
Mining Candidate Genes and Favorable Haplotypes for Flag Leaf Shape in Rice (Oryza sativa L.) Based on a Genome-Wide Association Study


Abstract
:1. Introduction
2. Materials and Methods
2.1. Material Distribution and Field Planting
2.2. Phenotypic Characterization
2.3. SNP Access
2.4. Population Genetic Analysis
2.5. Genome-Wide Association Mapping
2.6. Identification of the Candidate Genes and Haplotype Analysis
2.7. Prediction of Excellent Parents
3. Results
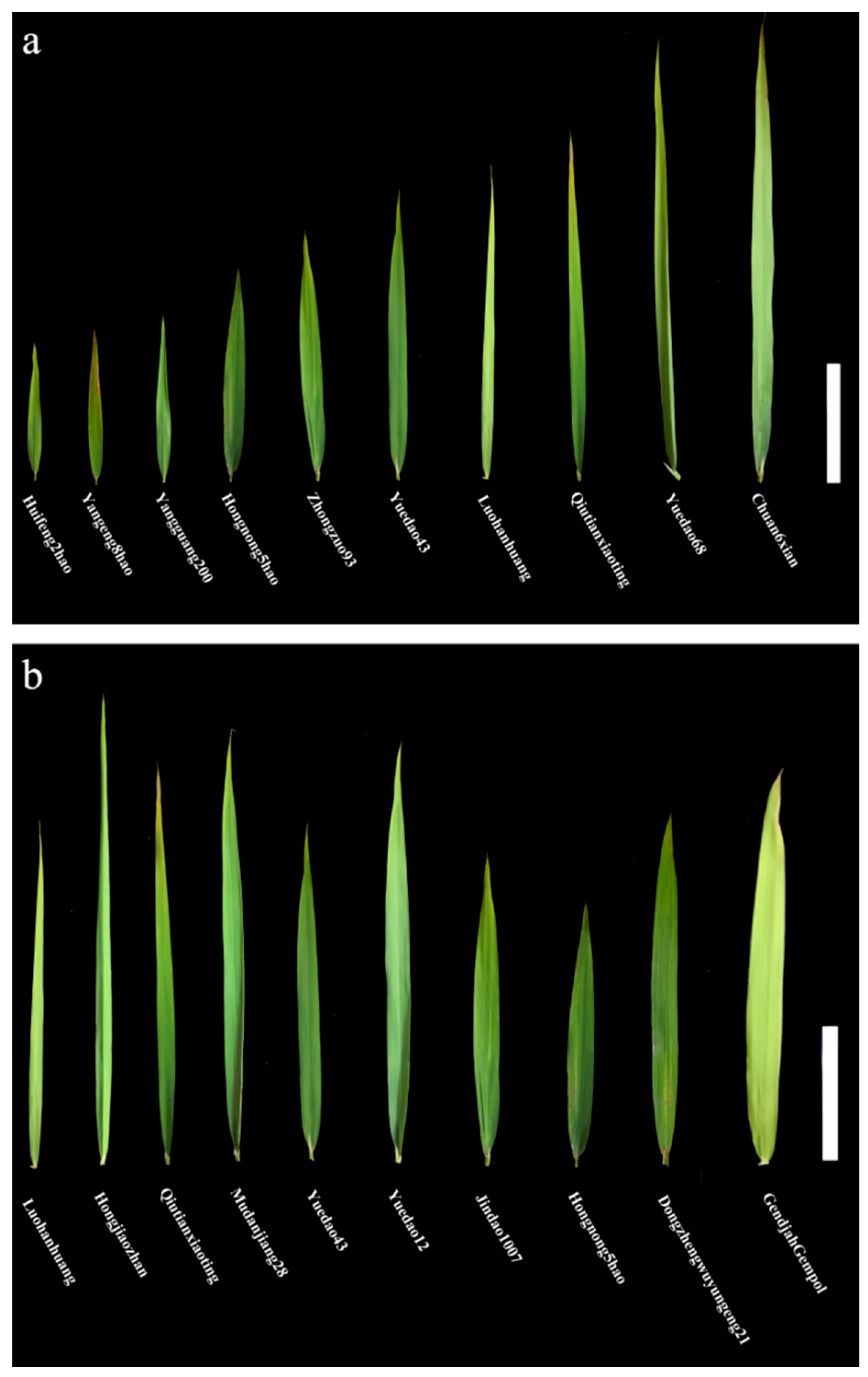
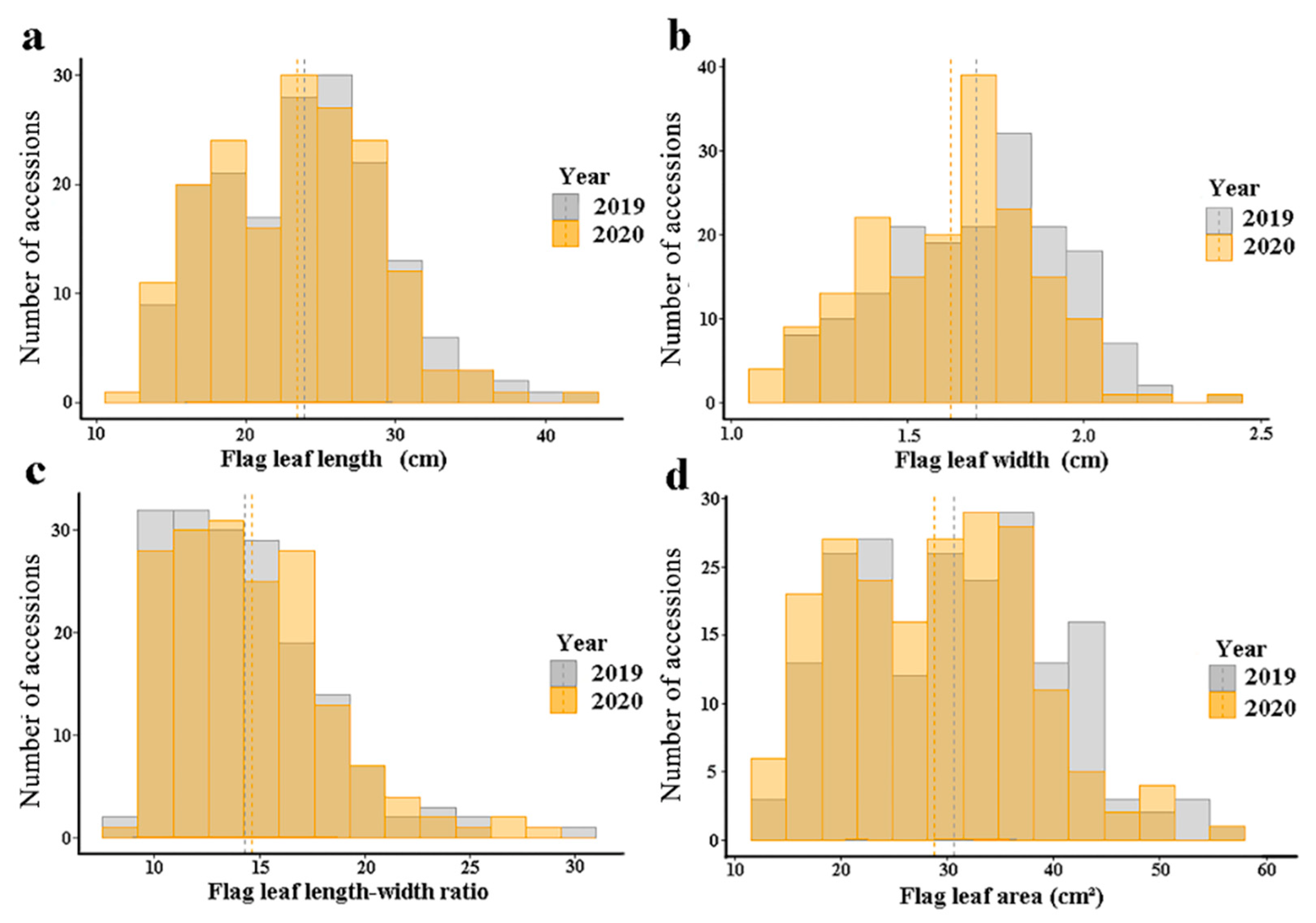
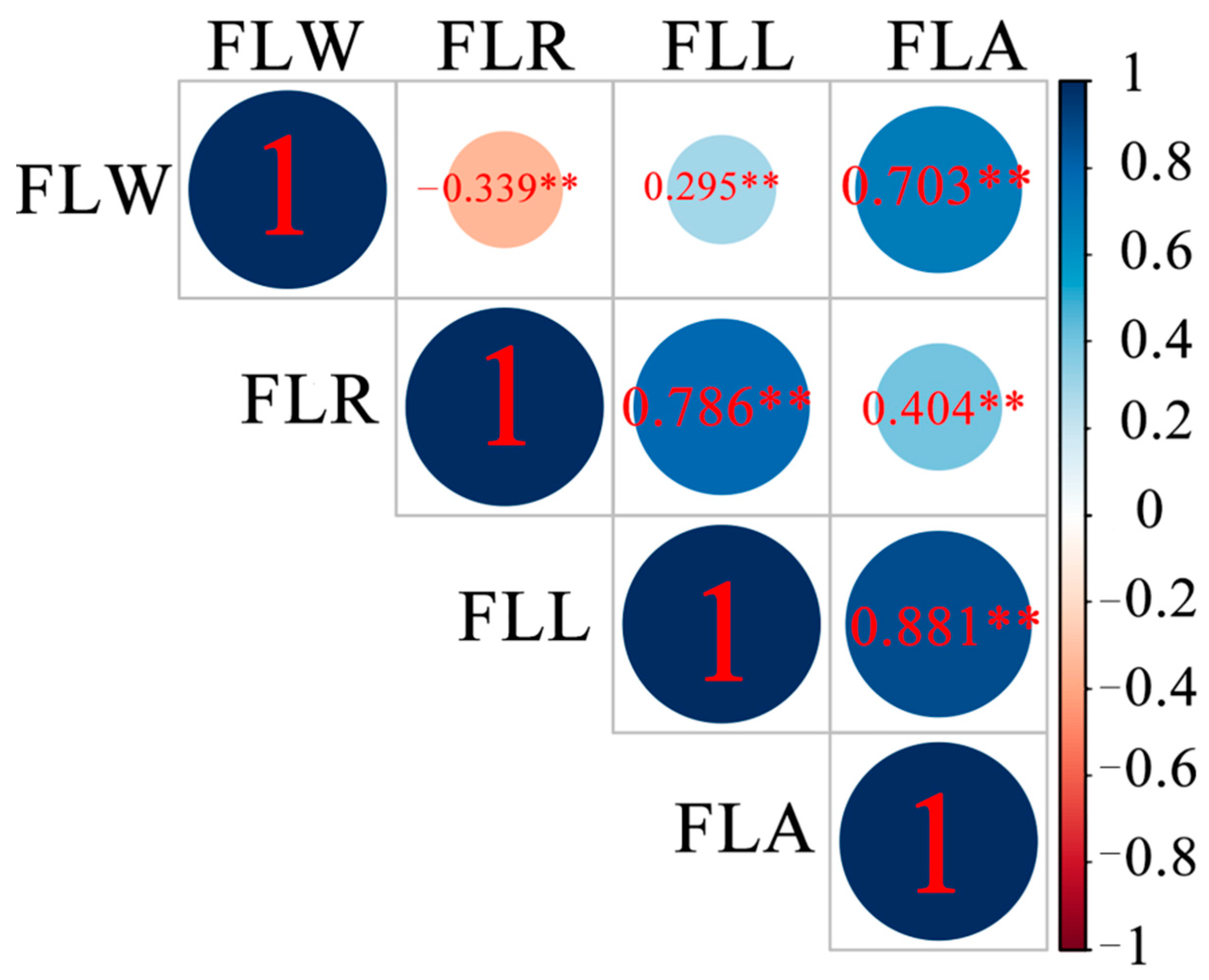
3.1. Phenotypic Variation of the Flag Leaf Shapes in the Natural Population
3.2. Genomic Variation at the SNP Level in the 173 Rice Accessions
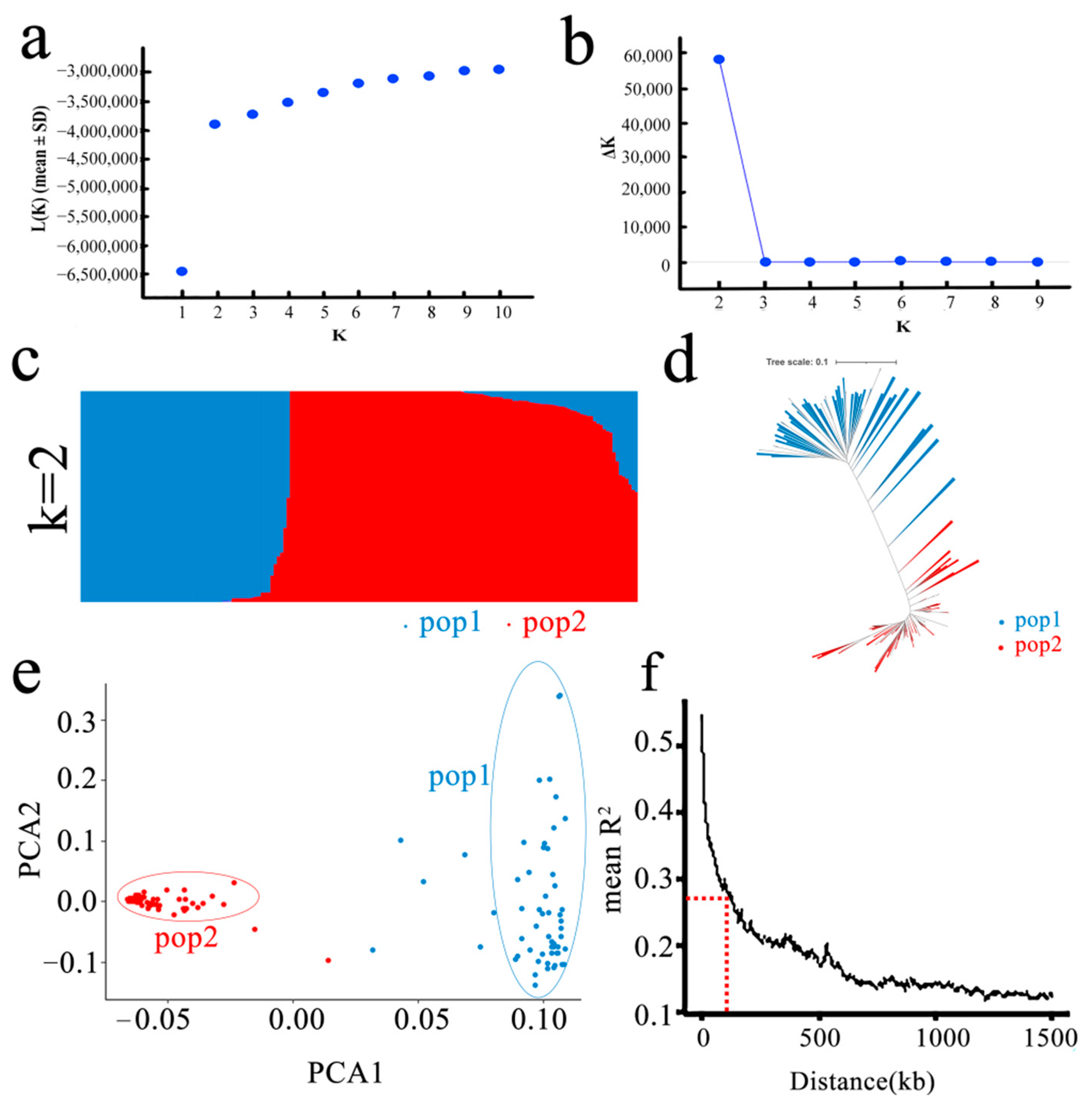
3.3. Population Structure and LD Analysis
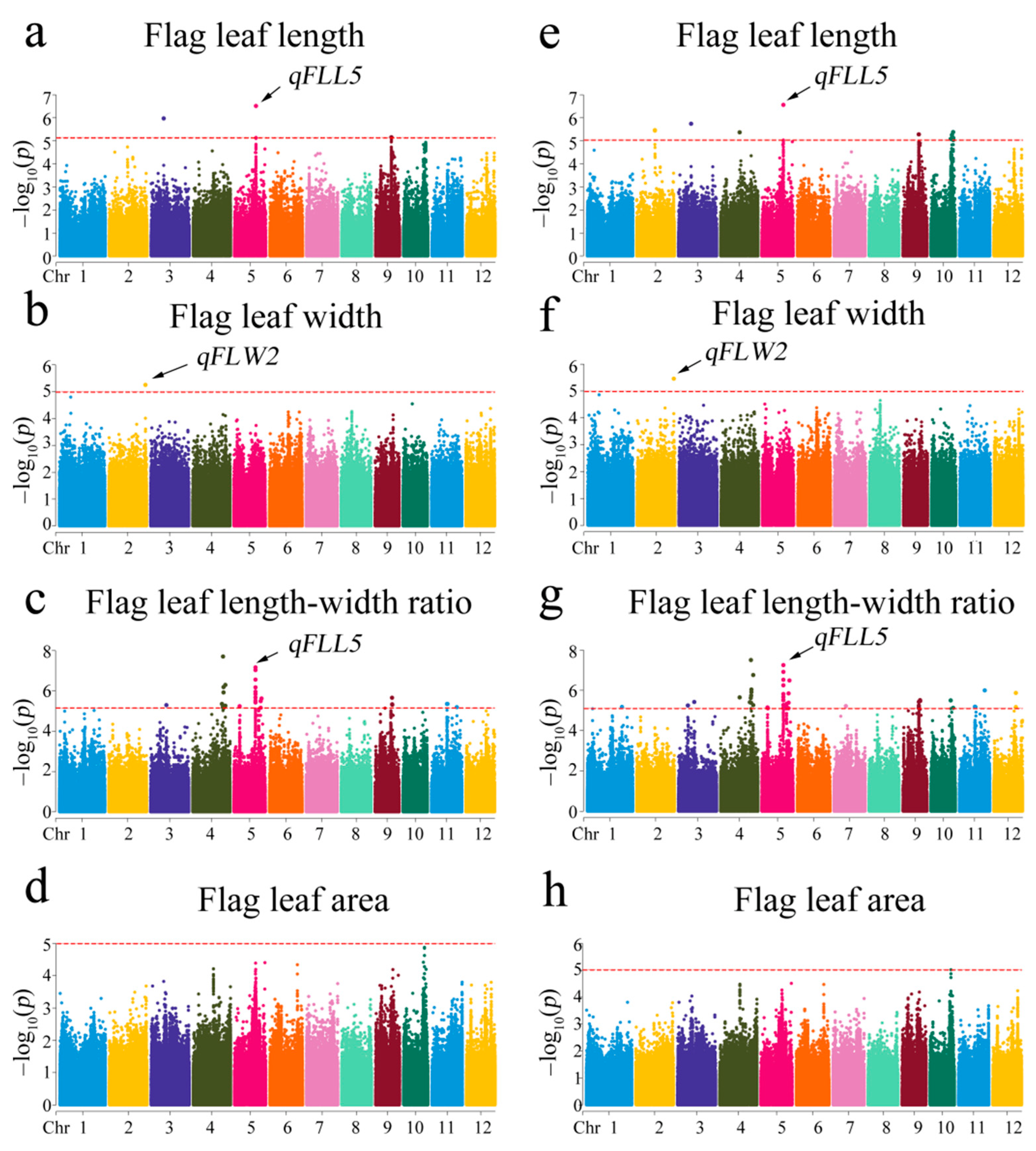
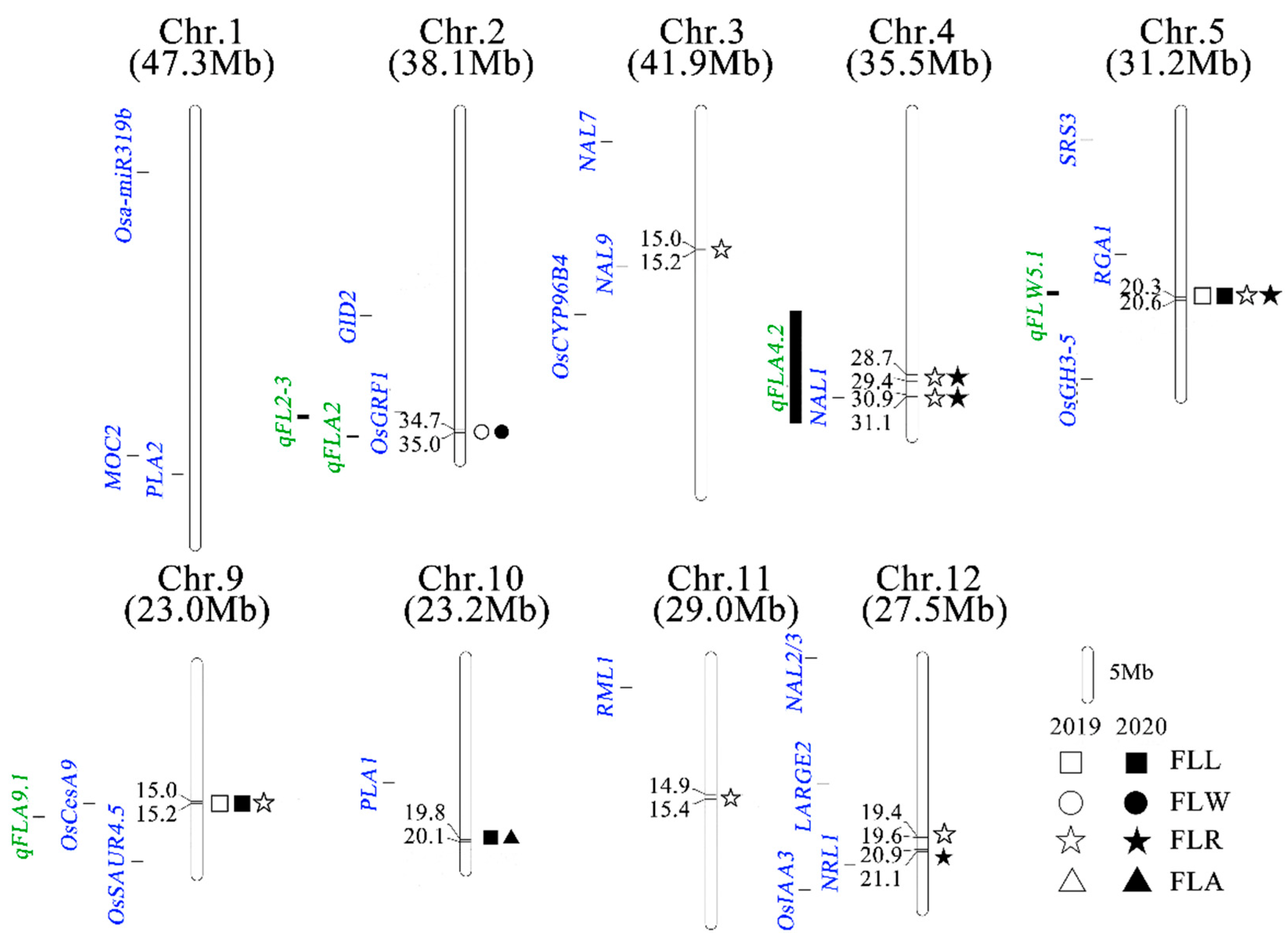
3.4. Genome-Wide Association Mapping for Flag Leaf Shapes
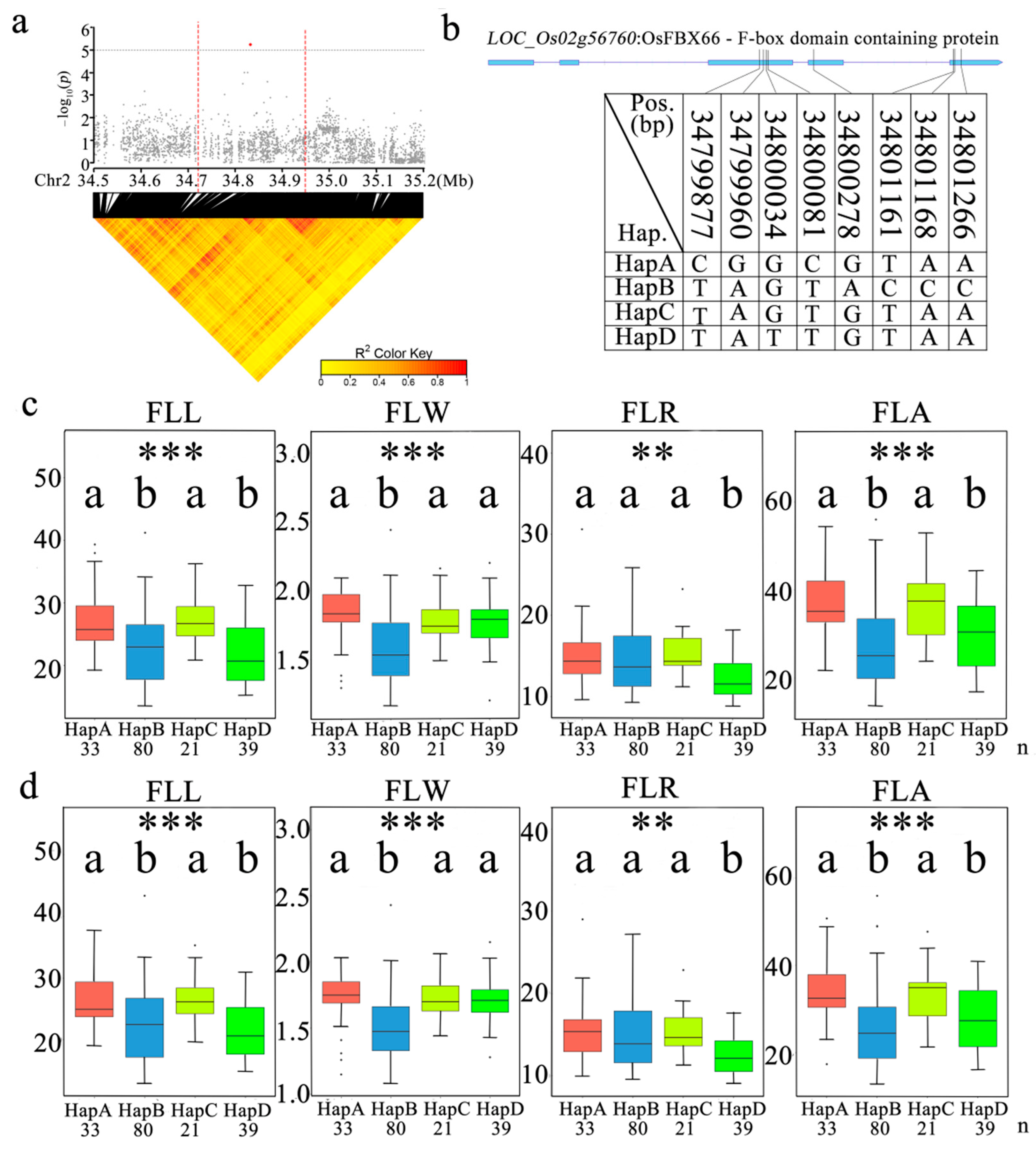
3.5. Identification of a Candidate Gene for qFLW2
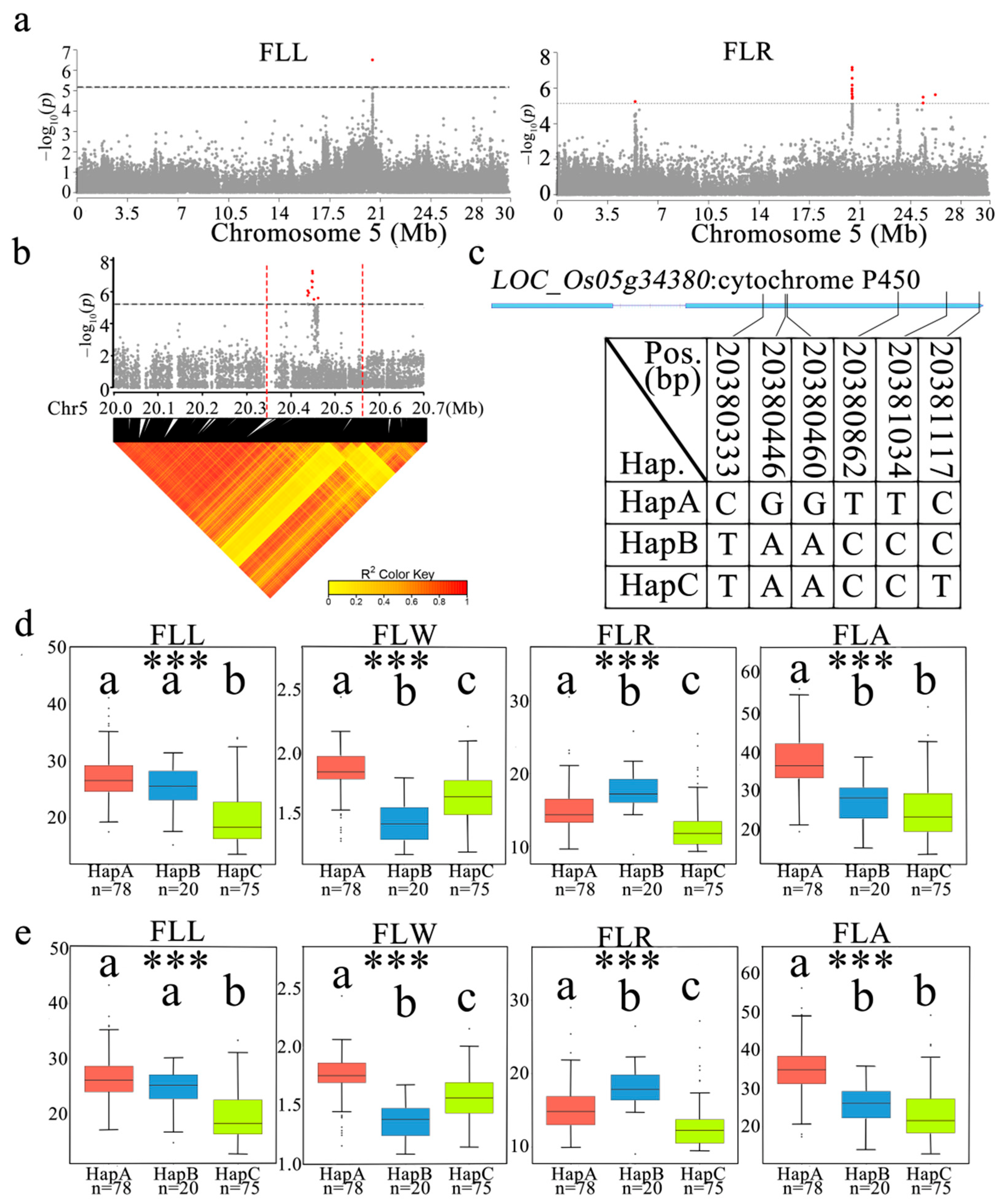
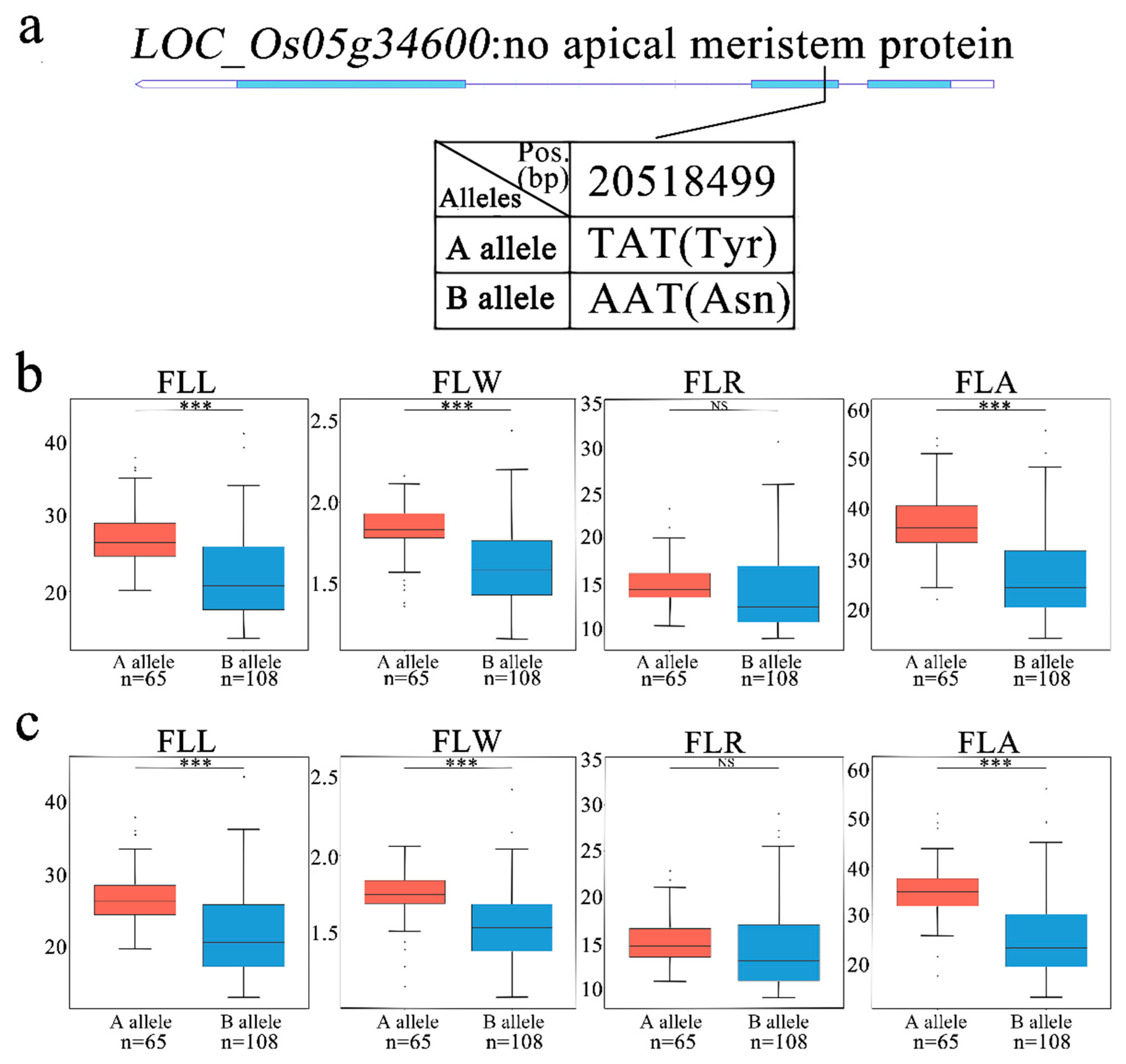
3.6. Identification of Candidate Genes for qFLL5 and qFLR5.1
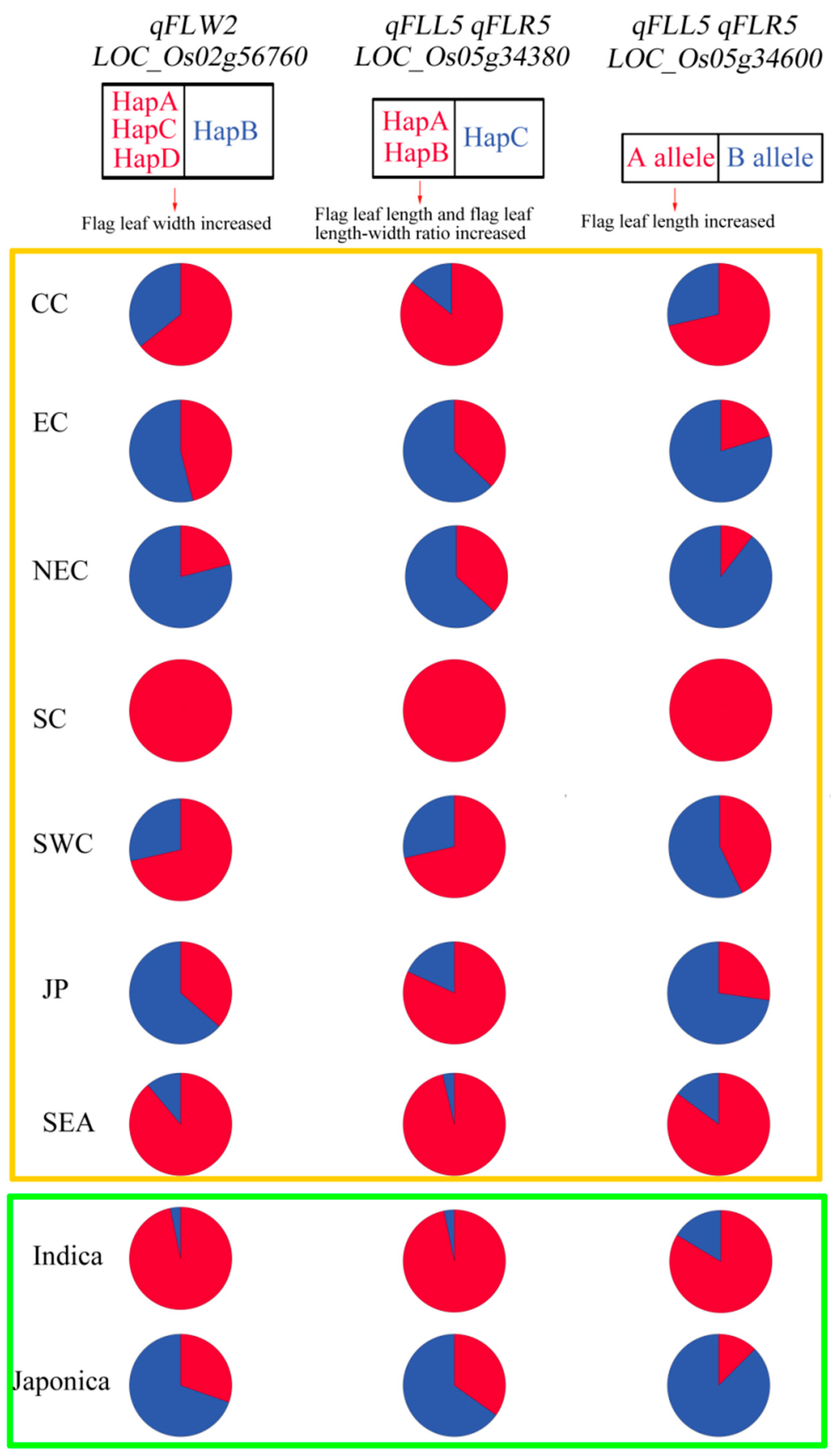
3.7. Haplotypes Distribution of Candidate Genes
3.8. Excellent Parental Combinations Predicted for Flag Leaf Shapes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fageria, N.K. Yield physiology of rice. J. Plant Nutr. 2007, 30, 843–879. [Google Scholar] [CrossRef]
- Yoshida, S. Fundamentals of Rice Crop Science; International Rice Research Institute: Manila, Philippines, 1981. [Google Scholar]
- Li, Z.; Pinson, S.R.; Stansel, J.W.; Paterson, A.H. Genetic dissection of the source-sink relationship affecting fecundity and yield in rice (shape Oryza sativa L.). Mol. Breed. 1998, 4, 419–426. [Google Scholar] [CrossRef]
- Yuan, L.; Denning, G.; Mew, T. Hybrid Rice Breeding for Super High Yield; Denning, G.L., Mew, T.W., Eds.; International Rice Research Institute: Manila, Philippines, 1998; pp. 10–12. [Google Scholar]
- Xu, J.; Zhao, Q.; Zhao, Y.; Zhu, L.; Xu, C.; Gu, M.; Han, B.; Liang, G. Mapping of QTLs for Flag Leaf Shape Using Whole-Genome Re-sequenced Chromosome Segment Substitution Lines in Rice. Chin. J. Rice Sci. 2011, 25, 483–487. [Google Scholar]
- Farooq, M.; Tagle, A.G.; Santos, R.E.; Ebron, L.A.; Fujita, D.; Kobayashi, N. Quantitative trait loci mapping for leaf length and leaf width in rice cv. IR64 derived lines. J. Integr. Plant Biol. 2010, 52, 578–584. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Zhao, Y.; Liu, F.; Chen, Q.; Qi, J. Narrow leaf 1 (NAL1) regulates leaf shape by affecting cell expansion in rice (Oryza sativa L.). Biochem. Biophys. Res. Commun. 2019, 516, 957–962. [Google Scholar] [CrossRef]
- Zhang, G.H.; Li, S.Y.; Wang, L.; Ye, W.J.; Zeng, D.L.; Rao, Y.C.; Peng, Y.L.; Hu, J.; Yang, Y.L.; Xu, J.; et al. LSCHL4 from Japonica Cultivar, Which Is Allelic to NAL1, Increases Yield of Indica Super Rice 93–11. Mol. Plant 2014, 7, 1350–1364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, J.; Zhang, M.; Guo, L.; Li, X.; Bao, J.; Ma, L. QTLs for rice flag leaf traits in doubled haploid populations in different environments. Genet. Mol. Res. 2015, 14, 6786–6795. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Ye, H.; Li, S.; Jiang, J.; Zhang, Y.; Pan, C.; Zhou, W.; Chen, W.; Wang, K.; Dai, G. QTL mapping and analysis of candidate genes in flag leaf morphology in rice. Sci. Sin. Vitae 2021, 51, 567–578. [Google Scholar] [CrossRef]
- Chen, M.; Luo, J.; Shao, G.; Wei, X.; Tang, S.; Sheng, Z.; Song, J.; Hu, P. Fine mapping of a major QTL for flag leaf width in rice, qFLW4, which might be caused by alternative splicing of NAL1. Plant Cell Rep. 2012, 31, 863–872. [Google Scholar] [CrossRef]
- Xiao, K.; Zuo, H.L.; Gong, Y.J.; Zhang, J.Z.; Zhang, Y.J.; Dong, Y.J. Locating quantitative trait loci affecting flag-leaf shape traits in rice (Oryza sativa L.). J. Shanghai Norm. Univ. Nat. Sci. 2007, 36, 66–70. [Google Scholar]
- Jiang, S.; Zhang, X.; Wang, J.; Chen, W.; Xu, Z. Fine mapping of the quantitative trait locus qFLL9 controlling flag leaf length in rice. Euphytica 2010, 176, 341–347. [Google Scholar] [CrossRef]
- Peng, M.; Yang, G.; Zhang, Q.; An, B.; Li, Y. QTL analysis for flag leaf morphological traits in rice (Oryza sativa L.) under different genetic backgrounds. Chin. J. Rice Sci. 2007, 21, 247–252. [Google Scholar]
- Wang, J.; Wang, T.; Wang, Q.; Tang, X.; Ren, Y.; Zheng, H.; Liu, K.; Yang, L.; Jiang, H.; Li, Y. QTL mapping and candidate gene mining of flag leaf size traits in Japonica rice based on linkage mapping and genome-wide association study. Mol. Biol. Rep. 2022, 49, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Gong, R.; Sun, W.; Zhang, C.; Yu, S. Genetic dissection and validation of candidate genes for flag leaf size in rice (Oryza sativa L.). Theor. Appl. Genet. 2018, 131, 801–815. [Google Scholar] [CrossRef]
- Li, W.; Wu, C.; Hu, G.; Xing, L.; Qian, W.; Si, H.; Sun, Z.; Wang, X.; Fu, Y.; Liu, W. Characterization and fine mapping of a novel rice narrow leaf mutant nal9. J. Integr. Plant Biol. 2013, 55, 1016–1025. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Murata, K.; Yamazaki, M.; Onosato, K.; Miyao, A.; Hirochika, H. Three distinct rice cellulose synthase catalytic subunit genes required for cellulose synthesis in the secondary wall. Plant Physiol. 2003, 133, 73–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, J.; Qian, Q.; Bu, Q.; Li, S.; Chen, Q.; Sun, J.; Liang, W.; Zhou, Y.; Chu, C.; Li, X. Mutation of the rice Narrow leaf1 gene, which encodes a novel protein, affects vein patterning and polar auxin transport. Plant Physiol. 2008, 147, 1947–1959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujino, K.; Matsuda, Y.; Ozawa, K.; Nishimura, T.; Koshiba, T.; Fraaije, M.W.; Sekiguchi, H. NARROW LEAF 7 controls leaf shape mediated by auxin in rice. Mol. Genet. Genom. 2008, 279, 499–507. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.X.; Xiao, M.Z.; Liu, Y.; Fu, J.L.; He, Y.; Jiang, D.A. The small auxin-up RNA OsSAUR45 affects auxin synthesis and transport in rice. Plant Mol. Biol. 2017, 94, 97–107. [Google Scholar] [CrossRef]
- Hu, J.; Zhu, L.; Zeng, D.; Gao, Z.; Guo, L.; Fang, Y.; Zhang, G.; Dong, G.; Yan, M.; Liu, J. Identification and characterization of NARROW AND ROLLED LEAF 1, a novel gene regulating leaf morphology and plant architecture in rice. Plant Mol. Mol. Biol. 2010, 73, 283–292. [Google Scholar] [CrossRef]
- Wu, C.; Fu, Y.; Hu, G.; Si, H.; Cheng, S.; Liu, W. Isolation and characterization of a rice mutant with narrow and rolled leaves. Planta 2010, 232, 313–324. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.H.; Yoo, S.C.; Zhang, H.; Pandeya, D.; Koh, H.J.; Hwang, J.Y.; Kim, G.T.; Paek, N.C. The rice narrow leaf2 and narrow leaf3 loci encode WUSCHEL-related homeobox 3 A (OsWOX3A) and function in leaf, spikelet, tiller and lateral root development. New Phytol. 2013, 198, 1071–1084. [Google Scholar] [CrossRef] [PubMed]
- Kang, L.; Xu, J. Genetic basis of grain yield potential in rice: A critical review on QTL studies. J. Plant Genet. Resour. 2008, 9, 545–550. [Google Scholar]
- Murray, M.G.; Thompson, W.F. Rapid isolation of high molecular weight plant DNA. Nucleic Acids Acids Res. 1980, 8, 4321–4325. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Genome Project Data, P. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Li, M.Y.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, 7. [Google Scholar] [CrossRef]
- Yang, J.; Lee, S.H.; Goddard, M.E.; Visscher, P.M. GCTA: A tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 2011, 88, 76–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pritchard, J.K.; Stephens, M.; Rosenberg, N.A.; Donnelly, P. Association mapping in structured populations. Am. J. Hum. Genet. 2000, 67, 170–181. [Google Scholar] [CrossRef] [Green Version]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; Bakker, P.I.W.D.; Daly, M.J. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [Green Version]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [Green Version]
- Earl, D.A.; Vonholdt, B.M. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Bradbury, P.J.; Zhang, Z.; Kroon, D.E.; Casstevens, T.M.; Ramdoss, Y.; Buckler, E.S. TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics 2007, 23, 2633–2635. [Google Scholar] [CrossRef] [PubMed]
- Mather, K.A.; Caicedo, A.L.; Polato, N.R.; Olsen, K.M.; McCouch, S.; Purugganan, M.D. The extent of linkage disequilibrium in rice (Oryza sativa L.). Genetics 2007, 177, 2223–2232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, J.H.; Blay, S.; McNeney, B.; Graham, J. LDheatmap: An R function for graphical display of pairwise linkage disequilibria between single nucleotide polymorphisms. J. Stat. Stat. Softw. 2006, 16, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.M.; Pressoir, G.; Briggs, W.H.; Bi, I.V.; Yamasaki, M.; Doebley, J.F.; McMullen, M.D.; Gaut, B.S.; Nielsen, D.M.; Holland, J.B.; et al. A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat. Genet. 2006, 38, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Baute, J.; Polyn, S.; De Block, J.; Blomme, J.; Van Lijsebettens, M.; Inzé, D. F-box protein FBX92 affects leaf size in Arabidopsis thaliana. Plant Cell Physiol. 2017, 58, 962–975. [Google Scholar] [CrossRef] [Green Version]
- Bak, S.; Tax, F.E.; Feldmann, K.A.; Galbraith, D.W.; Feyereisen, R. CYP83B1, a cytochrome P450 at the metabolic branch point in auxin and indole glucosinolate biosynthesis in Arabidopsis. Plant Cell 2001, 13, 101–111. [Google Scholar] [CrossRef] [Green Version]
- Mitsuda, N.; Seki, M.; Shinozaki, K.; Ohme-Takagi, M. The NAC transcription factors NST1 and NST2 of Arabidopsis regulate secondary wall thickenings and are required for anther dehiscence. Plant Cell 2005, 17, 2993–3006. [Google Scholar] [CrossRef] [Green Version]
- Peng, W.; Sun, P.; Pan, S.; Li, W. Mapping QTLs for grain shape, flag leaf traits, and plant height in rice variety Mowanggu. Acta Agron. Sin. 2018, 44, 1673–1680. [Google Scholar] [CrossRef]
- Li, J.; Tian, R.; Bai, T.; Zhu, C.; Song, J.; Tian, L.; Ma, S.; Lv, J.; Hu, H.; Wang, Z. Comprehensive Evaluation and QTL Analysis for Flag Leaf Traits Using a Backcross Population in Rice. Chin. J. Rice Sci. 2021, 35, 573–585. [Google Scholar]
- Zhu, S.; Lv, W.; He, L.; Xing, D.; Yang, L.; Qiu, X.; Xu, J. Genetic Dissection of Flag Leaf Related Traits and Grain Yield per Plant Using Genome-wide Association Analysis. J. Plant Genet. Resour. 2020, 21, 663–673. [Google Scholar]
- Rohila, J.S.; Edwards, J.D.; Tran, G.D.; Jackson, A.K.; McClung, A.M. Identification of Superior Alleles for Seedling Stage Salt Tolerance in the USDA Rice Mini-Core Collection. Plants 2019, 8, 472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Feng, F.; Lian, X.; Teng, X.; Wei, H.; Yu, H.; Xie, W.; Yan, M.; Fan, P.; Li, Y. Genome-wide Association Study (GWAS) of mesocotyl elongation based on re-sequencing approach in rice. BMC Plant Biol. 2015, 15, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.R.; Xu, X.; Vieira, F.G.; Xiao, Y.H.; Li, Z.K.; Wang, J.; Nielsen, R.; Chu, C.C. The Power of Inbreeding: NGS-Based GWAS of Rice Reveals Convergent Evolution during Rice Domestication. Mol. Plant 2016, 9, 975–985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamura, K.; Nei, M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol. Biol. Evol. 1993, 10, 512–526. [Google Scholar]
- Bruno, W.J.; Socci, N.D.; Halpern, A.L. Weighted neighbor joining: A likelihood-based approach to distance-based phylogeny reconstruction. Mol. Biol. Evol. 2000, 17, 189–197. [Google Scholar] [CrossRef] [Green Version]
- Sober, E. The contest between parsimony and likelihood. Syst. Biol. 2004, 53, 644–653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holder, M.; Lewis, P.O. Phylogeny estimation: Traditional and Bayesian approaches. Nat. Rev. Genet. 2003, 4, 275–284. [Google Scholar] [CrossRef]
- Price, A.L.; Patterson, N.J.; Plenge, R.M.; Weinblatt, M.E.; Shadick, N.A.; Reich, D. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 2006, 38, 904–909. [Google Scholar] [CrossRef]
- Liu, X.L.; Huang, M.; Fan, B.; Buckler, E.S.; Zhang, Z.W. Iterative Usage of Fixed and Random Effect Models for Powerful and Efficient Genome-Wide Association Studies. PLoS Genet. 2016, 12, e1005767. [Google Scholar] [CrossRef]
- Zhao, K.Y.; Aranzana, M.J.; Kim, S.; Lister, C.; Shindo, C.; Tang, C.L.; Toomajian, C.; Zheng, H.G.; Dean, C.; Marjoram, P.; et al. An Arabidopsis example of association mapping in structured samples. PLoS Genet. 2007, 3, e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, N.; Zheng, H.L.; Cui, J.N.; Wang, J.G.; Liu, H.L.; Sun, J.; Liu, T.T.; Zhao, H.W.; Lai, Y.C.; Zou, D.T. Genome-wide association study and candidate gene analysis of alkalinity tolerance in japonica rice germplasm at the seedling stage. Rice 2019, 12, 24. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Han, B. Natural variations and genome-wide association studies in crop plants. Annu. Rev. Plant Biol. 2014, 65, 531–551. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Meng, Y.; Zeng, J.; Luo, Y.; Feng, Z.; Bian, L.; Gao, S. Coordination between GROWTH-REGULATING FACTOR1 and GRF-INTERACTING FACTOR1 plays a key role in regulating leaf growth in rice. BMC Plant Biol. 2020, 20, 200. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Zhang, G.; Zhou, S.; Ren, Y.; Wang, W. The improvement of salt tolerance in transgenic tobacco by overexpression of wheat F-box gene TaFBA1. Plant Sci. 2017, 259, 71–85. [Google Scholar] [CrossRef]
- Dreher, K.; Callis, J. Ubiquitin, hormones and biotic stress in plants. Annals of botany 2007, 99, 787–822. [Google Scholar] [CrossRef] [PubMed]
- Lechner, E.; Achard, P.; Vansiri, A.; Potuschak, T.; Genschik, P. F-box proteins everywhere. Curr. Opin. Plant Biol. 2006, 9, 631–638. [Google Scholar] [CrossRef]
- Pinot, F.; Beisson, F. Cytochrome P450 metabolizing fatty acids in plants: Characterization and physiological roles. FEBS J. 2011, 278, 195–205. [Google Scholar] [CrossRef]
- Paddon, C.J.; Westfall, P.J.; Pitera, D.J.; Benjamin, K.; Fisher, K.; McPhee, D.; Leavell, M.; Tai, A.; Main, A.; Eng, D. High-level semi-synthetic production of the potent antimalarial artemisinin. Nature 2013, 496, 528–532. [Google Scholar] [CrossRef] [Green Version]
- Höfer, R.; Boachon, B.; Renault, H.; Gavira, C.; Miesch, L.; Iglesias, J.; Ginglinger, J.-F.; Allouche, L.; Miesch, M.; Grec, S. Dual function of the cytochrome P450 CYP76 family from Arabidopsis thaliana in the metabolism of monoterpenols and phenylurea herbicides. Plant Physiol. 2014, 166, 1149–1161. [Google Scholar] [CrossRef] [Green Version]
- Hull, A.K.; Vij, R.; Celenza, J.L. Arabidopsis cytochrome P450s that catalyze the first step of tryptophan-dependent indole-3-acetic acid biosynthesis. Proc. Natl. Acad. Sci. USA 2000, 97, 2379–2384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, H.; Cheng, H.Y.; Chen, C.; Yu, X.W.; Yang, J.N.; Gao, W.R.; Shi, Q.H.; Zhang, H.; Li, J.G.; Ma, H. A NAC transcription factor gene of Chickpea (Cicer arietinum), CarNAC3, is involved in drought stress response and various developmental processes. J. Plant Physiol. 2009, 166, 1934–1945. [Google Scholar] [CrossRef] [PubMed]
- Souer, E.; van Houwelingen, A.; Kloos, D.; Mol, J.; Koes, R. The no apical meristem gene of Petunia is required for pattern formation in embryos and flowers and is expressed at meristem and primordia boundaries. Cell 1996, 85, 159–170. [Google Scholar] [CrossRef] [Green Version]
- Sablowski, R.W.; Meyerowitz, E.M. A homolog of NO APICAL MERISTEM is an immediate target of the floral homeotic genes APETALA3/PISTILLATA. Cell 1998, 92, 93–103. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phenotype | Year | Mean ± SD (cm) | Range (cm) | CV (%) | HB2 (%) |
|---|---|---|---|---|---|
| FLL | 2019 | 23.86 ± 5.68 | 13.57–41.23 | 23.81 | 80.30 |
| 2020 | 23.38 ± 5.56 | 12.83–43.35 | 23.76 | 66.15 | |
| FLW | 2019 | 1.69 ± 0.25 | 1.16–2.44 | 14.79 | 91.82 |
| 2020 | 1.62 ± 0.24 | 1.08–2.42 | 14.73 | 77.70 | |
| FLR | 2019 | 14.31 ± 3.71 | 8.84–30.60 | 25.89 | 81.91 |
| 2020 | 14.65 ± 3.75 | 9.03–29.02 | 25.58 | 69.85 | |
| FLA | 2019 | 30.62 ± 9.46 | 13.66–55.99 | 30.90 | 84.09 |
| 2020 | 28.80 ± 8.91 | 12.88–55.95 | 30.95 | 68.44 |
| Trait | QTLs | Chr | SNP | Allele | 2019 | 2020 | Model | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| p-Value | FDR | PVE(%) | p-Value | FDR | PVE (%) | ||||||
| FLL | qFLL5 | 5 | 20452668 | G/A | 3.06 × 10−7 | 1.84 × 10−6 | 16.03 | 2.74 × 10−7 | 3.15 × 10−6 | 16.18 | MLM |
| 5 | 20452668 | G/A | 9.05 × 10−11 | 8.71 × 10−10 | 16.07 | 7.04 × 10−11 | 6.86 × 10−10 | 16.63 | GLM | ||
| qFLL9 | 9 | 15134940 | C/A | 6.96 × 10−6 | 8.51 × 10−6 | 14.38 | 5.22 × 10−6 | 8.58 × 10−6 | 14.76 | MLM | |
| 9 | 15134940 | C/A | 1.89 × 10−10 | 1.57 × 10−9 | 17.03 | 6.28 × 10−11 | 6.36 × 10−10 | 18.20 | GLM | ||
| FLW | qFLW2 | 2 | 34839350 | G/A | 5.57 × 10−6 | 5.57 × 10−6 | 15.09 | 3.48 × 10−6 | 3.48 × 10−6 | 15.71 | MLM |
| 2 | 34839350 | G/A | 2.93 × 10−8 | 2.93 × 10−8 | 14.49 | 1.64 × 10−8 | 4.09 × 10−8 | 15.14 | GLM | ||
| FLR | qFLR4.1 | 4 | 28859732 | C/T | 1.98 × 10−8 | 9.5 × 10−7 | 20.06 | 3.17 × 10−8 | 2.88 × 10−6 | 19.45 | MLM |
| 4 | 28859732 | C/T | 2.72 × 10−14 | 2.69 × 10−11 | 27.81 | 8.08 × 10−15 | 5.73 × 10−12 | 28.89 | GLM | ||
| qFLR4.2 | 4 | 29302234 | T/A | 6.53 × 10−7 | 4.03 × 10−6 | 17.92 | 9.22 × 10−7 | 9.32 × 10−6 | 17.47 | MLM | |
| 4 | 29302234 | T/A | 5.12 × 10−14 | 1.68 × 10−11 | 29.19 | 7.26 × 10−15 | 1.03 × 10−11 | 30.85 | GLM | ||
| qFLR4.3 | 4 | 31016407 | G/T | 5.04 × 10−7 | 4.03 × 10−6 | 15.77 | 1.74 × 10−7 | 3.17 × 10−6 | 17.18 | MLM | |
| 4 | 31016407 | G/T | 2.84 × 10−8 | 4.22 × 10−8 | 15.97 | 9.15 × 10−9 | 2.07 × 10−8 | 17.07 | GLM | ||
| qFLR5 | 5 | 20452668 | G/A | 6.83 × 10−8 | 1.09 × 10−6 | 18.40 | 5.61 × 10−8 | 1.7 × 10−6 | 18.68 | MLM | |
| 5 | 20452668 | G/A | 3.63 × 10−11 | 1.43 × 10−9 | 21.88 | 8.07 × 10−12 | 5.21 × 10−10 | 23.25 | GLM | ||
| Best Predicted Parents | Predicted FLL Improvement (cm) | Predicted FLW Improvement (cm) | Total Predicted FLA Improvement (cm2) | |
|---|---|---|---|---|
| LOC_Os05g34380 | LOC_Os05g34600 | LOC_Os02g56760 | ||
| Xiangchuanwuxinbaimi (japonica) | HapA (3.17) | HapA (2.96) | HapC (0.08) | 0.18 |
| Tijin (japonica) | HapA (3.17) | HapA (2.96) | HapD (0.09) | 0.21 |
| Xiangjing 9407 (japonica) | HapA (3.17) | HapA (2.96) | HapD (0.09) | 0.21 |
| Shengtangqing (japonica) | HapA (3.17) | HapA (2.96) | HapD (0.09) | 0.21 |
| Nannongjing 1R (japonica) | HapA (3.17) | HapA (2.96) | HapA (0.12) | 0.28 |
| Yuedao 68 (indica) | HapA (3.17) | HapA (2.96) | HapA (0.12) | 0.28 |
| Yuetai B (indica) | HapA (3.17) | HapA (2.96) | HapA (0.12) | 0.28 |
| Yuedao 37 (indica) | HapA (3.17) | HapA (2.96) | HapA (0.12) | 0.28 |
| Yuedao 61 (indica) | HapA (3.17) | HapA (2.96) | HapA (0.12) | 0.28 |
| Yuedao 9 (indica) | HapA (3.17) | HapA (2.96) | HapA (0.12) | 0.28 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Du, M.; Xiong, M.; Chang, Y.; Liu, Z.; Wang, R.; Lin, X.; Zhou, Z.; Lu, M.; Liu, C.; Liu, E. Mining Candidate Genes and Favorable Haplotypes for Flag Leaf Shape in Rice (Oryza sativa L.) Based on a Genome-Wide Association Study. Agronomy 2022, 12, 1814. https://doi.org/10.3390/agronomy12081814
Du M, Xiong M, Chang Y, Liu Z, Wang R, Lin X, Zhou Z, Lu M, Liu C, Liu E. Mining Candidate Genes and Favorable Haplotypes for Flag Leaf Shape in Rice (Oryza sativa L.) Based on a Genome-Wide Association Study. Agronomy. 2022; 12(8):1814. https://doi.org/10.3390/agronomy12081814
Chicago/Turabian StyleDu, Mingyu, Mengyuan Xiong, Yinping Chang, Zhengbo Liu, Rui Wang, Xingxing Lin, Zhenzhen Zhou, Mingwei Lu, Cuiping Liu, and Erbao Liu. 2022. "Mining Candidate Genes and Favorable Haplotypes for Flag Leaf Shape in Rice (Oryza sativa L.) Based on a Genome-Wide Association Study" Agronomy 12, no. 8: 1814. https://doi.org/10.3390/agronomy12081814
APA StyleDu, M., Xiong, M., Chang, Y., Liu, Z., Wang, R., Lin, X., Zhou, Z., Lu, M., Liu, C., & Liu, E. (2022). Mining Candidate Genes and Favorable Haplotypes for Flag Leaf Shape in Rice (Oryza sativa L.) Based on a Genome-Wide Association Study. Agronomy, 12(8), 1814. https://doi.org/10.3390/agronomy12081814