



Long-Term Organic Manure Application Alters Urease Activity and Ureolytic Microflora Structure in Agricultural Soils


Abstract
:1. Introduction
2. Materials and Methods
2.1. Experimental Site and Sampling
2.2. Soil Physicochemical Properties and Urease Activity
2.3. DNA Extraction
2.4. Quantification of ureC and 16S rRNA Genes
2.5. PCR Amplification of ureC Gene and Sequencing
2.6. Statistical Analysis
3. Results
3.1. Soil Physicochemical Properties
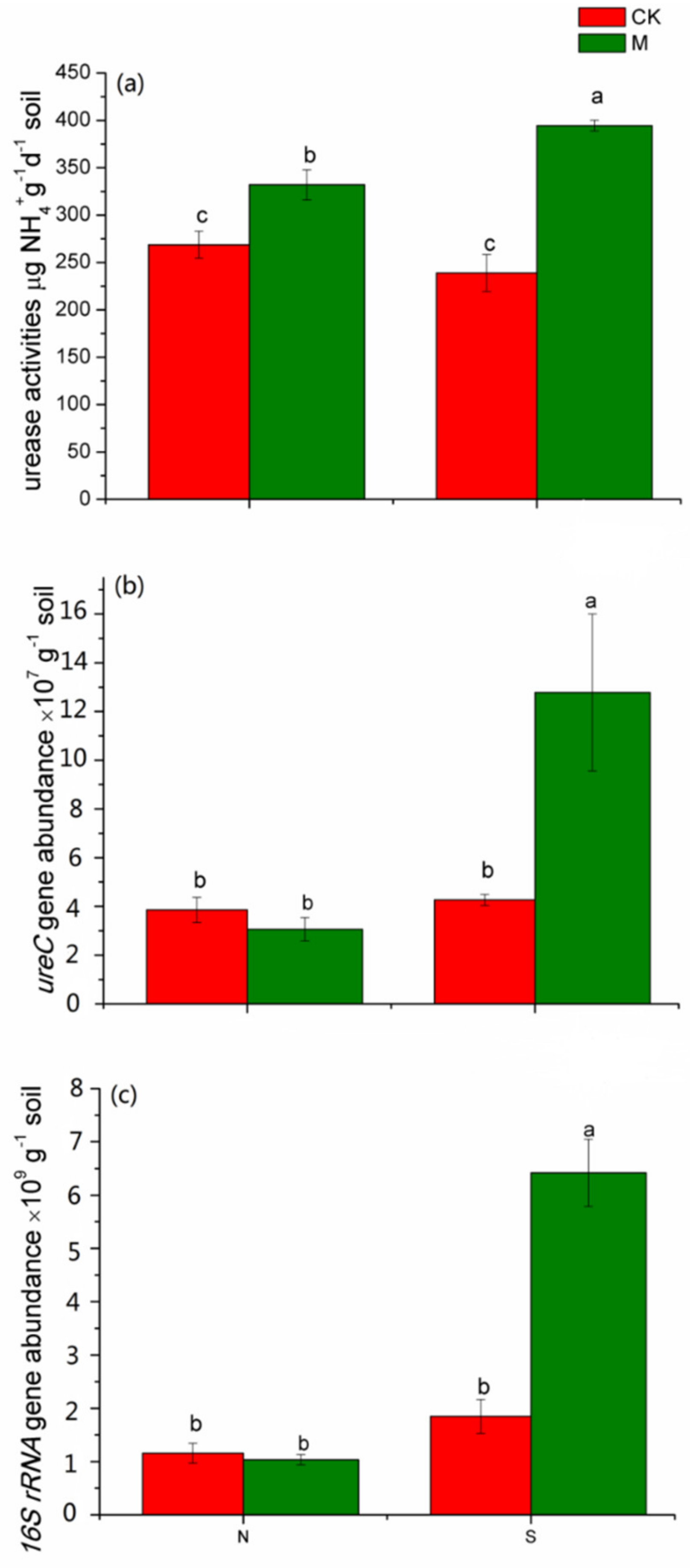
3.2. The Urease Activity and Abundance of the Ureolytic Microflora
3.3. The α-Diversity of the Ureolytic Microflora
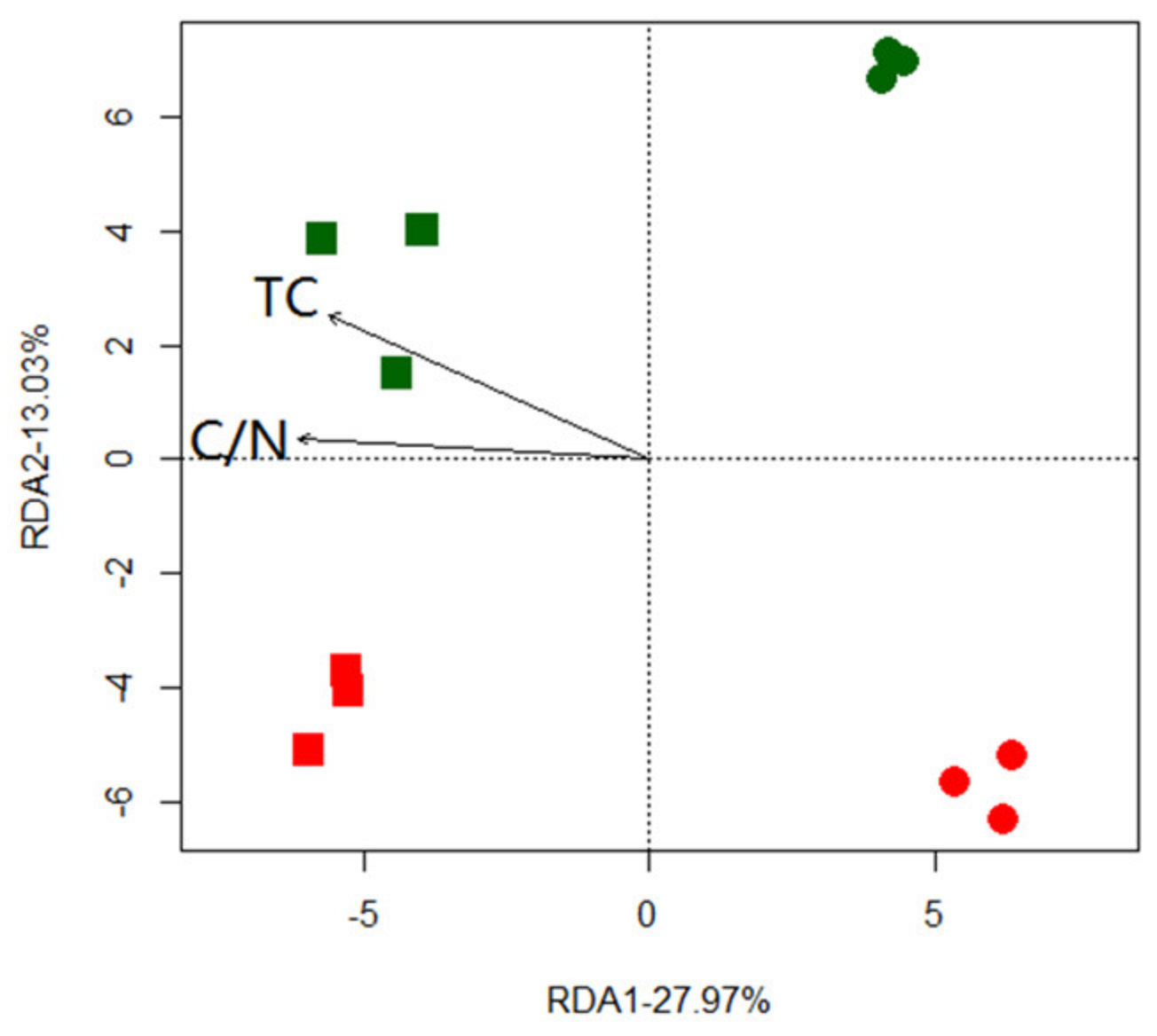
3.4. The Structure of Ureolytic Microflora and Drivers
3.5. The Composition of Ureolytic Microflora
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Compliance with Ethical Standards
References
- Vogels, G.; van der Drift, C. Degradation of purines and pyrimidines by microorganisms. Bacteriol. Rev. 1976, 40, 403–468. [Google Scholar] [CrossRef] [PubMed]
- Glibert, P.M.; Maranger, R.; Sobota, D.J. The Haber Bosch-harmful algal bloom (HB-HAB) link. Environ. Res. Lett. 2014, 9, 105001. [Google Scholar] [CrossRef] [Green Version]
- Mobley, H.L.T.; Hausinger, R.P. Microbial ureases: Significance, regulation, and molecular characterization. Microbiol. Mol. Biol. Rev. 1989, 53, 85–108. [Google Scholar] [CrossRef] [PubMed]
- Bolton, J.B.; Elliott, L.F.; Papendick, R.I.; Bezdicek, D.F. Soil microbial biomass and selected soil enzyme activities: Effect of fertilization and cropping practices. Soil Biol. Biochem. 1985, 17, 297–302. [Google Scholar] [CrossRef]
- Cetin, S.C.; Ekinci, H.; Kavdir, Y.; Yüksel, O. Using soil urease enzyme activity as soil quality indicator for reflecting fire influence in forest ecosystem. Fresenius Environ. Bull. 2009, 18, 2380–2387. [Google Scholar]
- García-Ruiz, R.; Ochoa, V.; Hinojosa, M.B.; Carreira, J.A. Suitability of enzyme activities for the monitoring of soil quality improvement in organic agricultural systems. Soil Biol. Biochem. 2008, 40, 2137–2145. [Google Scholar] [CrossRef]
- Lloyd, A.B.; Sheaffe, M.J. Urease activity in soils. Plant Soil 1973, 39, 71–80. [Google Scholar] [CrossRef]
- Reed, K.E. Restriction enzyme mapping of bacterial urease genes: Using degenerate primers to expand experimental outcomes. Biochem. Mol. Biol. Educ. 2001, 29, 239–244. [Google Scholar] [CrossRef]
- Greshan, T.L.; Sheridan, P.P.; Watwood, M.E.; Fujita, Y.; Collwell, F.S. Design and validation of ureC-based primers for groundwater detection of urea-hydrolyzing bacteria. Geomicrobiol. J. 2007, 24, 353–364. [Google Scholar] [CrossRef]
- Singh, B.K.; Nunan, N.; Millard, P. Response of fungal, bacterial and ureolytic communities to synthetic sheep urine deposition in a grassland soil. FEMS Microbiol. Ecol. 2009, 70, 109–117. [Google Scholar] [CrossRef] [Green Version]
- Collier, J.L.; Baker, K.M.; Bell, S.L. Diversity of urea-degrading microorganisms in open-ocean and estuarine planktonic communities. Environ. Microbiol. 2009, 11, 3118–3131. [Google Scholar] [CrossRef] [PubMed]
- Haynes, R.J.; Naidu, R. Influence of lime, fertilizer and manure applications on soil organic matter content and soil physical conditions: A review. Nutr. Cycl. Agroecosyst. 1998, 51, 123–137. [Google Scholar] [CrossRef]
- Kandeler, E.; Stemmer, M.; Klimanek, E.M. Response of soil microbial biomass, urease and xylanase within particle size fractions to long-term soil management. Soil Biol. Biochem. 1999, 31, 261–273. [Google Scholar] [CrossRef]
- Liang, Q.; Chen, H.; Gong, Y.; Yang, H.; Fan, M.; Kuzyakov, Y. Effects of 15 years of manure and mineral fertilizers on enzyme activities in particle-size fractions in a north China plain soil. Eur. J. Soil Biol. 2014, 60, 112–119. [Google Scholar] [CrossRef]
- Nayak, D.R.; Jagadeesh, B.Y.; Adhya, T.K. Long-term application of compost influences microbial biomass and enzyme activities in a tropical aeric endoaquept planted to rice under flooded condition. Soil Biol. Biochem. 2007, 39, 1897–1906. [Google Scholar] [CrossRef]
- Nelson, D.W.; Sommers, L.E. Total carbon, organic carbon and organic matter. In Methods of Soil Analysis; Page, A.L., Miller, R.H., Keeney, D.R., Eds.; America Society of Agronomy: Madison, WI, USA, 1982; pp. 539–579. [Google Scholar]
- Hoffmann, G. Eine photometrische Methode zur Bestimmung der Phosphatase-Aktivität in Böden. J. Plant Nutr. Soil Sci. 1968, 118, 161–172. [Google Scholar] [CrossRef]
- Griffiths, R.I.; Whitely, A.S.; O’Donnell, A.G.; Bailey, M.J. Rapid method for coextraction of DNA and RNA from natural environments for analysis of ribosomal DNA and rRNA based microbial community composition. Appl. Environ. Microbiol. 2000, 66, 5488–5491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fierer, N.; Jackson, J.A.; Vilgalys, R.; Jackson, R.B. Assessment of soil microbial community structure by use of taxon-specific quantitative PCR assays. Appl. Environ. Microbiol. 2005, 71, 4117–4120. [Google Scholar] [CrossRef] [Green Version]
- Anderson, M.J. A new method for non-parametric multivariate analysis of variance. Austral Ecol. 2001, 26, 32–46. [Google Scholar]
- MacArthur, R.; Wilson, E. The Theory of Island Biogeography; Princeton University Press: Princeton, NJ, USA, 1967. [Google Scholar]
- Meyer, O. Functional groups of microorganisms. In Biodiversity and Ecosystem Function; Schulze, E., Mooney, H., Eds.; Springer-Verlag: New York, NY, USA, 1994; pp. 67–96. [Google Scholar]
- Tate, R. Soil Microbiology; John Wiley and Sons: New York, NY, USA, 2000. [Google Scholar]
- Fazi, S.; Amalfitano, S.; Pernthaler, J.; Puddu, A. Bacterial communities associated with benthic organic matter in headwater stream microhabitats. Environ. Microbiol. 2005, 7, 1633–1640. [Google Scholar] [CrossRef]
- Kirchman, D.L. The ecology of Cytophaga–Flavobacteria in aquatic environments. FEMS Microbiol. Ecol. 2002, 39, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Simon, M.; Grossart, H.P.; Schweitzer, B.; Ploug, H. Microbial ecology of organic aggregates in aquatic ecosystems. Aquat. Microb. Ecol. 2002, 28, 175–211. [Google Scholar] [CrossRef]
- Ai, C.; Liang, G.; Sun, J.; Wang, X.; He, P.; Zhou, W.; He, X. Reduced dependence of rhizosphere microbiome on plant-derived carbon in 32-year long-term inorganic and organic fertilized soils. Soil Biol. Biochem. 2015, 80, 70–78. [Google Scholar] [CrossRef]
- Griffith, J.C.; Lee, W.G.; Orlovich, D.A.; Summerfield, T.C. Contrasting bacterial communities in two indigenous chionochloa (poaceae) grassland soils in new zealand. PLoS ONE 2017, 12, e0179652. [Google Scholar] [CrossRef] [Green Version]
- Fierer, N.; Bradford, M.A.; Jackson, R.B. Toward an ecological classification of soil bacteria. Ecology 2007, 88, 1354–1364. [Google Scholar] [CrossRef]
- Elser, J.J.; Sterner, R.W.; Galford, A.E.; Chrzanowski, T.H.; Findalay, D.L.; Mills, K.H.; Paterson, M.J.; Stainton, M.P.; Schindler, D.W. Pelagic C:N:P stoichiometry in a eutrophied lake: Responses to a whole-lake food-web manipulation. Ecosystems 2000, 3, 293–307. [Google Scholar] [CrossRef]
- Elser, J.J.; Fagan, W.F.; Denno, R.F.; Dobberfuhl, D.R.; Folarin, A.; Huberty, A.; Interlandi, S.; Kilham, S.S.; McCauley, E.; Schulz, K.L.; et al. Nutritional constrains in terrestrial and freshwater food webs. Nature 2000, 408, 578–580. [Google Scholar] [CrossRef] [PubMed]
- Elser, J.J.; Sterner, R.W.; Gorokhova, F.; Fagan, W.F.; Markov, T.A.; Cotner, J.B.; Harrison, J.F.; Hobbie, S.E.; Odell, G.M.; Weider, L.W. Biological stoichiometry from genes to ecosystem. Ecol. Lett. 2000, 5, 540–550. [Google Scholar] [CrossRef] [Green Version]
- Sterner, R.W.; Elser, J.J. Ecological Stoichiometry: The Biology of Elements from Molecules to the Biosphere; Princeton University Press: Princeton, NJ, USA, 2002. [Google Scholar]
- Delgado-Baquerizo, M.; Reich, P.B.; Khachane, A.N.; Campbell, C.D.; Thomas, N.; Freitag, T.E.; Abu Al-Soud, W.; Sørensen, S.; Bardgett, R.D.; Singh, B.K. It is elemental: Soil nutrient stoichiometry drives bacterial diversity. Environ. Microbiol. 2016, 19, 1176–1188. [Google Scholar] [CrossRef] [Green Version]
- Räsänen, N.; Kankaala, P.; Tahvanainen, T.; Akkanen, J.; Saarnio, S. Short-term effects of phosphorus addition and pH rise on bacterial utilization and biodegradation of dissolved organic carbon (DOC) from boreal mires. Aquat. Ecol. 2014, 48, 435–446. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Treatment | pH | TC | TN | C/N | NH4+-N | SOC | TP |
|---|---|---|---|---|---|---|---|
| (g/kg) | (g/kg) | (mg/kg) | (g/kg) | (mg/kg) | |||
| NCK | 6.33 ± 0.01 a | 14.49 ± 0.04 b | 1.12 ± 0.02 b | 12.93 ± 0.22 a | 5.11 ± 0.58 b | 8.19 ± 0.53 a | 385.43 ± 0.67 ab |
| NM | 6.23 ± 0.12 a | 16.73 ± 0.31 a | 1.37 ± 0.06 a | 12.23 ± 0.8 a | 4.87 ± 0.15 b | 9.56 ± 1.02 a | 465.59 ± 25.91 ab |
| SCK | 5.06 ± 0.05 b | 4.04 ± 0.28 d | 0.73 ± 0.01 c | 5.53 ± 0.43 c | 9.44 ± 2.40 a | 3.91 ± 0.42 b | 300.90 ± 9.07 c |
| SM | 6.35 ± 0.06 a | 10.67 ± 0.14 c | 1.34 ± 0.01 a | 7.78 ± 0.13 b | 10.54 ± 1.18 a | 8.27 ± 0.09 a | 1138.61 ± 95.11 a |
| Sample | No. of Reads | No. of Even Depth Reads | No. of OTUs | Chao1 | ACE | Shannon | Simpson |
|---|---|---|---|---|---|---|---|
| NCK-1 | 58,498 | 28,306 | 2855 | ||||
| NCK-2 | 82,375 | 28,281 | 2836 | ||||
| NCK-3 | 69,621 | 28,227 | 2849 | 3042 ± 21 a | 3148 ± 23 a | 9.54 ± 0.21 a | 0.994 ± 0.002 a |
| NM-1 | 62,208 | 28,275 | 2432 | ||||
| NM-2 | 56,324 | 28,576 | 3025 | ||||
| NM-3 | 105,332 | 28,135 | 2846 | 2950 ± 353 a | 3032 ± 371 a | 9.39 ± 0.41 a | 0.992 ± 0.004 ab |
| SCK-1 | 43,249 | 28,575 | 2101 | ||||
| SCK-2 | 43,883 | 28,660 | 1387 | ||||
| SCK-3 | 45,248 | 28,643 | 1965 | 1963 ± 418 b | 1945 ± 422 b | 8.25 ± 0.68 b | 0.976 ± 0.010 b |
| SM-1 | 33,378 | 28,686 | 1653 | ||||
| SM-2 | 35,007 | 28,759 | 1666 | ||||
| SM-3 | 32,167 | 28,777 | 1684 | 1670 ± 16 b | 1670 ± 16 b | 8.73 ± 0.23 ab | 0.987 ± 0.007 ab |
| Treatment | Betaproteobacteria | Alphaproteobacteria | Gammaproteobacteria | Deltaproteobacteria | Actinobacteria | Nitrospirae | Verrucomicrobia | Bacteroidetes |
|---|---|---|---|---|---|---|---|---|
| NCK | 42.3 ± 3.2% a | 13.0 ± 2.0% b | 12.7 ± 2.1% a | 11.3 ± 2.3% a | 5.0 ± 1.0% c | 5.7 ± 2.5% a | 4.0 ± 1.0% a | 3.0 ± 1.7% ab |
| NM | 36.0 ± 2.6% a | 13.3 ± 2.9% b | 14.0 ± 2.0% a | 10.0 ± 2.0% a | 4.7 ± 1.5% c | 8.7 ± 3.1% a | 4.3 ± 1.2% a | 4.7 ± 2.9% a |
| SCK | 24.7 ± 6.8% b | 38.0 ± 8.7% a | 9.0 ± 1.0% b | 6.3 ± 0.6% b | 18.0 ± 3.5% a | 0.7 ± 0.6% b | 2.3 ± 1.5% a | 0.3 ± 0.6% b |
| SM | 32.3 ± 3.2% ab | 34.3 ± 3.2% a | 7.3 ± 0.6% b | 13.0 ± 1.7% a | 10.0 ± 1.0% b | 0.0 ± 0.0% b | 2.0 ± 0.0% a | 0.0 ± 0.0% b |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, L.; Xiong, X. Long-Term Organic Manure Application Alters Urease Activity and Ureolytic Microflora Structure in Agricultural Soils. Agronomy 2022, 12, 3018. https://doi.org/10.3390/agronomy12123018
Wang L, Xiong X. Long-Term Organic Manure Application Alters Urease Activity and Ureolytic Microflora Structure in Agricultural Soils. Agronomy. 2022; 12(12):3018. https://doi.org/10.3390/agronomy12123018
Chicago/Turabian StyleWang, Li, and Xiang Xiong. 2022. "Long-Term Organic Manure Application Alters Urease Activity and Ureolytic Microflora Structure in Agricultural Soils" Agronomy 12, no. 12: 3018. https://doi.org/10.3390/agronomy12123018
APA StyleWang, L., & Xiong, X. (2022). Long-Term Organic Manure Application Alters Urease Activity and Ureolytic Microflora Structure in Agricultural Soils. Agronomy, 12(12), 3018. https://doi.org/10.3390/agronomy12123018