
DArTseq Genotypic and Phenotypic Diversity of Barley Landraces Originating from Different Countries




Abstract
:1. Introduction
2. Materials and Methods
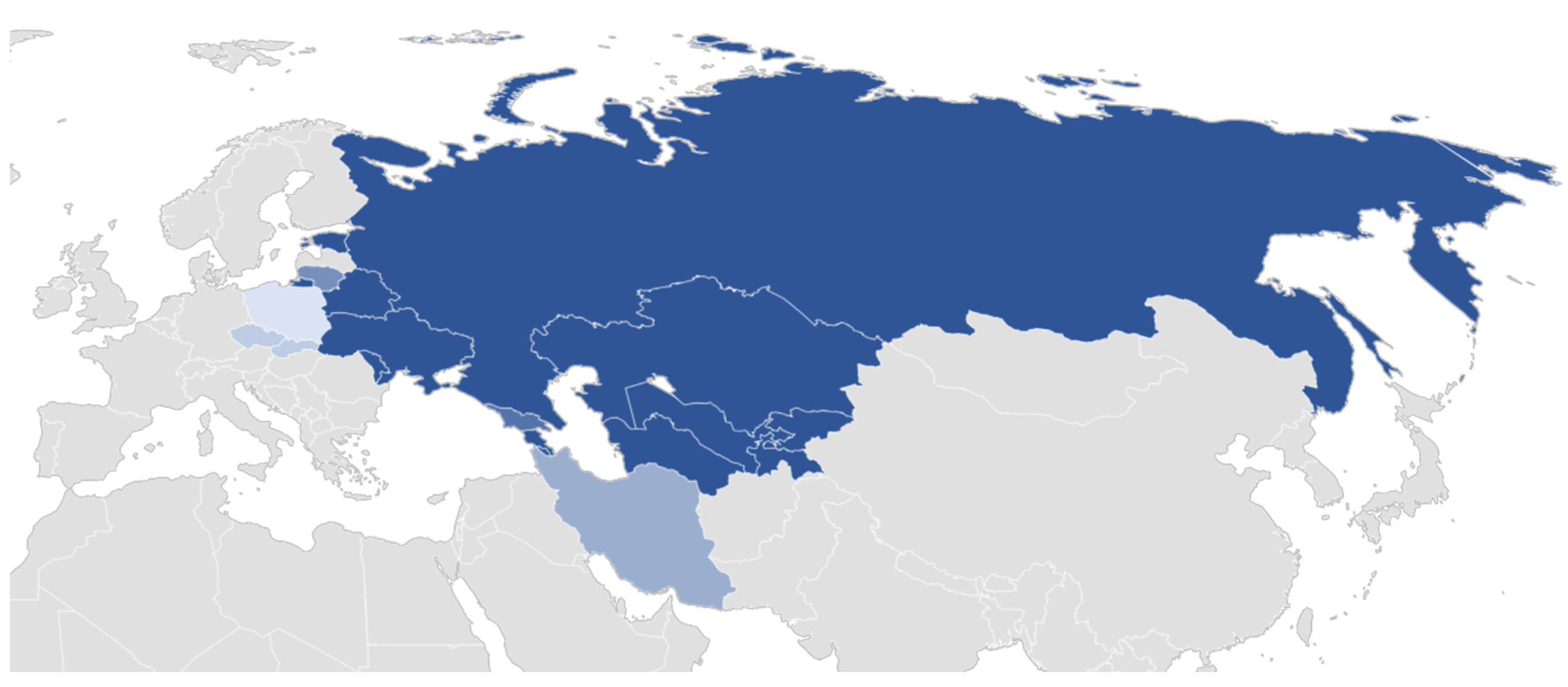
2.1. Plant Material
2.2. Agro-Morphological Features
2.3. DNA Isolation
2.4. DArTseq Genotyping
2.5. ISSR Analysis
2.6. Marker Data Analysis
3. Results
3.1. Agro-Morphological Diversity
3.2. Genetic Analysis
3.2.1. Data Quality Analysis
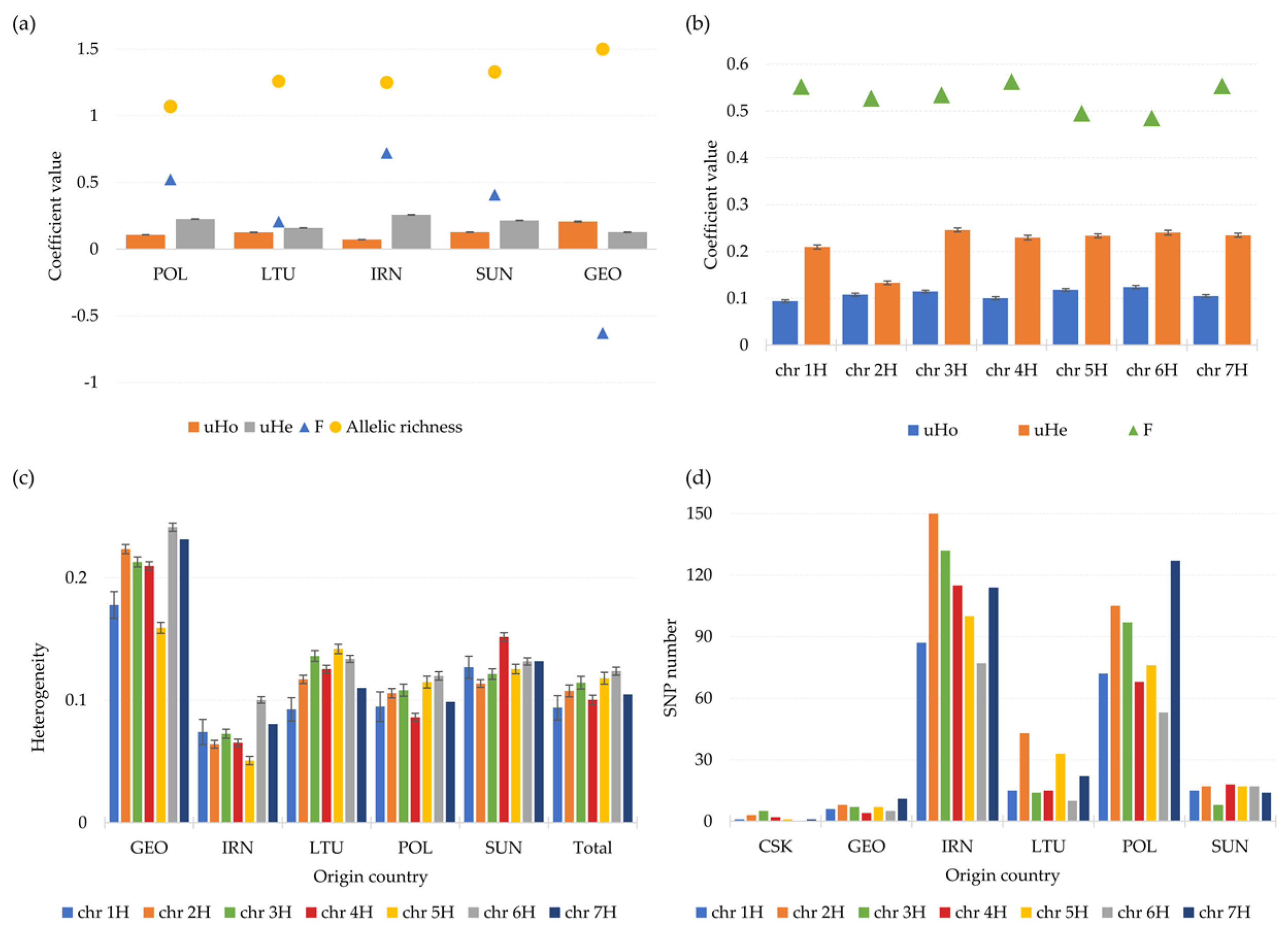
3.2.2. Genetic Diversity
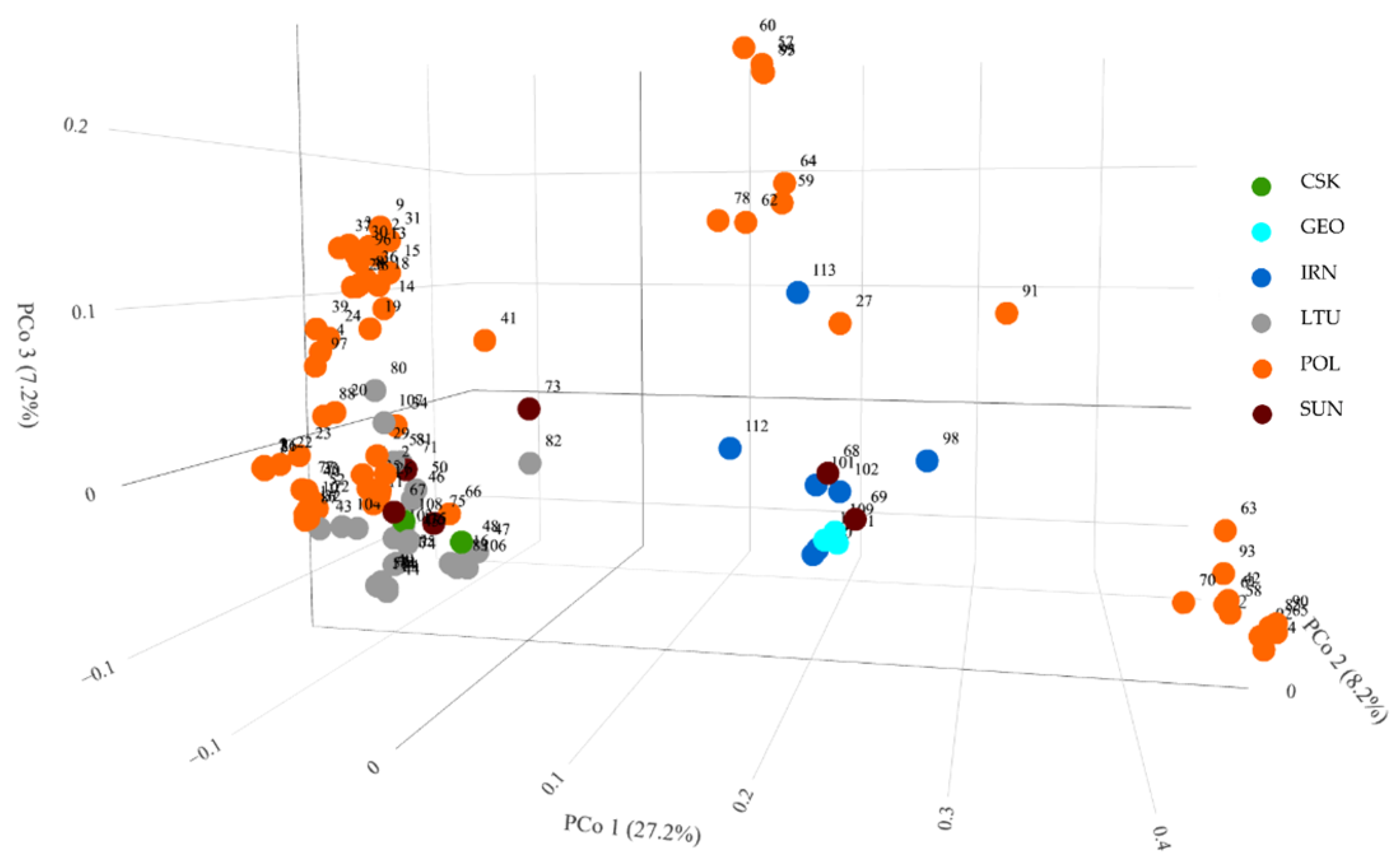
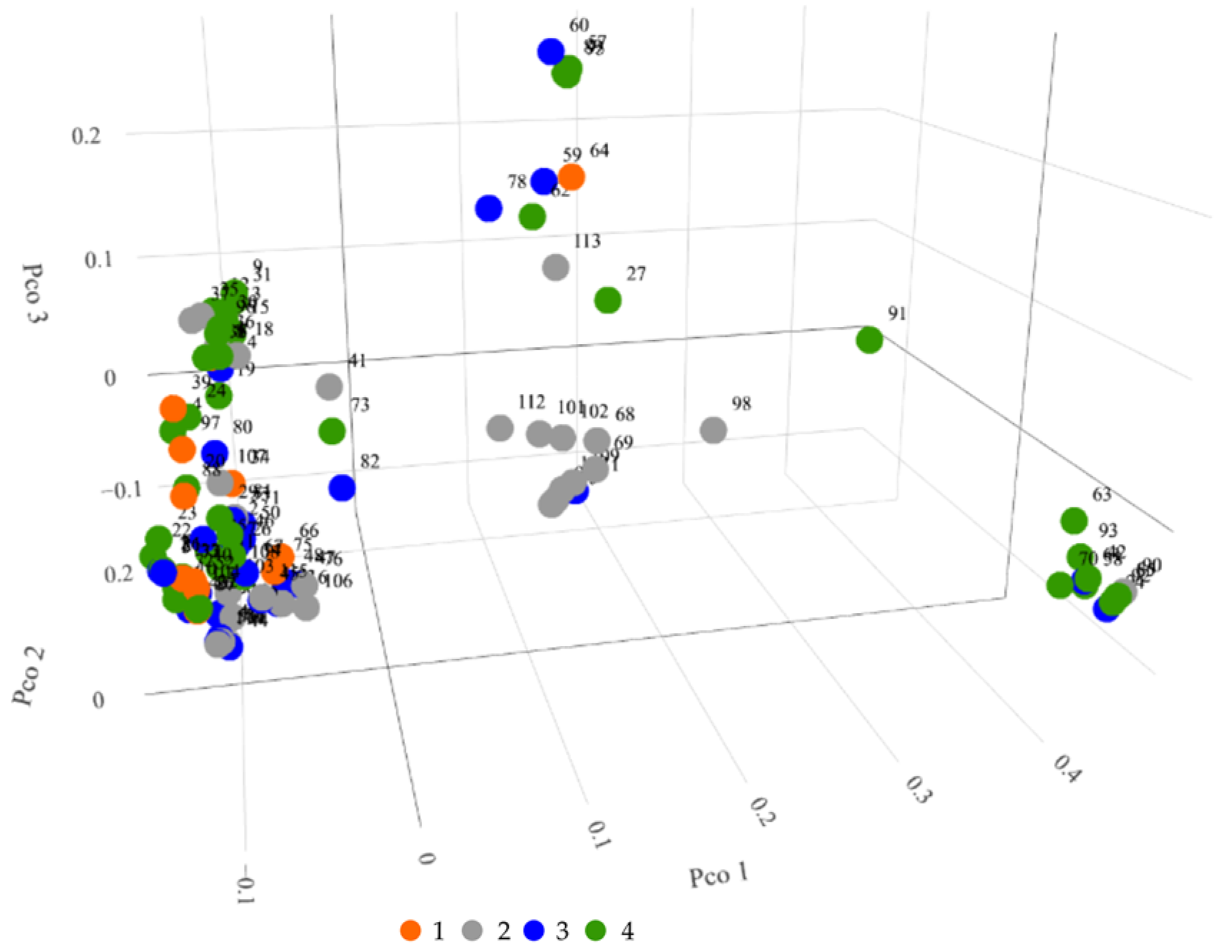
3.2.3. Principal Coordinate Analysis
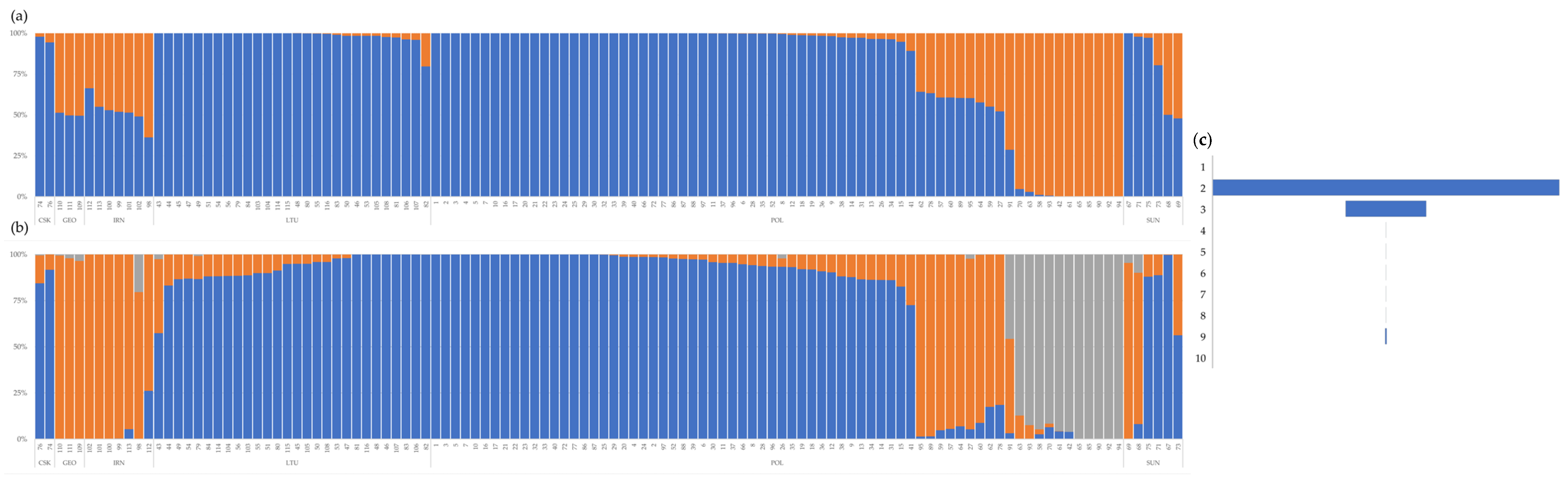
3.2.4. Population Structure
3.3. Joint Analysis
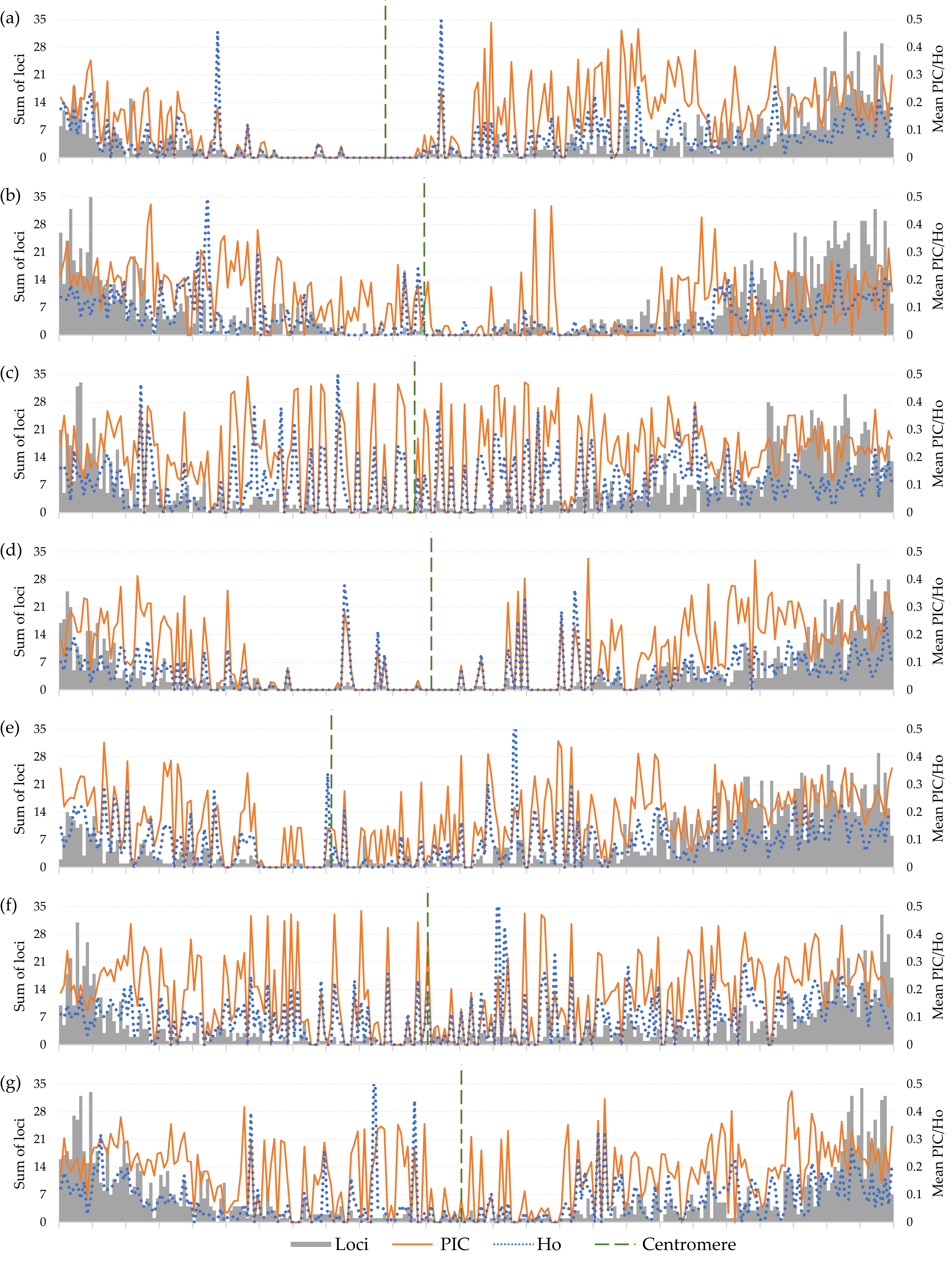
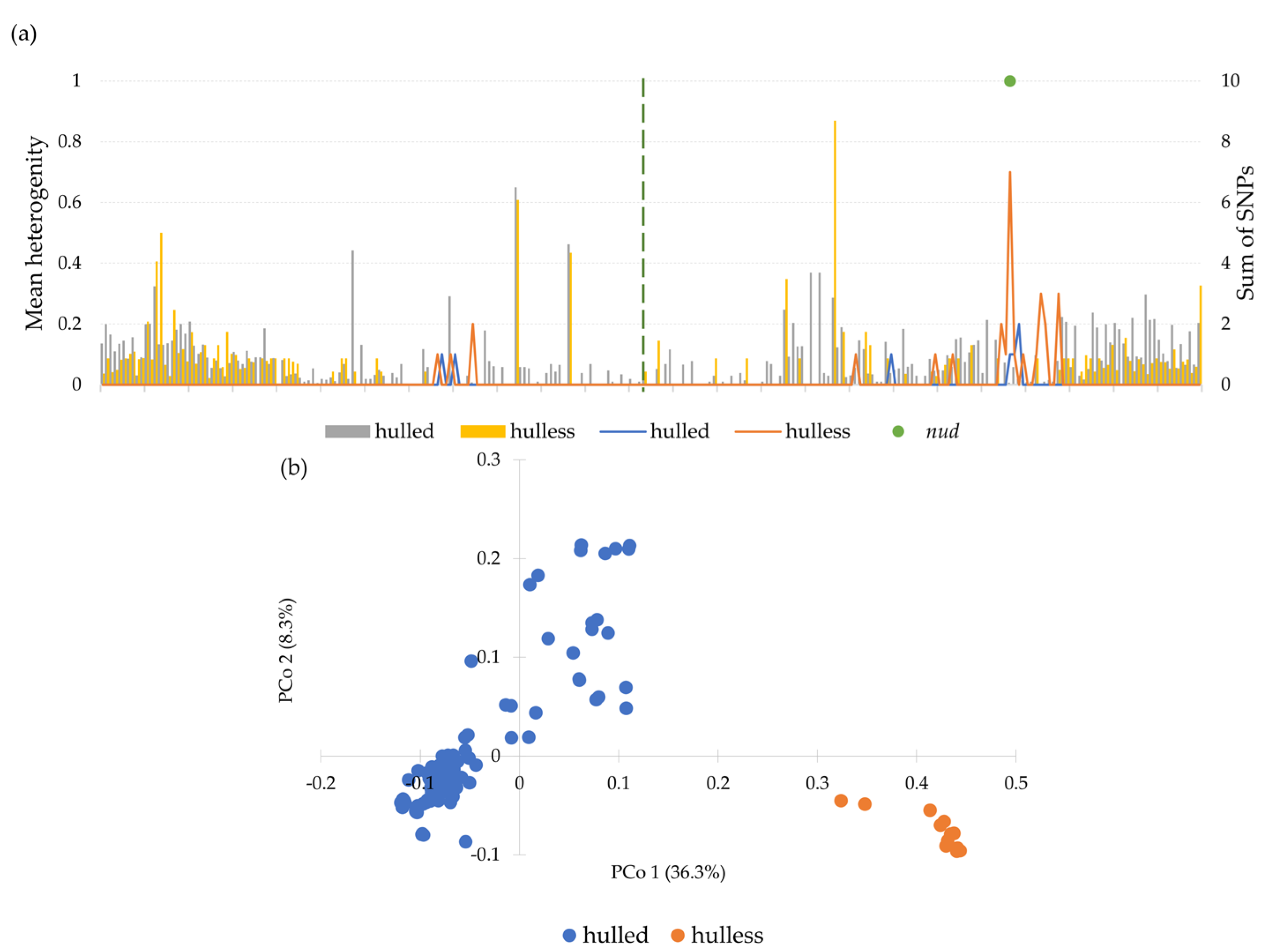
3.4. Diversity of Chromosome 7H
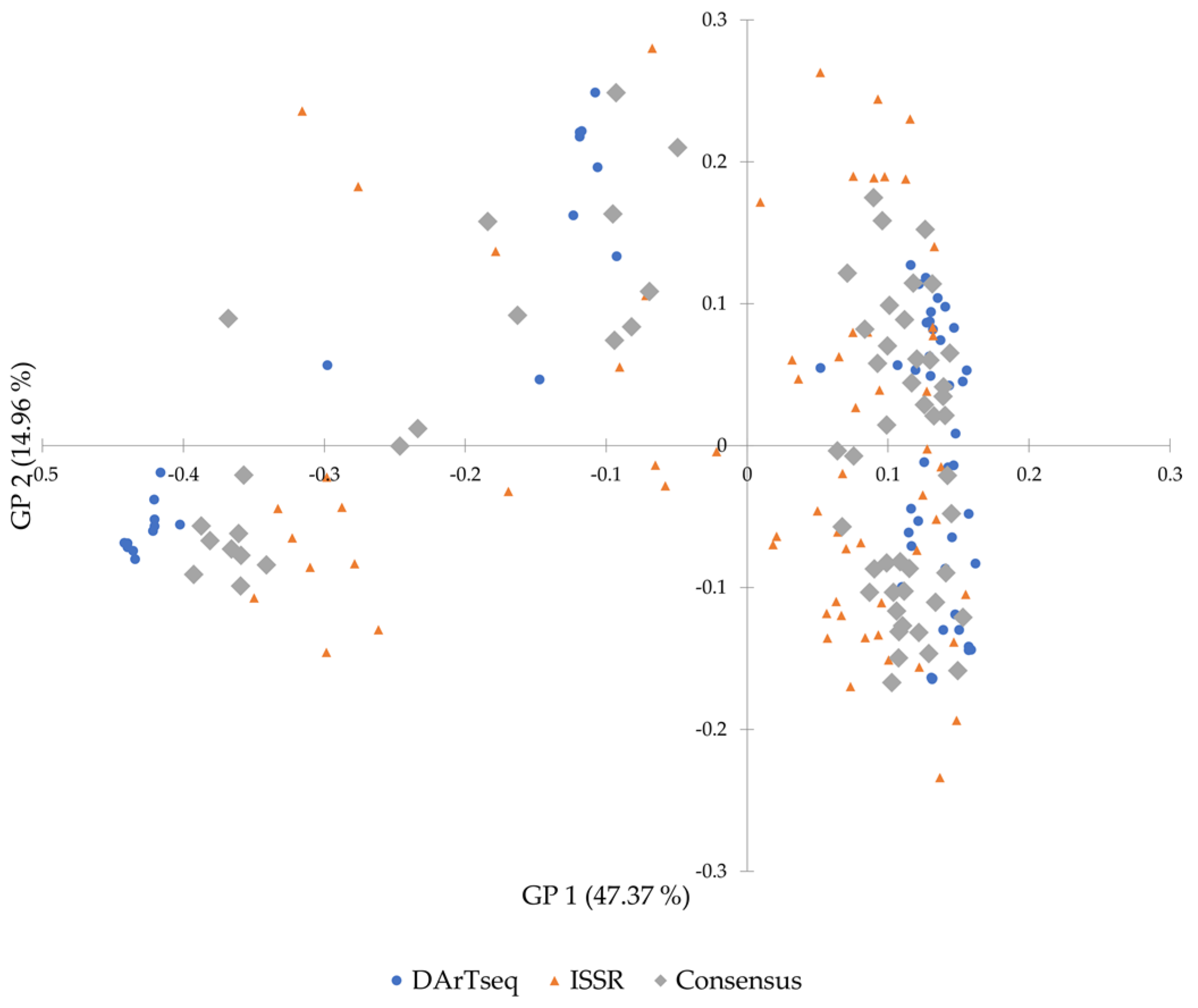
3.5. Comparison between DArTseq and ISSR Data
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Veteläinen, M.; Negri, V.; Maxted, N. European Landraces: On-Farm Conservation, Management and Use; Bioversity International: Rome, Italy, 2009; pp. 1–22. [Google Scholar]
- Marone, D.; Russo, M.A.; Mores, A.; Ficco, D.; Laidò, G.; Mastrangelo, A.M.; Borrelli, G.M. Importance of Landraces in Cereal Breeding for Stress Tolerance. Plants 2021, 10, 1267. [Google Scholar] [CrossRef] [PubMed]
- FAO. Voluntary Guidelines for the Conservation and Sustainable Use of Farmers’ Varieties/Landraces; FAO: Rome, Italy, 2017. [Google Scholar]
- Maxted, N.; Dulloo, M.E.; Ford-Lloyd, B.V. Enhancing Crop Genepool Use: Capturing Wild Relative and Landrace Diversity for Crop Improvement; CABI: Boston, MA, USA, 2016. [Google Scholar]
- Doebley, J.F.; Gaut, B.S.; Smith, B.D. The molecular genetics of crop domestication. Cell 2006, 127, 1309–1321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kilian, B.; Özkan, H.; Kohl, J.; von Haeseler, A.; Barale, F.; Deusch, O.; Brandolini, A.; Yucel, C.; Martin, W.; Salamini, F. Haplotype structure at seven barley genes: Relevance to gene pool bottlenecks, phylogeny of ear type and site of barley domestication. Mol. Genet. Genom. 2006, 276, 230–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boczkowska, M.; Łapiński, B.; Kordulasińska, I.; Dostatny, D.F.; Czembor, J.H. Promoting the use of common oat genetic resources through diversity analysis and core collection construction. PLoS ONE 2016, 11, e0167855. [Google Scholar]
- Ellis, R.P.; Forster, B.P.; Robinson, D.; Handley, L.; Gordon, D.C.; Russell, J.R.; Powell, W. Wild barley: A source of genes for crop improvement in the 21st century? J. Exp. Bot. 2000, 51, 9–17. [Google Scholar] [CrossRef]
- Sharma, S.; Upadhyaya, H.D.; Varshney, R.K.; Gowda, C. Pre-breeding for diversification of primary gene pool and genetic enhancement of grain legumes. Front. Plant Sci. 2013, 4, 309. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Verma, R.P.S.; Singh, A.; Sharma, H.K.; Devi, G. Barley landraces: Ecological heritage for edaphic stress adaptations and sustainable production. Environ. Sustain. Indic. 2020, 6, 100035. [Google Scholar] [CrossRef]
- Pasam, R.K.; Sharma, R.; Walther, A.; Özkan, H.; Graner, A.; Kilian, B. Genetic diversity and population structure in a legacy collection of spring barley landraces adapted to a wide range of climates. PLoS ONE 2014, 9, e116164. [Google Scholar] [CrossRef] [Green Version]
- Ceccarelli, S. Positive interpretation of genotype by environment interactions in relation to sustainability and biodiversity. In Plant Adaptation and Crop Improvement; IRRI: Manila, Philippines, 1996; pp. 467–486. [Google Scholar]
- Grausgruber, H.; Bointner, H.; Tumpold, R.; Ruckenbauer, P.; Fischbeck, G. Genetic improvement of agronomic and qualitative traits of spring barley. Plant Breed. 2002, 121, 411–416. [Google Scholar] [CrossRef]
- Rasmusson, D.; Phillips, R. Plant breeding progress and genetic diversity from de novo variation and elevated epistasis. Crop Sci. 1997, 37, 303–310. [Google Scholar] [CrossRef]
- Okada, Y.; Kanatani, R.; Arai, S.; Ito, K. Interaction between barley yellow mosaic disease-resistance genes rym1 and rym5, in the response to BaYMV strains. Breed. Sci. 2004, 54, 319–325. [Google Scholar] [CrossRef] [Green Version]
- Steffenson, B.J. Analysis of durable resistance to stem rust in barley. Euphytica 1992, 63, 153–167. [Google Scholar] [CrossRef]
- Bregitzer, P.; Mornhinweg, D.; Hammon, R.; Stack, M.; Baltensperger, D.; Hein, G.; O’neill, M.; Whitmore, J.; Fiedler, D. Registration of ‘Burton’ barley. Crop Sci. 2005, 45, 1166–1168. [Google Scholar] [CrossRef]
- Burnett, P.; Comeau, A.; Qualset, C. Host plant tolerance or resistance for control of barley yellow dwarf. In Barley Yellow Dwarf: 40 Years of Progress; D’Arcy, C.J., Burnett, P.A., Eds.; APS Press: Saint Paul, MN, USA, 1995; pp. 321–343. [Google Scholar]
- Piffanelli, P.; Ramsay, L.; Waugh, R.; Benabdelmouna, A.; D’Hont, A.; Hollricher, K.; Jørgensen, J.H.; Schulze-Lefert, P.; Panstruga, R. A barley cultivation-associated polymorphism conveys resistance to powdery mildew. Nature 2004, 430, 887–891. [Google Scholar] [CrossRef] [Green Version]
- Dawson, I.K.; Russell, J.; Powell, W.; Steffenson, B.; Thomas, W.T.; Waugh, R. Barley: A translational model for adaptation to climate change. New Phytol. 2015, 206, 913–931. [Google Scholar] [CrossRef] [PubMed]
- Pietrusińska, A.; Żurek, M.; Piechota, U.; Słowacki, P.; Smolińska, K. Searching for diseases resistance sources in old cultivars, landraces and wild relatives of cereals. A review. Agron. Sci. 2018, 73, 45–60. [Google Scholar] [CrossRef]
- Hernandez, J.; Meints, B.; Hayes, P. Introgression breeding in barley: Perspectives and case studies. Front. Plant Sci. 2020, 11, 761. [Google Scholar] [CrossRef]
- Jaradat, A.; Shahid, M. Population and multilocus isozyme structures in a barley landrace. Plant Genet. Resour. 2006, 4, 108–116. [Google Scholar] [CrossRef] [Green Version]
- Bjørnstad, A.; Demissie, A.; Kilian, A.; Kleinhofs, A. The distinctness and diversity of Ethiopian barleys. Theor. Appl. Genet. 1997, 94, 514. [Google Scholar] [CrossRef]
- Demissie, A.; Bjørnstad, Å.; Kleinhofs, A. Restriction Fragment Length Polymorphisms in Landrace Barleys from Ethiopia in Relation to Geographic, Altitude, and Agro-Ecological Factors. Crop Sci. 1998, 38, 237–243. [Google Scholar] [CrossRef]
- Manjunatha, T.; Bisht, I.; Bhat, K.; Singh, B. Genetic diversity in barley (Hordeum vulgare L. ssp. vulgare) landraces from Uttaranchal Himalaya of India. Genet. Resour. Crop Evol. 2007, 54, 55–65. [Google Scholar] [CrossRef]
- Abdellaoui, R.; M’Hamed, H.C.; Naceur, M.B.; Bettaieb-Kaab, L.; Hamida, J.B. Morpho-physiological and molecular characterization of some Tunisian barley ecotypes. Asian J. Plant Sci. 2007, 6, 26–268. [Google Scholar] [CrossRef]
- Yahiaoui, S.; Igartua, E.; Moralejo, M.; Ramsay, L.; Molina-Cano, J.L.; Lasa, J.; Gracia, M.; Casas, A. Patterns of genetic and eco-geographical diversity in Spanish barleys. Theor. Appl. Genet. 2008, 116, 271–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, Z.-Y.; Zhang, L.-L.; Zhang, Y.-Z.; Ling, H.-Q. Genetic diversity and geographical differentiation of cultivated six-rowed naked barley landraces from the Qinghai-Tibet plateau of China detected by SSR analysis. Genet. Mol. Biol. 2006, 29, 330–338. [Google Scholar] [CrossRef] [Green Version]
- Hamza, S.; Hamida, W.B.; Rebaï, A.; Harrabi, M. SSR-based genetic diversity assessment among Tunisian winter barley and relationship with morphological traits. Euphytica 2004, 135, 107–118. [Google Scholar] [CrossRef]
- Van Treuren, R.; Tchoudinova, I.; Van Soest, L.; van Hintum, T.J. Marker-assisted acquisition and core collection formation: A case study in barley using AFLPs and pedigree data. Genet. Resour. Crop Evol. 2006, 53, 43–52. [Google Scholar] [CrossRef] [Green Version]
- Dziurdziak, J.; Bolc, P.; Wlodarczyk, S.; Puchta, M.; Gryziak, G.; Podyma, W.; Boczkowska, M. Multifaceted Analysis of Barley Landraces Collected during Gene Bank Expeditions in Poland at the End of the 20th Century. Agronomy 2020, 10, 1958. [Google Scholar] [CrossRef]
- Varshney, R.; Baum, M.; Guo, P.; Grando, S.; Ceccarelli, S.; Graner, A. Features of SNP and SSR diversity in a set of ICARDA barley germplasm collection. Mol. Breed. 2010, 26, 229–242. [Google Scholar] [CrossRef]
- Varshney, R.; Thiel, T.; Sretenovic-Rajicic, T.; Baum, M.; Valkoun, J.; Guo, P.; Grando, S.; Ceccarelli, S.; Graner, A. Identification and validation of a core set of informative genic SSR and SNP markers for assaying functional diversity in barley. Mol. Breed. 2008, 22, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Russell, J.R.; Ellis, R.P.; Thomas, W.T.; Waugh, R.; Provan, J.; Booth, A.; Fuller, J.; Lawrence, P.; Young, G.; Powell, W. A retrospective analysis of spring barley germplasm development fromfoundation genotypes’ to currently successful cultivars. Mol. Breed. 2000, 6, 553–568. [Google Scholar] [CrossRef]
- Nandha, P.S.; Singh, J. Comparative assessment of genetic diversity between wild and cultivated barley using g SSR and EST-SSR markers. Plant Breed. 2014, 133, 28–35. [Google Scholar] [CrossRef]
- Mohammadi, S.A.; Sisi, N.A.; Sadeghzadeh, B. The influence of breeding history, origin and growth type on population structure of barley as revealed by SSR markers. Sci. Rep. 2020, 10, 19165. [Google Scholar] [CrossRef]
- Lister, D.L.; Jones, H.; Jones, M.K.; O’Sullivan, D.M.; Cockram, J. Analysis of DNA polymorphism in ancient barley herbarium material: Validation of the KASP SNP genotyping platform. Taxon 2013, 62, 779–789. [Google Scholar] [CrossRef]
- Close, T.J.; Bhat, P.R.; Lonardi, S.; Wu, Y.; Rostoks, N.; Ramsay, L.; Druka, A.; Stein, N.; Svensson, J.T.; Wanamaker, S. Development and implementation of high-throughput SNP genotyping in barley. BMC Genom. 2009, 10, 582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bayer, M.M.; Rapazote-Flores, P.; Ganal, M.; Hedley, P.E.; Macaulay, M.; Plieske, J.; Ramsay, L.; Russell, J.; Shaw, P.D.; Thomas, W. Development and evaluation of a barley 50k iSelect SNP array. Front. Plant Sci. 2017, 8, 1792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Comadran, J.; Kilian, B.; Russell, J.; Ramsay, L.; Stein, N.; Ganal, M.; Shaw, P.; Bayer, M.; Thomas, W.; Marshall, D. Natural variation in a homolog of Antirrhinum CENTRORADIALIS contributed to spring growth habit and environmental adaptation in cultivated barley. Nat. Genet. 2012, 44, 1388–1392. [Google Scholar] [CrossRef]
- Torkamaneh, D.; Laroche, J.; Bastien, M.; Abed, A.; Belzile, F. Fast-GBS: A new pipeline for the efficient and highly accurate calling of SNPs from genotyping-by-sequencing data. BMC Bioinform. 2017, 18, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milner, S.G.; Jost, M.; Taketa, S.; Mazón, E.R.; Himmelbach, A.; Oppermann, M.; Weise, S.; Knüpffer, H.; Basterrechea, M.; König, P. Genebank genomics highlights the diversity of a global barley collection. Nat. Genet. 2019, 51, 319–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wenzl, P.; Carling, J.; Kudrna, D.; Jaccoud, D.; Huttner, E.; Kleinhofs, A.; Kilian, A. Diversity Arrays Technology (DArT) for whole-genome profiling of barley. Proc. Natl. Acad. Sci. USA 2004, 101, 9915–9920. [Google Scholar] [CrossRef] [Green Version]
- Bengtsson, T.; Manninen, O.; Jahoor, A.; Orabi, J. Genetic diversity, population structure and linkage disequilibrium in Nordic spring barley (Hordeum vulgare L. subsp. vulgare). Genet. Resour. Crop Evol. 2017, 64, 2021–2033. [Google Scholar] [CrossRef] [Green Version]
- Moragues, M.; Comadran, J.; Waugh, R.; Milne, I.; Flavell, A.; Russell, J.R. Effects of ascertainment bias and marker number on estimations of barley diversity from high-throughput SNP genotype data. Theor. Appl. Genet. 2010, 120, 1525–1534. [Google Scholar] [CrossRef] [PubMed]
- NCPGR. EGISET. Available online: https://wyszukiwarka.ihar.edu.pl/pl (accessed on 15 August 2021).
- Doyle, J.J.; Doyle, J.L. Isolation ofplant DNA from fresh tissue. Focus 1990, 12, 39–40. [Google Scholar]
- Edwards, K.; Johnstone, C.; Thompson, C. A simple and rapid method for the preparation of plant genomic DNA for PCR analysis. Nucleic Acids Res. 1991, 19, 1349. [Google Scholar] [CrossRef] [PubMed]
- Wójcik-Jagła, M.; Rapacz, M.; Barcik, W.; Janowiak, F. Differential regulation of barley (Hordeum distichon) HVA1 and SRG6 transcript accumulation during the induction of soil and leaf water deficit. Acta Physiol. Plant. 2012, 34, 2069–2078. [Google Scholar] [CrossRef] [Green Version]
- Mascher, M.; Gundlach, H.; Himmelbach, A.; Beier, S.; Twardziok, S.O.; Wicker, T.; Radchuk, V.; Dockter, C.; Hedley, P.E.; Russell, J. A chromosome conformation capture ordered sequence of the barley genome. Nature 2017, 544, 427–433. [Google Scholar] [CrossRef] [Green Version]
- Leberg, P. Estimating allelic richness: Effects of sample size and bottlenecks. Mol. Ecol. 2002, 11, 2445–2449. [Google Scholar] [CrossRef]
- Hubisz, M.J.; Falush, D.; Stephens, M.; Pritchard, J.K. Inferring weak population structure with the assistance of sample group information. Mol. Ecol. Resour. 2009, 9, 1322–1332. [Google Scholar] [CrossRef] [Green Version]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the Number of Clusters of Individuals Using the Software Structure: A Simulation Study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [Green Version]
- Gower, J.C. Generalized procrustes analysis. Psychometrika 1975, 40, 33–51. [Google Scholar] [CrossRef]
- Peakall, R.; Smouse, P.E. Genalex 6: Genetic analysis in Excel. Population genetic software for teaching and research. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef] [Green Version]
- Kalinowski, S.T. hp-rare 1.0: A computer program for performing rarefaction on measures of allelic richness. Mol. Ecol. Notes 2005, 5, 187–189. [Google Scholar] [CrossRef]
- Kopelman, N.M.; Mayzel, J.; Jakobsson, M.; Rosenberg, N.A.; Mayrose, I. Clumpak: A program for identifying clustering modes and packaging population structure inferences across K. Mol. Ecol. Resour. 2015, 15, 1179–1191. [Google Scholar] [CrossRef] [Green Version]
- Zietkiewicz, E.; Rafalski, A.; Labuda, D. Genome fingerprinting by simple sequence repeat (SSR)-anchored polymerase chain reaction amplification. Genomics 1994, 20, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Villa, T.C.C.; Maxted, N.; Scholten, M.; Ford-Lloyd, B. Defining and identifying crop landraces. Plant Genet. Resour. 2005, 3, 373–384. [Google Scholar] [CrossRef] [Green Version]
- Podyma, W.; Boczkowska, M.; Wolko, B.; Dostatny, D.F. Morphological, isoenzymatic and ISSRs-based description of diversity of eight sand oat (Avena strigosa Schreb.) landraces. Genet. Resour. Crop Evol. 2017, 64, 1661–1674. [Google Scholar] [CrossRef]
- Ávila, C.M.; Requena-Ramírez, M.D.; Rodríguez-Suárez, C.; Flores, F.; Sillero, J.C.; Atienza, S.G. Genome-Wide Association Analysis for Stem Cross Section Properties, Height and Heading Date in a Collection of Spanish Durum Wheat Landraces. Plants 2021, 10, 1123. [Google Scholar] [CrossRef]
- Hahn, V.; Würschum, T. Molecular genetic characterization of Central European soybean breeding germplasm. Plant Breed. 2014, 133, 748–755. [Google Scholar] [CrossRef]
- Maccaferri, M.; Harris, N.S.; Twardziok, S.O.; Pasam, R.K.; Gundlach, H.; Spannagl, M.; Ormanbekova, D.; Lux, T.; Prade, V.M.; Milner, S.G. Durum wheat genome highlights past domestication signatures and future improvement targets. Nat. Genet. 2019, 51, 885–895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bustos-Korts, D.; Dawson, I.K.; Russell, J.; Tondelli, A.; Guerra, D.; Ferrandi, C.; Strozzi, F.; Nicolazzi, E.L.; Molnar-Lang, M.; Ozkan, H. Exome sequences and multi-environment field trials elucidate the genetic basis of adaptation in barley. Plant J. 2019, 99, 1172–1191. [Google Scholar] [CrossRef] [Green Version]
- Amezrou, R.; Gyawali, S.; Belqadi, L.; Chao, S.; Arbaoui, M.; Mamidi, S.; Rehman, S.; Sreedasyam, A.; Verma, R.P.S. Molecular and phenotypic diversity of ICARDA spring barley (Hordeum vulgare L.) collection. Genet. Resour. Crop Evol. 2018, 65, 255–269. [Google Scholar] [CrossRef]
- Chen, F.; Chen, D.; Vallés, M.-P.; Gao, Z.; Chen, X. Analysis of diversity in Chinese cultivated barley with simple sequence repeats: Differences between eco-geographic populations. Biochem. Genet. 2010, 48, 44–56. [Google Scholar] [CrossRef]
- Lasa, J.; Igartua, E.; Ciudad, F.; Codesal, P.; Garcíaa, E.; Gracia, M.; Medina, B.; Romagosa, I.; Molina-Cano, J.; Montoya, J. Morphological and agronomical diversity patterns in the Spanish barley core collection. Hereditas 2001, 135, 217–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zohary, D.; Hopf, M.; Weiss, E. Domestication of Plants in the Old World: The Origin and Spread of Domesticated Plants in Southwest Asia, Europe, and the Mediterranean Basin; Oxford University Press on Demand: Oxford, UK, 2012. [Google Scholar]
- Taketa, S.; Kikuchi, S.; Awayama, T.; Yamamoto, S.; Ichii, M.; Kawasaki, S. Monophyletic origin of naked barley inferred from molecular analyses of a marker closely linked to the naked caryopsis gene (nud). Theor. Appl. Genet. 2004, 108, 1236–1242. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Guo, Y.; Xu, Q.; Mascher, M.; Guo, G.; Li, S.; Mao, L.; Liu, Q.; Xia, Z.; Zhou, J. Origin and evolution of qingke barley in Tibet. Nat. Commun. 2018, 9, 5433. [Google Scholar] [CrossRef]
- Kikuchi, S.; Taketa, S.; Ichii, M.; Kawasaki, S. Efficient fine mapping of the naked caryopsis gene (nud) by HEGS (High Efficiency Genome Scanning)/AFLP in barley. Theor. Appl. Genet. 2003, 108, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Taketa, S.; Amano, S.; Tsujino, Y.; Sato, T.; Saisho, D.; Kakeda, K.; Nomura, M.; Suzuki, T.; Matsumoto, T.; Sato, K. Barley grain with adhering hulls is controlled by an ERF family transcription factor gene regulating a lipid biosynthesis pathway. Proc. Natl. Acad. Sci. USA 2008, 105, 4062–4067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, S.; Long, H.; Deng, G.; Pan, Z.; Liang, J.; Zeng, X.; Tang, Y.; Tashi, N.; Yu, M. A single nucleotide polymorphism of Nud converts the caryopsis type of barley (Hordeum vulgare L.). Plant Mol. Biol. Report. 2016, 34, 242–248. [Google Scholar] [CrossRef] [Green Version]
- Boros, D.; Rek-Cieply, B.; Cyran, M. A note on the composition and nutritional value of hulless barley. J. Anim. Feed Sci. 1996, 5, 417–424. [Google Scholar] [CrossRef] [Green Version]
- Brennan, C.S. Dietary fibre, glycaemic response, and diabetes. Mol. Nutr. Food Res. 2005, 49, 560–570. [Google Scholar] [CrossRef]
- Jood, S.; Kalra, S. Chemical composition and nutritional characteristics of some hull less and hulled barley cultivars grown in India. Food/Nahr. 2001, 45, 35–39. [Google Scholar] [CrossRef]
- Owen, B.; Farmer, M.; Sosuski, F.; Wu, K. Variation in mineral content of Saskatchewan feed grains. Can. J. Anim. Sci. 1977, 57, 679–687. [Google Scholar] [CrossRef] [Green Version]
- Yadav, R.; Gautam, S.; Palikhey, E.; Joshi, B.; Ghimire, K.; Gurung, R.; Adhikari, A.; Pudasaini, N.; Dhakal, R. Agro-morphological diversity of Nepalese naked barley landraces. Agric. Food Secur. 2018, 7, 86. [Google Scholar] [CrossRef]
- Muñoz-Amatriaín, M.; Cuesta-Marcos, A.; Endelman, J.B.; Comadran, J.; Bonman, J.M.; Bockelman, H.E.; Chao, S.; Russell, J.; Waugh, R.; Hayes, P.M. The USDA barley core collection: Genetic diversity, population structure, and potential for genome-wide association studies. PLoS ONE 2014, 9, e94688. [Google Scholar]
- Barabaschi, D.; Francia, E.; Tondelli, A.; Gianinetti, A.; Stanca, A.M.; Pecchioni, N. Effect of the nud gene on grain yield in barley. Czech J. Genet. Plant Breed. 2012, 48, 10–22. [Google Scholar] [CrossRef] [Green Version]
- Gerasimova, S.V.; Hertig, C.; Korotkova, A.M.; Kolosovskaya, E.V.; Otto, I.; Hiekel, S.; Kochetov, A.V.; Khlestkina, E.K.; Kumlehn, J. Conversion of hulled into naked barley by Cas endonuclease-mediated knockout of the NUD gene. BMC Plant Biol. 2020, 20, 255. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Sun, G.; Ren, X.; Du, B.; Cheng, Y.; Wang, Y.; Li, C.; Sun, D. Dissecting the genetic basis of grain size and weight in barley (Hordeum vulgare L.) by QTL and comparative genetic analyses. Front. Plant Sci. 2019, 10, 469. [Google Scholar] [CrossRef]
- Tondelli, A.; Francia, E.; Visioni, A.; Comadran, J.; Mastrangelo, A.; Akar, T.; Al-Yassin, A.; Ceccarelli, S.; Grando, S.; Benbelkacem, A. QTLs for barley yield adaptation to Mediterranean environments in the ‘Nure’×’Tremois’ biparental population. Euphytica 2014, 197, 73–86. [Google Scholar] [CrossRef]
- Gong, X.; Wheeler, R.; Bovill, W.D.; McDonald, G.K. QTL mapping of grain yield and phosphorus efficiency in barley in a Mediterranean-like environment. Theor. Appl. Genet. 2016, 129, 1657–1672. [Google Scholar] [CrossRef]
- Rey, J.; Hayes, P.; Petrie, S.; Corey, A.; Flowers, M.; Ohm, J.-B.; Ong, C.; Rhinhart, K.; Ross, A. Production of dryland barley for human food: Quality and agronomic performance. Crop Sci. 2009, 4, 347–355. [Google Scholar] [CrossRef] [Green Version]
- Shoeva, O.Y.; Mock, H.-P.; Kukoeva, T.V.; Börner, A.; Khlestkina, E.K. Regulation of the flavonoid biosynthesis pathway genes in purple and black grains of Hordeum vulgare. PLoS ONE 2016, 11, e0163782. [Google Scholar]
- Aastrup, S.; Outtrup, H.; Erdal, K. Location of the proanthocyanidins in the barley grain. Carlsberg Res. Commun. 1984, 49, 105–109. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.-J.; Hyun, J.-N.; Kim, J.-A.; Park, J.-C.; Kim, M.-Y.; Kim, J.-G.; Lee, S.-J.; Chun, S.-C.; Chung, I.-M. Relationship between phenolic compounds, anthocyanins content and antioxidant activity in colored barley germplasm. J. Agric. Food Chem. 2007, 55, 4802–4809. [Google Scholar] [CrossRef]
- Harlan, H.V. Some Distinctions in Our Cultivated Barleys with Reference to Their Use in Plant Breeding; US Department of Agriculture: Washington, DC, USA, 1914.
- Yao, X.; Wu, K.; Yao, Y.; Bai, Y.; Ye, J.; Chi, D. Construction of a high-density genetic map: Genotyping by sequencing (GBS) to map purple seed coat color (Psc) in hulless barley. Hereditas 2018, 155, 37. [Google Scholar] [CrossRef] [Green Version]
- Larson, R.L.; Bussard, J.B. Microsomal flavonoid 3′-monooxygenase from maize seedlings. Plant Physiol. 1986, 80, 483–486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, X.; Li, J.; Fu, F.; Jin, M.; Chen, L.; Liu, L. Co-location of seed oil content, seed hull content and seed coat color QTL in three different environments in Brassica napus L. Euphytica 2009, 170, 355–364. [Google Scholar] [CrossRef]
- Abdel-Aal, E.-S.M.; Young, J.C.; Rabalski, I. Anthocyanin composition in black, blue, pink, purple, and red cereal grains. J. Agric. Food Chem. 2006, 54, 4696–4704. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trait | Unit | % of Evaluated Accessions | Mean | Min. | Max. | Variation (Cv) | |
|---|---|---|---|---|---|---|---|
| Phenological | days to heading | days | 95% | 69.7 | 52.0 (PL 42389) | 93.0 (2 access.) | 7% |
| grain filling duration | days | 85% | 35.4 | 22.0 (PL 505304) | 44.0 (PL 40554) | 13% | |
| days to maturity | days | 85% | 105.5 | 81.7 (PL 42768) | 117.0 (PL 41532) | 10% | |
| Metrical | plant length | cm | 96% | 81.3 | 52.0 (PL 502172) | 112.0 (PL 36315) | 13% |
| ear length | cm | 76% | 8.6 | 4.6 (PL 502171) | 11.8 (PL 501971) | 16% | |
| number of grains per ear | no. | 64% | 16.8 | 7.7 (PL 40553) | 43.6 (PL 502170) | 36% | |
| thousand seed weight | g | 95% | 47.7 | 30.5 (PL 36315) | 58.8 (PL 502169) | 11% | |
| Bonitations | lodging resistance | scale 1 | 85% | 7.7 | 2.0 (PL 41267) | 9.0 (42 access.) | 19% |
| powdery mildew resistance | scale 1 | 88% | 6.9 | 1.0 (3 access.) | 9.0 (21 access.) | 28% | |
| net blotch resistance | scale 1 | 91% | 8.5 | 5.0 (PL 501968.) | 9.0 (71 access.) | 10% | |
| stem rust resistance | scale 1 | 61% | 8.8 | 7.0 (5 access.) | 9.0 (65 access.) | 6% | |
| scald resistance | scale 1 | 67% | 8.7 | 6.0 (PL 505304) | 9.0 (63 access.) | 8% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dziurdziak, J.; Gryziak, G.; Groszyk, J.; Podyma, W.; Boczkowska, M. DArTseq Genotypic and Phenotypic Diversity of Barley Landraces Originating from Different Countries. Agronomy 2021, 11, 2330. https://doi.org/10.3390/agronomy11112330
Dziurdziak J, Gryziak G, Groszyk J, Podyma W, Boczkowska M. DArTseq Genotypic and Phenotypic Diversity of Barley Landraces Originating from Different Countries. Agronomy. 2021; 11(11):2330. https://doi.org/10.3390/agronomy11112330
Chicago/Turabian StyleDziurdziak, Joanna, Grzegorz Gryziak, Jolanta Groszyk, Wiesław Podyma, and Maja Boczkowska. 2021. "DArTseq Genotypic and Phenotypic Diversity of Barley Landraces Originating from Different Countries" Agronomy 11, no. 11: 2330. https://doi.org/10.3390/agronomy11112330
APA StyleDziurdziak, J., Gryziak, G., Groszyk, J., Podyma, W., & Boczkowska, M. (2021). DArTseq Genotypic and Phenotypic Diversity of Barley Landraces Originating from Different Countries. Agronomy, 11(11), 2330. https://doi.org/10.3390/agronomy11112330