
An Integration of Transcriptomic Data and Modular Gene Co-Expression Network Analysis Uncovers Drought Stress-Related Hub Genes in Transgenic Rice Overexpressing OsAbp57

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Preparation and RNA Extraction
2.2. Gene Expression Profiling, Differentially Expressed Genes (DEGs), and Enrichment Analyses
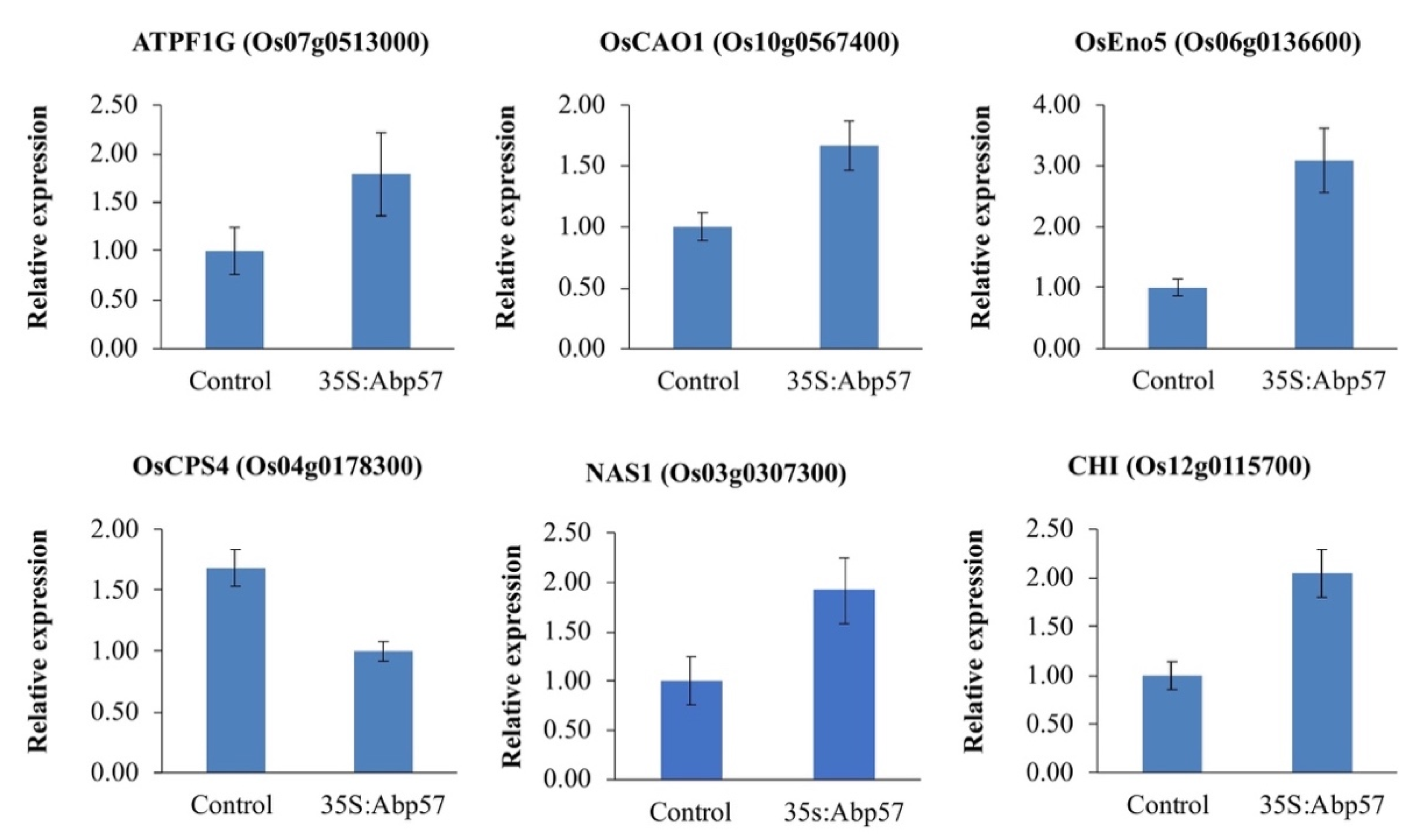
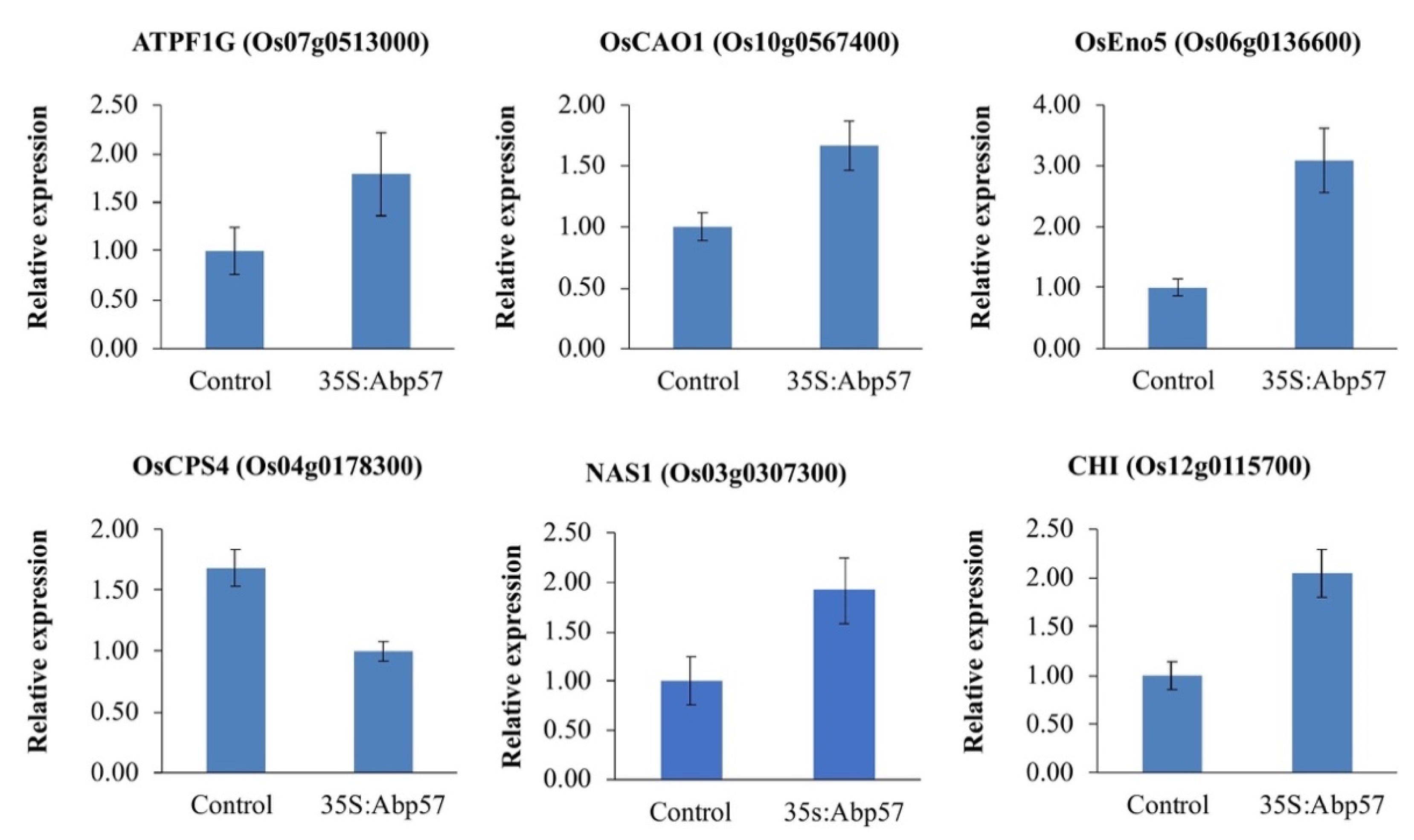
2.3. Expression Analyses of Randomly Selected Expressed Genes by qRT-PCR
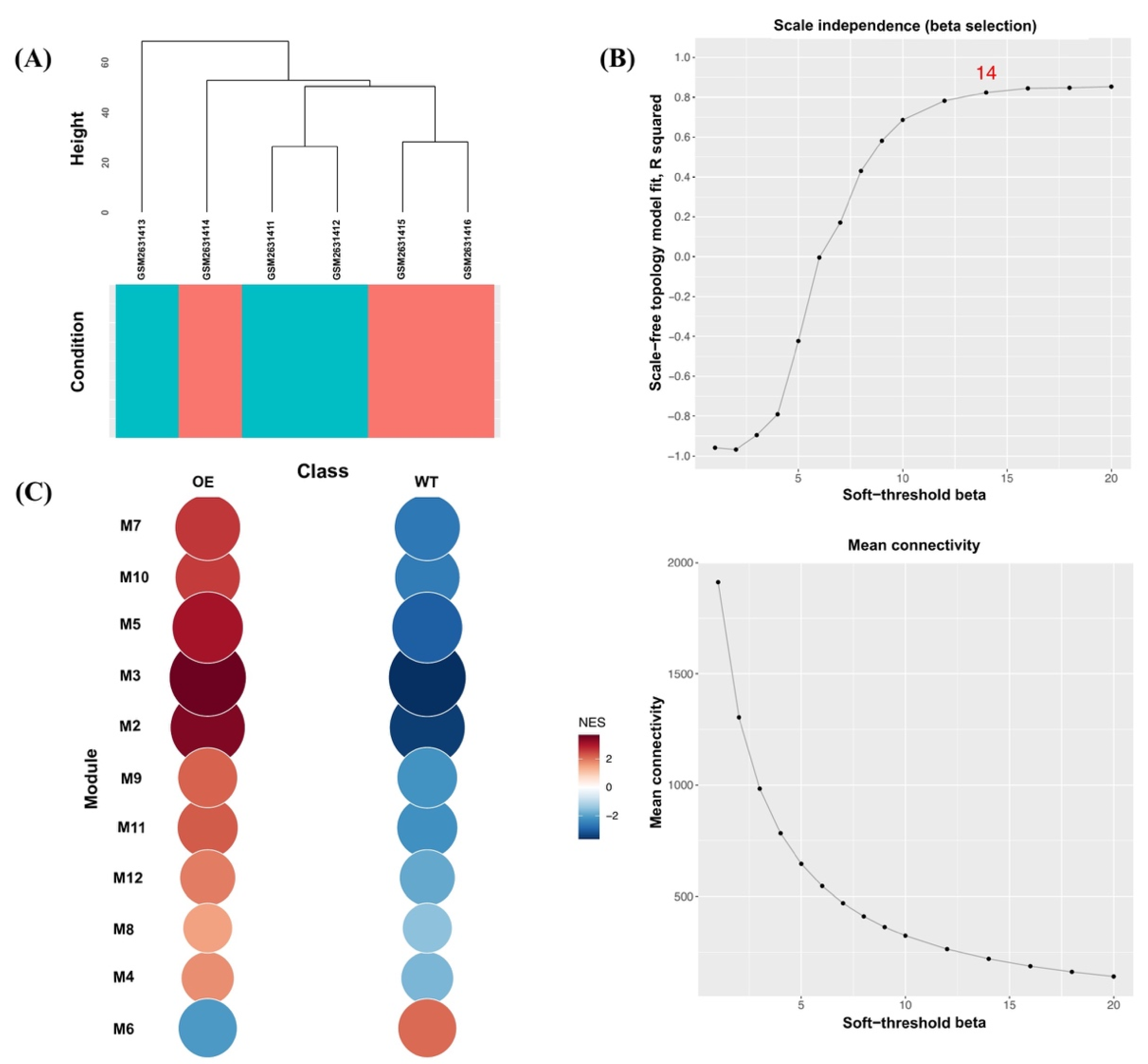
2.4. Construction of Gene Co-Expression Network Modules
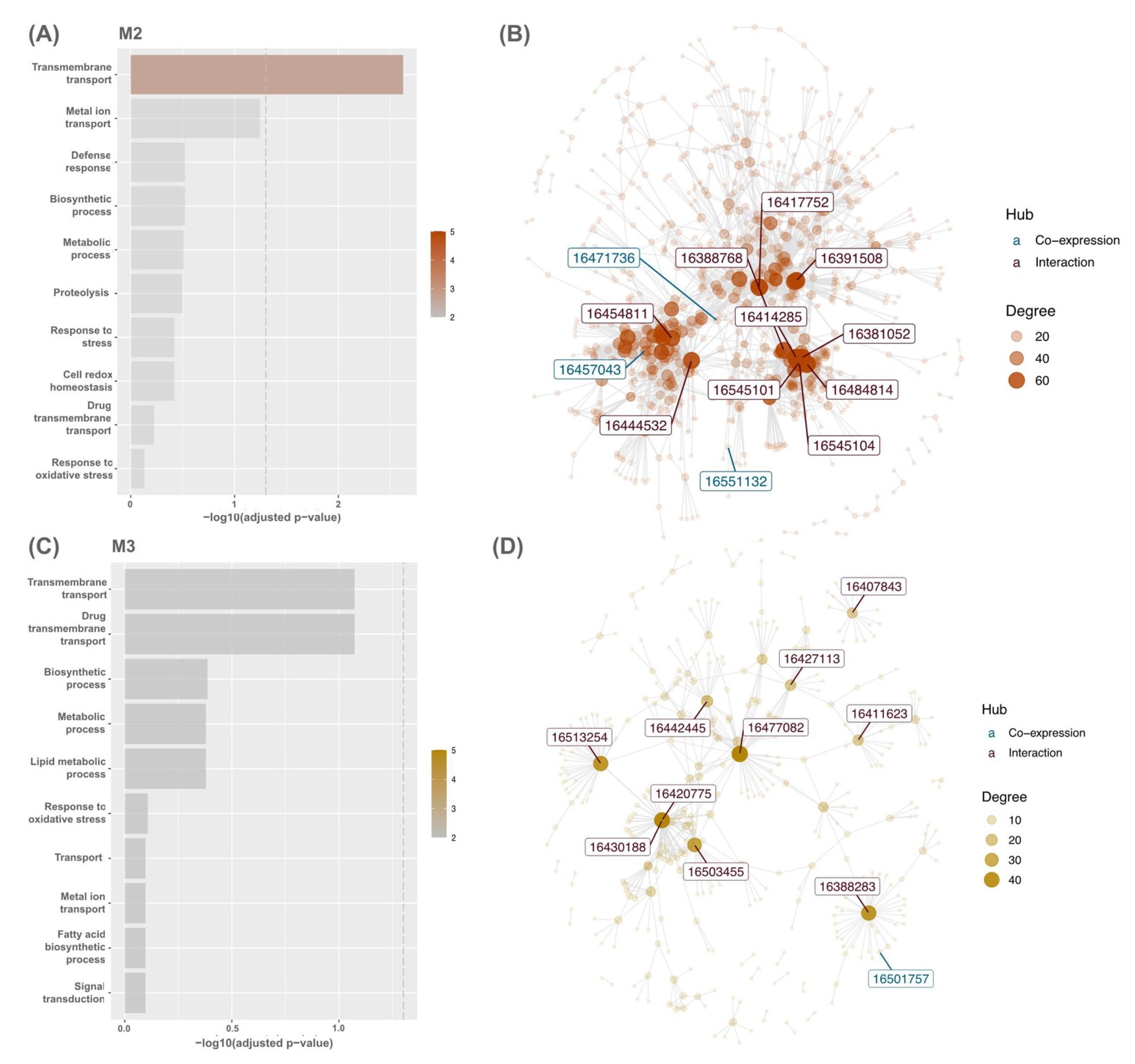
2.5. Gene Set Enrichment and Overrepresentation Analysis of Modules
2.6. Identification and Validation of Hub Genes
2.7. KEGG Pathway Mapping Analysis
3. Results
3.1. Global Gene Expression Profiling of the Abp57-OE Line
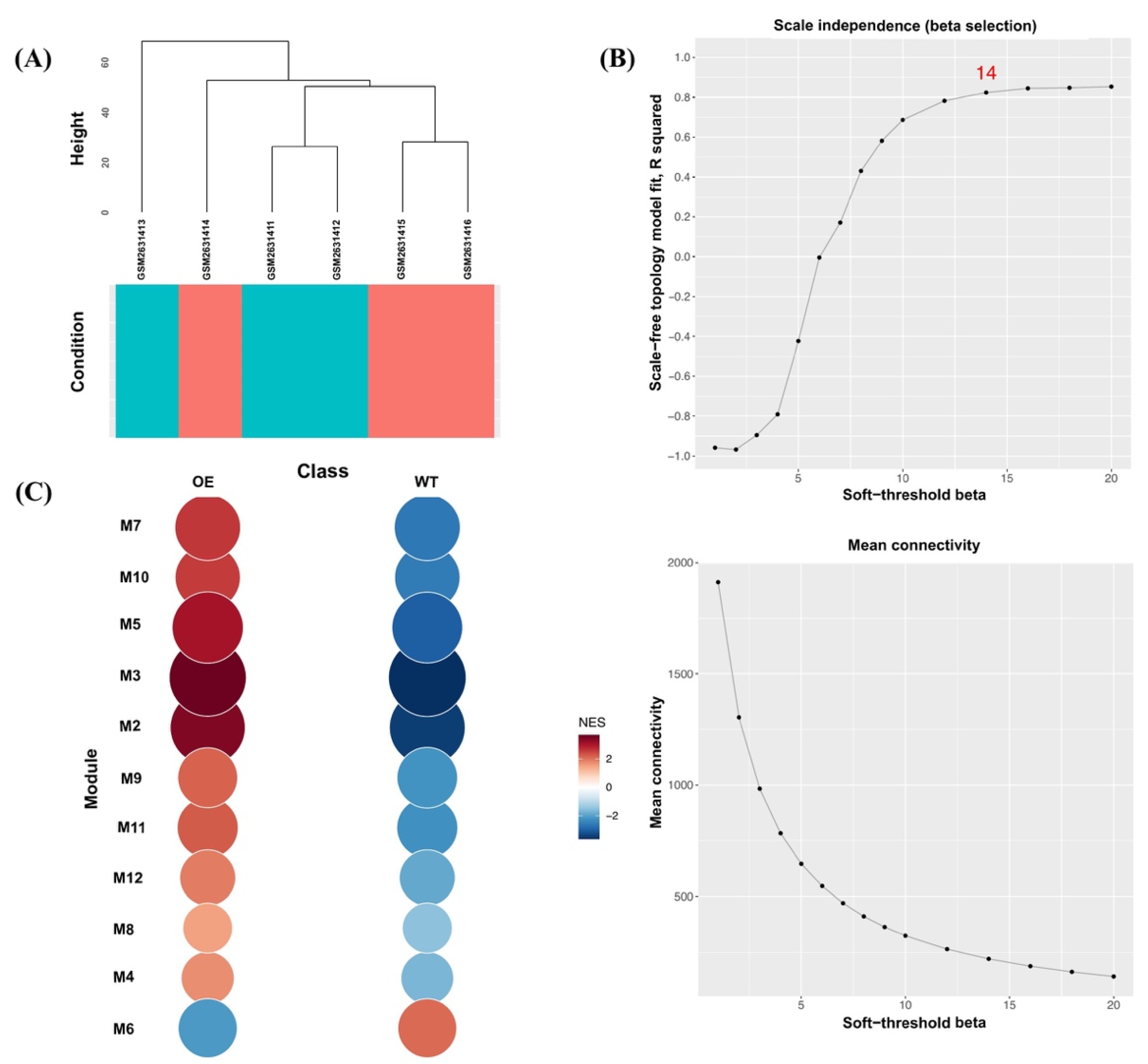
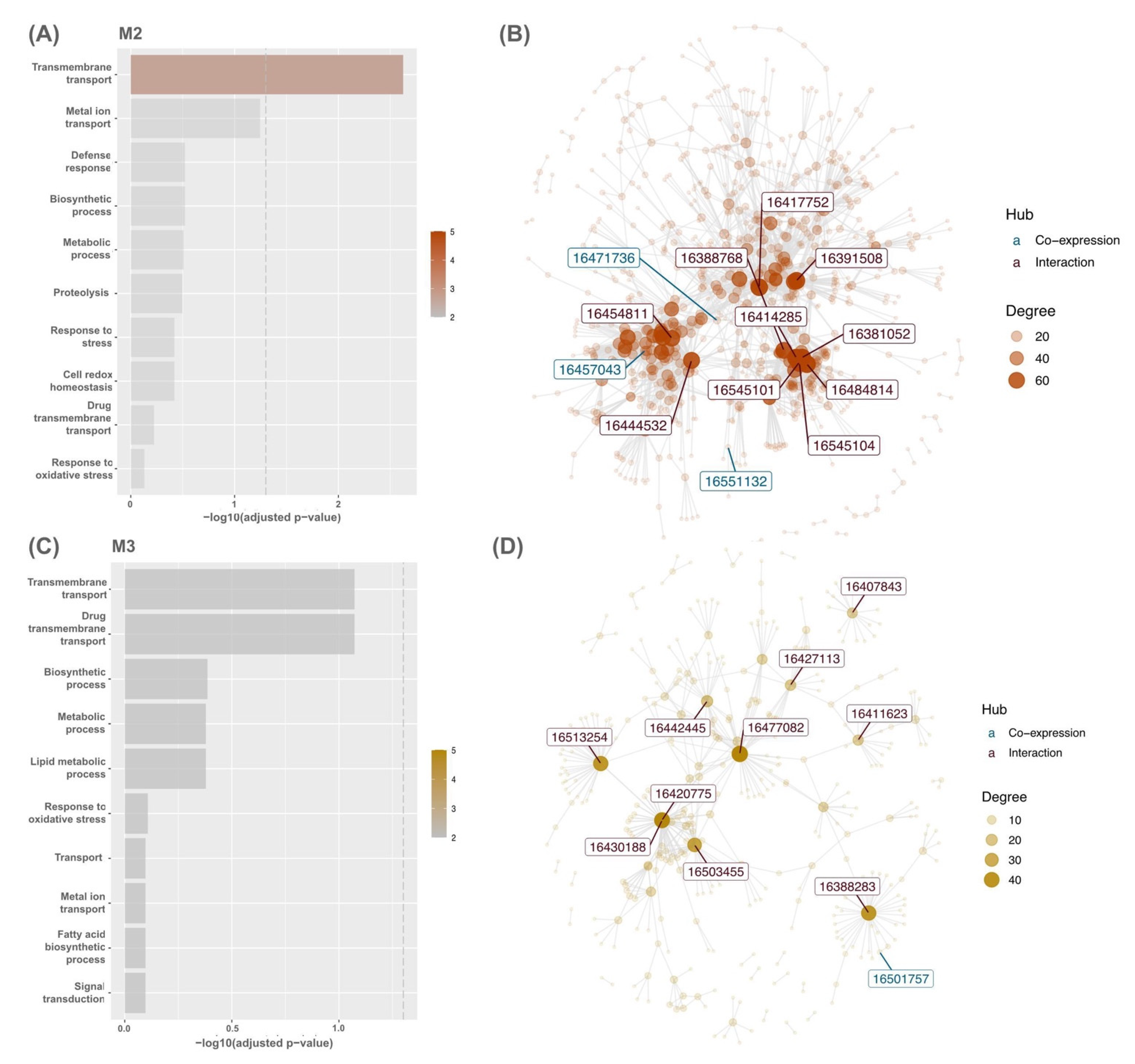
3.2. Modular Gene Co-Expression Analyses of the OsAbp57-OE Line
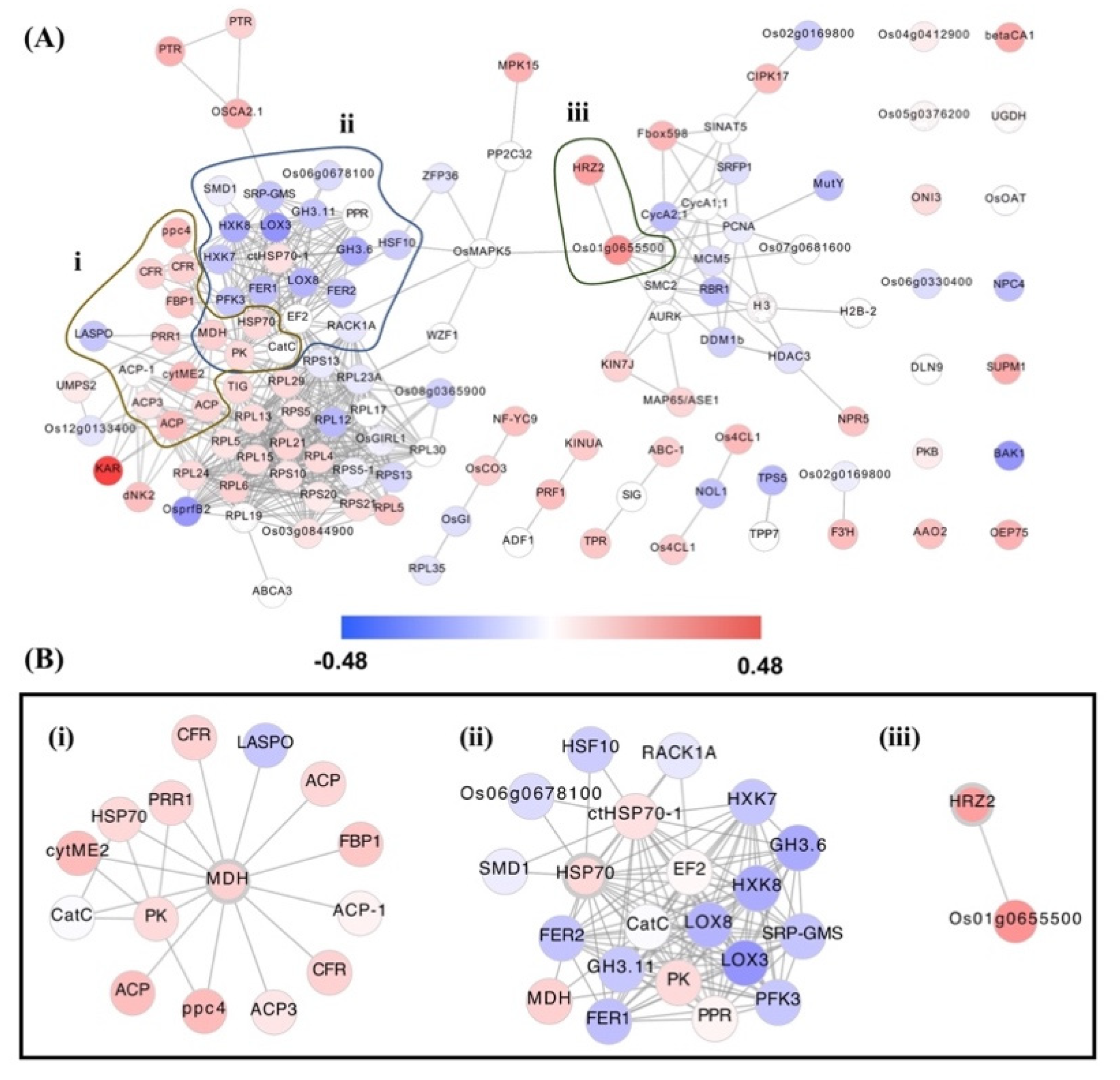
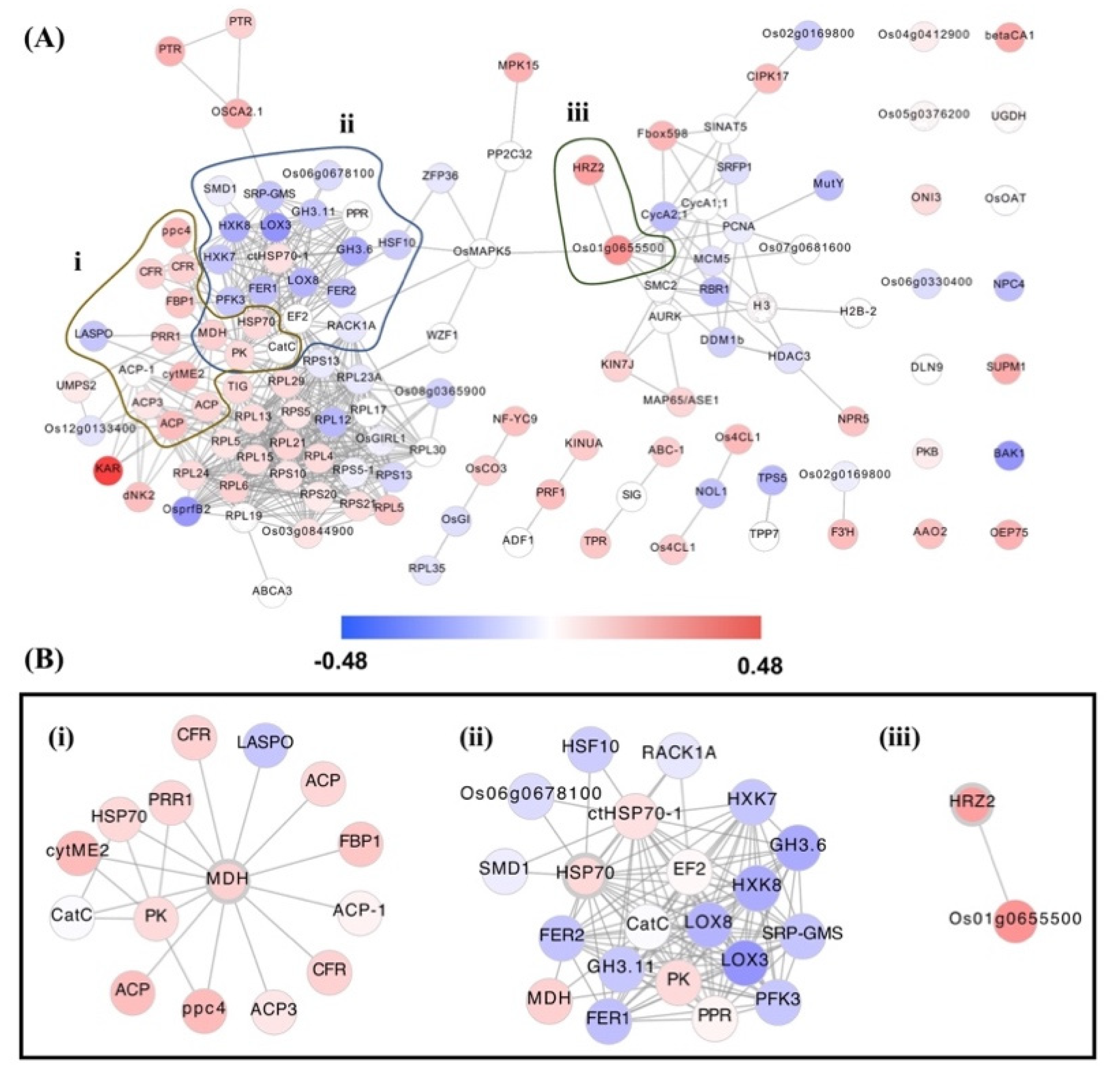
3.3. Hub Genes’ Identification in OsAbp57-OE Associated with Drought Stress
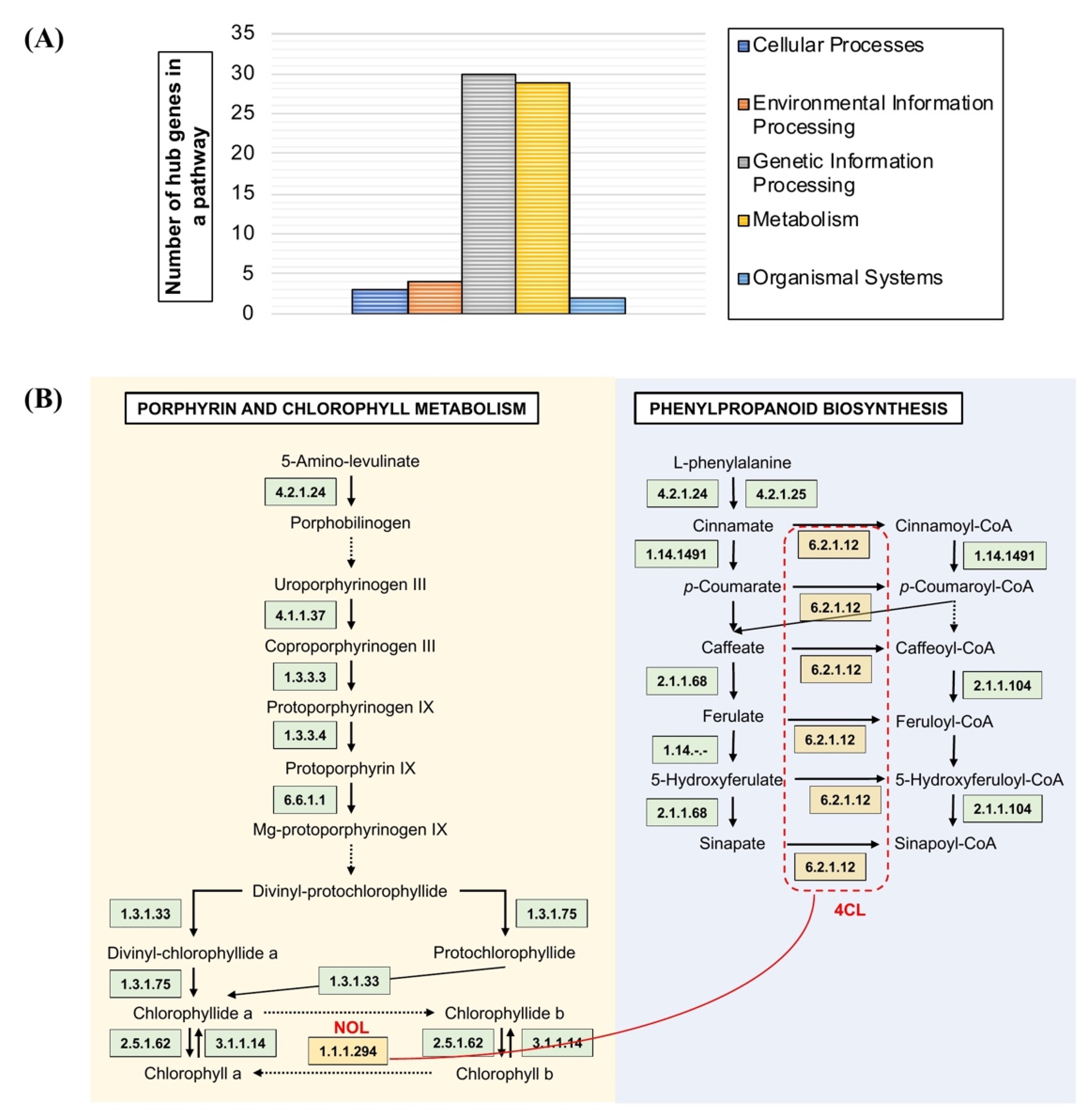
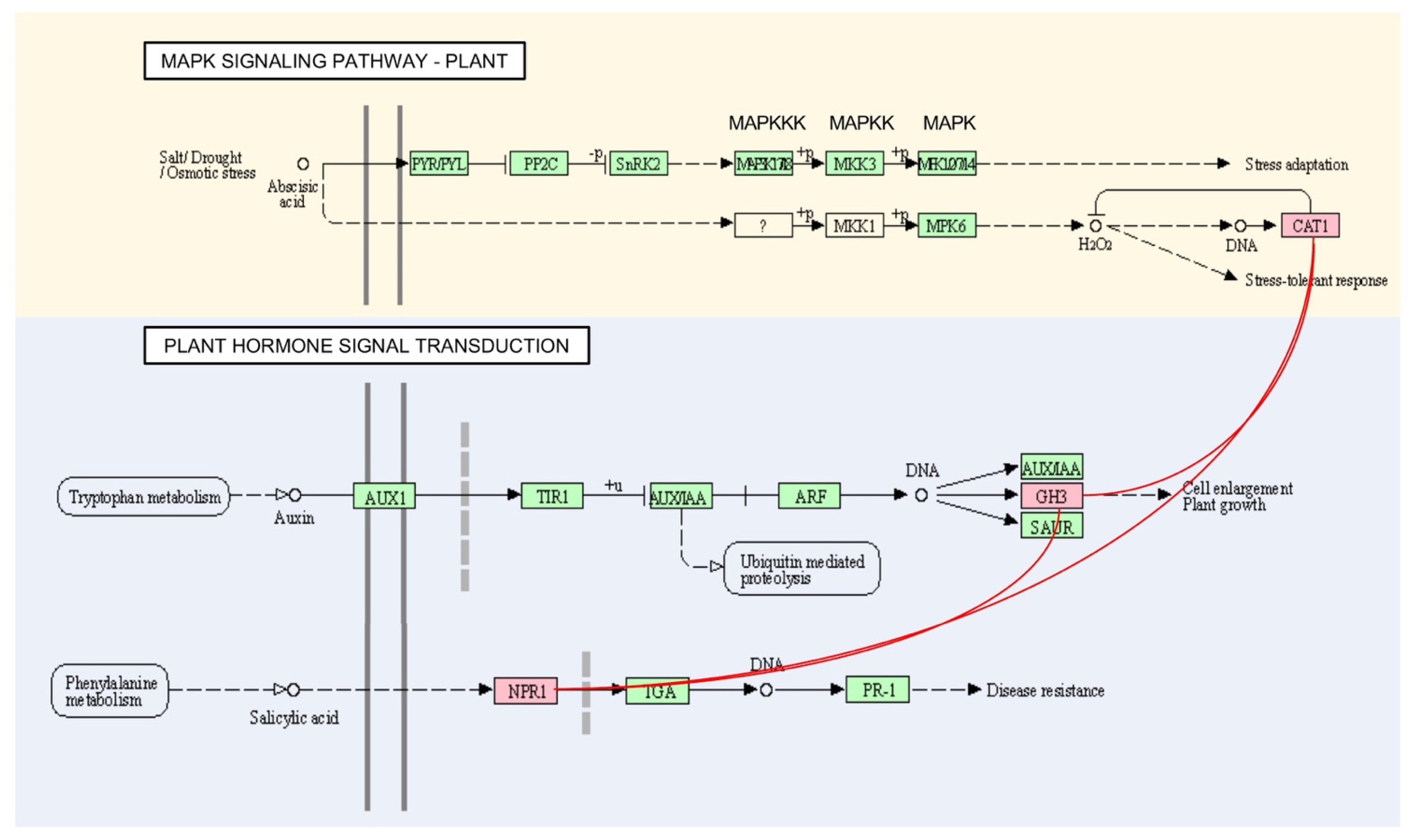
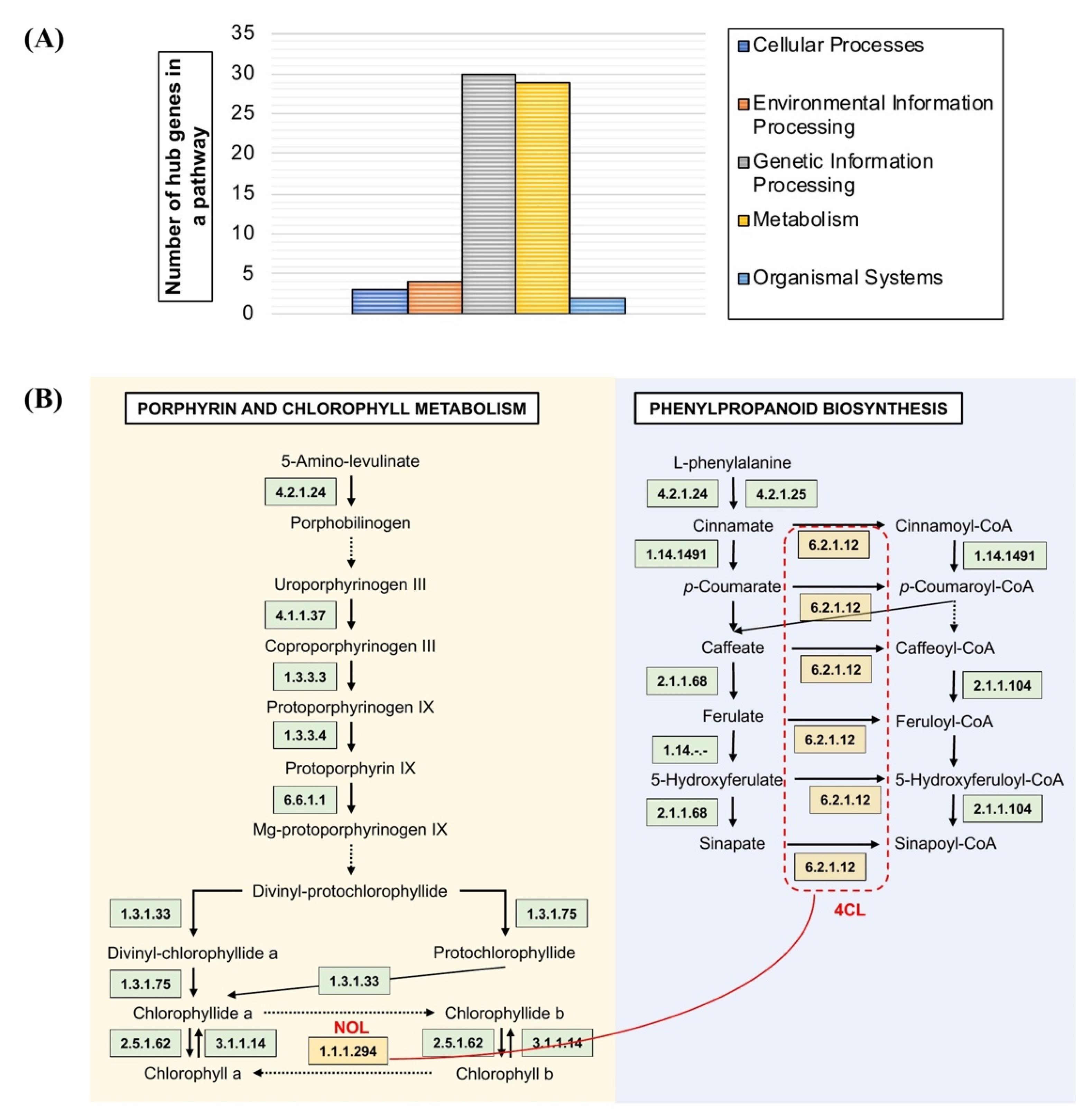
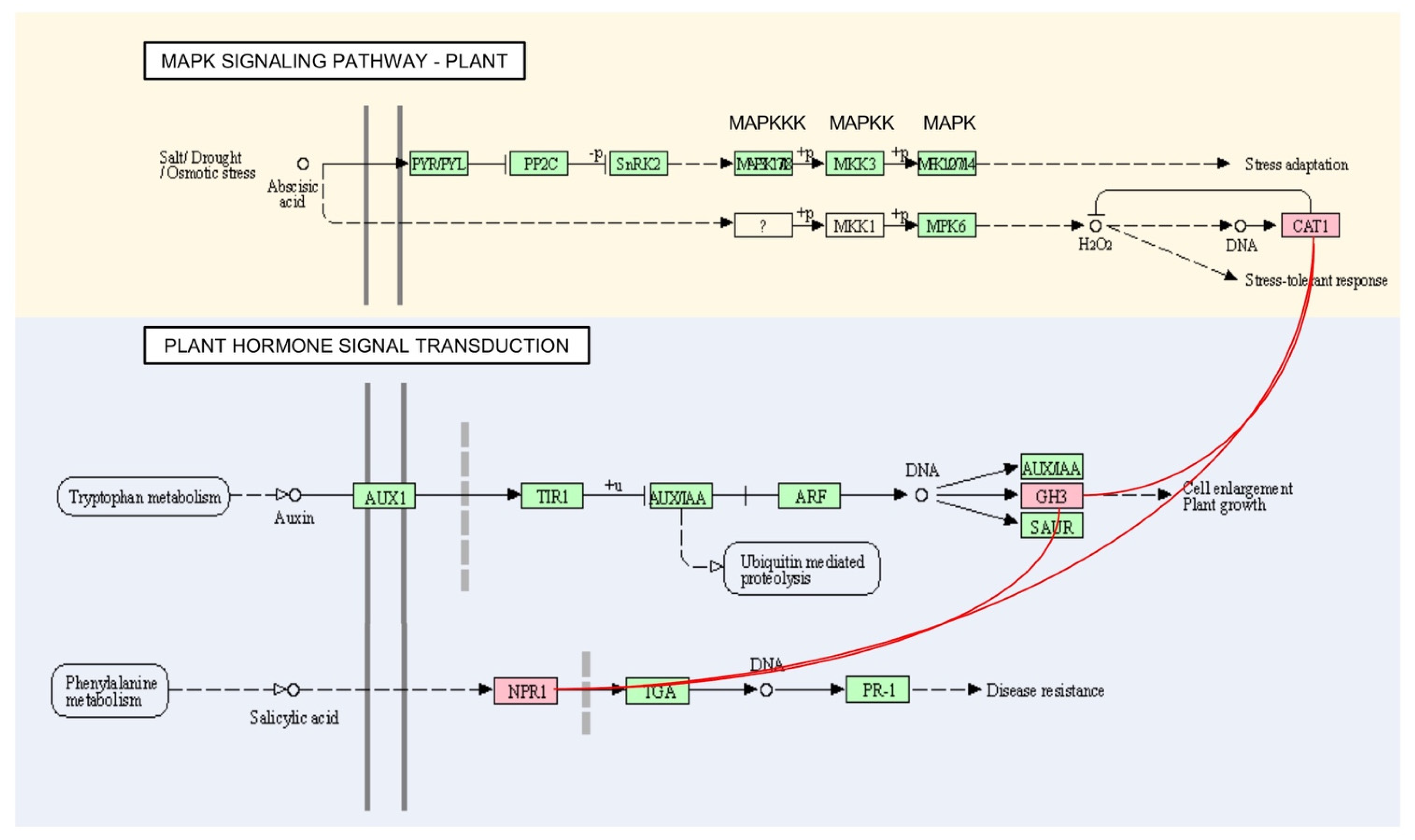
3.4. Pathway Mapping Analysis of OsAbp57-OE
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ray, D.K.; Mueller, N.D.; West, P.C.; Foley, J.A. Yield trends are insufficient to double global crop production by 2050. PLoS ONE 2013, 8, e66428. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, W.; Ma, L.; Wu, L.; Shen, J.; Davies, W.J.; Oenema, O.; Zhang, F.; Dou, Z. An analysis of China’s grain production: Looking back and looking forward. Food Energy Secur. 2014, 3, 19–32. [Google Scholar] [CrossRef]
- Pandey, S.; Bhandari, H. Drought: Economic costs and research implications. In Drought Frontiers in Rice: Crop Improvement for Increased Rainfed Production; Serraj, R., Bennett, J., Hardy, B., Eds.; World Scientific: Singapore, 2007; pp. 3–17. [Google Scholar]
- Swain, P.; Raman, A.; Singh, S.P.; Kumar, A. Breeding drought tolerant rice for shallow rainfed ecosystem of eastern India. Field Crops Res. 2017, 209, 168–178. [Google Scholar] [CrossRef] [PubMed]
- Woodward, A.W.; Bartel, B. Auxin: Regulation, action, and interaction. Ann. Bot. 2005, 95, 707–735. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Wang, L.; Xiong, L. Comprehensive expression profiling analysis of OsIAA gene family in developmental processes and in response to phytohormone and stress treatments. Planta 2009, 229, 577–591. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.; Khurana, J.P. Transcript profiling reveals diverse roles of auxin-responsive genes during reproductive development and abiotic stress in rice. FEBS J. 2009, 276, 3148–3162. [Google Scholar] [CrossRef]
- Kim, J.I.; Baek, D.; Park, H.C.; Chun, H.J.; Oh, D.H.; Lee, M.K.; Cha, J.Y.; Kim, W.Y.; Kim, M.C.; Chung, W.S.; et al. Overexpression of Arabidopsis YUCCA6 in potato results in high-auxin developmental phenotypes and enhanced resistance to water deficit. Mol. Plant. 2013, 6, 337–349. [Google Scholar] [CrossRef]
- Jung, H.; Lee, D.K.; Choi, Y.D.; Kim, J.K. OsIAA6, a member of the rice Aux/IAA gene family, is involved in drought tolerance and tiller outgrowth. Plant Sci. 2015, 236, 304–312. [Google Scholar] [CrossRef]
- Zhang, S.W.; Li, C.H.; Cao, J.; Zhang, Y.C.; Zhang, S.Q.; Xia, Y.F.; Sun, D.Y.; Sun, Y. Altered architecture and enhanced drought tolerance in rice via the down-regulation of indole-3-acetic acid by TLD1/OsGH3.13 activation. Plant Physiol. 2009, 151, 1889–1901. [Google Scholar] [CrossRef]
- Zhang, Q.; Li, J.; Zhang, W.; Yan, S.; Wang, R.; Zhao, J.; Li, Y.; Qi, Z.; Sun, Z.; Zhu, Z. The putative auxin efflux carrier OsPIN3t is involved in the drought stress response and drought tolerance. Plant J. 2012, 72, 805–816. [Google Scholar] [CrossRef]
- Kim, Y.S.; Kim, D.; Jung, J. Isolation of a novel auxin receptor from soluble fractions of rice (Oryza sativa L.) shoots. FEBS Lett. 1998, 438, 241–244. [Google Scholar] [CrossRef]
- Kim, Y.S.; Min, J.K.; Kim, D.; Jung, J. A soluble auxin-binding protein, ABP57. Purification with anti-bovine serum albumin antibody and characterization of its mechanistic role in the auxin effect on plant plasma membrane H+-ATPase. J. Biol. Chem. 2001, 276, 10730–10736. [Google Scholar] [CrossRef]
- Kim, Y.S.; Kim, D.; Jung, J. Two isoforms of soluble auxin receptor in rice (Oryza sativa L.) plants: Binding property for auxin and interaction with plasma membrane H+-ATPase. Plant Growth Regul. 2000, 32, 143–150. [Google Scholar] [CrossRef]
- Kim, D.H.; Lee, K.P.; Kim, M.I.; Kwon, Y.J.; Kim, Y.S. Gene encoding auxin receptor protein derived from rice and use thereof. US Patent 12/935,483, 24 May 2012. [Google Scholar]
- Kamarudin, Z.S.; Shamsudin, N.A.A.; Othman, M.H.C.; Shakri, T.; Tan, L.W.; Sukiran, N.L.; Isa, N.M.; Rahman, Z.A.; Zainal, Z. Morpho-physiology and antioxidant enzyme activities of transgenic rice plant overexpressing ABP57 under reproductive stage drought condition. Agronomy 2020, 10, 1530. [Google Scholar] [CrossRef]
- Hansen, B.O.; Vaid, N.; Musialak-Lange, M.; Janowski, M.; Mutwil, M. Elucidating gene function and function evolution through comparison of co-expression networks of plants. Front. Plant Sci. 2014, 19, 394. [Google Scholar] [CrossRef]
- Usadel, B.; Obayashi, T.; Mutwil, M.; Giorgi, F.M.; Bassel, G.W.; Tanimoto, M.; Chow, A.; Steinhauser, D.; Persson, S.; Provart, N.J. Co-expression tools for plant biology: Opportunities for hypothesis generation and caveats. Plant Cell Environ. 2009, 32, 1633–1651. [Google Scholar] [CrossRef]
- Allocco, D.J.; Kohane, I.S.; Butte, A.J. Quantifying the relationship between co-expression, co-regulation and gene function. BMC Bioinform. 2004, 5, 18. [Google Scholar] [CrossRef]
- Tan, L.W.; Rahman, Z.A.; Goh, H.H.; Hwang, D.J.; Ismail, I.; Zainal, Z. Production of transgenic rice (indica cv. MR219) overexpressing Abp57 gene through Agrobacterium-mediated transformation. Sains Malays 2017, 46, 703–711. [Google Scholar] [CrossRef]
- Tan, L.W.; Tan, C.S.; Rahman, Z.A.; Goh, H.H.; Ismail, I.; Zainal, Z. Microarray dataset of transgenic rice overexpressing Abp57. Data Brief 2017, 14, 267–271. [Google Scholar] [CrossRef]
- Barrett, T.; Edgar, R. Gene expression omnibus: Microarray data storage, submission, retrieval, and analysis. Methods Enzymol. 2006, 411, 352–369. [Google Scholar]
- Sayers, E.W.; Beck, J.; Bolton, E.E.; Bourexis, D.; Brister, J.R.; Canese, K.; Comeau, D.C.; Funk, K.; Kim, S.; Klimke, W.; et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2021, 49, D10–D17. [Google Scholar] [CrossRef]
- UniProt Consortium. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef]
- Kawahara, Y.; de la Bastide, M.; Hamilton, J.P.; Kanamori, H.; McCombie, W.R.; Ouyang, S.; Schwartz, D.C.; Tanaka, T.; Wu, J.; Zhou, S.; et al. Improvement of the Oryza sativa Nipponbare reference genome using next generation sequence and optical map data. Rice 2013, 6, 4. [Google Scholar] [CrossRef]
- Ohyanagi, H.; Tanaka, T.; Sakai, H.; Shigemoto, Y.; Yamaguchi, K.; Habara, T.; Fujii, Y.; Antonio, B.A.; Nagamura, Y.; Imanishi, T.; et al. The Rice Annotation Project Database (RAP-DB): Hub for Oryza sativa ssp. japonica genome information. Nucleic Acids Res. 2006, 34, D741–D744. [Google Scholar] [CrossRef]
- Zhao, W.; Wang, J.; He, X.; Huang, X.; Jiao, Y.; Dai, M.; Wei, S.; Fu, J.; Chen, Y.; Ren, X.; et al. BGI-RIS: An integrated information resource and comparative analysis workbench for rice genomics. Nucleic Acids Res. 2004, 32, D377–D382. [Google Scholar] [CrossRef]
- Tian, T.; Liu, Y.; Yan, H.; You, Q.; Yi, X.; Du, Z.; Xu, W.; Su, Z. agriGO v2.0: A GO analysis toolkit for the agricultural community, 2017 update. Nucleic Acids Res. 2017, 45, W122–W129. [Google Scholar] [CrossRef]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef]
- Russo, P.S.T.; Ferreira, G.R.; Cardozo, L.E.; Bürger, M.C.; Arias-Carrasco, R.; Maruyama, S.R.; Hirata, T.D.C.; Lima, D.S.; Passos, F.M.; Fukutani, K.F.; et al. CEMiTool: A Bioconductor package for performing comprehensive modular co-expression analyses. BMC Bioinform. 2018, 19, 56. [Google Scholar] [CrossRef]
- Korotkevich, G.; Sukhov, V.; Budin, N.; Shpak, B.; Artyomov, M.N.; Sergushichev, A. Fast gene set enrichment analysis. bioRxiv 2021. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. Omics 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Goodstein, D.M.; Shu, S.; Howson, R.; Neupane, R.; Hayes, R.D.; Fazo, J.; Mitros, T.; Dirks, W.; Hellsten, U.; Putnam, N.; et al. Phytozome: A comparative platform for green plant genomics. Nucleic Acids Res. 2012, 40, D1178–D1186. [Google Scholar] [CrossRef]
- Lin, C.T.; Xu, T.; Xing, S.L.; Zhao, L.; Sun, R.Z.; Liu, Y.; Moore, J.P.; Deng, X. Weighted Gene Co-expression Network Analysis (WGCNA) Reveals the Hub Role of Protein Ubiquitination in the Acquisition of Desiccation Tolerance in Boea hygrometrica. Plant Cell Physiol. 2019, 60, 2707–2719. [Google Scholar] [CrossRef] [PubMed]
- Doncheva, N.T.; Morris, J.H.; Gorodkin, J.; Jensen, L.J. Cytoscape StringApp: Network Analysis and Visualization of Proteomics Data. J. Proteome Res. 2019, 18, 623–632. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Sato, Y.; Kawashima, M. KEGG mapping tools for uncovering hidden features in biological data. Protein Sci. 2022, 31, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Guberman, J.M.; Ai, J.; Arnaiz, O.; Baran, J.; Blake, A.; Baldock, R.; Chelala, C.; Croft, D.; Cros, A.; Cutts, R.J.; et al. BioMart Central Portal: An open database network for the biological community. Database 2011, 2011, bar041. [Google Scholar] [CrossRef]
- Jan, A.; Maruyama, K.; Todaka, D.; Kidokoro, S.; Abo, M.; Yoshimura, E.; Shinozaki, K.; Nakashima, K.; Yamaguchi-Shinozaki, K. OsTZF1, a CCCH-tandem zinc finger protein, confers delayed senescence and stress tolerance in rice by regulating stress-related genes. Plant Physiol. 2013, 161, 1202–1216. [Google Scholar] [CrossRef] [PubMed]
- Sakuraba, Y.; Kim, Y.S.; Han, S.H.; Lee, B.D.; Paek, N.C. The Arabidopsis Transcription Factor NAC016 Promotes Drought Stress Responses by Repressing AREB1 Transcription through a Trifurcate Feed-Forward Regulatory Loop Involving NAP. Plant Cell 2015, 27, 1771–1787. [Google Scholar] [CrossRef]
- Liu, J.; Shen, J.; Xu, Y.; Li, X.; Xiao, J.; Xiong, L. Ghd2, a CONSTANS-like gene, confers drought sensitivity through regulation of senescence in rice. J. Exp. Bot. 2016, 67, 5785–5798. [Google Scholar] [CrossRef]
- Comas, L.H.; Becker, S.R.; Cruz, V.M.; Byrne, P.F.; Dierig, D.A. Root traits contributing to plant productivity under drought. Front. Plant Sci. 2013, 4, 442. [Google Scholar] [CrossRef]
- Fageria, N.K.; Moreira, A. The role of mineral nutrition on root growth of crop plants. In Advances in Agronomy; Sparks, D.L., Ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2011; Volume 110, pp. 251–331. [Google Scholar]
- Kim, H.; Lee, K.; Hwang, H.; Bhatnagar, N.; Kim, D.Y.; Yoon, I.S.; Byun, M.O.; Kim, S.T.; Jung, K.H.; Kim, B.G. Overexpression of PYL5 in rice enhances drought tolerance, inhibits growth, and modulates gene expression. J. Exp. Bot. 2014, 65, 453–464. [Google Scholar] [CrossRef]
- Nakashima, K.; Tran, L.S.; Van Nguyen, D.; Fujita, M.; Maruyama, K.; Todaka, D.; Ito, Y.; Hayashi, N.; Shinozaki, K.; Yamaguchi-Shinozaki, K. Functional analysis of a NAC-type transcription factor OsNAC6 involved in abiotic and biotic stress-responsive gene expression in rice. Plant J. 2007, 51, 617–630. [Google Scholar] [CrossRef]
- Lee, D.K.; Chung, P.J.; Jeong, J.S.; Jang, G.; Bang, S.W.; Jung, H.; Kim, Y.S.; Ha, S.H.; Choi, Y.D.; Kim, J.K. The rice OsNAC6 transcription factor orchestrates multiple molecular mechanisms involving root structural adaptions and nicotianamine biosynthesis for drought tolerance. Plant Biotechnol. J. 2017, 15, 754–764. [Google Scholar] [CrossRef]
- Cruz de Carvalho, M.H. Drought stress and reactive oxygen species: Production, scavenging and signaling. Plant Signal. Behav. 2008, 3, 156–165. [Google Scholar] [CrossRef]
- Xia, T.; Chen, H.; Dong, S.; Ma, Z.; Ren, H.; Zhu, X.; Fang, X.; Chen, F. OsWUS promotes tiller bud growth by establishing weak apical dominance in rice. Plant J. 2020, 104, 1635–1647. [Google Scholar] [CrossRef]
- Zawaski, C.; Busov, V.B. Roles of gibberellin catabolism and signaling in growth and physiological response to drought and short-day photoperiods in Populus trees. PLoS ONE 2014, 9, e86217. [Google Scholar] [CrossRef]
- Wang, B.; Wei, H.; Xue, Z.; Zhang, W.H. Gibberellins regulate iron deficiency-response by influencing iron transport and translocation in rice seedlings (Oryza sativa). Ann. Bot. 2017, 119, 945–956. [Google Scholar] [CrossRef]
- Nieves-Cordones, M.; García-Sánchez, F.; Pérez-Pérez, J.G.; Colmenero-Flores, J.M.; Rubio, F.; Rosales, M.A. Coping with Water Shortage: An Update on the Role of K+, Cl−, and Water Membrane Transport Mechanisms on Drought Resistance. Front. Plant Sci. 2019, 10, 1619. [Google Scholar] [CrossRef]
- Tripathi, D.K.; Singh, S.; Gaur, S.; Singh, S.; Yadav, V.; Liu, S.; Singh, V.P.; Sharma, S.; Srivastava, P.; Prasad, S.M.; et al. Acquisition and homeostasis of iron in higher plants and their probable role in abiotic stress tolerance. Front. Environ. Sci. 2018, 5, 86. [Google Scholar] [CrossRef]
- You, J.; Chan, Z. ROS Regulation during Abiotic Stress Responses in Crop Plants. Front. Plant Sci. 2015, 6, 1092. [Google Scholar] [CrossRef]
- Kobayashi, T.; Nagasaka, S.; Senoura, T.; Itai, R.N.; Nakanishi, H.; Nishizawa, N.K. Iron-binding haemerythrin RING ubiquitin ligases regulate plant iron responses and accumulation. Nat. Commun. 2013, 4, 2792. [Google Scholar] [CrossRef]
- Kobayashi, T.; Itai, R.N.; Senoura, T.; Oikawa, T.; Ishimaru, Y.; Ueda, M.; Nakanishi, H.; Nishizawa, N.K. Jasmonate signaling is activated in the very early stages of iron deficiency responses in rice roots. Plant Mol. Biol. 2016, 91, 533–547. [Google Scholar] [CrossRef] [PubMed]
- Aung, M.S.; Kobayashi, T.; Masuda, H.; Nishizawa, N.K. Rice HRZ ubiquitin ligases are crucial for response to excess iron. Physiol. Plant 2018, 163, 282–296. [Google Scholar] [CrossRef] [PubMed]
- Batth, R.; Singh, K.; Kumari, S.; Mustafiz, A. Transcript Profiling Reveals the Presence of Abiotic Stress and Developmental Stage Specific Ascorbate Oxidase Genes in Plants. Front. Plant Sci. 2017, 17, 198. [Google Scholar] [CrossRef] [PubMed]
- You, J.; Hu, H.; Xiong, L. An ornithine δ-aminotransferase gene OsOAT confers drought and oxidative stress tolerance in rice. Plant Sci. 2012, 197, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.G.; Lee, J.S.; Bae, H.H.; Kim, J.T.; Son, B.Y.; Kim, S.L.; Baek, S.-B.; Shin, S.; Jeon, W.-T. Physiological and proteomic analyses of Korean F1 maize (Zea mays L.) hybrids under water-deficit stress during flowering. Appl. Biol. Chem. 2019, 62, 32. [Google Scholar] [CrossRef]
- Rodrigues, F.A.; de Laia, M.L.; Zingaretti, S.M. Analysis of gene expression profiles under water stress in tolerant and sensitive sugarcane plants. Plant Sci. 2009, 176, 286–302. [Google Scholar] [CrossRef]
- Ahn, J.C.; Kim, D.W.; You, Y.N.; Seok, M.S.; Park, J.M.; Hwang, H.; Kim, B.G.; Luan, S.; Park, H.S.; Cho, H.S. Classification of rice (Oryza sativa L. Japonica nipponbare) immunophilins (FKBPs, CYPs) and expression patterns under water stress. BMC Plant Biol. 2010, 10, 253. [Google Scholar] [CrossRef]
- Newstead, S. Molecular insights into proton coupled peptide transport in the PTR family of oligopeptide transporters. Biochim. Biophys. Acta 2015, 1850, 488–499. [Google Scholar] [CrossRef]
- Tang, Z.; Chen, Y.; Chen, F.; Ji, Y.; Zhao, F.J. OsPTR7 (OsNPF8.1), a Putative Peptide Transporter in Rice, is Involved in Dimethylarsenate Accumulation in Rice Grain. Plant Cell Physiol. 2017, 58, 904–913. [Google Scholar] [CrossRef]
- Yang, X.; Xia, X.; Zeng, Y.; Nong, B.; Zhang, Z.; Wu, Y.; Tian, Q.; Zeng, W.; Gao, J.; Zhou, W.; et al. Genome-wide identification of the peptide transporter family in rice and analysis of the PTR expression modulation in two near-isogenic lines with different nitrogen use efficiency. BMC Plant Biol. 2020, 20, 193. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Wang, X.; He, Y.; Xu, H.; Wang, L. Clock component OsPRR73 positively regulates rice salt tolerance by modulating OsHKT2;1-mediated sodium homeostasis. EMBO J. 2021, 40, e105086. [Google Scholar] [CrossRef] [PubMed]
- Cho, E.K.; Choi, Y.J. A nuclear-localized HSP70 confers thermoprotective activity and drought-stress tolerance on plants. Biotechnol. Lett. 2009, 31, 597–606. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, N.K.; Kundnani, P.; Grover, A. Functional analysis of Hsp70 superfamily proteins of rice (Oryza sativa). Cell Stress Chaperones 2013, 18, 427–437. [Google Scholar] [CrossRef]
- Pan, L.; Zhang, X.; Wang, J.; Ma, X.; Zhou, M.; Huang, L.; Nie, G.; Wang, P.; Yang, Z.; Li, J. Transcriptional Profiles of Drought-Related Genes in Modulating Metabolic Processes and Antioxidant Defenses in Lolium multiflorum. Front. Plant Sci. 2016, 7, 519. [Google Scholar] [CrossRef]
- Xing, H.; Fu, X.; Yang, C.; Tang, X.; Guo, L.; Li, C.; Xu, C.; Luo, K. Genome-wide investigation of pentatricopeptide repeat gene family in poplar and their expression analysis in response to biotic and abiotic stresses. Sci. Rep. 2018, 8, 2817. [Google Scholar] [CrossRef]
- Jiang, S.C.; Mei, C.; Liang, S.; Yu, Y.T.; Lu, K.; Wu, Z.; Wang, X.F.; Zhang, D.P. Crucial roles of the pentatricopeptide repeat protein SOAR1 in Arabidopsis response to drought, salt and cold stresses. Plant Mol. Biol. 2015, 88, 369–385. [Google Scholar] [CrossRef]
- Bashir, K.; Hanada, K.; Shimizu, M.; Seki, M.; Nakanishi, H.; Nishizawa, N.K. Transcriptomic analysis of rice in response to iron deficiency and excess. Rice 2014, 7, 18. [Google Scholar] [CrossRef]
- Yang, X.; Liu, X.; Zhou, Y.; Zhang, F.; Huang, L.; Wang, J.; Song, J.; Qiu, L. New insights on the function of plant acyl carrier proteins from comparative and evolutionary analysis. Genomics 2021, 113, 1155–1165. [Google Scholar] [CrossRef]
- Masud, A.J.; Kastaniotis, A.J.; Rahman, M.T.; Autio, K.J.; Hiltunen, J.K. Mitochondrial acyl carrier protein (ACP) at the interface of metabolic state sensing and mitochondrial function. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 118540. [Google Scholar] [CrossRef]
- Huang, J.; Xue, C.; Wang, H.; Wang, L.; Schmidt, W.; Shen, R.; Lan, P. Genes of ACYL CARRIER PROTEIN Family Show Different Expression Profiles and Overexpression of ACYL CARRIER PROTEIN 5 Modulates Fatty Acid Composition and Enhances Salt Stress Tolerance in Arabidopsis. Front. Plant Sci. 2017, 8, 987. [Google Scholar] [CrossRef]
- Chatterjee, J.; Patra, B.; Mukherjee, R.; Basak, P.; Mukherjee, S.; Ray, S.; Bhattacharyya, S.; Maitra, S.; GhoshDastidar, K.; Ghosh, S.; et al. Cloning, characterization and expression of a chloroplastic fructose-1, 6-bisphosphatase from Porteresia coarctata conferring salt-tolerance in transgenic tobacco. Plant Cell Tissue Organ Cult. 2013, 114, 395–409. [Google Scholar] [CrossRef]
- Shen, J.; Zou, Z.; Zhang, X.; Zhou, L.; Wang, Y.; Fang, W.; Zhu, X. Metabolic analyses reveal different mechanisms of leaf color change in two purple-leaf tea plant (Camellia sinensis L.) cultivars. Hortic. Res. 2018, 5, 7. [Google Scholar] [CrossRef]
- Ye, N.; Zhu, G.; Liu, Y.; Li, Y.; Zhang, J. ABA controls H2O2 accumulation through the induction of OsCATB in rice leaves under water stress. Plant Cell Physiol. 2011, 52, 689–698. [Google Scholar] [CrossRef]
- Greco, M.; Chiappetta, A.; Bruno, L.; Bitonti, M.B. In Posidonia oceanica, cadmium induces changes in DNA methylation and chromatin patterning. J. Exp. Bot. 2012, 63, 695–709. [Google Scholar] [CrossRef]
- Pandey, P.; Irulappan, V.; Bagavathiannan, M.V.; Senthil-Kumar, M. Impact of Combined Abiotic and Biotic Stresses on Plant Growth and Avenues for Crop Improvement by Exploiting Physio-morphological Traits. Front. Plant Sci. 2017, 8, 537. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GO Term | Ontology | Description | Number of DEGs | FDR |
|---|---|---|---|---|
| GO:0050896 | P | Response to stimulus | 32 | 2.20 × 10−24 |
| GO:0051234 | P | Establishment of localisation | 30 | 6.00 × 10−21 |
| GO:0051179 | P | Localisation | 30 | 6.00 × 10−21 |
| GO:0006810 | P | Transport | 29 | 4.60 × 10−20 |
| GO:0009987 | P | Cellular process | 51 | 8.40 × 10−18 |
| GO:0009607 | P | Response to biotic stimulus | 14 | 8.50 × 10−18 |
| GO:0006811 | P | Ion transport | 17 | 3.20 × 10−16 |
| GO:0008152 | P | Metabolic process | 47 | 1.70 × 10−15 |
| GO:0006950 | P | Response to stress | 17 | 5.80 × 10−12 |
| GO:0042592 | P | Homeostatic process | 10 | 1.00 × 10−11 |
| GO:0051704 | P | Multi-organism process | 10 | 2.30 × 10−10 |
| GO:0003824 | F | Catalytic activity | 50 | 6.30 × 10−14 |
| GO:0022857 | F | Transmembrane transporter activity | 17 | 7.60 × 10−14 |
| GO:0005488 | F | Binding | 48 | 2.60 × 10−12 |
| GO:0015075 | F | Ion transmembrane transporter activity | 13 | 2.60 × 10−12 |
| GO:0005215 | F | Transporter activity | 17 | 5.00 × 10−11 |
| GO:0022891 | F | Substrate-specific transmembrane transporter activity | 13 | 5.00 × 10−11 |
| GO:0046872 | F | Metal ion binding | 25 | 6.60 × 10−11 |
| GO:0043167 | F | Ion binding | 25 | 1.20 × 10−10 |
| GO:0043169 | F | Cation binding | 25 | 1.20 × 10−10 |
| GO:0022892 | F | Substrate-specific transporter activity | 13 | 2.80 × 10−10 |
| Probe ID | Gene ID | Gene Name | Description | Gene Ontology | Trait Ontology | Log2FC | Up or Down |
| 16381052 | Os01g0100700 | RPS5-1 | 40S ribosomal protein S5-1 | Defence response to fungus, response to H2O2, ABA, salt stress, water deprivation | Oxidative stress, sheath blight disease resistance, drought and salt tolerance, ABA sensitivity | 0.04197709 | Down |
| 16388768 | Os01g0686800 | RACK1A | WD repeat-containing protein | Shoot and root development, seed germination, circadian rhythm, detection of redox state, defence response to fungus, ET- and SA-activated signalling pathway, SA-mediated signalling pathway, photoperiodism, response to SA, ET, salt stress, H2O2, and Cd2+ | ABA content, blast disease, disease resistance, ET sensitivity, photoperiod sensitivity, salt tolerance | 0.0606787 | Down |
| 16391508 | Os01g0840100 | HSP70 | Heat shock protein Hsp70 | Response to Cd2+ and stress | – | 0.09918908 | Up |
| 16454811 | Os02g0797400 | MCM5 | MCM family protein | Cell cycle, DNA replication | – | 0.0735361 | Down |
| 16444532 | Os02g0805200 | PCNA | Proliferating cell nuclear antigen | Regulation of DNA replication, double-stranded break repair, response to gamma ray, DNA damage stimulus, UV, radiation, and H2O2 | UV light sensitivity | 0.0467894 | Down |
| 16457043 | Os03g0152900 | KINUA | Kinesin-like protein KIN-UA | Microtubule-based movement | – | 0.13444718 | Up |
| 16471736 | Os03g0308800 | TPR | Tetratricopeptide-like helical domain containing protein | RNA processing, regulation of translation | – | 0.1440686 | Up |
| 16484814 | Os04g0598200 | RPL12 | 60S ribosomal protein L12 | Translation, ribosomal large subunit assembly, defence response to bacterium, response to heat, cold, SA, JA, H2O2, and water deprivation | Heat, cold & drought tolerance, JA sensitivity, bacterial blight disease resistance, oxidative stress | 0.1768968 | Down |
| 16545101 | Os08g0117200 | RPS13 | 40S ribosomal protein S13 | Translation, defence response to fungus, response to water deprivation, salt stress, ABA, H2O2 | Drought & salt tolerance, oxidative stress, sheath blight disease resistance, ABA sensitivity. | 0.93240096 | Down |
| 16545104 | Os08g0117300 | RPS13 | 40S ribosomal protein S13 | Translation | – | 0.0503529 | Down |
| 16551132 | Os09g0250700 | ABC-1 | ABC-1 domain containing protein. | – | – | 0.1204065 | Up |
| 16414285 | Os10g0466700 | RPL17 | 60S ribosomal protein L17 | Translation | – | 0.0262824 | Down |
| 16417752 | Os11g0216000 | PK | Pyruvate kinase family protein | Glycolysis | – | 0.09241819 | Up |
| 16388283 | Os01g0655500 | Os01g0655500 | Protein kinase, core domain containing protein. | Protein phosphorylation | – | 0.27208653 | Up |
| 16442445 | Os02g0618200 | PRR1 | Two-component response regulator-like PRR1 (Pseudo-response regulator 1) | Phosphorelay signal transduction system, regulation of circadian rhythm | – | 0.10612373 | Up |
| 16477082 | Os03g0773800 | MDH | Malate dehydrogenase | Response to ROS, metabolic processes of malate and carbohydrate, cell redox homeostasis, TCA cycle | – | 0.11776116 | Up |
| 16503455 | Os05g0143800 | GH3.6 | GH3 auxin-responsive promoter domain containing protein | Response to IAA, light stimulus, salt stress | Salt tolerance | 0.2140365 | Down |
| 16501757 | Os05g0551000 | HRZ2 | Hemerythrin motif-containing RING- and Zn-finger protein2 | Response to Fe3+ starvation, JA-mediated signalling pathway | Fe3+ sensitivity | 0.24291249 | Up |
| 16513254 | Os06g0308000 | TIG | Trigger factor-like protein | Protein folding and transport | – | 0.09959792 | Up |
| 16411623 | Os10g0111700 | PTR | POT family protein, peptide transporter | Oligopeptide transport | – | 0.20793006 | Up |
| 16407843 | Os10g0323600 | PRF1 | Profilin A | Cytoskeleton organisation, sequestering of actin monomers | – | 0.16372392 | Up |
| 16420775 | Os11g0106700 | FER1 | FERRITIN 1 | Response to Fe3+, Zn2+, H2O2, ABA, bacterdium and cold; leaf and flower development; Fe3+ transport and homeostasis; photosynthesis | – | 0.1615892 | Down |
| 16430188 | Os12g0106000 | FER2 | FERRITIN 2 | Response to Fe3+, Zn2+, Cu2+, H2O2, cold, bacterium, ABA; leaf and flower development; Fe3+ transport and homeostasis; photosynthesis | Temperature response trait, Cu2+ sensitivity | 0.1529454 | Down |
| 16427113 | Os12g0242700 | KAR | Beta-ketoacyl reductase | Fatty acid elongation | – | 0.48300481 | Up |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abdullah-Zawawi, M.-R.; Tan, L.-W.; Ab Rahman, Z.; Ismail, I.; Zainal, Z. An Integration of Transcriptomic Data and Modular Gene Co-Expression Network Analysis Uncovers Drought Stress-Related Hub Genes in Transgenic Rice Overexpressing OsAbp57. Agronomy 2022, 12, 1959. https://doi.org/10.3390/agronomy12081959
Abdullah-Zawawi M-R, Tan L-W, Ab Rahman Z, Ismail I, Zainal Z. An Integration of Transcriptomic Data and Modular Gene Co-Expression Network Analysis Uncovers Drought Stress-Related Hub Genes in Transgenic Rice Overexpressing OsAbp57. Agronomy. 2022; 12(8):1959. https://doi.org/10.3390/agronomy12081959
Chicago/Turabian StyleAbdullah-Zawawi, Muhammad-Redha, Lay-Wen Tan, Zuraida Ab Rahman, Ismanizan Ismail, and Zamri Zainal. 2022. "An Integration of Transcriptomic Data and Modular Gene Co-Expression Network Analysis Uncovers Drought Stress-Related Hub Genes in Transgenic Rice Overexpressing OsAbp57" Agronomy 12, no. 8: 1959. https://doi.org/10.3390/agronomy12081959
APA StyleAbdullah-Zawawi, M.-R., Tan, L.-W., Ab Rahman, Z., Ismail, I., & Zainal, Z. (2022). An Integration of Transcriptomic Data and Modular Gene Co-Expression Network Analysis Uncovers Drought Stress-Related Hub Genes in Transgenic Rice Overexpressing OsAbp57. Agronomy, 12(8), 1959. https://doi.org/10.3390/agronomy12081959