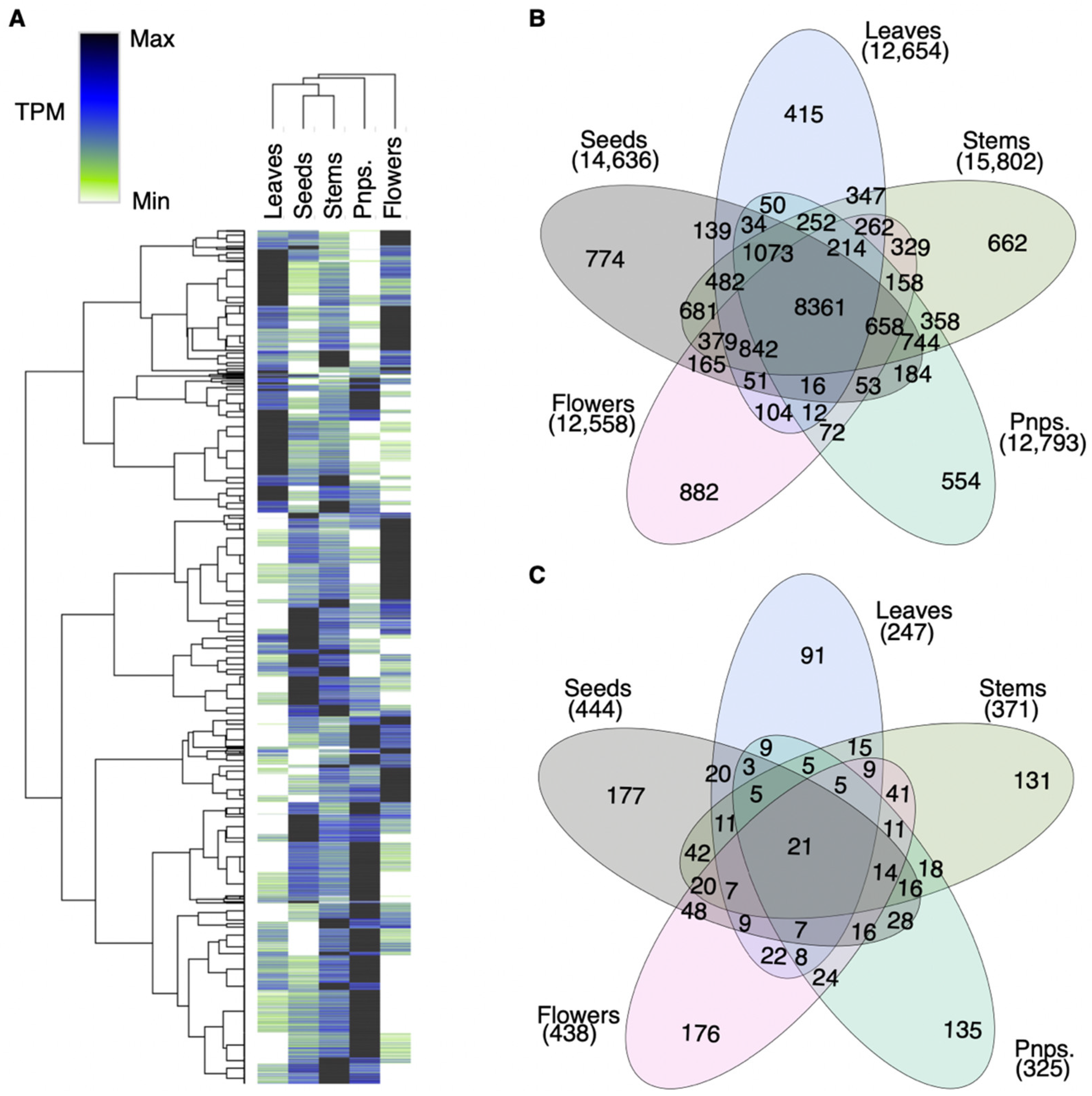
2.5. Intersection Analyses
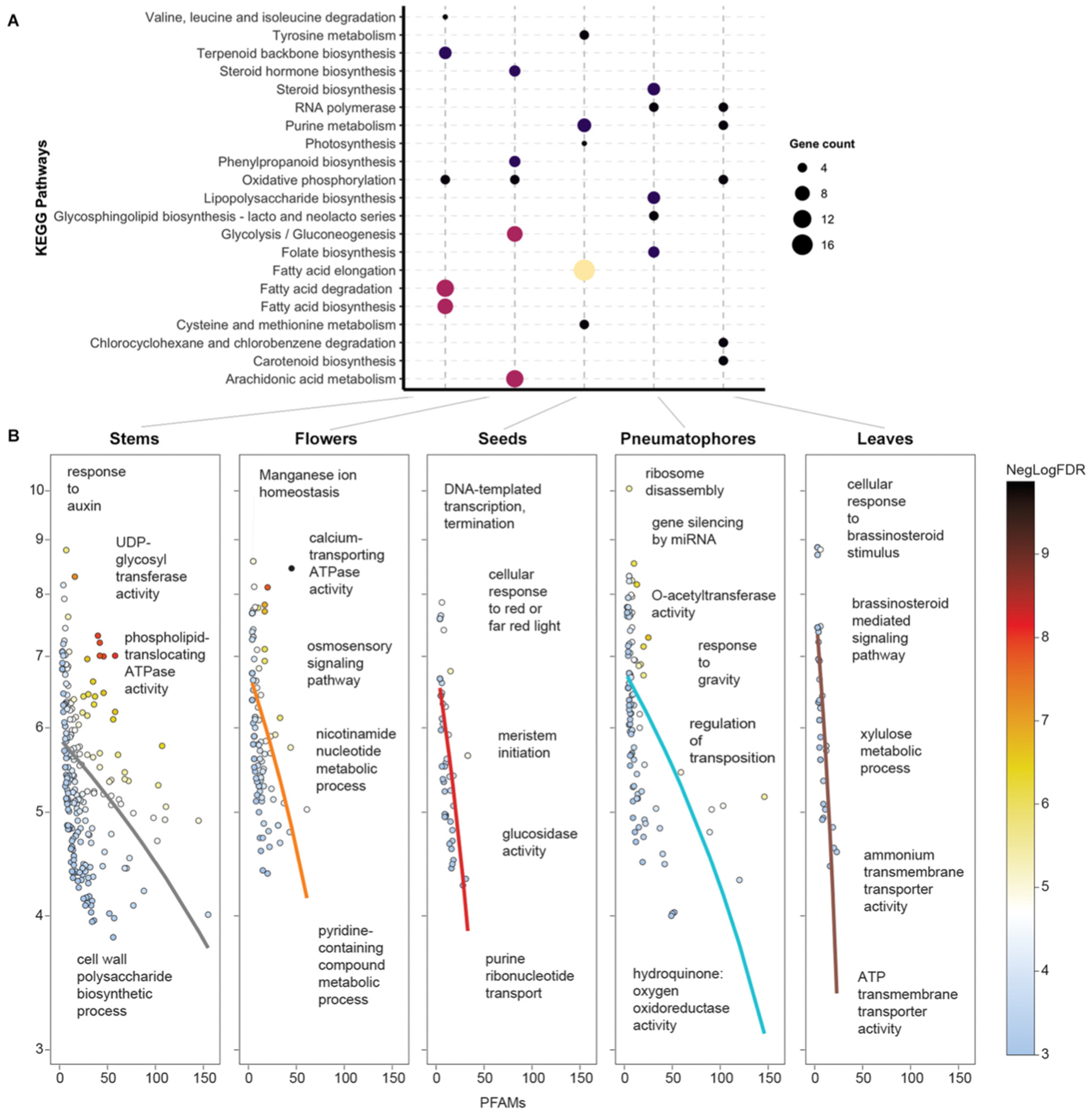
2.6. Domain-Centric GO Enrichment Analysis
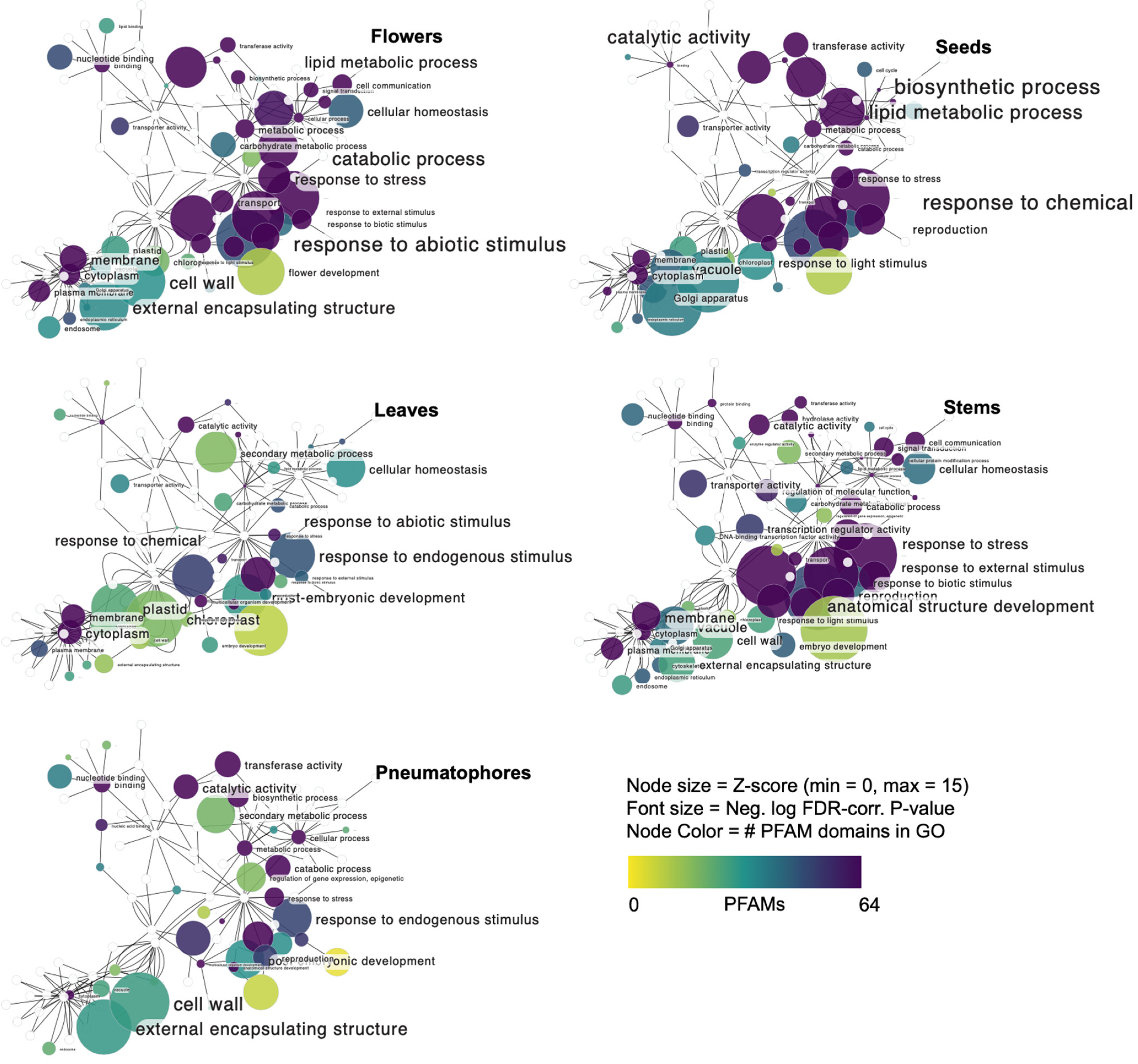
2.7. GO Network
2.8. Metabolic Map Reconstruction
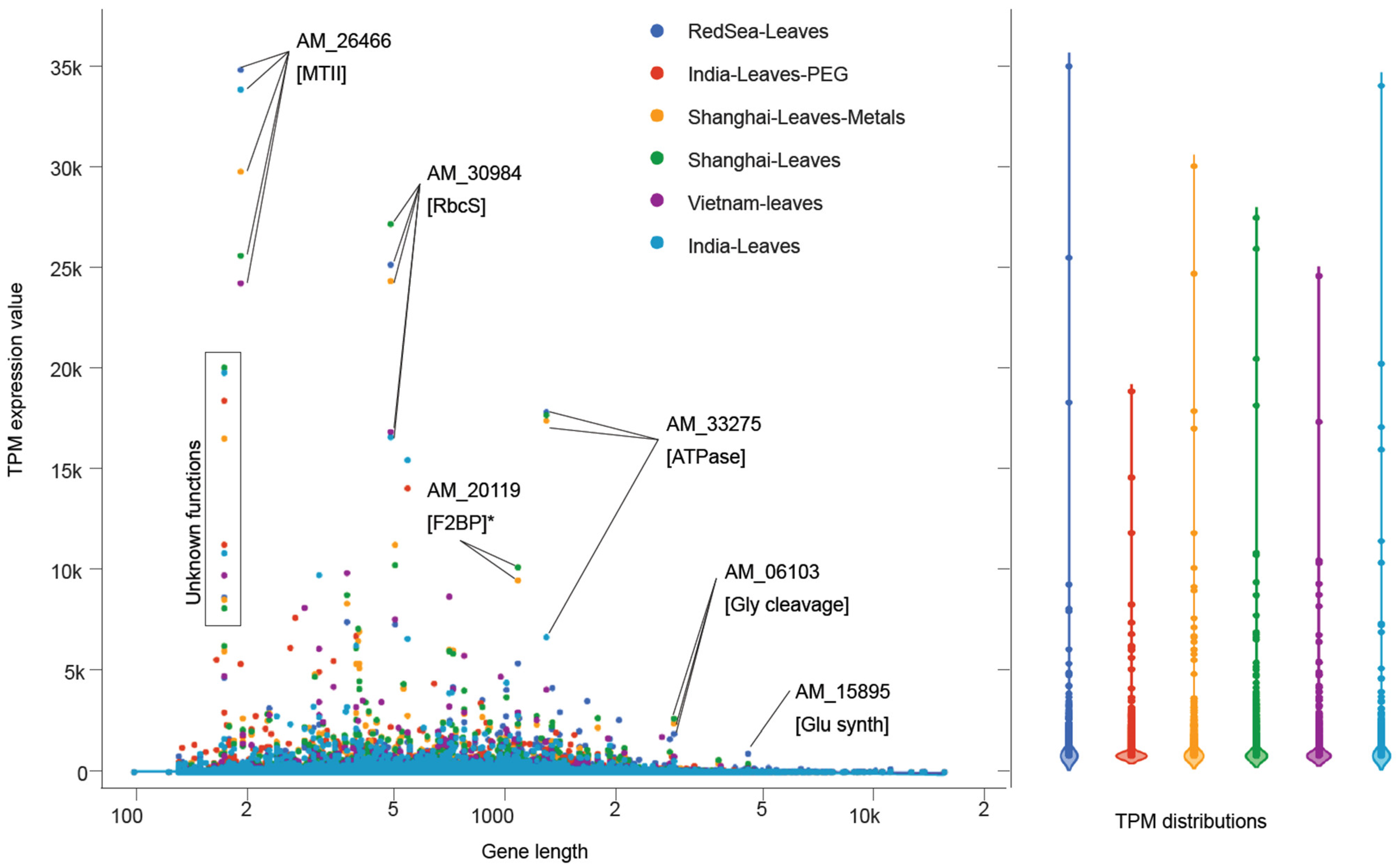
2.9. Manhattan and Violin Plots
3. Results and Discussion
3.1. A. marina Coding Potential and Transcriptome
3.2. A. marina Tissue-Specific Transcript Expression
3.3. Flower-Specific Transcript Expression
3.4. Seed-Specific Transcript Expression
3.5. Leaf-Specific Transcript Expression
3.6. Stem-Specific Transcript Expression
3.7. Pneumatophore-Specific Transcript Expression
3.8. Comparison of A. marina Transcriptomes across Geographies and Climates
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cavanaugh, K.C.; Dangremond, E.M.; Doughty, C.L.; Williams, A.P.; Parker, J.D.; Hayes, M.A.; Rodriguez, W.; Feller, I.C. Climate-driven regime shifts in a mangrove-salt marsh ecotone over the past 250 years. Proc. Natl. Acad. Sci. USA 2019, 116, 21602–21608. [Google Scholar] [CrossRef]
- Charles, S.P.; Kominoski, J.S.; Armitage, A.R.; Guo, H.; Weaver, C.A.; Pennings, S.C. Quantifying how changing mangrove cover affects ecosystem carbon storage in coastal wetlands. Ecology 2019, 101, e02916. [Google Scholar] [CrossRef] [PubMed]
- Inoue, T.; Shimono, A.; Akaji, Y.; Baba, S.; Takenaka, A.; Tuck Chan, H. Mangrove-diazotroph relationships at the root, tree and forest scales: Diazotrophic communities create high soil nitrogenase activities in Rhizophora stylosa rhizospheres. Ann. Bot. 2019, 125, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Sasmito, S.D.; Taillardat, P.; Clendenning, J.N.; Cameron, C.; Friess, D.A.; Murdiyarso, D.; Hutley, L.B. Effect of land-use and land-cover change on mangrove blue carbon: A systematic review. Glob. Change Biol. 2019, 25, 4291–4302. [Google Scholar] [CrossRef] [PubMed]
- Theuerkauff, D.; Rivera-Ingraham, G.A.; Lambert, S.; Mercky, Y.; Lejeune, M.; Lignot, J.H.; Sucre, E. Wastewater bioremediation by mangrove ecosystems impacts crab ecophysiology: In-situ caging experiment. Aquat. Toxicol. 2019, 218, 105358. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Li, X.; Yang, M.; Wang, X.; Zhong, C.; Duke, N.C.; Wu, C.-I.; Shi, S. Speciation with gene flow via cycles of isolation and migration: Insights from multiple mangrove taxa. Natl. Sci. Rev. 2019, 6, 275–288. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Xu, S.; Zhang, Z.; Guo, W.; Lyu, H.; Zhong, C.; Boufford, D.E.; Duke, N.C.; Shi, S.; The International Mangrove Consortium. Convergent adaptation of the genomes of woody plants at the land–sea interface. Natl. Sci. Rev. 2020, 7, 978–993. [Google Scholar] [CrossRef]
- Arnaud-Haond, S.; Teixeira, S.; Massa, S.I.; Billot, C.; Saenger, P.; Coupland, G.; Duarte, C.M.; Serrão, E.A. Genetic structure at range edge: Low diversity and high inbreeding in Southeast Asian mangrove (Avicennia marina) populations. Mol. Ecol. 2006, 15, 3515–3525. [Google Scholar] [CrossRef]
- Dodd, R.S.; Afzal-Rafii, Z.; Kashani, N.; Budrick, J. Land barriers and open oceans: Effects on gene diversity and population structure in Avicennia germinans L. (Avicenniaceae). Mol. Ecol. 2002, 11, 1327–1338. [Google Scholar] [CrossRef]
- Hazarika, D.; Thangaraj, M.; Sahu, S.K.; Kathiresan, K. Genetic diversity in three populations of Avicennia marina along the eastcoast of India by RAPD markers. J. Environ. Biol. 2013, 34, 663–666. [Google Scholar]
- Maguire, T.L.; Peakall, R.; Saenger, P. Comparative analysis of genetic diversity in the mangrove species Avicennia marina (Forsk.) Vierh. (Avicenniaceae) detected by AFLPs and SSRs. Theor. Appl. Genet. 2002, 104, 388–398. [Google Scholar] [CrossRef]
- Friis, G.; Burt, J.A. Evolution of mangrove research in an extreme environment: Historical trends and future opportunities in Arabia. Ocean Coast. Manag. 2020, 195, 105288. [Google Scholar] [CrossRef]
- Almahasheer, H.; Duarte, C.M.; Irigoien, X. Phenology and Growth dynamics of Avicennia marina in the Central Red Sea. Sci. Rep. 2016, 6, 37785. [Google Scholar] [CrossRef]
- Alzubaidy, H.; Essack, M.; Malas, T.B.; Bokhari, A.; Motwalli, O.; Kamanu, F.K.; Jamhor, S.A.; Mokhtar, N.A.; Antunes, A.; Simoes, M.F.; et al. Rhizosphere microbiome metagenomics of gray mangroves (Avicennia marina) in the Red Sea. Gene 2016, 576, 626–636. [Google Scholar] [CrossRef]
- Chen, C.; Han, S.; Zhu, Z.; Fu, G.; Wang, R.; Zhang, Q.; Ye, Y.; Ren, Y.; Yan, C.; Xu, L.; et al. Idiomarina mangrovi sp. nov., isolated from rhizosphere soil of a mangrove Avicennia marina forest. Int. J. Syst. Evol. Microbiol. 2019, 69, 1662–1668. [Google Scholar] [CrossRef]
- Clarke, P.J.; Allaway, W.G. The regeneration niche of the grey mangrove (Avicennia marina): Effects of salinity, light and sediment factors on establishment, growth and survival in the field. Oecologia 1993, 93, 548–556. [Google Scholar] [CrossRef]
- Ganesan, G.; Sankararamasubramanian, H.; Narayanan, J.M.; Sivaprakash, K.; Parida, A. Transcript level characterization of a cDNA encoding stress regulated NAC transcription factor in the mangrove plant Avicennia marina. Plant Physiol. Biochem. 2008, 46, 928–934. [Google Scholar] [CrossRef]
- Huang, G.Y.; Wang, Y.S. Expression and characterization analysis of type 2 metallothionein from grey mangrove species (Avicennia marina) in response to metal stress. Aquat. Toxicol. 2010, 99, 86–92. [Google Scholar] [CrossRef]
- Khraiwesh, B.; Pugalenthi, G.; Fedoroff, N.V. Identification and analysis of red sea mangrove (Avicennia marina) microRNAs by high-throughput sequencing and their association with stress responses. PloS ONE 2013, 8, e60774. [Google Scholar] [CrossRef]
- Schmitz, N.; Robert, E.M.; Verheyden, A.; Kairo, J.G.; Beeckman, H.; Koedam, N. A patchy growth via successive and simultaneous cambia: Key to success of the most widespread mangrove species Avicennia marina? Ann. Bot. 2008, 101, 49–58. [Google Scholar] [CrossRef]
- Simoes, M.F.; Antunes, A.; Ottoni, C.A.; Amini, M.S.; Alam, I.; Alzubaidy, H.; Mokhtar, N.A.; Archer, J.A.; Bajic, V.B. Soil and Rhizosphere Associated Fungi in Gray Mangroves (Avicennia marina) from the Red Sea--A Metagenomic Approach. Genom. Proteom. Bioinform. 2015, 13, 310–320. [Google Scholar] [CrossRef] [PubMed]
- Usman, A.R.; Alkredaa, R.S.; Al-Wabel, M.I. Heavy metal contamination in sediments and mangroves from the coast of Red Sea: Avicennia marina as potential metal bioaccumulator. Ecotoxicol. Environ. Saf. 2013, 97, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Mildenhall, D.C.; Brown, L.J. An early Holocene occurrence of the mangrove Avicennia marina in Poverty Bay, North Island, New Zealand: Its climatic and geological implications. N. Z. J. Bot. 1987, 25, 281–294. [Google Scholar] [CrossRef]
- Schmitz, N.; Verheyden, A.; Kairo, J.G.; Beeckman, H.; Koedam, N. Successive cambia development in Avicennia marina (Forssk.) Vierh. is not climatically driven in the seasonal climate at Gazi Bay, Kenya. Dendrochronologia 2007, 25, 87–96. [Google Scholar] [CrossRef]
- Cheng, B.; Li, H. Impact of climate change and human activities on economic values produced by ecosystem service functions of rivers in water shortage area of Northwest China. Environ. Sci. Pollut. Res. Int. 2020, 27, 26570–26578. [Google Scholar] [CrossRef]
- Cheng, H.; Inyang, A.; Li, C.D.; Fei, J.; Zhou, Y.W.; Wang, Y.S. Salt tolerance and exclusion in the mangrove plant Avicennia marina in relation to root apoplastic barriers. Ecotoxicology 2020, 29, 676–683. [Google Scholar] [CrossRef]
- Jyothi-Prakash, P.A.; Mohanty, B.; Wijaya, E.; Lim, T.-M.; Lin, Q.; Loh, C.-S.; Kumar, P.P. Identification of salt gland-associated genes and characterization of a dehydrin from the salt secretor mangrove Avicennia officinalis. BMC Plant Biol. 2014, 14, 291. [Google Scholar] [CrossRef]
- Song, J.; Wang, B. Using euhalophytes to understand salt tolerance and to develop saline agriculture: Suaeda salsa as a promising model. Ann. Bot. 2015, 115, 541–553. [Google Scholar] [CrossRef]
- Tan, W.-K.; Lin, Q.; Lim, T.-M.; Kumar, P.; Loh, C.-S. Dynamic secretion changes in the salt glands of the mangrove tree species Avicennia officinalis in response to a changing saline environment. Plant Cell Environ. 2013, 36, 1410–1422. [Google Scholar] [CrossRef]
- Krishnamurthy, P.; Mohanty, B.; Wijaya, E.; Lee, D.Y.; Lim, T.M.; Lin, Q.; Xu, J.; Loh, C.S.; Kumar, P.P. Transcriptomics analysis of salt stress tolerance in the roots of the mangrove Avicennia officinalis. Sci. Rep. 2017, 7, 10031. [Google Scholar] [CrossRef]
- Kryger, L.; Lee, S.K. Effects of Mangrove Soil Ageing on the Accumulation of Hydrogen Sulphide in Roots of Avicennia spp. Biogeochemistry 1996, 35, 367–375. [Google Scholar] [CrossRef]
- Arshad, M.; Eid, E.M.; Hasan, M. Mangrove health along the hyper-arid southern Red Sea coast of Saudi Arabia. Environ. Monit. Assess. 2020, 192, 189. [Google Scholar] [CrossRef]
- Masoud, M.S.; Abdel-Halim, A.M.; El Ashmawy, A.A. Seasonal variation of nutrient salts and heavy metals in mangrove (Avicennia marina) environment, Red Sea, Egypt. Environ. Monit. Assess. 2019, 191, 425. [Google Scholar] [CrossRef]
- Gao, X.; Liu, Y.; Sun, B. Water shortage risk assessment considering large-scale regional transfers: A copula-based uncertainty case study in Lunan, China. Environ. Sci. Pollut. Res. Int. 2018, 25, 23328–23341. [Google Scholar] [CrossRef]
- Kummu, M.; Guillaume, J.H.; de Moel, H.; Eisner, S.; Florke, M.; Porkka, M.; Siebert, S.; Veldkamp, T.I.; Ward, P.J. The world’s road to water scarcity: Shortage and stress in the 20th century and pathways towards sustainability. Sci. Rep. 2016, 6, 38495. [Google Scholar] [CrossRef]
- Nabi, G.; Ali, M.; Khan, S.; Kumar, S. The crisis of water shortage and pollution in Pakistan: Risk to public health, biodiversity, and ecosystem. Environ. Sci. Pollut. Res. Int. 2019, 26, 10443–10445. [Google Scholar] [CrossRef]
- Xu, Z.; Cai, X.; Yin, X.; Su, M.; Wu, Y.; Yang, Z. Is water shortage risk decreased at the expense of deteriorating water quality in a large water supply reservoir? Water Res. 2019, 165, 114984. [Google Scholar] [CrossRef]
- Hayat, K.; Zhou, Y.; Menhas, S.; Bundschuh, J.; Hayat, S.; Ullah, A.; Wang, J.; Chen, X.; Zhang, D.; Zhou, P. Pennisetum giganteum: An emerging salt accumulating/tolerant non-conventional crop for sustainable saline agriculture and simultaneous phytoremediation. Environ. Pollut. 2020, 265, 114876. [Google Scholar] [CrossRef]
- Illera-Vives, M.; Lopez-Mosquera, M.E.; Salas-Sanjuan Mdel, C.; Lopez-Fabal, A. Leaching techniques for saline wastes composts used as growing media in organic agriculture: Assessment and modelling. Environ. Sci. Pollut. Res. Int. 2015, 22, 6854–6863. [Google Scholar] [CrossRef]
- Jin, Y.; Weining, S.; Nevo, E. A MAPK gene from Dead Sea fungus confers stress tolerance to lithium salt and freezing-thawing: Prospects for saline agriculture. Proc. Natl. Acad. Sci. USA 2005, 102, 18992–18997. [Google Scholar] [CrossRef]
- Qadir, M.; Oster, J.D. Crop and irrigation management strategies for saline-sodic soils and waters aimed at environmentally sustainable agriculture. Sci. Total Environ. 2004, 323, 1–19. [Google Scholar] [CrossRef]
- Qin, S.; Liu, Y.; Han, Y.; Xu, G.; Wan, S.; Cui, F.; Li, G. Aquaporins and their function in root water transport under salt stress conditions in Eutrema salsugineum. Plant Sci. 2019, 287, 110199. [Google Scholar] [CrossRef]
- Guo, R.Z.; Yan, H.Y.; Li, X.X.; Zou, X.X.; Zhang, X.J.; Yu, X.N.; Ci, D.W.; Wang, Y.F.; Si, T. Green leaf volatile (Z)-3-hexeny-1-yl acetate reduces salt stress in peanut by affecting photosynthesis and cellular redox homeostasis. Physiol. Plant 2020, 10, 785. [Google Scholar] [CrossRef]
- Lekklar, C.; Suriya-Arunroj, D.; Pongpanich, M.; Comai, L.; Kositsup, B.; Chadchawan, S.; Buaboocha, T. Comparative genomic analysis of rice with contrasting photosynthesis and grain production under salt stress. Genes 2019, 10, 562. [Google Scholar] [CrossRef]
- Nounjan, N.; Chansongkrow, P.; Charoensawan, V.; Siangliw, J.L.; Toojinda, T.; Chadchawan, S.; Theerakulpisut, P. High Performance of Photosynthesis and Osmotic Adjustment Are Associated With Salt Tolerance Ability in Rice Carrying Drought Tolerance QTL: Physiological and Co-expression Network Analysis. Front. Plant Sci. 2018, 9, 1135. [Google Scholar] [CrossRef]
- Wang, Z.; Yang, H.; Ramesh, A.; Roberts, L.J., 2nd; Zhou, L.; Lin, X.; Zhao, Y.; Guo, Z. Overexpression of Cu/Zn-superoxide dismutase and/or catalase accelerates benzo(a)pyrene detoxification by upregulation of the aryl hydrocarbon receptor in mouse endothelial cells. Free Radic. Biol. Med. 2009, 47, 1221–1229. [Google Scholar] [CrossRef]
- Wasai, S.; Kanno, N.; Matsuura, K.; Haruta, S. Increase of Salt Tolerance in Carbon-Starved Cells of Rhodopseudomonas palustris Depending on Photosynthesis or Respiration. Microorganisms 2018, 6, 4. [Google Scholar] [CrossRef]
- Zhang, Z.; Li, X.; Cui, L.; Meng, S.; Ye, N.; Peng, X. Catalytic and functional aspects of different isozymes of glycolate oxidase in rice. BMC Plant Biol. 2017, 17, 135. [Google Scholar] [CrossRef]
- Chang, B.; Yang, L.; Cong, W.; Zu, Y.; Tang, Z. The improved resistance to high salinity induced by trehalose is associated with ionic regulation and osmotic adjustment in Catharanthus roseus. Plant Physiol. Biochem. 2014, 77, 140–148. [Google Scholar] [CrossRef]
- Frank, W.; Ratnadewi, D.; Reski, R. Physcomitrella patens is highly tolerant against drought, salt and osmotic stress. Planta 2005, 220, 384–394. [Google Scholar] [CrossRef]
- Pires, R.M.O.; Avila, M.A.B.; Leite, D.G.; Santos, H.O.; Souza, G.A.; Von Pinho, E.V.R. Physiological and enzymatic alterations in sesame seeds submitted to different osmotic potentials. Genet. Mol. Res. 2017, 16, gmr16039425. [Google Scholar] [CrossRef] [PubMed]
- Empitu, M.A.; Kadariswantiningsih, I.N. Modelling salt transport disorders of human kidney in zebrafish: The grain of salt. J. Physiol. 2019, 597, 5529–5530. [Google Scholar] [CrossRef] [PubMed]
- Grosell, M.; Heuer, R.M.; Wu, N.C.; Cramp, R.L.; Wang, Y.; Mager, E.M.; Dwyer, R.G.; Franklin, C.E. Salt-water acclimation of the estuarine crocodile Crocodylus porosus involves enhanced ion transport properties of the urodaeum and rectum. J. Exp. Biol. 2020, 223, jeb210732. [Google Scholar] [CrossRef] [PubMed]
- Stolting, G.; Fahlke, C. Chloride channels in renal salt and water transport. Acta Physiol. 2017, 219, 11–13. [Google Scholar] [CrossRef]
- Wu, H.; Li, Z. The Importance of Cl− Exclusion and Vacuolar Cl− Sequestration: Revisiting the Role of Cl− Transport in Plant Salt Tolerance. Front. Plant Sci. 2019, 10, 1418. [Google Scholar] [CrossRef]
- Zhang, Y.; Fang, J.; Wu, X.; Dong, L. Na+/K+ Balance and Transport Regulatory Mechanisms in Weedy and Cultivated Rice (Oryza sativa L.) Under Salt Stress. BMC Plant Biol. 2018, 18, 375. [Google Scholar] [CrossRef]
- Zhou, Y.; Tao, J.; Ahammed, G.J.; Li, J.; Yang, Y. Genome-wide identification and expression analysis of aquaporin gene family related to abiotic stress in watermelon. Genome 2019, 62, 643–656. [Google Scholar] [CrossRef]
- Severi, E.; Hosie, A.H.; Hawkhead, J.A.; Thomas, G.H. Characterization of a novel sialic acid transporter of the sodium solute symporter (SSS) family and in vivo comparison with known bacterial sialic acid transporters. FEMS Microbiol. Lett. 2010, 304, 47–54. [Google Scholar] [CrossRef]
- Friis, G.; Vizueta, J.; Smith, E.G.; Nelson, D.R.; Khraiwesh, B.; Qudeimat, E.; Salehi-Ashtiani, K.; Ortega, A.; Marshell, A.; Duarte, C.M.; et al. A high-quality genome assembly and annotation of the gray mangrove, Avicennia marina. G3 Genes Genomes Genet. 2021, 11, jkaa025. [Google Scholar] [CrossRef]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Meth. 2008, 5, 621–628. [Google Scholar] [CrossRef]
- Conesa, A.; Madrigal, P.; Tarazona, S.; Gomez-Cabrero, D.; Cervera, A.; McPherson, A.; Szczesniak, M.W.; Gaffney, D.J.; Elo, L.L.; Zhang, X.; et al. A survey of best practices for RNA-seq data analysis. Genome Biol. 2016, 17, 13. [Google Scholar] [CrossRef]
- Stark, R.; Grzelak, M.; Hadfield, J. RNA sequencing: The teenage years. Nat. Rev. Genet. 2019, 20, 631–656. [Google Scholar] [CrossRef]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef]
- Lee, S.; Seo, C.H.; Lim, B.; Yang, J.O.; Oh, J.; Kim, M.; Lee, S.; Lee, B.; Kang, C.; Lee, S. Accurate quantification of transcriptome from RNA-Seq data by effective length normalization. Nucleic Acids Res. 2011, 39, e9. [Google Scholar] [CrossRef]
- Li, P.; Piao, Y.; Shon, H.S.; Ryu, K.H. Comparing the normalization methods for the differential analysis of Illumina high-throughput RNA-Seq data. BMC Bioinform. 2015, 16, 347. [Google Scholar] [CrossRef]
- Wu, P.Y.; Phan, J.H.; Zhou, F.; Wang, M.D. Evaluation of Normalization Methods for RNA-Seq Gene Expression Estimation. In Proceedings of the 2011 IEEE International Conference on Bioinformatics and Biomedicine Workshops (BIBMW), Atlanta, GA, USA, 12–15 November 2011; pp. 50–57. [Google Scholar] [CrossRef]
- Zyprych-Walczak, J.; Szabelska, A.; Handschuh, L.; Gorczak, K.; Klamecka, K.; Figlerowicz, M.; Siatkowski, I. The Impact of Normalization Methods on RNA-Seq Data Analysis. Biomed. Res. Int. 2015, 2015, 621690. [Google Scholar] [CrossRef]
- McInnes, L.; Healy, J.; Melville, J. UMAP: Uniform Manifold Approximation and Projection for Dimension Reduction. arXiv 2018, arXiv:1802.03426. [Google Scholar]
- Warnes, G.R.; Bolker, B.; Bonebakker, L.; Gentleman, R.; Huber, W.; Liaw, A.; Lumley, T.; Maechler, M.; Magnusson, A.; Moeller, S. gplots: Various R Programming Tools for Plotting Data. R Package Version 2009, 2. Available online: https://scholar.google.com/citations?view_op=view_citation&hl=en&user=o7GsfJwAAAAJ&citation_for_view=o7GsfJwAAAAJ:u-x6o8ySG0sC (accessed on 3 July 2022).
- Heberle, H.; Meirelles, G.V.; da Silva, F.R.; Telles, G.P.; Minghim, R. InteractiVenn: A web-based tool for the analysis of sets through Venn diagrams. BMC Bioinform. 2015, 16, 169. [Google Scholar] [CrossRef]
- Conway, J.R.; Lex, A.; Gehlenborg, N. UpSetR: An R package for the visualization of intersecting sets and their properties. Bioinformatics 2017, 33, 2938–2940. [Google Scholar] [CrossRef]
- Fang, H.; Gough, J. A domain-centric solution to functional genomics via dcGO Predictor. BMC Bioinform. 2013, 14 (Suppl. S3), S9. [Google Scholar] [CrossRef]
- Krzywinski, M.I.; Schein, J.E.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef]
- Sonnhammer, E.L.; Eddy, S.R.; Durbin, R. Pfam: A comprehensive database of protein domain families based on seed alignments. Proteins 1997, 28, 405–420. [Google Scholar] [CrossRef]
- Mistry, J.; Chuguransky, S.; Williams, L.; Qureshi, M.; Salazar, G.A.; Sonnhammer, E.L.L.; Tosatto, S.C.E.; Paladin, L.; Raj, S.; Richardson, L.J.; et al. Pfam: The protein families database in 2021. Nucleic Acids Res. 2021, 49, D412–D419. [Google Scholar] [CrossRef]
- Landry, C.L. Pollinator-mediated competition between two co-flowering Neotropical mangrove species, Avicennia germinans (Avicenniaceae) and Laguncularia racemosa (Combretaceae). Ann. Bot. 2013, 111, 207–214. [Google Scholar] [CrossRef]
- Liu, L.; Guo, Z.; Zhong, C.; Shi, S. DNA barcoding reveals insect diversity in the mangrove ecosystems of Hainan Island, China. Genome 2018, 61, 797–806. [Google Scholar] [CrossRef]
- De Vetten, N.; ter Horst, J.; van Schaik, H.P.; de Boer, A.; Mol, J.; Koes, R. A cytochrome b5 is required for full activity of flavonoid 3', 5'-hydroxylase, a cytochrome P450 involved in the formation of blue flower colors. Proc. Natl. Acad. Sci. USA 1999, 96, 778–783. [Google Scholar] [CrossRef]
- Holton, T.A.; Brugliera, F.; Lester, D.R.; Tanaka, Y.; Hyland, C.D.; Menting, J.G.; Lu, C.Y.; Farcy, E.; Stevenson, T.W.; Cornish, E.C. Cloning and expression of cytochrome P450 genes controlling flower colour. Nature 1993, 366, 276–279. [Google Scholar] [CrossRef]
- Liu, Z.; Boachon, B.; Lugan, R.; Tavares, R.; Erhardt, M.; Mutterer, J.; Demais, V.; Pateyron, S.; Brunaud, V.; Ohnishi, T.; et al. A Conserved Cytochrome P450 Evolved in Seed Plants Regulates Flower Maturation. Mol. Plant. 2015, 8, 1751–1765. [Google Scholar] [CrossRef]
- Martsinkovskaya, A.I.; Poghosyan, Z.P.; Haralampidis, K.; Murphy, D.J.; Hatzopoulos, P. Temporal and spatial gene expression of cytochrome B5 during flower and fruit development in olives. Plant Mol. Biol. 1999, 40, 79–90. [Google Scholar] [CrossRef] [PubMed]
- Petkova-Andonova, M.; Imaishi, H.; Ohkawa, H. CYP92B1, A cytochrome P450, expressed in petunia flower buds, that catalyzes monooxidation of long-chain fatty acids. Biosci. Biotechnol. Biochem. 2002, 66, 1819–1828. [Google Scholar] [CrossRef] [PubMed]
- Shahak, Y.; Posner, H.B.; Avron, M. Evidence for a Block between Plastoquinone and Cytochrome f in a Photosynthetic Mutant of Lemna with Abnormal Flowering Behavior. Plant Physiol. 1976, 57, 577–579. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ito, A.; Saito, T.; Nishijima, T.; Moriguchi, T. Effect of extending the photoperiod with low-intensity red or far-red light on the timing of shoot elongation and flower-bud formation of 1-year-old Japanese pear (Pyrus pyrifolia). Tree Physiol. 2014, 34, 534–546. [Google Scholar] [CrossRef]
- Koornneef, M. Plant development: Timing when to flower. Curr. Biol. 1997, 7, R651–652. [Google Scholar] [CrossRef]
- Lord, E.M.; Crone, W.; Hill, J.P. Timing of events during flower organogenesis: Arabidopsis as a model system. Curr. Top Dev. Biol. 1994, 29, 325–356. [Google Scholar] [CrossRef] [PubMed]
- Wagner, D. Flower morphogenesis: Timing is key. Dev. Cell. 2009, 16, 621–622. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Davis, E.J.; Ballif, J.; Liang, M.; Bushman, E.; Haroldsen, V.; Torabinejad, J.; Wu, Y. Mutant identification and characterization of the laccase gene family in Arabidopsis. J. Exp. Bot. 2006, 57, 2563–2569. [Google Scholar] [CrossRef] [PubMed]
- Salekeen, R.; Mou, S.N.; Islam, M.E.; Ahmed, A.; Billah, M.M.; Rahman, S.M.M.; Islam, K.M.D. Predicting multi-enzyme inhibition in the arachidonic acid metabolic network by Heritiera fomes extracts. J. Biomol. Struct. Dyn. 2022, 40, 4259–4272. [Google Scholar] [CrossRef]
- Aubert, D.; Chevillard, M.; Dorne, A.M.; Arlaud, G.; Herzog, M. Expression patterns of GASA genes in Arabidopsis thaliana: The GASA4 gene is up-regulated by gibberellins in meristematic regions. Plant Mol. Biol. 1998, 36, 871–883. [Google Scholar] [CrossRef]
- Roxrud, I.; Lid, S.E.; Fletcher, J.C.; Schmidt, E.D.; Opsahl-Sorteberg, H.G. GASA4, one of the 14-member Arabidopsis GASA family of small polypeptides, regulates flowering and seed development. Plant Cell Physiol. 2007, 48, 471–483. [Google Scholar] [CrossRef]
- Rubinovich, L.; Weiss, D. The Arabidopsis cysteine-rich protein GASA4 promotes GA responses and exhibits redox activity in bacteria and in planta. Plant J. Cell Mol. Biol. 2010, 64, 1018–1027. [Google Scholar] [CrossRef]
- Carvalho, A.T.; Dotterl, S.; Schlindwein, C. An aromatic volatile attracts oligolectic bee pollinators in an interdependent bee-plant relationship. J. Chem. Ecol. 2014, 40, 1126–1134. [Google Scholar] [CrossRef]
- Ilc, T.; Parage, C.; Boachon, B.; Navrot, N.; Werck-Reichhart, D. Monoterpenol Oxidative Metabolism: Role in Plant Adaptation and Potential Applications. Front. Plant Sci. 2016, 7, 509. [Google Scholar] [CrossRef]
- Awadasseid, A.; Li, W.; Liu, Z.; Qiao, C.; Pang, J.; Zhang, G.; Luo, Y. Characterization of Camptotheca acuminata 10-Hydroxygeraniol oxidoreductase and iridoid synthase and their application in biological preparation of nepetalactol in Escherichia coli featuring NADP+-NADPH cofactors recycling. Int. J. Biol. Macromol. 2020, 162, 1076–1085. [Google Scholar] [CrossRef]
- Carbonezi, C.A.; Martins, D.; Young, M.C.; Lopes, M.N.; Furlan, M.; Rodriguez Filho, E.; Bolzani Vda, S. Iridoid and seco-iridoid glucosides from Chioccoca alba (Rubiaceae). Phytochemistry 1999, 51, 781–785. [Google Scholar] [CrossRef]
- Hofer, R.; Dong, L.; Andre, F.; Ginglinger, J.F.; Lugan, R.; Gavira, C.; Grec, S.; Lang, G.; Memelink, J.; Van der Krol, S.; et al. Geraniol hydroxylase and hydroxygeraniol oxidase activities of the CYP76 family of cytochrome P450 enzymes and potential for engineering the early steps of the (seco)iridoid pathway. Metab. Eng. 2013, 20, 221–232. [Google Scholar] [CrossRef]
- Miettinen, K.; Dong, L.; Navrot, N.; Schneider, T.; Burlat, V.; Pollier, J.; Woittiez, L.; van der Krol, S.; Lugan, R.; Ilc, T.; et al. The seco-iridoid pathway from Catharanthus roseus. Nat. Commun. 2014, 5, 3606. [Google Scholar] [CrossRef] [PubMed]
- Valletta, A.; Trainotti, L.; Santamaria, A.R.; Pasqua, G. Cell-specific expression of tryptophan decarboxylase and 10-hydroxygeraniol oxidoreductase, key genes involved in camptothecin biosynthesis in Camptotheca acuminata Decne (Nyssaceae). BMC Plant Biol. 2010, 10, 69. [Google Scholar] [CrossRef]
- Willems, M. Quantitative Determination of Seco-Iridoid Glucosides from the Fruits of Ligustrum vulgare by HPLC. Planta Med. 1988, 54, 66–68. [Google Scholar] [CrossRef]
- Do, B.T.N.; Koedam, N.; Triest, L. Avicennia marina maintains genetic structure whereas Rhizophora stylosa connects mangroves in a flooded, former inner sea (Vietnam). Estuar. Coast. Shelf Sci. 2019, 222, 195–204. [Google Scholar] [CrossRef]
- Geng, Q.; Wang, Z.; Tao, J.; Kimura, M.K.; Liu, H.; Hogetsu, T.; Lian, C. Ocean Currents Drove Genetic Structure of Seven Dominant Mangrove Species Along the Coastlines of Southern China. Front. Genet. 2021, 12, 615911. [Google Scholar] [CrossRef] [PubMed]
- Rademacher, S.; Sprunck, S. Downregulation of egg cell-secreted EC1 is accompanied with delayed gamete fusion and polytubey. Plant Signal. Behav. 2013, 8, e27377. [Google Scholar] [CrossRef] [PubMed]
- Sprunck, S.; Rademacher, S.; Vogler, F.; Gheyselinck, J.; Grossniklaus, U.; Dresselhaus, T. Egg cell-secreted EC1 triggers sperm cell activation during double fertilization. Science 2012, 338, 1093–1097. [Google Scholar] [CrossRef] [PubMed]
- Deveson, I.W.; Holleley, C.E.; Blackburn, J.; Marshall Graves, J.A.; Mattick, J.S.; Waters, P.D.; Georges, A. Differential intron retention in Jumonji chromatin modifier genes is implicated in reptile temperature-dependent sex determination. Sci. Adv. 2017, 3, e1700731. [Google Scholar] [CrossRef] [PubMed]
- Gan, E.S.; Xu, Y.; Wong, J.Y.; Goh, J.G.; Sun, B.; Wee, W.Y.; Huang, J.; Ito, T. Jumonji demethylases moderate precocious flowering at elevated temperature via regulation of FLC in Arabidopsis. Nat. Commun. 2014, 5, 5098. [Google Scholar] [CrossRef]
- Zheng, S.; Hu, H.; Ren, H.; Yang, Z.; Qiu, Q.; Qi, W.; Liu, X.; Chen, X.; Cui, X.; Li, S.; et al. The Arabidopsis H3K27me3 demethylase JUMONJI 13 is a temperature and photoperiod dependent flowering repressor. Nat Commun 2019, 10, 1303. [Google Scholar] [CrossRef]
- Leon, K.E.; Aird, K.M. Jumonji C Demethylases in Cellular Senescence. Genes 2019, 10, 33. [Google Scholar] [CrossRef]
- Bentz, A.B.; Thomas, G.W.C.; Rusch, D.B.; Rosvall, K.A. Tissue-specific expression profiles and positive selection analysis in the tree swallow (Tachycineta bicolor) using a de novo transcriptome assembly. Sci. Rep. 2019, 9, 15849. [Google Scholar] [CrossRef]
- Klepikova, A.V.; Penin, A.A. Gene Expression Maps in Plants: Current State and Prospects. Plants 2019, 8, 309. [Google Scholar] [CrossRef]
- Kubínová, Z.; Janáček, J.; Lhotáková, Z.; Kubínová, L.; Albrechtová, J. Unbiased estimation of chloroplast number in mesophyll cells: Advantage of a genuine three-dimensional approach. J. Exp. Bot. 2014, 65, 609–620. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Lunde, C.; Drew, D.P.; Jacobs, A.K.; Tester, M. Exclusion of Na+ via sodium ATPase (PpENA1) ensures normal growth of Physcomitrella patens under moderate salt stress. Plant Physiol. 2007, 144, 1786–1796. [Google Scholar] [CrossRef][Green Version]
- Qudeimat, E.; Frank, W. Ca2+ signatures: The role of Ca2+-ATPases. Plant Signal. Behav. 2009, 4, 350–352. [Google Scholar] [CrossRef][Green Version]
- Cui, L.-L.; Lu, Y.-S.; Li, Y.; Yang, C.; Peng, X.-X. Overexpression of Glycolate Oxidase Confers Improved Photosynthesis under High Light and High Temperature in Rice. Front. Plant Sci. 2016, 7, 1165. [Google Scholar] [CrossRef]
- Fernandez, L.R.; Vandenbussche, G.; Roosens, N.; Govaerts, C.; Goormaghtigh, E.; Verbruggen, N. Metal binding properties and structure of a type III metallothionein from the metal hyperaccumulator plant Noccaea caerulescens. Biochim. Biophys. Acta 2012, 1824, 1016–1023. [Google Scholar] [CrossRef]
- Babaei-Bondarti, Z.; Shahpiri, A. A metallothionein type 2 from Avicennia marina binds to iron and mediates hydrogen peroxide balance by activation of enzyme catalase. Phytochemistry 2020, 176, 112396. [Google Scholar] [CrossRef]
- Freisinger, E. Plant MTs-long neglected members of the metallothionein superfamily. Dalton. Trans. 2008, 6663–6675. [Google Scholar] [CrossRef]
- Pirzadeh, S.; Shahpiri, A. Functional characterization of a type 2 metallothionein isoform (OsMTI-2b) from rice. Int. J. Biol. Macromol. 2016, 88, 491–496. [Google Scholar] [CrossRef]
- Cabral, A.C.S.; Jakovleska, J.; Deb, A.; Penner-Hahn, J.E.; Pecoraro, V.L.; Freisinger, E. Further insights into the metal ion binding abilities and the metalation pathway of a plant metallothionein from Musa acuminata. J. Biol. Inorg. Chem. 2018, 23, 91–107. [Google Scholar] [CrossRef]
- Rice, J.M.; Zweifach, A.; Lynes, M.A. Metallothionein regulates intracellular zinc signaling during CD4+ T cell activation. BMC Immunol. 2016, 17, 13. [Google Scholar] [CrossRef] [PubMed]
- Singh, G.; Tripathi, S.; Shanker, K.; Sharma, A. Cadmium-induced conformational changes in type 2 metallothionein of medicinal plant Coptis japonica: Insights from molecular dynamics studies of apo, partially and fully metalated forms. J. Biomol. Struct. Dyn. 2019, 37, 1520–1533. [Google Scholar] [CrossRef] [PubMed]
- Patankar, H.V.; Al-Harrasi, I.; Al Kharusi, L.; Jana, G.A.; Al-Yahyai, R.; Sunkar, R.; Yaish, M.W. Overexpression of a Metallothionein 2A Gene from Date Palm Confers Abiotic Stress Tolerance to Yeast and Arabidopsis thaliana. Int. J. Mol. Sci. 2019, 20, 2871. [Google Scholar] [CrossRef] [PubMed]
- Hamelet, J.; Seltzer, V.; Petit, E.; Noll, C.; Andreau, K.; Delabar, J.M.; Janel, N. Cystathionine beta synthase deficiency induces catalase-mediated hydrogen peroxide detoxification in mice liver. Biochim. Biophys. Acta 2008, 1782, 482–488. [Google Scholar] [CrossRef] [PubMed]
- Pheomphun, P.; Treesubsuntorn, C.; Jitareerat, P.; Thiravetyan, P. Contribution of Bacillus cereus ERBP in ozone detoxification by Zamioculcas zamiifolia plants: Effect of ascorbate peroxidase, catalase and total flavonoid contents for ozone detoxification. Ecotoxicol. Environ. Saf. 2019, 171, 805–812. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, R.; Bhattacharjee, A.; Majumdar, U.; Ray, S.S.; Dutta, T.; Ghosh, S. A novel role of catalase in detoxification of peroxynitrite in S. cerevisiae. Biochem. Biophys. Res. Commun. 2009, 385, 507–511. [Google Scholar] [CrossRef] [PubMed]
- Stührwohldt, N.; Bühler, E.; Sauter, M.; Schaller, A. Phytosulfokine (PSK) precursor processing by subtilase SBT3.8 and PSK signaling improve drought stress tolerance in Arabidopsis. J. Exp. Bot. 2021, 72, 3427–3440. [Google Scholar] [CrossRef]
- Koonin, E.V.; Tatusov, R.L. Computer analysis of bacterial haloacid dehalogenases defines a large superfamily of hydrolases with diverse specificity. Application of an iterative approach to database search. J. Mol. Biol. 1994, 244, 125–132. [Google Scholar] [CrossRef]
- Wang, Y.; Zheng, X.; He, X.; Lü, Q.; Qian, X.; Xiao, Q.; Lin, R. Effects of Pseudomonas TCd-1 on rice (Oryza sativa) cadmium uptake, rhizosphere soils enzyme activities and cadmium bioavailability under cadmium contamination. Ecotoxicol. Environ. Saf. 2021, 218, 112249. [Google Scholar] [CrossRef]
- Pandey, B.K.; Mehra, P.; Verma, L.; Bhadouria, J.; Giri, J. OsHAD1, a Haloacid Dehalogenase-Like APase, Enhances Phosphate Accumulation. Plant Physiol. 2017, 174, 2316–2332. [Google Scholar] [CrossRef]
- Hanin, M.; Brini, F.; Ebel, C.; Toda, Y.; Takeda, S.; Masmoudi, K. Plant dehydrins and stress tolerance: Versatile proteins for complex mechanisms. Plant Signal. Behav. 2011, 6, 1503–1509. [Google Scholar] [CrossRef]
- Hinniger, C.; Caillet, V.; Michoux, F.; Ben Amor, M.; Tanksley, S.; Lin, C.; McCarthy, J. Isolation and characterization of cDNA encoding three dehydrins expressed during Coffea canephora (Robusta) grain development. Ann. Bot. 2006, 97, 755–765. [Google Scholar] [CrossRef][Green Version]
- Nath, B.; Birch, G.; Chaudhuri, P. Assessment of sediment quality in Avicennia marina-dominated embayments of Sydney Estuary: The potential use of pneumatophores (aerial roots) as a bio-indicator of trace metal contamination. Sci. Total Environ. 2014, 472, 1010–1022. [Google Scholar] [CrossRef]
- Alishahi, M.; Kamali, R. Forced diffusion of water molecules through aquaporin-5 biomembrane; a molecular dynamics study. Biophys. Physicobiol. 2018, 15, 255–262. [Google Scholar] [CrossRef]
- Merlaen, B.; De Keyser, E.; Ding, L.; Leroux, O.; Chaumont, F.; Van Labeke, M.C. Physiological responses and aquaporin expression upon drought and osmotic stress in a conservative vs prodigal Fragaria x ananassa cultivar. Plant Physiol. Biochem. 2019, 145, 95–106. [Google Scholar] [CrossRef]
- Sahebi, S.; Fadaie, N.; Mirshekar, L.; Kamarehie, B.; Mohammadi, T. Assessing biomimetic aquaporin membrane for forward osmosis desalination process: A dataset. Data Brief 2019, 26, 104482. [Google Scholar] [CrossRef]
- Wei, X.; Jin, X.; Ndayambaza, B.; Min, X.; Zhang, Z.; Wang, Y.; Liu, W. Transcriptome-wide characterization and functional identification of the aquaporin gene family during drought stress in common Vetch. DNA Cell Biol. 2019, 38, 374–384. [Google Scholar] [CrossRef]
- Zou, Z.; Yang, J. Genome-wide comparison reveals divergence of cassava and rubber aquaporin family genes after the recent whole-genome duplication. BMC Genom. 2019, 20, 380. [Google Scholar] [CrossRef]
- Krishnamurthy, P.; Tan, X.F.; Lim, T.K.; Lim, T.-M.; Kumar, P.P.; Loh, C.-S.; Lin, Q. Proteomic analysis of plasma membrane and tonoplast from the leaves of mangrove plant Avicennia officinalis. Proteomics 2014, 14, 2545–2557. [Google Scholar] [CrossRef]
- Sadhukhan, A.; Kobayashi, Y.; Nakano, Y.; Iuchi, S.; Kobayashi, M.; Sahoo, L.; Koyama, H. Genome-wide Association Study Reveals that the Aquaporin NIP1;1 Contributes to Variation in Hydrogen Peroxide Sensitivity in Arabidopsis thaliana. Mol. Plant 2017, 10, 1082–1094. [Google Scholar] [CrossRef]
- Yusupov, M.; Razzokov, J.; Cordeiro, R.M.; Bogaerts, A. Transport of reactive oxygen and nitrogen species across aquaporin: A molecular level picture. Oxid. Med. Cell Longev. 2019, 2019, 2930504. [Google Scholar] [CrossRef] [PubMed]
- Nickerson, N.H.; Thibodeau, F.R. Association between Pore Water Sulfide Concentrations and the Distribution of Mangroves. Biogeochemistry 1985, 1, 183–192. [Google Scholar] [CrossRef]
- Kreuzwieser, J.; Rennenberg, H. Molecular and physiological responses of trees to waterlogging stress: Responses of tree to waterlogging. Plant Cell Environ. 2014, 37, 2245–2259. [Google Scholar] [CrossRef] [PubMed]
- Amala, M.; Rajamanikandan, S.; Prabhu, D.; Surekha, K.; Jeyakanthan, J. Identification of anti-filarial leads against aspartate semialdehyde dehydrogenase of Wolbachia endosymbiont of Brugia malayi: Combined molecular docking and molecular dynamics approaches. J. Biomol. Struct. Dyn. 2019, 37, 394–410. [Google Scholar] [CrossRef]
- Cohen, G.N. Aspartate-semialdehyde dehydrogenase from Escherichia coli. Methods Enzymol. 1985, 113, 600–602. [Google Scholar] [CrossRef]
- Cox, R.J.; Gibson, J.S.; Hadfield, A.T. Design, synthesis and analysis of inhibitors of bacterial aspartate semialdehyde dehydrogenase. Chembiochem 2005, 6, 2255–2260. [Google Scholar] [CrossRef]
- Dahal, G.P.; Viola, R.E. Structural insights into inhibitor binding to a fungal ortholog of aspartate semialdehyde dehydrogenase. Biochem. Biophys. Res. Commun. 2018, 503, 2848–2854. [Google Scholar] [CrossRef]
- Evitt, A.S.; Cox, R.J. Synthesis and evaluation of conformationally restricted inhibitors of aspartate semialdehyde dehydrogenase. Mol. Biosyst. 2011, 7, 1564–1575. [Google Scholar] [CrossRef]
- Gayler, K.R.; Morgan, W.R. An NADP-dependent Glutamate Dehydrogenase in Chloroplasts from the Marine Green Alga Caulerpa simpliciuscula. Plant Physiol. 1976, 58, 283–287. [Google Scholar] [CrossRef]
- Born, T.L.; Blanchard, J.S. Structure/function studies on enzymes in the diaminopimelate pathway of bacterial cell wall biosynthesis. Curr. Opin. Chem. Biol. 1999, 3, 607–613. [Google Scholar] [CrossRef]
- Mengin-Lecreulx, D.; van Heijenoort, J.; Park, J.T. Identification of the mpl gene encoding UDP-N-acetylmuramate: L-alanyl-gamma-D-glutamyl-meso-diaminopimelate ligase in Escherichia coli and its role in recycling of cell wall peptidoglycan. J. Bacteriol. 1996, 178, 5347–5352. [Google Scholar] [CrossRef]
- Park, J.T.; Raychaudhuri, D.; Li, H.; Normark, S.; Mengin-Lecreulx, D. MppA, a periplasmic binding protein essential for import of the bacterial cell wall peptide L-alanyl-gamma-D-glutamyl-meso-diaminopimelate. J. Bacteriol. 1998, 180, 1215–1223. [Google Scholar] [CrossRef]
- Wehrmann, A.; Phillipp, B.; Sahm, H.; Eggeling, L. Different modes of diaminopimelate synthesis and their role in cell wall integrity: A study with Corynebacterium glutamicum. J. Bacteriol. 1998, 180, 3159–3165. [Google Scholar] [CrossRef]
- Villa, S.T.; Xu, Q.; Downie, A.B.; Clarke, S.G. Arabidopsis protein repair l-isoaspartyl methyltransferases: Predominant activities at lethal temperatures. Physiol. Plant. 2006, 128, 581–592. [Google Scholar] [CrossRef]
- Gonzalez-Mendoza, D.; Moreno, A.Q.; Zapata-Perez, O. Coordinated responses of phytochelatin synthase and metallothionein genes in black mangrove, Avicennia germinans, exposed to cadmium and copper. Aquat. Toxicol. 2007, 83, 306–314. [Google Scholar] [CrossRef]
- Nelson, D.R.; Chaiboonchoe, A.; Fu, W.; Hazzouri, K.M.; Huang, Z.; Jaiswal, A.; Daakour, S.; Mystikou, A.; Arnoux, M.; Sultana, M.; et al. Potential for Heightened Sulfur-Metabolic Capacity in Coastal Subtropical Microalgae. iScience 2019, 11, 450–465. [Google Scholar] [CrossRef]
- Cotelle, N.; Moreau, S.; Cotelle, P.; Catteau, J.P.; Bernier, J.L.; Henichart, J.P. Generation of free radicals by simple prenylated hydroquinone derivatives, natural antitumor agents from the marine urochordate Aplidium californicum. Chem. Res. Toxicol. 1991, 4, 300–305. [Google Scholar] [CrossRef]
- Naik, M.M.; Pandey, A.; Dubey, S.K. Pseudomonas aeruginosa strain WI-1 from Mandovi estuary possesses metallothionein to alleviate lead toxicity and promotes plant growth. Ecotoxicol. Environ. Saf. 2012, 79, 129–133. [Google Scholar] [CrossRef]
- Attia, O.E.; Ghrefat, H. Assessing heavy metal pollution in the recent bottom sediments of Mabahiss Bay, North Hurghada, Red Sea, Egypt. Environ. Monit. Assess. 2013, 185, 9925–9934. [Google Scholar] [CrossRef]
- Idris, A.M.; Said, T.O.; Omran, A.A.; Fawy, K.F. Combining multivariate analysis and human risk indices for assessing heavy metal contents in muscle tissues of commercially fish from Southern Red Sea, Saudi Arabia. Environ. Sci. Pollut. Res. Int. 2015, 22, 17012–17021. [Google Scholar] [CrossRef]
- Mohamed, S.A.; Elshal, M.F.; Kumosani, T.A.; Mal, A.O.; Ahmed, Y.M.; Almulaiky, Y.Q.; Asseri, A.H.; Zamzami, M.A. Heavy Metal Accumulation is Associated with Molecular and Pathological Perturbations in Liver of Variola louti from the Jeddah Coast of Red Sea. Int. J. Environ. Res. Public Health 2016, 13, 342. [Google Scholar] [CrossRef]
- Saleh, Y.S.; Marie, M.A. Assessment of metal contamination in water, sediment, and tissues of Arius thalassinus fish from the Red Sea coast of Yemen and the potential human risk assessment. Environ. Sci. Pollut. Res. Int. 2015, 22, 5481–5490. [Google Scholar] [CrossRef]
- Euceda, N.; Jahnke, J.; Espinal, A.; Louis, M.F.; Bashkin, E.; Roccanova, P.; Espaillat, A.; Fuentes, G.V.; Nieto, F.; Gao, R. Thioguanine restoration through type I photosensitization-superoxide oxidation-glutathione reduction cycles. Physical. Chem. Chem. Phys. 2021, 23, 5069–5073. [Google Scholar] [CrossRef]
- Simunkova, M.; Barbierikova, Z.; Jomova, K.; Hudecova, L.; Lauro, P.; Alwasel, S.H.; Alhazza, I.; Rhodes, C.J.; Valko, M. Antioxidant vs. Prooxidant Properties of the Flavonoid, Kaempferol, in the Presence of Cu(II) Ions: A ROS-Scavenging Activity, Fenton Reaction and DNA Damage Study. Int. J. Mol. Sci. 2021, 22, 1619. [Google Scholar] [CrossRef]
- Tsang, T.; Davis, C.I.; Brady, D.C. Copper biology. Curr. Biol. 2021, 31, R421–R427. [Google Scholar] [CrossRef]
- Mir, G. A plant type 2 metallothionein (MT) from cork tissue responds to oxidative stress. J. Exp. Bot. 2004, 55, 2483–2493. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nelson, D.R.; Chaiboonchoe, A.; Hazzouri, K.M.; Khraiwesh, B.; Alzahmi, A.; Jaiswal, A.; Friis, G.; Burt, J.A.; Amiri, K.M.A.; Salehi-Ashtiani, K. Tissue-Specific Transcriptomes Outline Halophyte Adaptive Strategies in the Gray Mangrove (Avicennia marina). Agronomy 2022, 12, 2030. https://doi.org/10.3390/agronomy12092030
Nelson DR, Chaiboonchoe A, Hazzouri KM, Khraiwesh B, Alzahmi A, Jaiswal A, Friis G, Burt JA, Amiri KMA, Salehi-Ashtiani K. Tissue-Specific Transcriptomes Outline Halophyte Adaptive Strategies in the Gray Mangrove (Avicennia marina). Agronomy. 2022; 12(9):2030. https://doi.org/10.3390/agronomy12092030
Chicago/Turabian StyleNelson, David R., Amphun Chaiboonchoe, Khaled M. Hazzouri, Basel Khraiwesh, Amnah Alzahmi, Ashish Jaiswal, Guillermo Friis, John A. Burt, Khaled M. A. Amiri, and Kourosh Salehi-Ashtiani. 2022. "Tissue-Specific Transcriptomes Outline Halophyte Adaptive Strategies in the Gray Mangrove (Avicennia marina)" Agronomy 12, no. 9: 2030. https://doi.org/10.3390/agronomy12092030
APA StyleNelson, D. R., Chaiboonchoe, A., Hazzouri, K. M., Khraiwesh, B., Alzahmi, A., Jaiswal, A., Friis, G., Burt, J. A., Amiri, K. M. A., & Salehi-Ashtiani, K. (2022). Tissue-Specific Transcriptomes Outline Halophyte Adaptive Strategies in the Gray Mangrove (Avicennia marina). Agronomy, 12(9), 2030. https://doi.org/10.3390/agronomy12092030