
Conversion and Validation of Uniplex SNP Markers for Selection of Resistance to Cassava Mosaic Disease in Cassava Breeding Programs


 , , , ,
, , , ,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Background to Marker Discovery
2.2. Development of Allele-Specific PCR Markers Linked to CMD Resistance
2.3. Predictive Performance of Markers
2.3.1. Study Populations
2.3.2. Phenotyping
2.3.3. Genotyping
2.4. Data Analysis
2.4.1. Phenotypic Data Analysis
2.4.2. Marker Prediction Analysis Using Logistic Regression
2.4.3. Within-Family Prediction Analysis
2.4.4. Estimation of Biological Metrics
3. Results
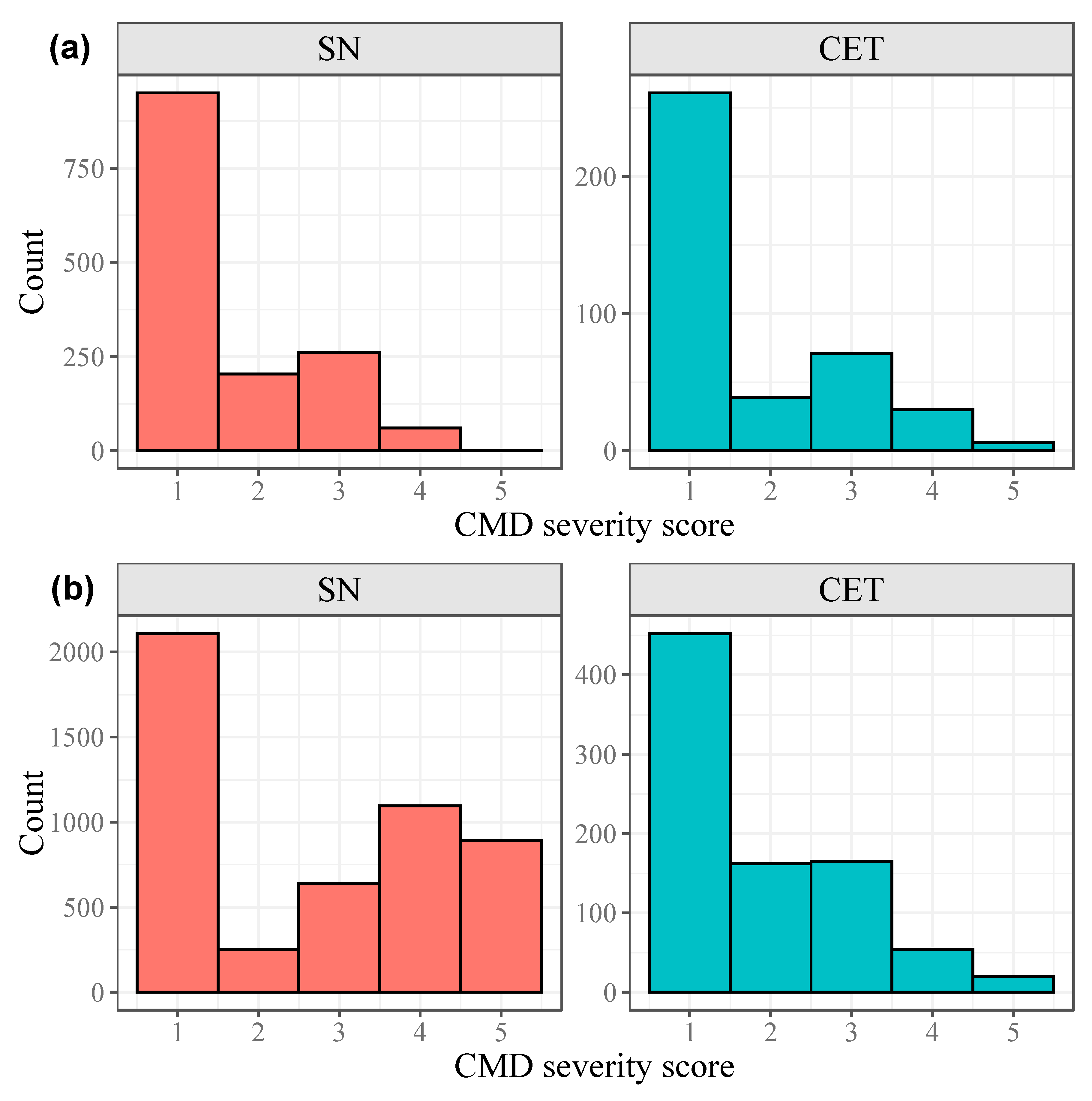
3.1. Phenotypic Variation for Resistance to CMD
3.2. Performance of Markers Linked to CMD Resistance
3.2.1. Favorable Allele and Genotype Frequencies
3.2.2. Marker Effects on CMD Resistance
3.2.3. Effect of Resistance-Linked Alleles on Yield Traits
3.2.4. Population-Level Marker Performance
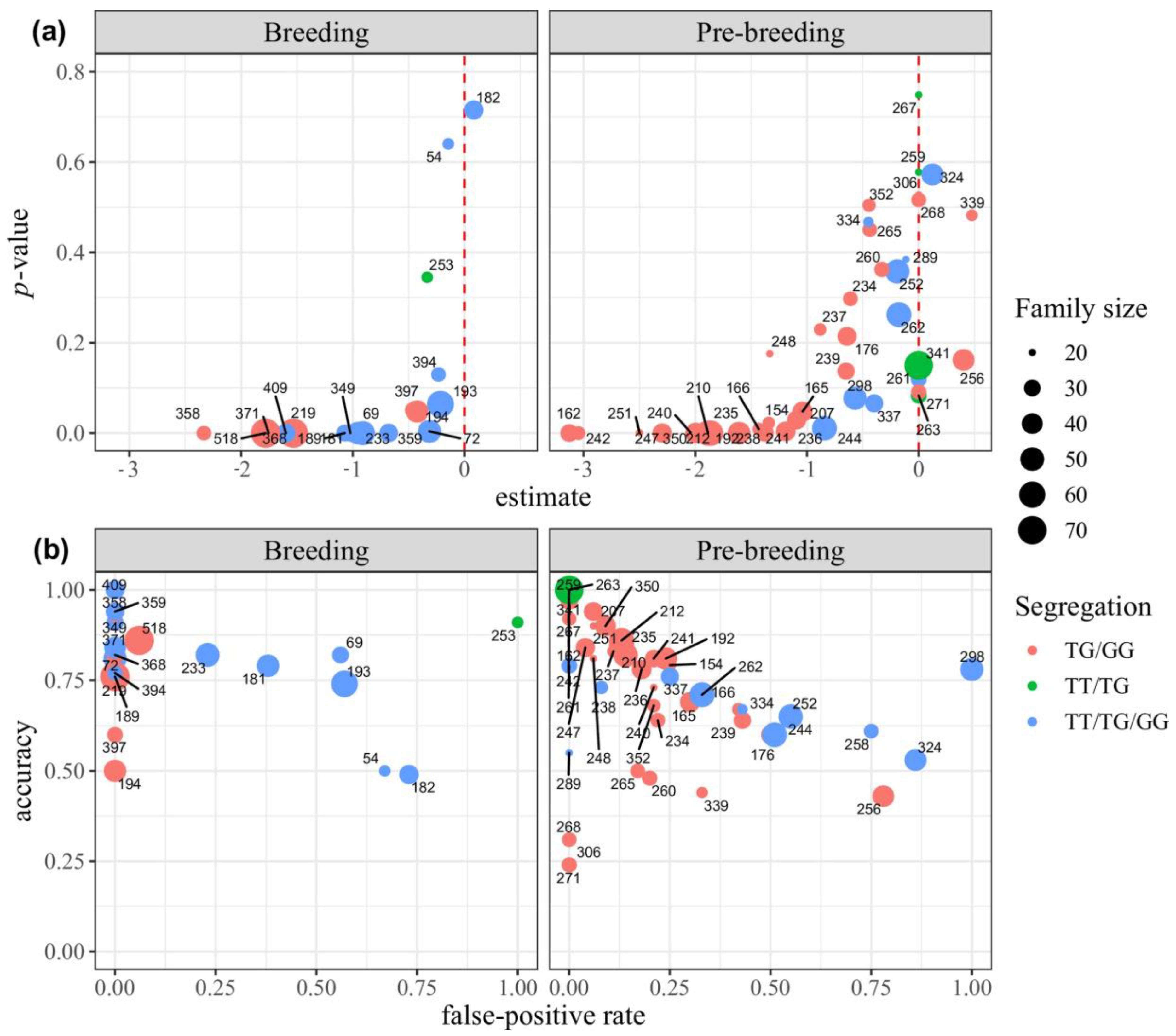
3.2.5. Performance Metrics of Marker S12_7926132 within the Families
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Montagnac, J.A.; Davis, C.R.; Tanumihardjo, S.A. Nutritional Value of Cassava for Use as a Staple Food and Recent Advances for Improvement. Compr. Rev. Food Sci. Food Saf. 2009, 8, 181–194. [Google Scholar] [CrossRef] [PubMed]
- FAOSTAT. Food and Agriculture Organization of the United Nations Statistics. Available online: https://www.fao.org/faostat/en/#data/QC (accessed on 1 December 2020).
- Tan, S.L. Cassava-silently, the tuber fills: The lowly cassava, regarded as a poor man’s crop, may help save the world from the curse of plastic pollution. Utar Agric. Sci. J. 2015, 1, 12–24. [Google Scholar]
- Burns, A.; Gleadow, R.; Cliff, J.; Zacarias, A.; Cavagnaro, T. Cassava: The Drought, War and Famine Crop in a Changing World. Sustainability 2010, 2, 3572–3607. [Google Scholar] [CrossRef]
- Balagopalan, C. Cassava utilization in food, feed and industry. In Cassava: Biology, Production and Utilization; CABI Publishing: Kerala, India, 2009; pp. 301–318. [Google Scholar]
- Patil, B.L.; Kanju, E.; Legg, J.P.; Fauquet, C.M. Cassava brown streak disease: A threat to food security in Africa. J. Gen. Virol. 2015, 96, 956–968. [Google Scholar] [CrossRef]
- Masinde, E.A.; Kimata, B.; Ogendo, J.O.; Mulwa, R.M.; Mkamillo, G.; Maruthi, M.N. Developing dual resistant cassava to the two major viral diseases. Crop. Sci. 2020. [Google Scholar] [CrossRef]
- Thottappilly, G.; Thresh, J.M.; Calvert, L.A.; Winter, S. Cassava. In Virus and Virus-like Diseases of Major Crops in Developing Countries; Loebenstein, G., Thottappilly, G., Eds.; Springer Science & Business Media: Dordrecht, The Netherlands, 2003; pp. 107–165. [Google Scholar]
- Alabi, O.J.; Mulenga, R.M.; Legg, J.P. Cassava mosaic. In Virus diseases of tropical and subtropical crops; Tennant, P., Fermin, G., Eds.; CAB International: Wallingford, UK, 2015; pp. 56–72. [Google Scholar]
- Wang, H.L.; Cui, X.Y.; Wang, X.W.; Liu, S.S.; Zhang, Z.H.; Zhou, X.P. First Report of Sri Lankan cassava mosaic virus Infecting Cassava in Cambodia. Plant Dis. 2016, 100, 1029. [Google Scholar] [CrossRef]
- Minato, N.; Sok, S.; Chen, S.; Delaquis, E.; Phirun, I.; Le, V.X.; Burra, D.D.; Newby, J.C.; Wyckhuys, K.A.G.; De Haan, S. Surveillance for Sri Lankan cassava mosaic virus (SLCMV) in Cambodia and Vietnam one year after its initial detection in a single plantation in 2015. PLoS ONE 2019, 14, e0212780. [Google Scholar] [CrossRef]
- Wang, D.; Yao, X.M.; Huang, G.X.; Shi, T.; Wang, G.F.; Ye, J. First Report of Sri Lankan Cassava Mosaic Virus Infected Cassava in China. Plant Dis. 2019, 103, 1437. [Google Scholar] [CrossRef]
- Owor, B.; Legg, J.P.; Okao-Okuja, G.; Obonyo, R.; Ogenga-Latigo, M.W. The effect of cassava mosaic geminiviruses on symptom severity, growth and root yield of a cassava mosaic virus disease-susceptible cultivar in Uganda. Ann. Appl. Biol. 2004, 145, 331–337. [Google Scholar] [CrossRef]
- Legg, J.; Owor, B.; Sseruwagi, P.; Ndunguru, J. Cassava Mosaic Virus Disease in East and Central Africa: Epidemiology and Management of a Regional Pandemic. Adv. Virus Res. 2006, 67, 355–418. [Google Scholar] [CrossRef]
- Storey, H.H.; Nichols, R.F.W. Studies of the Mosaic Diseases of Cassava. Ann. Appl. Biol. 1938, 25, 790–806. [Google Scholar] [CrossRef]
- CABI Invasive Species Compendium: UK. Available online: https://www.cabi.org/isc/datasheet/2747 (accessed on 11 December 2020).
- Akano, A.; Dixon, A.; Mba, C.; Barrera, E.; Fregene, M. Genetic mapping of a dominant gene conferring resistance to cassava mosaic disease. Theor. Appl. Genet. 2002, 105, 521–525. [Google Scholar] [CrossRef] [PubMed]
- Rabbi, I.Y.; Hamblin, M.T.; Kumar, P.L.; Gedil, M.A.; Ikpan, A.S.; Jannink, J.-L.; Kulakow, P.A. High-resolution mapping of resistance to cassava mosaic geminiviruses in cassava using genotyping-by-sequencing and its implications for breeding. Virus Res. 2014, 186, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Jennings, D.L. Breeding for resistance to African cassava mosaic disease: Progress and prospects. In African Cassava Mosaic; Barry, L.N., Ed.; International Development Research Centre: Ottawa, ON, Canada, 1976; p. 42. [Google Scholar]
- Wolfe, M.D.; Bauchet, G.J.; Chan, A.W.; Lozano, R.; Ramu, P.; Egesi, C.; Kawuki, R.; Kulakow, P.; Rabbi, I.; Jannink, J.-L. Historical Introgressions from a Wild Relative of Modern Cassava Improved Important Traits and May Be Under Balancing Selection. Genetics 2019, 213, 1237–1253. [Google Scholar] [CrossRef] [PubMed]
- Fregene, M.; Okogbenin, E.; Mba, C.; Angel, F.; Suarez, M.C.; Janneth, G.; Chavarriaga, P.; Roca, W.; Bonierbale, M.; Tohme, J. Genome mapping in cassava improvement: Challenges, achievements and opportunities. Euphytica 2001, 120, 159–165. [Google Scholar] [CrossRef]
- Lokko, Y.; Danquah, E.Y.; Offei, S.K.; Dixon, A.G.O.; Gedil, M.A. Molecular markers associated with a new source of resistance to the cassava mosaic disease. Afr. J. Biotechnol. 2005, 4. [Google Scholar] [CrossRef]
- Okogbenin, E.; Egesi, C.N.; Olasanmi, B.; Ogundapo, O.; Kahya, S.S.; Hurtado, P.; Marin, J.; Akinbo, O.; Mba, C.; Gomez, H.; et al. Molecular Marker Analysis and Validation of Resistance to Cassava Mosaic Disease in Elite Cassava Genotypes in Nigeria. Crop. Sci. 2012, 52, 2576–2586. [Google Scholar] [CrossRef]
- Wolfe, M.D.; Rabbi, I.Y.; Egesi, C.; Hamblin, M.; Kawuki, R.; Kulakow, P.; Lozano, R.; Del Carpio, D.P.; Ramu, P.; Jannink, J. Genome-Wide Association and Prediction Reveals Genetic Architecture of Cassava Mosaic Disease Resistance and Prospects for Rapid Genetic Improvement. Plant Genome 2016, 9, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Rabbi, I.Y.; Kayondo, S.I.; Bauchet, G.; Yusuf, M.; Aghogho, C.I.; Ogunpaimo, K.; Uwugiaren, R.; Smith, I.A.; Peteti, P.; Agbona, A.; et al. Genome-wide association analysis reveals new insights into the genetic architecture of defensive, agro-morphological and quality-related traits in cassava. Plant Mol. Biol. 2020, 1–19. [Google Scholar] [CrossRef]
- Ceballos, H.; Kawuki, R.S.; Gracen, V.E.; Yencho, G.C.; Hershey, C.H. Conventional breeding, marker-assisted selection, genomic selection and inbreeding in clonally propagated crops: A case study for cassava. Theor. Appl. Genet. 2015, 128, 1647–1667. [Google Scholar] [CrossRef] [PubMed]
- Chagné, D.; Vanderzande, S.; Kirk, C.; Profitt, N.; Weskett, R.; Gardiner, S.E.; Peace, C.P.; Volz, R.K.; Bassil, N.V. Validation of SNP markers for fruit quality and disease resistance loci in apple (Malus × domestica Borkh.) using the OpenArray® platform. Hortic. Res. 2019, 6, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Collard, B.C.; Mackill, D.J. Marker-assisted selection: An approach for precision plant breeding in the twenty-first century. Philos. Trans. R. Soc. B Biol. Sci. 2007, 363, 557–572. [Google Scholar] [CrossRef] [PubMed]
- Platten, J.D.; Cobb, J.N.; Zantua, R.E. Criteria for evaluating molecular markers: Comprehensive quality metrics to improve marker-assisted selection. PLoS ONE 2019, 14, e0210529. [Google Scholar] [CrossRef] [PubMed]
- Ceballos, H.; Kulakow, P.; Hershey, C. Cassava Breeding: Current Status, Bottlenecks and the Potential of Biotechnology Tools. Trop. Plant Biol. 2012, 5, 73–87. [Google Scholar] [CrossRef]
- Ceballos, H.; Rojanaridpiched, C.; Phumichai, C.; Becerra, L.A.; Kittipadakul, P.; Iglesias, C.; Gracen, V.E. Excellence in Cas-sava Breeding: Perspectives for the Future. CBGG 2020, 2. Available online: https://cbgg.hapres.com/UpLoad/PdfFile/CBGG_1209.pdf (accessed on 28 December 2020).
- Ferguson, M.; Rabbi, I.; Kim, D.-J.; Gedil, M.; López-Lavalle, L.A.B.; Okogbenin, E. Molecular Markers and Their Application to Cassava Breeding: Past, Present and Future. Trop. Plant Biol. 2011, 5, 95–109. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Abass, A.B.; Towo, E.; Mukuka, I.; Okechukwu, R.U.; Ranaivoson, R.; Tarawali, G.; Kanju, E. Growing Cassava: A Training Manual from Production to Postharvest; International Institute of Tropical Agriculture: Ibadan, Nigeria, 2014; pp. 1–29. [Google Scholar]
- Atser, G.; Dixon, A.; Ekeleme, F.; Chikoye, D.; Dashiell, K.E.; Ayankanmi, T.G.; Hauser, S.; Agada, M.; Okwusi, M.; Sokoya, G. The ABC of Weed Management in Cassava Production in Nigeria: A Training Manual; International Institute of Tropical Agriculture: Ibadan, Nigeria, 2017; pp. 1–30. [Google Scholar]
- Genomics LGC. KASP Genotyping Chemistry User Guide and Manual; LGC Limited: Teddington, UK, 2013; Available online: https://www.biosearchtech.com/ (accessed on 17 July 2018).
- Bates, D.; Mächler, M.; Bolker, B.; Walker, S. Fitting Linear Mixed-Effects Models Usinglme4. J. Stat. Softw. 2015, 67, 1–48. [Google Scholar] [CrossRef]
- R Development Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2015; Available online: http://www.R-project.org/ (accessed on 21 February 2021).
- Kling, J. Introduction to Augmented Designs. Oregon State University. National Association of Plant Breeders. 2011. Available online: https://pbgworks.org/sites/pbgworks.org/files/AugmentedDesignsFINAL.pdf (accessed on 24 December 2020).
- Kuhn, M.; Wickham, H. Tidymodels: Easily Install and Load the’Tidymodels’ Packages, R package version 0.1.0; 2020; Available online: https://tidymodels.tidymodels.org/ (accessed on 21 February 2021).
- Ray, S.; Satya, P. Next generation sequencing technologies for next generation plant breeding. Front. Plant Sci. 2014, 5, 367. [Google Scholar] [CrossRef]
- Win, K.T.; Vegas, J.; Zhang, C.; Song, K.; Lee, S. QTL mapping for downy mildew resistance in cucumber via bulked segregant analysis using next-generation sequencing and conventional methods. Theor. Appl. Genet. 2017, 130, 199–211. [Google Scholar] [CrossRef]
- Esuma, W.; Herselman, L.; Labuschagne, M.T.; Ramu, P.; Lu, F.; Baguma, Y.; Buckler, E.S.; Kawuki, R.S. Genome-wide association mapping of provitamin A carotenoid content in cassava. Euphytica 2016, 212, 97–110. [Google Scholar] [CrossRef]
- Rabbi, I.Y.; Udoh, L.I.; Wolfe, M.; Parkes, E.Y.; Gedil, M.A.; Dixon, A.; Ramu, P.; Jannink, J.; Kulakow, P. Genome-Wide Association Mapping of Correlated Traits in Cassava: Dry Matter and Total Carotenoid Content. Plant Genome 2017, 10. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Chen, X.; Lu, C.; Ye, J.; Zou, M.; Lu, K.; Feng, S.; Pei, J.; Liu, C.; Zhou, X.; et al. Genome-Wide Association Studies of 11 Agronomic Traits in Cassava (Manihot esculenta Crantz). Front. Plant Sci. 2018, 9, 503. [Google Scholar] [CrossRef] [PubMed]
- Neelam, K.; Brown-Guedira, G.; Huang, L. Development and validation of a breeder-friendly KASPar marker for wheat leaf rust resistance locus Lr21. Mol. Breed. 2012, 31, 233–237. [Google Scholar] [CrossRef]
- Li, L.; Tacke, E.; Hofferbert, H.-R.; Lübeck, J.; Strahwald, J.; Draffehn, A.M.; Walkemeier, B.; Gebhardt, C. Validation of candidate gene markers for marker-assisted selection of potato cultivars with improved tuber quality. Theor. Appl. Genet. 2013, 126, 1039–1052. [Google Scholar] [CrossRef]
- Rasheed, A.; Wen, W.; Gao, F.; Zhai, S.; Jin, H.; Liu, J.; Guo, Q.; Zhang, Y.; Dreisigacker, S.; Xia, X.; et al. Development and validation of KASP assays for genes underpinning key economic traits in bread wheat. Theor. Appl. Genet. 2016, 129, 1843–1860. [Google Scholar] [CrossRef] [PubMed]
- Chandra, S.; Singh, D.; Pathak, J.; Kumari, S.; Kumar, M.; Poddar, R.; Balyan, H.S.; Prabhu, K.V.; Gupta, P.K.; Mukhopadhyay, K. SNP discovery from next-generation transcriptome sequencing data and their validation using KASP assay in wheat (Triticum aestivum L.). Mol. Breed. 2017, 37, 92. [Google Scholar] [CrossRef]
- Udoh, L.I.; Parkes, E.Y.; Kulakow, P.; Adesoye, A.; Nwuba, C.; Rabbi, I.Y.; Gedil, M. Candidate gene sequencing and validation of SNP markers linked to carotenoid content in cassava (Manihot esculenta Crantz). Mol. Breed. 2017, 37, 123. [Google Scholar] [CrossRef]
- Javid, M.; Rosewarne, G.M.; Sudheesh, S.; Kant, P.; Leonforte, A.; Lombardi, M.; Kennedy, P.R.; Cogan, N.O.I.; Slater, A.T.; Kaur, S. Validation of molecular markers associated with boron tolerance, powdery mildew resistance and salinity tolerance in field peas. Front. Plant Sci. 2015, 6, 917. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.-T.; Assanga, S.; Zhang, G.; Rudd, J.C.; Haley, S.D.; Xue, Q.; Ibrahim, A.; Bai, G.; Zhang, X.; Byrne, P.; et al. Development and Validation of KASP Markers for Wheat Streak Mosaic Virus Resistance Gene Wsm2. Crop. Sci. 2016, 57, 340–349. [Google Scholar] [CrossRef]
- Trebbi, D.; Ravi, S.; Broccanello, C.; Chiodi, C.; Francis, G.; Oliver, J.; Mulpuri, S.; Srinivasan, S.; Stevanato, P. Identification and validation of SNP markers linked to seed toxicity in Jatropha curcas L. Sci. Rep. 2019, 9, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Zhang, P.; Wu, R.; Lu, K.; Zhou, H. Identifying the Best Marker Combination in CEA, CA125, CY211, NSE, and SCC for Lung Cancer Screening by Combining ROC Curve and Logistic Regression Analyses: Is It Feasible? Dis. Markers 2018, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Ceballos, H.; Gracen, V.E. A Dominant Inhibitor Gene Inhibits the Expression of Ht2 against Exserohilum turcicum Race 2 in Corn Inbred Lines Related to B14. Plant Breed. 1989, 102, 35–44. [Google Scholar] [CrossRef]
- Pita, J.S.; Fondong, V.N.; Sangaré, A.; Otim-Nape, G.W.; Ogwal, S.; Fauquet, C.M. Recombination, pseudorecombination and synergism of geminiviruses are determinant keys to the epidemic of severe cassava mosaic disease in Uganda. J. Gen. Virol. 2001, 82, 655–665. [Google Scholar] [CrossRef]
- Zinga, I.; Chiroleu, F.; Legg, J.; Lefeuvre, P.; Komba, E.K.; Semballa, S.; Yandia, S.P.; Mandakombo, N.B.; Reynaud, B.; Lett, J.-M. Epidemiological assessment of cassava mosaic disease in Central African Republic reveals the importance of mixed viral infection and poor health of plant cuttings. Crop. Prot. 2013, 44, 6–12. [Google Scholar] [CrossRef]
- Mukiibi, D.R.; Alicai, T.; Kawuki, R.; Okao-Okuja, G.; Tairo, F.; Sseruwagi, P.; Ndunguru, J.; Ateka, E.M. Resistance of advanced cassava breeding clones to infection by major viruses in Uganda. Crop. Prot. 2019, 115, 104–112. [Google Scholar] [CrossRef]
- Mulamba, N.N.; Hallauer, A.R.; Smith, O.S. Recurrent Selection for Grain Yield in a Maize Population 1. Crop. Sci. 1983, 23, 536–540. [Google Scholar] [CrossRef]
- Iglesias, C.A.; Hallauer, A.R. S2 recurrent selection in maize populations with exotic germplasm. Maydica 1989, 34, 133–140. [Google Scholar]
- Acquaah, G. Principles of Plant. Genetics and Breeding; Blackwell Publishing Limited: Malden, MA, USA, 2007; pp. 371, 374. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Markers | Chr. | Location (bp) | Allele 1 | Allele 2 | β | SE | p-Value |
|---|---|---|---|---|---|---|---|
| S12_7926132 | 12 | 7926132 | G | T * | 0.89 | 0.02 | p ≈ 0 |
| S12_7926163 | 12 | 7926163 | A | G * | 0.89 | 0.02 | p ≈ 0 |
| S14_4626854 | 14 | 4626854 | A * | G | −0.23 | 0.03 | 1.00 × 10–14 |
| Markers | SNP and 100 bp Flanking Sequences | Forward Primer Allele 1 and Allele 2 | Primer Common | Reference |
|---|---|---|---|---|
| S12_7926132 | CTGCACACTCAAAGCTGCATCCTATTTTCCATGTTTCCACCCTCAAATG(G/T)TATCACAAAGGACAAGATTCTTGTACTCCAATGCTGCCACCAACTCCACC | Allele 1: TTCCATGTTTCCACCCTCAAATGG; Allele 2: TTCCATGTTTCCACCCTCAAATGT | GGAGTACAAGAATCTTGTCCTTTGTGATA | [25] |
| S12_7926163 | TGTTTCCACCCTCAAATGGTATCACAAAGGACAAGATTCTTGTACTCCA(A/G)TGCTGCCACCAACTCCACCTGATGTTCCTCTTCAACCTCTGGCTGTTTTA | Allele 1: ACAAAGGACAAGATTCTTGTACTCCAA; Allele 2: CAAAGGACAAGATTCTTGTACTCCAG | GTTGAAGAGGAACATCAGGTGGAGTT | [25] |
| S14_4626854 | ACCACTGCATCTTGTGCTCATGAGCCATTGCACGCTGCACCTCTTCATT(G/A)ATCGCTCATTTGCATCCCACCTTTGGATAGCGCGACTATGAGCTGCATCA | Allele 1: GCACGCTGCACCTCTTCATTA; Allele 2: GCACGCTGCACCTCTTCATTG | CAAAGGTGGGATGCAAATGAGCGAT | [25] |
| Breeding Population | Pre-Breeding Population | |||||
|---|---|---|---|---|---|---|
| Traits | σ2g | σ2e | H2 | σ2g | σ2e | H2 |
| CMD severity score | 1.15 | 0.13 | 0.90 | 0.98 | 0.18 | 0.84 |
| Root number | 13.02 | 50.02 | 0.21 | 76.96 | 97.66 | 0.44 |
| Root weight | 5.51 | 11.04 | 0.33 | 41.54 | 49.52 | 0.46 |
| Breeding | Pre-Breeding | |||||||
|---|---|---|---|---|---|---|---|---|
| cmd1s_CET | cmd3s_CET | rtno_CET | rtwt_CET | cmd1s_CET | cmd3s_CET | rtno_CET | rtwt_CET | |
| cmd3s_SN | 0.75 | 0.76 | −0.19 | −0.28 | 0.55 | 0.50 | −0.25 | −0.25 |
| cmd1s_CET | 0.84 | −0.21 | −0.31 | 0.68 | −0.31 | −0.35 | ||
| cmd3s_CET | −0.18 | −0.26 | −0.29 | −0.33 | ||||
| rtno_CET | 0.75 | 0.73 | ||||||
| Population | N | Accuracy | Standard Error | AUC | Standard Error | |
|---|---|---|---|---|---|---|
| Breeding | Training set | 1351 | 0.80 | 0.003 | 0.80 | 0.005 |
| Testing set | 450 | 0.80 | 0.80 | |||
| Pre-breeding | Training set | 2574 | 0.76 | 0.003 | 0.82 | 0.005 |
| Testing set | 857 | 0.78 | 0.86 |
| Truth | ||||||
|---|---|---|---|---|---|---|
| Population | Prediction | Resistant | Susceptible | FPR (%) | FNR (%) | Misclassification (%) |
| Breeding | Resistant | 242 | 54 | 33 | 16 | 22.0 |
| Susceptible | 45 | 108 | ||||
| Pre-breeding | Resistant | 269 | 72 | 15 | 29 | 21.2 |
| Susceptible | 109 | 405 | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ige, A.D.; Olasanmi, B.; Mbanjo, E.G.N.; Kayondo, I.S.; Parkes, E.Y.; Kulakow, P.; Egesi, C.; Bauchet, G.J.; Ng, E.; Lopez-Lavalle, L.A.B.; et al. Conversion and Validation of Uniplex SNP Markers for Selection of Resistance to Cassava Mosaic Disease in Cassava Breeding Programs. Agronomy 2021, 11, 420. https://doi.org/10.3390/agronomy11030420
Ige AD, Olasanmi B, Mbanjo EGN, Kayondo IS, Parkes EY, Kulakow P, Egesi C, Bauchet GJ, Ng E, Lopez-Lavalle LAB, et al. Conversion and Validation of Uniplex SNP Markers for Selection of Resistance to Cassava Mosaic Disease in Cassava Breeding Programs. Agronomy. 2021; 11(3):420. https://doi.org/10.3390/agronomy11030420
Chicago/Turabian StyleIge, Adenike D., Bunmi Olasanmi, Edwige Gaby Nkouaya Mbanjo, Ismail S. Kayondo, Elizabeth Y. Parkes, Peter Kulakow, Chiedozie Egesi, Guillaume J. Bauchet, Enghwa Ng, Luis Augusto Becerra Lopez-Lavalle, and et al. 2021. "Conversion and Validation of Uniplex SNP Markers for Selection of Resistance to Cassava Mosaic Disease in Cassava Breeding Programs" Agronomy 11, no. 3: 420. https://doi.org/10.3390/agronomy11030420
APA StyleIge, A. D., Olasanmi, B., Mbanjo, E. G. N., Kayondo, I. S., Parkes, E. Y., Kulakow, P., Egesi, C., Bauchet, G. J., Ng, E., Lopez-Lavalle, L. A. B., Ceballos, H., & Rabbi, I. Y. (2021). Conversion and Validation of Uniplex SNP Markers for Selection of Resistance to Cassava Mosaic Disease in Cassava Breeding Programs. Agronomy, 11(3), 420. https://doi.org/10.3390/agronomy11030420