Differential Gene Expression Analysis of Wheat Breeding Lines Reveal Molecular Insights in Yellow Rust Resistance under Field Conditions

 , ,
, , 
Abstract
1. Introduction
2. Materials and Methods
2.1. Field Experiments and Sampling
2.2. RNAseq Library Preparation Sequencing and Quality Control
2.3. De Novo and Reference Transcriptome Assembly and Validation
2.4. Identification of Differentially Expressed Genes (DEGs), Annotation and Gene Ontology (GO) Analysis
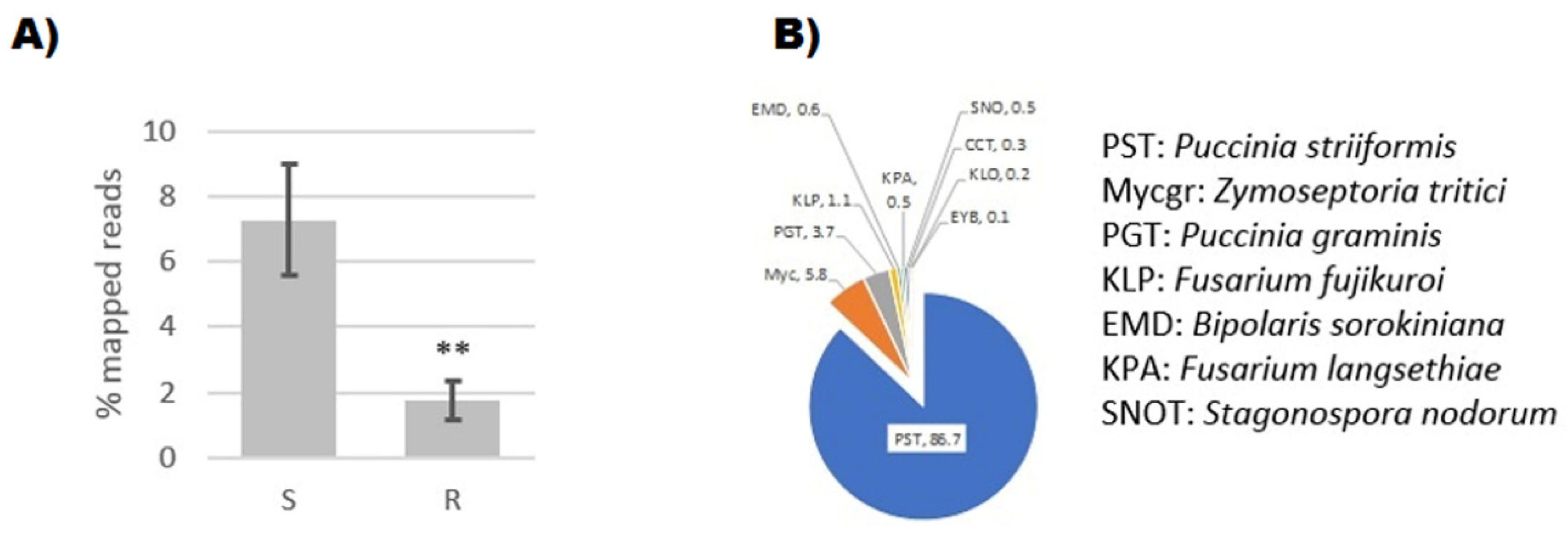
2.5. Metagenomic Analysis of Selected Wheat Pathogens
3. Results
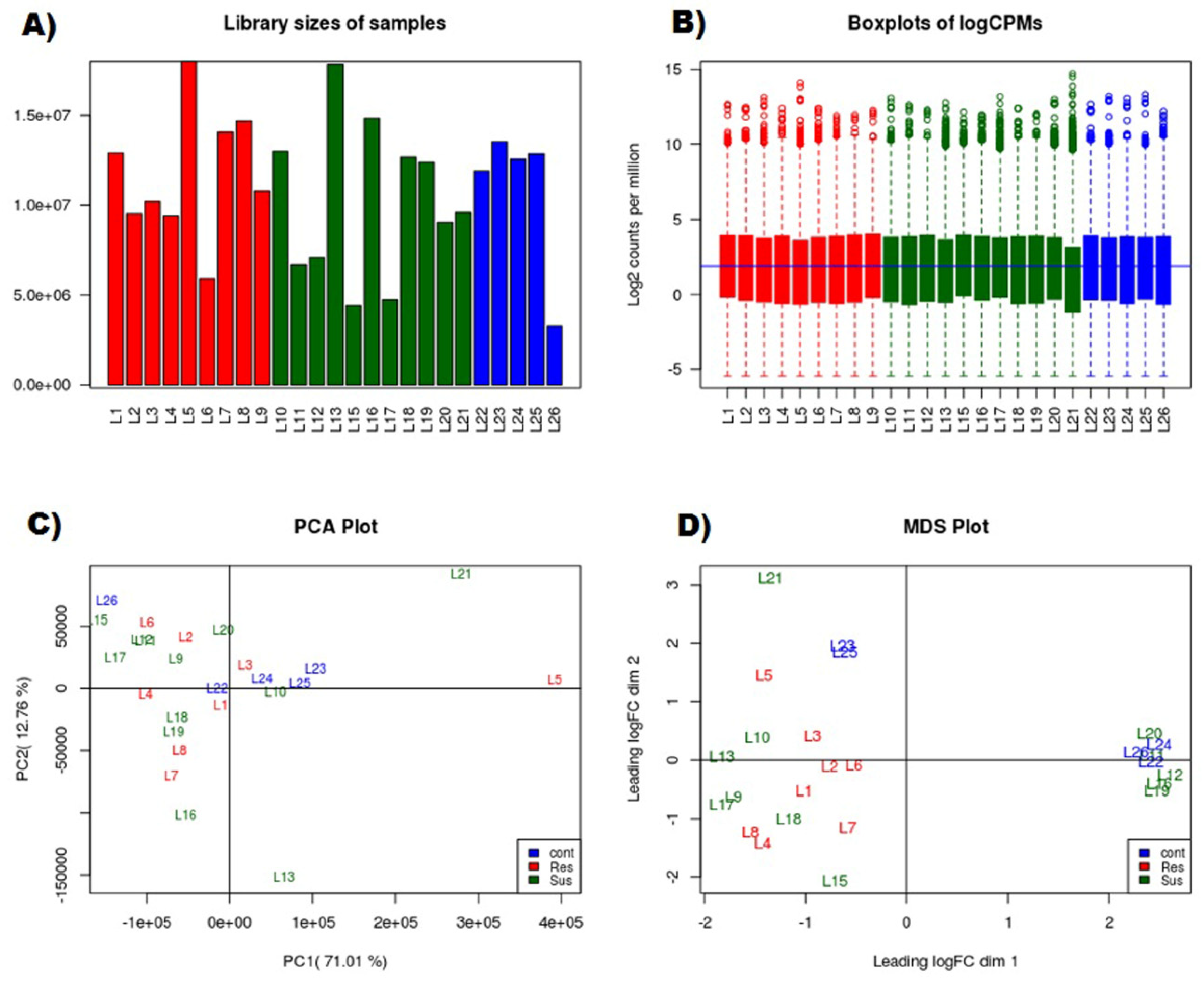
3.1. RNAseq Data Analyses
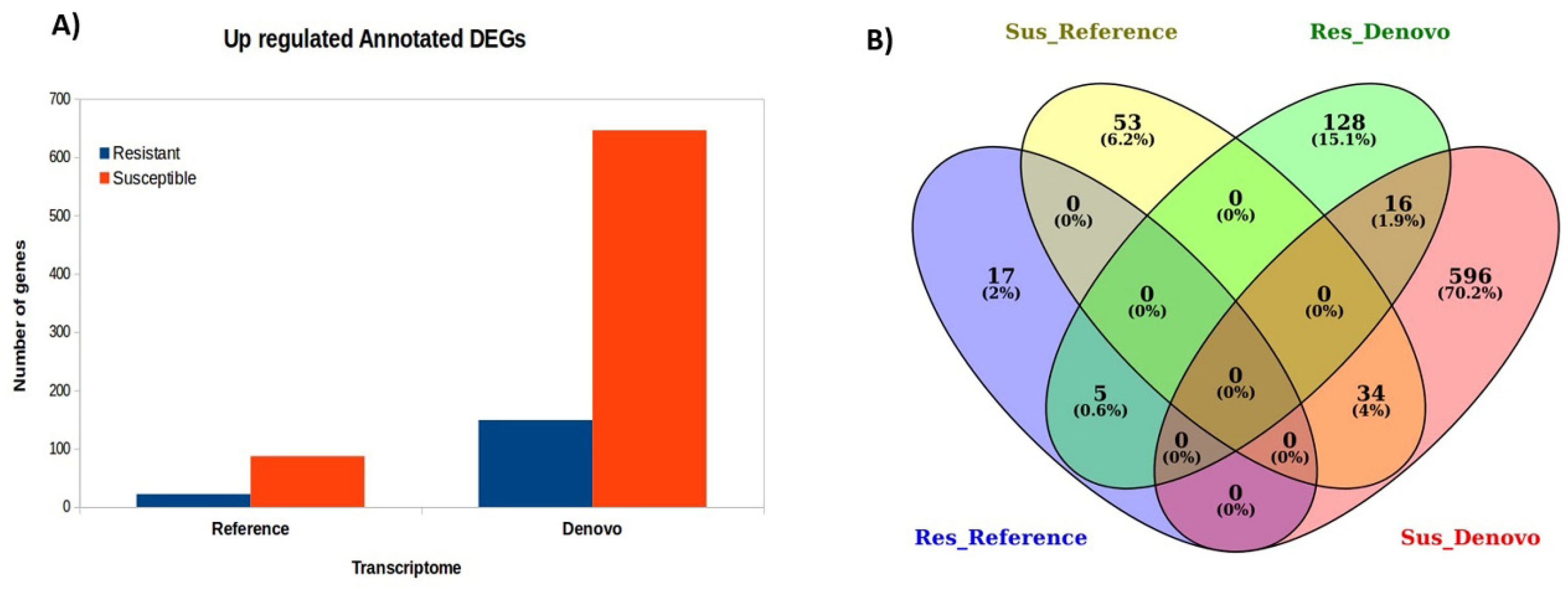
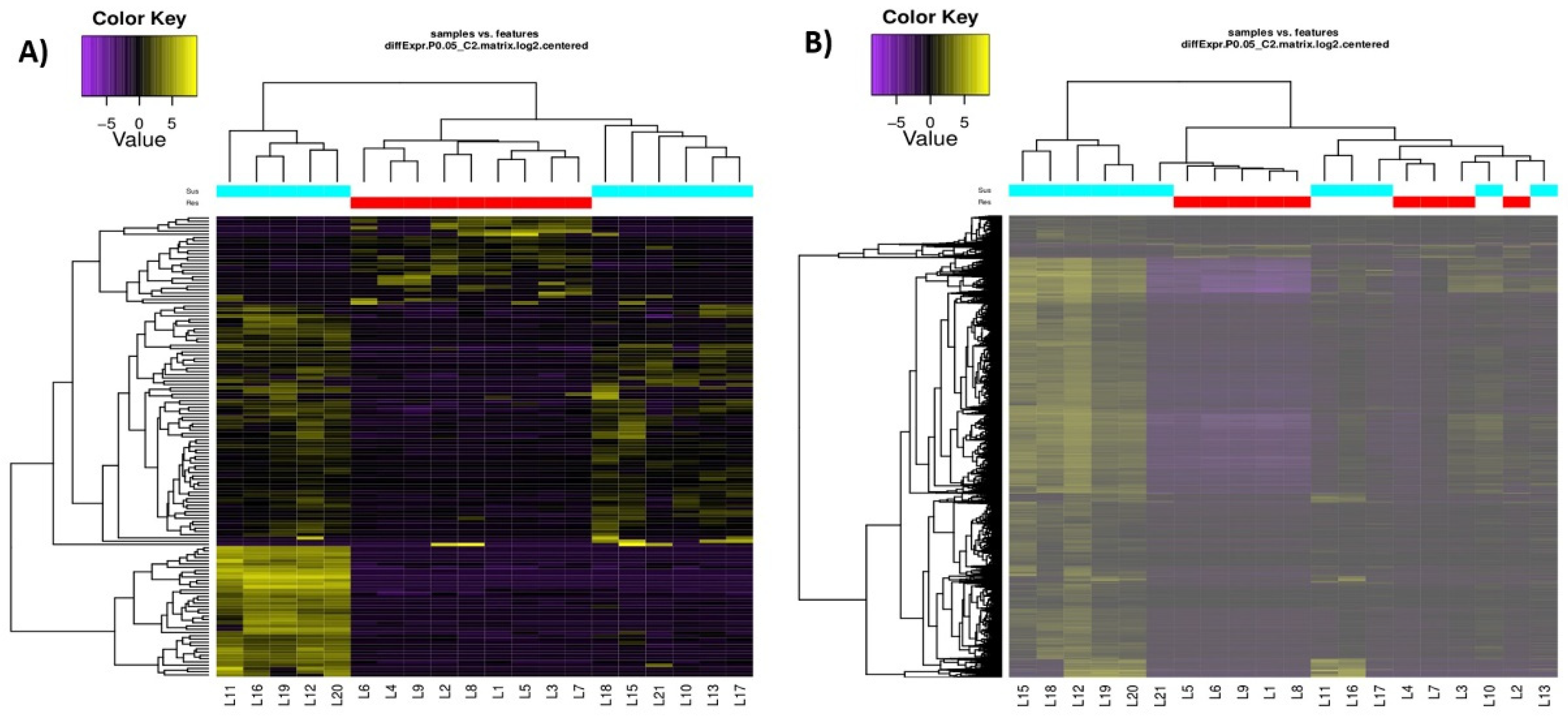
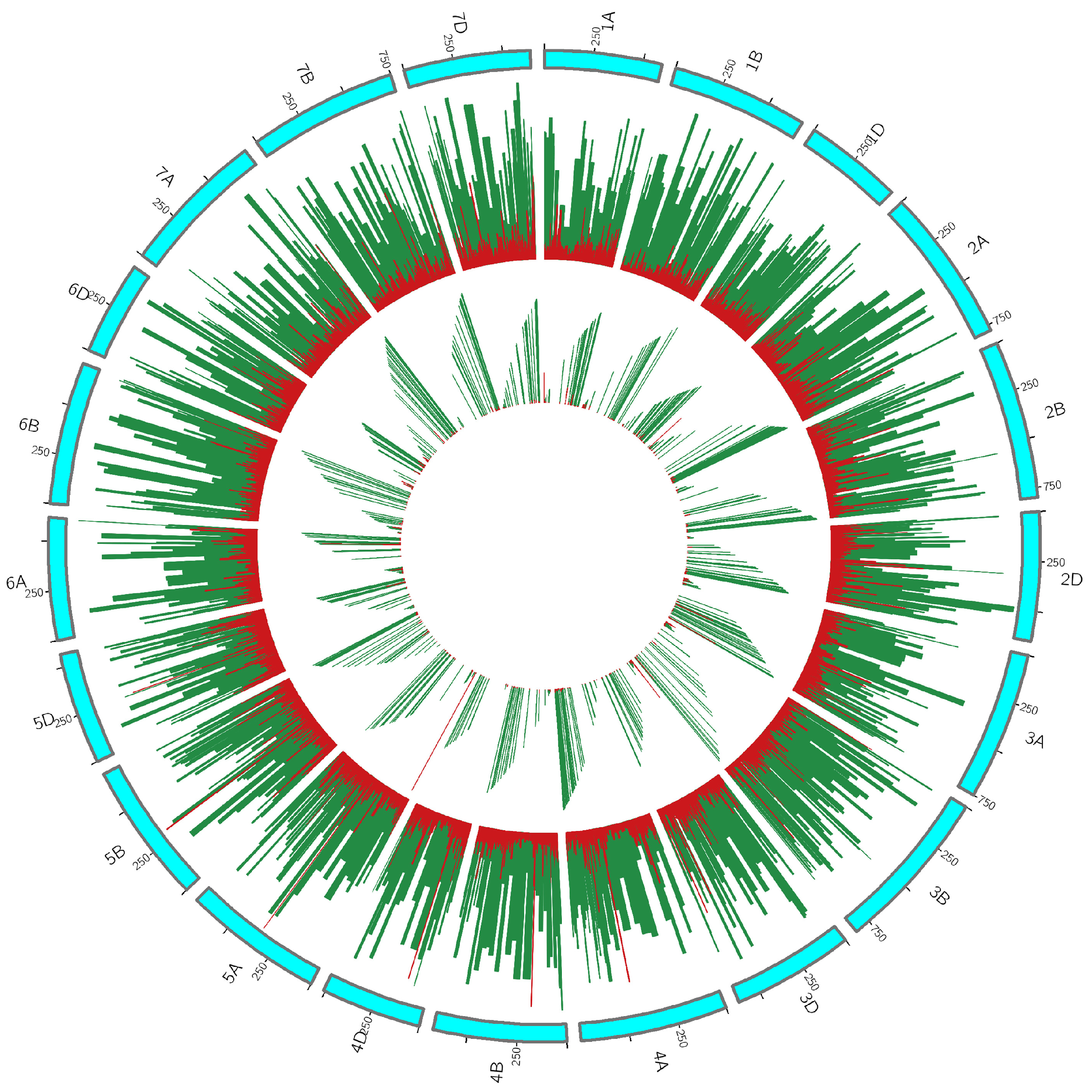
3.2. DEGs Identified by De Novo and Reference-Based Methods
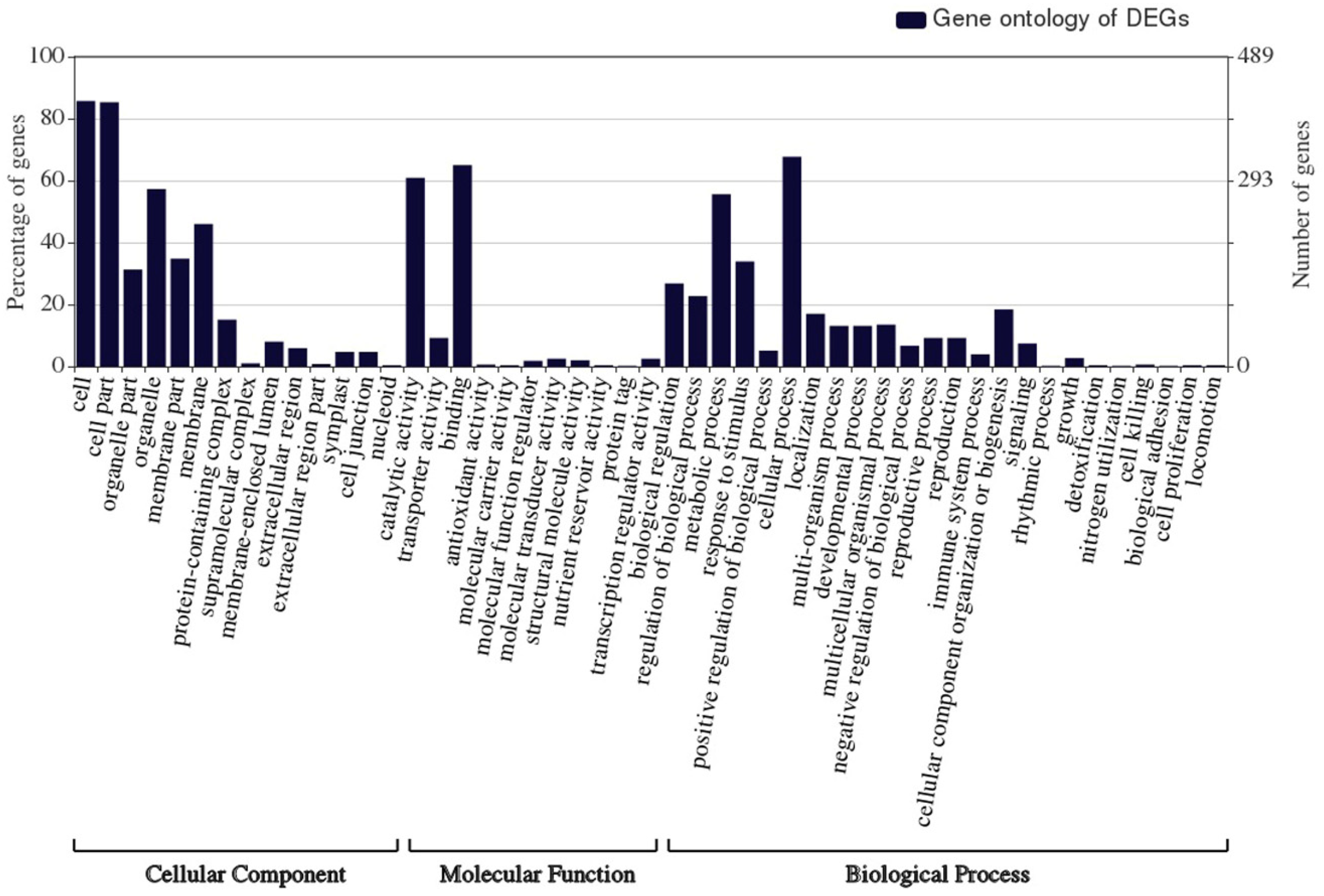
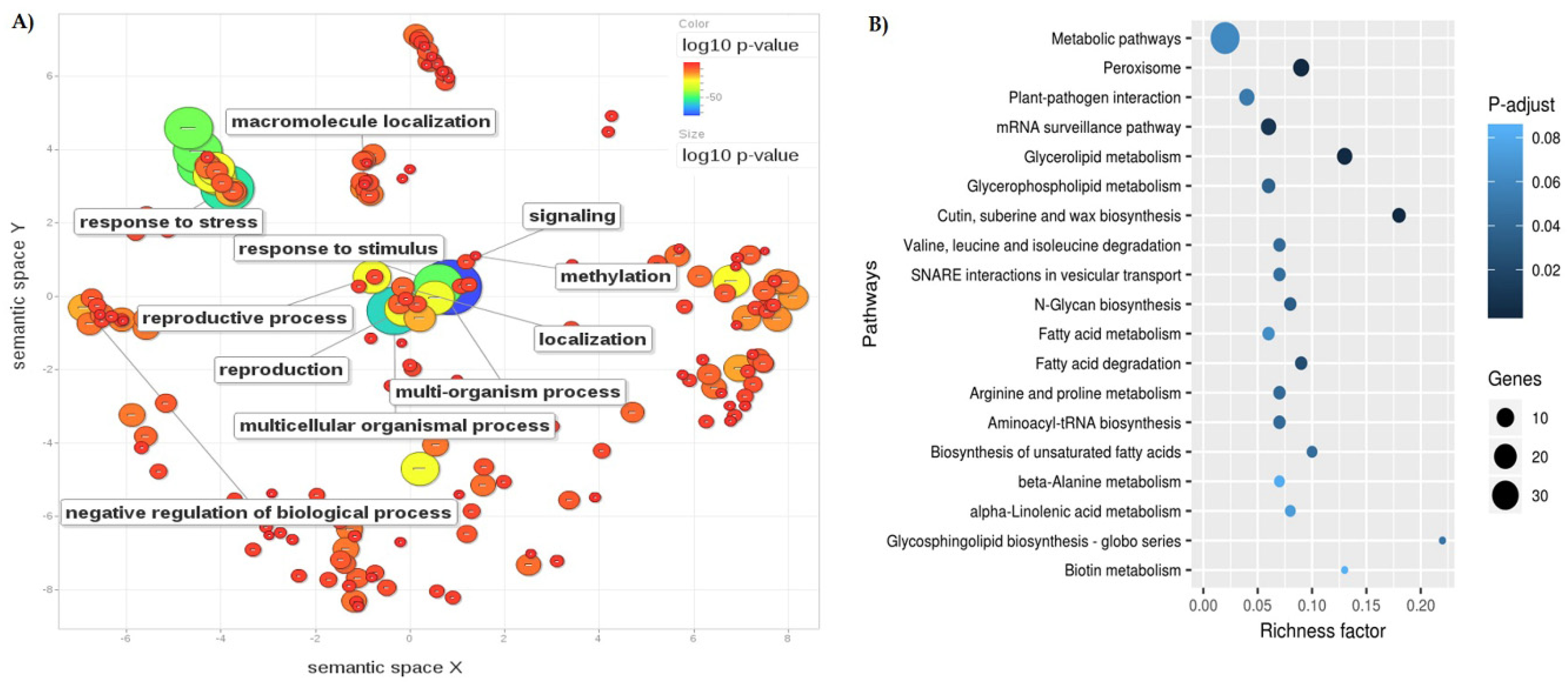
3.3. Differentially Expressed Genes Annotation and GO Enrichment
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chen, X.M. Challenges and solutions for stripe rust control in the United States. Aust. J. Agric. Res. 2007, 58, 648–655. [Google Scholar] [CrossRef]
- Brown, J.K.M.; Hovmøller, M.S. Aerial Dispersal of Pathogens on the Global and Continental Scales and Its Impact on Plant Disease. Science 2002, 297, 537–541. [Google Scholar] [CrossRef] [PubMed]
- Hovmøller, M.S.; Walter, S.; Bayles, R.A.; Hubbard, A.; Flath, K.; Sommerfeldt, N.; Leconte, M.; Czembor, P.; Rodriguez-Algaba, J.; Thach, T.; et al. Replacement of the European wheat yellow rust population by new races from the centre of diversity in the near-Himalayan region. Plant Pathol. 2016, 65, 402–411. [Google Scholar] [CrossRef]
- Jones, J.D.G.; Dangl, J.L. The plant immune system. Nature 2006, 444, 323–329. [Google Scholar] [CrossRef] [PubMed]
- Schwessinger, B. Fundamental wheat stripe rust research in the 21st century. New Phytol. 2017, 213, 1625–1631. [Google Scholar] [CrossRef]
- Bux, H.; Ashraf, M.; Chen, X. Expression of high-temperature adult-plant (HTAP) resistance against stripe rust (Puccinia striiformis f. sp. tritici) in Pakistan wheat landraces. Can. J. Plant Pathol. 2012, 34, 68–74. [Google Scholar] [CrossRef]
- Chen, W.; Wellings, C.; Chen, X.; Kang, Z.; Liu, T. Wheat stripe (yellow) rust caused by Puccinia striiformis f. sp. tritici. Mol. Plant Pathol. 2014, 15, 433–446. [Google Scholar] [CrossRef]
- Jamil, S.; Shahzad, R.; Ahmad, S.; Fatima, R.; Zahid, R.; Anwar, M.; Iqbal, M.Z.; Wang, X. Role of Genetics, Genomics, and Breeding Approaches to Combat Stripe Rust of Wheat. Front. Nutr. 2020, 7, 173. [Google Scholar] [CrossRef]
- Xu, L.S.; Wang, M.N.; Cheng, P.; Kang, Z.S.; Hulbert, S.H.; Chen, X.M. Molecular mapping of Yr53, a new gene for stripe rust resistance in durum wheat accession PI 480148 and its transfer to common wheat. Theor. Appl. Genet. 2013, 126, 523–533. [Google Scholar] [CrossRef]
- Rosewarne, G.M.; Herrera-Foessel, S.A.; Singh, R.P.; Huerta-Espino, J.; Lan, C.X.; He, Z.H. Quantitative trait loci of stripe rust resistance in wheat. Theor. Appl. Genet. 2013, 126, 2427–2449. [Google Scholar] [CrossRef]
- Lindlöf, A.; Chawade, A.; Sikora, P.; Olsson, O. Comparative Transcriptomics of Sijung and Jumli Marshi Rice during Early Chilling Stress Imply Multiple Protective Mechanisms. PLoS ONE 2015, 10, e0125385. [Google Scholar] [CrossRef] [PubMed]
- Yaari, G.; Bolen, C.R.; Thakar, J.; Kleinstein, S.H. Quantitative set analysis for gene expression: A method to quantify gene set differential expression including gene-gene correlations. Nucleic Acids Res. 2013, 41, e170. [Google Scholar] [CrossRef] [PubMed]
- Chawade, A.; Alexandersson, E.; Bengtsson, T.; Andreasson, E.; Levander, F. Targeted Proteomics Approach for Precision Plant Breeding. J. Proteome Res. 2016, 15, 638–646. [Google Scholar] [CrossRef]
- Chawade, A.; Armoniené, R.; Berg, G.; Brazauskas, G.; Frostgård, G.; Geleta, M.; Gorash, A.; Henriksson, T.; Himanen, K.; Ingver, A.; et al. A transnational and holistic breeding approach is needed for sustainable wheat production in the Baltic Sea region. Physiol. Plant. 2018, 164, 442–451. [Google Scholar] [CrossRef] [PubMed]
- Coram, T.E.; Huang, X.; Zhan, G.; Settles, M.L.; Chen, X. Meta-analysis of transcripts associated with race-specific resistance to stripe rust in wheat demonstrates common induction of blue copper-binding protein, heat-stress transcription factor, pathogen-induced WIR1A protein, and ent-kaurene synthase transcripts. Funct. Integr. Genom. 2010, 10, 383–392. [Google Scholar]
- Hulbert, S.H.; Bai, J.; Fellers, J.P.; Pacheco, M.G.; Bowden, R.L. Gene Expression Patterns in Near Isogenic Lines for Wheat Rust Resistance Gene Lr34/Yr18. Phytopathology 2007, 97, 1083–1093. [Google Scholar] [CrossRef]
- Chen, X.; Coram, T.; Huang, X.; Wang, M.; Dolezal, A. Understanding Molecular Mechanisms of Durable and Non-durable Resistance to Stripe Rust in Wheat Using a Transcriptomics Approach. Curr. Genom. 2013, 14, 111–126. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Yang, Y.; Wang, C.; Liu, M.; Li, H.; Fu, Y.; Wang, Y.; Nie, Y.; Liu, X.; Ji, W. Large-scale transcriptome comparison reveals distinct gene activations in wheat responding to stripe rust and powdery mildew. BMC Genom. 2014, 15, 898. [Google Scholar] [CrossRef]
- Hao, Y.; Wang, T.; Wang, K.; Wang, X.; Fu, Y.; Huang, L.; Kang, Z. Transcriptome Analysis Provides Insights into the Mechanisms Underlying Wheat Plant Resistance to Stripe Rust at the Adult Plant Stage. PLoS ONE 2016, 11, e0150717. [Google Scholar] [CrossRef]
- Krattinger, S.G.; Lagudah, E.S.; Spielmeyer, W.; Singh, R.P.; Huerta-Espino, J.; McFadden, H.; Bossolini, E.; Selter, L.L.; Keller, B. A Putative ABC Transporter Confers Durable Resistance to Multiple Fungal Pathogens in Wheat. Science 2009, 323, 1360. [Google Scholar] [CrossRef]
- Fu, D.; Uauy, C.; Distelfeld, A.; Blechl, A.; Epstein, L.; Chen, X.; Sela, H.; Fahima, T.; Dubcovsky, J. A kinase-START gene confers temperature-dependent resistance to wheat stripe rust. Science 2009, 323, 1357–1360. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Frick, M.; Huel, R.; Nykiforuk, C.L.; Wang, X.; Gaudet, D.A.; Eudes, F.; Conner, R.L.; Kuzyk, A.; Chen, Q.; et al. The Stripe Rust Resistance Gene Yr10 Encodes an Evolutionary-Conserved and Unique CC–NBS–LRR Sequence in Wheat. Mol. Plant 2014, 7, 1740–1755. [Google Scholar] [CrossRef] [PubMed]
- FastQC: A Quality Control Tool for High Throughput Sequence Data. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 1 November 2019).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Best Practices for De Novo Transcriptome Assembly with Trinity. Available online: https://informatics.fas.harvard.edu/best-practices-for-de-novo-transcriptome-assembly-with-trinity.html (accessed on 1 November 2019).
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650. [Google Scholar] [CrossRef] [PubMed]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef]
- DESeq2 Package. Available online: http://www.bioconductor.org/packages/release/bioc/html/DESeq2.html (accessed on 1 December 2019).
- Pheatmap: A Function to Draw Clustered Heatmaps. Available online: https://www.rdocumentation.org/packages/pheatmap/versions/1.0.12/topics/pheatmap (accessed on 1 December 2019).
- Ye, J.; Fang, L.; Zheng, H.; Zhang, Y.; Chen, J.; Zhang, Z.; Wang, J.; Li, S.; Li, R.; Bolund, L.; et al. WEGO: A web tool for plotting GO annotations. Nucleic Acids Res. 2006, 34, W293–W297. [Google Scholar] [CrossRef]
- Du, Z.; Zhou, X.; Ling, Y.; Zhang, Z.; Su, Z. Agrigo: A GO analysis toolkit for the agricultural community. Nucleic Acids Res. 2010, 38, W64–W70. [Google Scholar] [CrossRef]
- Supek, F.; Bošnjak, M.; Škunca, N.; Šmuc, T. REVIGO Summarizes and Visualizes Long Lists of Gene Ontology Terms. PLoS ONE 2011, 6, e21800. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.-Y.; Wei, L. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, W316–W322. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.A.; Wang, Z. Next-generation transcriptome assembly. Nat. Rev. Genet. 2011, 12, 671–682. [Google Scholar] [CrossRef] [PubMed]
- Kovi, M.R.; Abdelhalim, M.; Kunapareddy, A.; Ergon, Å.; Tronsmo, A.M.; Brurberg, M.B.; Hofgaard, I.S.; Asp, T.; Rognli, O.A. Global transcriptome changes in perennial ryegrass during early infection by pink snow mould. Sci. Rep. 2016, 6, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Ward, J.A.; Ponnala, L.; Weber, C.A. Strategies for transcriptome analysis in nonmodel plants. Am. J. Bot. 2012, 99, 267–276. [Google Scholar] [CrossRef]
- Zhang, W.; Duan, S.; Kistner, E.O.; Bleibel, W.K.; Huang, R.S.; Clark, T.A.; Chen, T.X.; Schweitzer, A.C.; Blume, J.E.; Cox, N.J.; et al. Evaluation of genetic variation contributing to differences in gene expression between populations. Am. J. Hum. Genet. 2008, 82, 631–640. [Google Scholar] [CrossRef]
- Kimura, S.; Hunter, K.; Vaahtera, L.; Tran, H.C.; Citterico, M.; Vaattovaara, A.; Rokka, A.; Stolze, S.C.; Harzen, A.; Meißner, L.; et al. CRK2 and C-terminal Phosphorylation of NADPH Oxidase RBOHD Regulate Reactive Oxygen Species Production in Arabidopsis. Plant Cell 2020, 32, 1063. [Google Scholar] [CrossRef]
- Chern, M.; Xu, Q.; Bart, R.S.; Bai, W.; Ruan, D.; Sze-To, W.H.; Canlas, P.E.; Jain, R.; Chen, X.; Ronald, P.C. A Genetic Screen Identifies a Requirement for Cysteine-Rich–Receptor-Like Kinases in Rice NH1 (OsNPR1)-Mediated Immunity. PLoS Genet. 2016, 12, e1006049. [Google Scholar]
- Escobar-Sepúlveda, H.F.; Trejo-Téllez, L.I.; Pérez-Rodríguez, P.; Hidalgo-Contreras, J.V.; Gómez-Merino, F.C. Diacylglycerol Kinases Are Widespread in Higher Plants and Display Inducible Gene Expression in Response to Beneficial Elements, Metal, and Metalloid Ions. Front. Plant Sci. 2017, 8, 129. [Google Scholar] [CrossRef]
- McHale, L.; Tan, X.; Koehl, P.; Michelmore, R.W. Plant NBS-LRR proteins: Adaptable guards. Genome Biol. 2006, 7, 1–11. [Google Scholar] [CrossRef]
- Yamaguchi, K.; Yamada, K.; Ishikawa, K.; Yoshimura, S.; Hayashi, N.; Uchihashi, K.; Ishihama, N.; Kishi-Kaboshi, M.; Takahashi, A.; Tsuge, S.; et al. A Receptor-like Cytoplasmic Kinase Targeted by a Plant Pathogen Effector Is Directly Phosphorylated by the Chitin Receptor and Mediates Rice Immunity. Cell Host Microbe 2013, 13, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Lin, G.; Langdon, W.Y.; Tao, L.; Zhang, J. Regulation of C-Type Lectin Receptor-Mediated Antifungal Immunity. Front. Immunol. 2018, 9, 123. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.-R.; Brurberg, M.B.; Elameen, A.; Klemsdal, S.S.; Martinussen, I. Expression of resistance gene analogs in woodland strawberry (Fragaria vesca) during infection with Phytophthora cactorum. Mol. Genet. Genom. 2016, 291, 1967–1978. [Google Scholar] [CrossRef]
- Ye, H.; Du, H.; Tang, N.; Li, X.; Xiong, L. Identification and expression profiling analysis of TIFY family genes involved in stress and phytohormone responses in rice. Plant Mol. Biol. 2009, 71, 291–305. [Google Scholar] [CrossRef] [PubMed]
- Wagner, T.A.; Kohorn, B.D. Wall-associated kinases are expressed throughout plant development and are required for cell expansion. Plant Cell 2001, 13, 303–318. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Transcriptome | Contigs | N50 | Avg. Length | Assembly Size | Longest Contig Size | Contig Size >10 KB | Contig Size >1 KB |
|---|---|---|---|---|---|---|---|
| Reference | 219,435 | 1835 | 1477 | 324,000,551 | 22,462 | 58 | 140,664 |
| De novo | 511,926 | 1420 | 953 | 488,058,762 | 15,387 | 30 | 175,517 |
| BUSCO Description | Reference | De Novo |
|---|---|---|
| Complete BUSCOs (S+D) | 1302 (94.7%) | 1079 (78.5%) |
| Complete and single-copy BUSCOs (S) | 69 (5.0%) | 229 (16.7%) |
| Complete and duplicated BUSCOs (D) | 1233 (D: 89.7%) | 850 (D: 61.8%) |
| Fragmented BUSCOs (F) | 61 (4.4%) | 213 (15.5%) |
| Missing BUSCOs (M) | 12 (0.9%) | 83 (6.0%) |
| Reference/De Novo | log2FC | Annotation | UniprotIds | IWGSC | Signal |
|---|---|---|---|---|---|
| Transcript | Chromosome Location | ||||
| TRINITY_DN51078_c2_g4_i2 | 805.981 | Cysteine-rich receptor-like protein kinase | CRK6_ORYSJ | 2D:189356990:189359834:1 | Y |
| TRINITY_DN46223_c0_g1_i25 | 98.303 | Cysteine-rich receptor-like protein kinase | CRK6_ORYSJ | 5A:546234176:546238163:−1 | Y |
| TRINITY_DN46223_c0_g1_i31 | 82.836 | Cysteine-rich receptor-like protein kinase | CRK6_ORYSJ | 5A:546238637:546241834:1 | Y |
| TRINITY_DN46223_c0_g1_i17 | 52.416 | Cysteine-rich receptor-like protein kinase | CRK6_ORYSJ | 5A:546238637:546241834:1 | Y |
| TRINITY_DN38661_c1_g1_i1 | 38.481 | Cysteine-rich receptor-like protein kinase | CRK6_ORYSJ | 2B:245784381:245787768:1 | Y |
| TRINITY_DN45544_c0_g1_i24 | 16.282 | Cysteine-rich receptor-like protein kinase | CRK6_ORYSJ | 2D:20432763:20439635:1 | Y |
| TRINITY_DN37687_c0_g2_i5 | 11.964 | Cysteine-rich receptor-like protein kinase | CRK7_ARATH | 5A:546306021:546309203:−1 | Y |
| TRINITY_DN49928_c0_g1_i18 | 11.545 | Diacylglycerol kinase 5 OS = Arabidopsis thaliana | DGK5_ARATH | 2A:748963953:748967651:−1 | N |
| TRINITY_DN35364_c0_g1_i3 | 2.971 | Disease resistance protein RGA2 OS = Solanum | RGA2_SOLBU | 1D:455752931:455755683:1 | N |
| TRINITY_DN44057_c1_g5_i1 | 7.574 | Disease resistance protein RPP8 OS = Arabidopsis | RPP8_ARATH | 6B:662028638:662032091:−1 | N |
| TRINITY_DN39548_c1_g3_i2 | 11.392 | Leaf rust 10 disease-resistance locus receptor-like | LRL28_ARATH | 3B:29721933:29735756:1 | Y |
| TRINITY_DN35553_c1_g1_i1 | 5.456 | Leaf rust 10 disease-resistance locus receptor-like | LRL28_ARATH | 6D:2047133:2050237:−1 | Y |
| TRINITY_DN38577_c0_g1_i1 | 12.655 | LRR receptor-like serine/threonine-protein | FLS2_ARATH | 1D:466386615:466388390:−1 | Y |
| TRINITY_DN38835_c1_g4_i13 | 4.145 | LRR receptor-like serine/threonine-protein | FLS2_ORYSJ | 6D:380568621:380572398:1 | Y |
| TraesCS2B02G608600.1 | 59.366 | Probable LRR receptor-like serine/threonine-protein | Y3475_ARATH | 2B:788840706:788842030:−1 | N |
| TRINITY_DN41813_c2_g5_i3 | 28.015 | Probable LRR receptor-like serine/threonine-protein | Y3475_ARATH | 2D:646488160:646491914:1 | N |
| TRINITY_DN51069_c0_g2_i8 | 2.348 | Protein TIFY 6b OS = Oryza sativa subsp. | TIF6B_ORYSJ | 5B:369635031:369638011:−1 | N |
| TRINITY_DN35330_c0_g3_i8 | 3.364 | Putative disease resistance protein RGA4 | RGA4_SOLBU | Un:47532810:47545586:1 | N |
| TRINITY_DN51664_c0_g1_i11 | 3.269 | Putative disease resistance protein RGA4 | RGA4_SOLBU | Un:95706705:95715329:1 | N |
| TRINITY_DN35364_c0_g4_i1 | 3.209 | Putative disease resistance protein RGA4 | RGA4_SOLBU | Un:234394428:234397967:1 | N |
| TRINITY_DN51887_c1_g1_i1 | 4.127 | Putative disease resistance RPP13-like | R13L2_ARATH | 7D:11663563:11672717:1 | N |
| TRINITY_DN52456_c0_g2_i15 | 8.426 | Rust resistance kinase Lr10 OS = Triticum | LRK10_WHEAT | 1A:9359231:9363721:1 | N |
| TraesCS6B02G091700.1 | 4.309 | S-(+)-linalool synthase, chloroplastic OS = Oryza | LINS_ORYSJ | 6B:67408283:67411671:−1 | N |
| TRINITY_DN51301_c1_g1_i10 | 11.432 | Vesicle-associated protein 1-1 OS = Arabidopsis | VAP11_ARATH | 7B:717630687:717634406:−1 | N |
| TRINITY_DN46935_c4_g2_i7 | 5.042 | Vesicle-associated protein 1-1 OS = Arabidopsis | VAP11_ARATH | 7B:717785488:717796197:1 | N |
| TRINITY_DN52084_c1_g1_i8 | 19.697 | Wall-associated receptor kinase 1 OS = Arabidopsis | WAK1_ARATH | 5A:464158592:464170977:−1 | Y |
| TRINITY_DN50322_c1_g1_i11 | 5.429 | Wall-associated receptor kinase 3 OS = Arabidopsis | WAK3_ARATH | 2B:657850507:657854763:−1 | Y |
| TRINITY_DN39541_c5_g2_i2 | 11.016 | Wall-associated receptor kinase 5 OS = Arabidopsis | WAK5_ARATH | 6D:467856066:467860898:1 | Y |
| TRINITY_DN38381_c2_g3_i6 | 5.209 | Wall-associated receptor kinase 5 OS = Arabidopsis | WAK5_ARATH | 6B:713458733:713462914:1 | Y |
| TRINITY_DN52648_c2_g2_i1 | 7.132 | Wall-associated receptor kinase-like 9 | WAKLH_ARATH | 6D:467856066:467860898:1 | Y |
| TRINITY_DN51423_c2_g1_i4 | 12.889 | Receptor-like cytoplasmic kinase 185 OS = Oryza | RK185_ORYSJ | 2B:104817628:104821546:−1 | N |
| TRINITY_DN50690_c0_g4_i1 | 5.46 | G-type lectin S-receptor-like serine/threonine-protein | Y1130_ARATH | 2D:642197592:642202812:1 | N |
| TRINITY_DN35935_c1_g1_i3 | 5.033 | G-type lectin S-receptor-like serine/threonine-protein | CE101_ARATH | 5B:690469413:690490270:−1 | N |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kumar Kushwaha, S.; Vetukuri, R.R.; Odilbekov, F.; Pareek, N.; Henriksson, T.; Chawade, A. Differential Gene Expression Analysis of Wheat Breeding Lines Reveal Molecular Insights in Yellow Rust Resistance under Field Conditions. Agronomy 2020, 10, 1888. https://doi.org/10.3390/agronomy10121888
Kumar Kushwaha S, Vetukuri RR, Odilbekov F, Pareek N, Henriksson T, Chawade A. Differential Gene Expression Analysis of Wheat Breeding Lines Reveal Molecular Insights in Yellow Rust Resistance under Field Conditions. Agronomy. 2020; 10(12):1888. https://doi.org/10.3390/agronomy10121888
Chicago/Turabian StyleKumar Kushwaha, Sandeep, Ramesh R. Vetukuri, Firuz Odilbekov, Nidhi Pareek, Tina Henriksson, and Aakash Chawade. 2020. "Differential Gene Expression Analysis of Wheat Breeding Lines Reveal Molecular Insights in Yellow Rust Resistance under Field Conditions" Agronomy 10, no. 12: 1888. https://doi.org/10.3390/agronomy10121888
APA StyleKumar Kushwaha, S., Vetukuri, R. R., Odilbekov, F., Pareek, N., Henriksson, T., & Chawade, A. (2020). Differential Gene Expression Analysis of Wheat Breeding Lines Reveal Molecular Insights in Yellow Rust Resistance under Field Conditions. Agronomy, 10(12), 1888. https://doi.org/10.3390/agronomy10121888