
Synthesis of Vinyl-Containing Polydimethylsiloxane in An Active Medium

 , ,
, ,  , , and
, , and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Characterization
2.3. Synthesis of PDMS-50-Vin (MeVin/Me2 = 1/1) by Simultaneous Mixing
2.4. Synthesis of PDMS-75-Vin (MeVin/Me2 = 3/1) by Simultaneous Mixing
2.5. Synthesis of PDMS-25-Vin (MeVin/Me2 = 1/3) by Simultaneous Mixing
2.6. Synthesis of PDMS-100-Vin (MeVin/Me2 = 1/0) by Simultaneous Mixing
2.7. Synthesis of PDMS-50-Vin (MeVin/Me2 = 1/1) by Slow Injection
2.8. Synthesis of PDMS-75-Vin (MeVin/Me2 = 3/1) by Slow Injection
2.9. Synthesis of PDMS-25-Vin (MeVin/Me2 = 1/3) by Slow Injection
2.10. Synthesis of PDMS-100-Vin (MeVin/Me2 = 1/0) by Slow Injection
2.11. Blocking of Condensation Products
2.12. Synthesis of PDMS-50-Vin (MeVin/Me2 = 1/1) by Slow Introduction with Rate of 0.3 mL/min
2.13. Synthesis of PDMS-75-Vin (MeVin/Me2 = 3/1) by Slow Introduction with Rate of 0.3 mL/min
2.14. Synthesis of PDMS-25-Vin (MeVin/Me2 = 1/3) by Slow Introduction with Rate of 0.3 mL/min
2.15. Oligo(methylvinyl)(dimethyl)siloxane Thermal Condensation
2.16. Oligo(methylvinyl)(dimethyl)siloxane Thermal Condensation with AcOK
2.17. The Analysis of the Microstructure of Copolymers
3. Results and Discussion
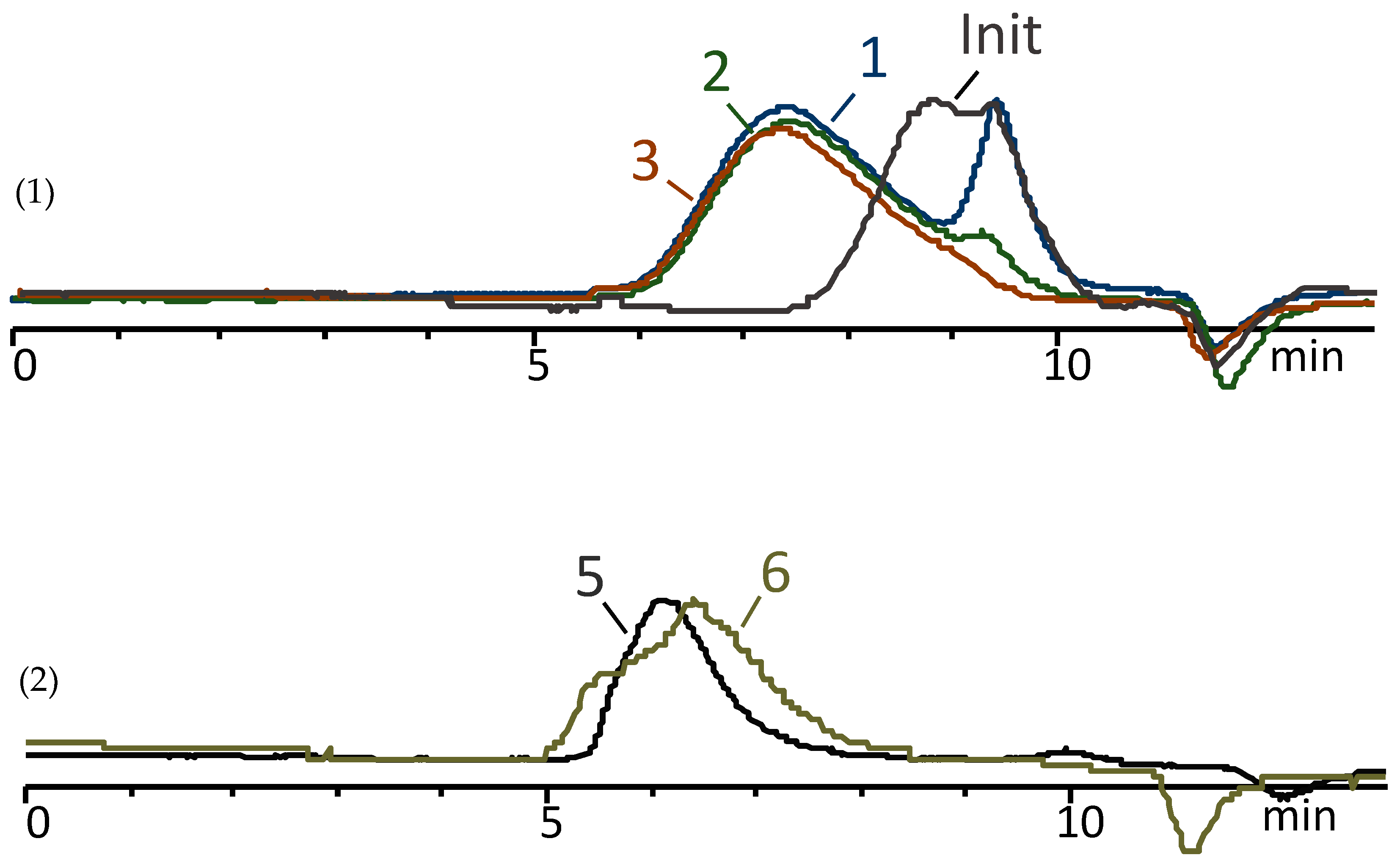
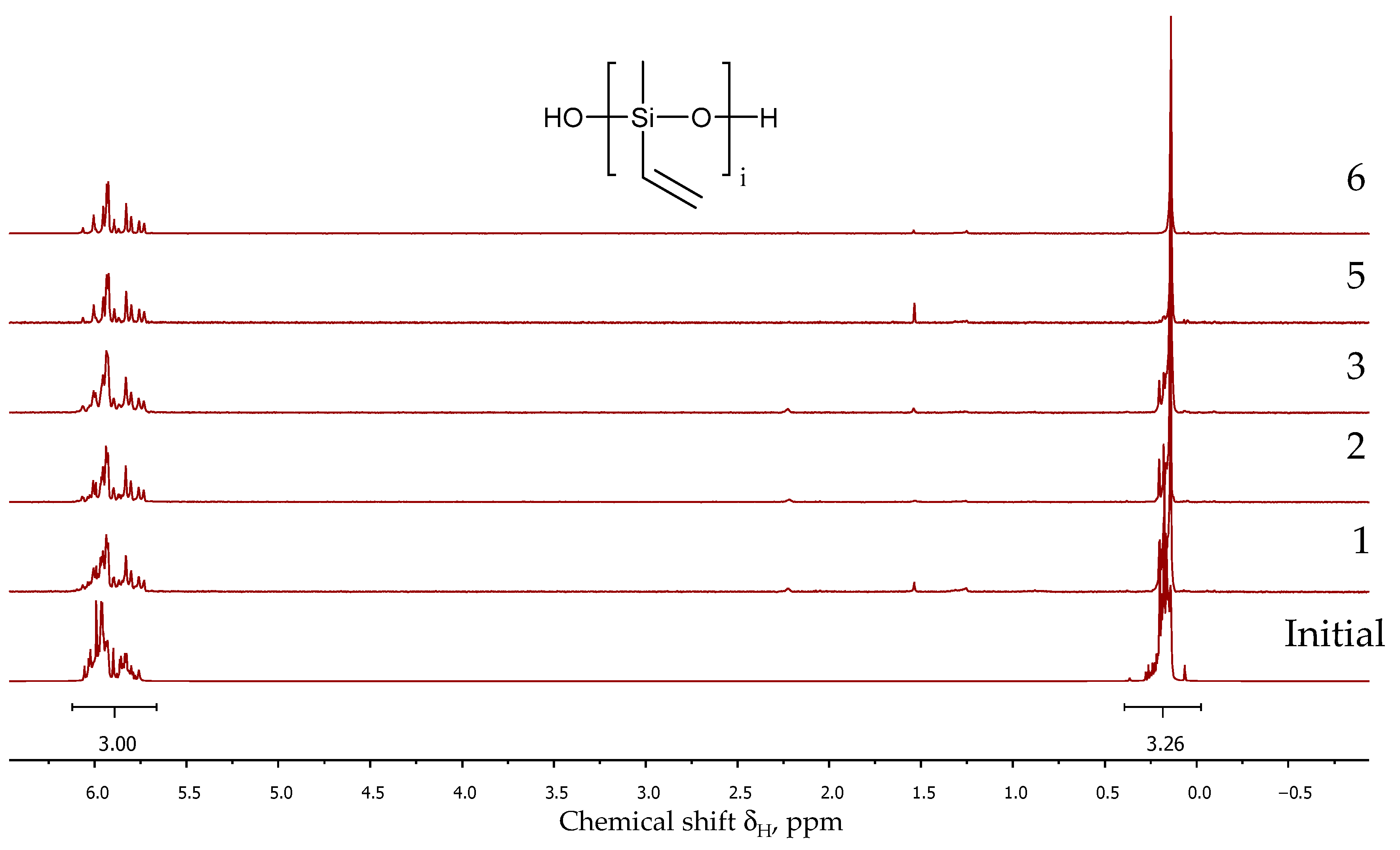
3.1. Copolycondensation of Dimethyldiethoxysilane and Methylvinyldimethoxysilane in Active Medium
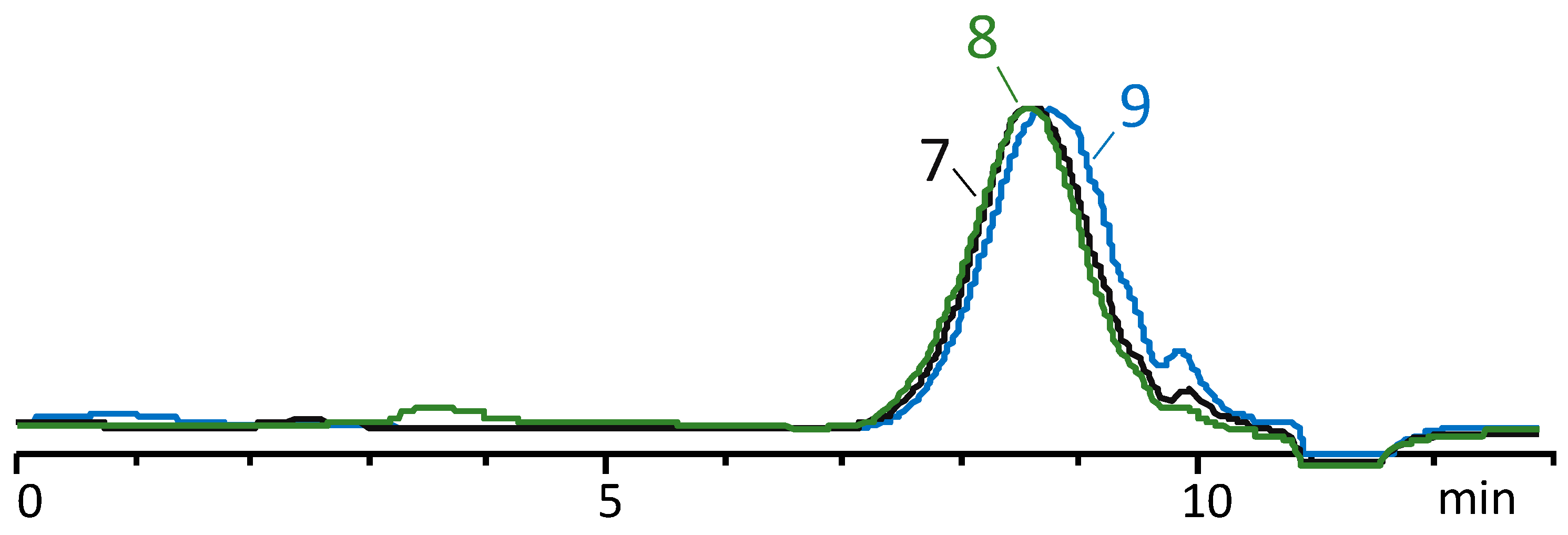
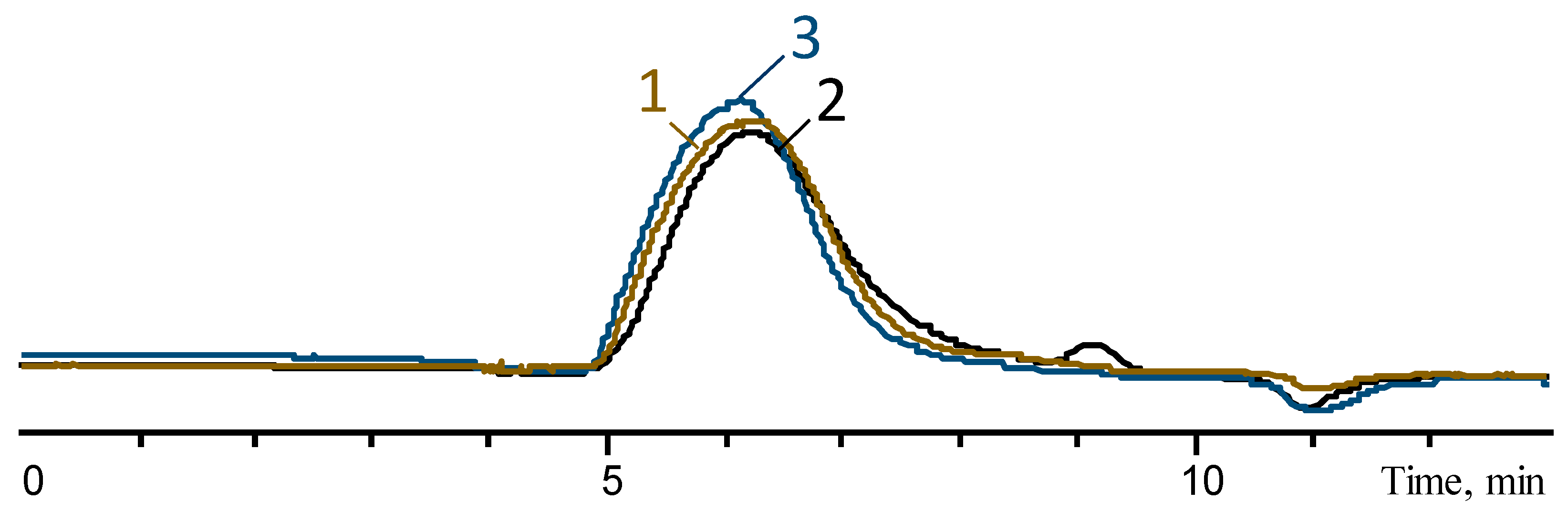
3.2. Enhancing the Molecular Weight of Oligodimethylmethylvinylsiloxanes
- -
- Only in the presence of a catalyst for the condensation of silanol groups—AcOK in the case of methylvinylsiloxane oligomers PDMS-100-Vin;
- -
- Both in the presence of a catalyst and without it in the of case oligomers with a mixed composition. Simultaneously with postcondensation, the cycles formed at the first stage of polycondensation in the active medium are distilled off, which makes it possible to obtain a final polymer that does not contain low-molecular-weight cyclic impurities (according to GPCs).
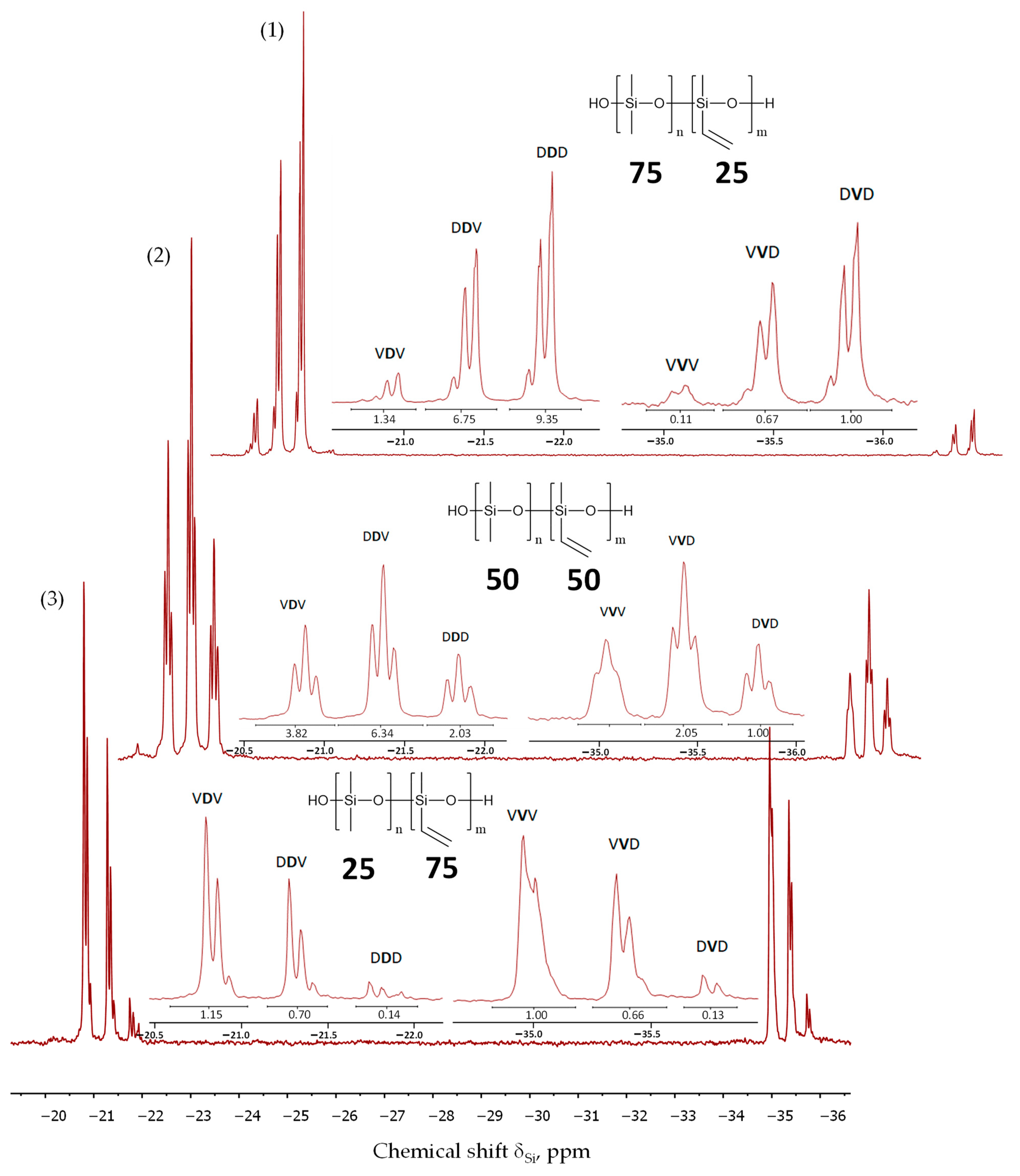
3.3. The Study of Polymer Microstructure
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bezsudnov, I.V.E.; Khmelnitskaia, A.G.; Kalinina, A.A.; Ponomarenko, S.A. Dielectric elastomer actuators: Materials and design. Russ. Chem. Rev. 2023, 92, RCR5070. [Google Scholar] [CrossRef]
- Mancuso, A.; Tarsitano, M.; Udongo, B.P.; Cristiano, M.C.; Torella, D.; Paolino, D.; Fresta, M. A comparison between silicone-free and silicone-based emulsions: Technological features and in vivo evaluation. Int. J. Cosmet. Sci. 2022, 44, 514. [Google Scholar] [CrossRef] [PubMed]
- Olejnik, A.; Sztorch, B.; Brząkalski, D.; Przekop, R.E. Silsesquioxanes in the Cosmetics Industry—Applications and Perspectives. Materials 2022, 15, 1126. [Google Scholar] [CrossRef]
- Segal, M. Material history: Learning from silicon. Nature 2012, 483, S43. [Google Scholar] [CrossRef]
- Miranda, I.; Souza, A.; Sousa, P.; Ribeiro, J.; Castanheira, E.M.; Lima, R.; Minas, G. Properties and applications of PDMS for biomedical engineering: A review. J. Funct. Biomater. 2021, 13, 2. [Google Scholar] [CrossRef]
- Opris, D.M. Polar elastomers as novel materials for electromechanical actuator applications. Adv. Mater. 2018, 30, 1703678. [Google Scholar] [CrossRef]
- Wagner, S.; Bauer, S. Materials for Stretchable Electronics. MRS Bull. 2012, 37, 207. [Google Scholar] [CrossRef]
- Wang, D.P.; Zhao, Z.H.; Li, C.H.; Zuo, J.L. An ultrafast self-healing polydimethylsiloxane elastomer with persistent sealing performance. Mater. Chem. Front. 2019, 3, 1411. [Google Scholar] [CrossRef]
- Ariati, R.; Sales, F.; Souza, A.; Lima, R.A.; Ribeiro, J. Polydimethylsiloxane composites characterization and its applications: A review. Polymers 2021, 13, 4258. [Google Scholar] [CrossRef]
- Kumar, V.; Alam, M.N.; Manikkavel, A.; Song, M.; Lee, D.J.; Park, S.S. Silicone Rubber Composites Reinforced by Carbon Nanofillers and Their Hybrids for Various Applications: A Review. Polymers 2021, 13, 2322. [Google Scholar] [CrossRef]
- Li, J.; Shepelin, N.A.; Sherrell, P.C.; Ellis, A.V. Poly (dimethylsiloxane) for Triboelectricity: From Mechanisms to Practical Strategies. Chem. Mater. 2021, 33, 4304. [Google Scholar] [CrossRef]
- Obrezkova, M.A.; Gorodov, V.V.; Khanin, D.A.; Buzin, M.I.; Bobrova, O.V.; Gorshkov, A.V.; Muzafarov, A.M. Synthesis of a Frost-Resistant Siloxane Rubber and an Elastomer on its Basis. INEOS Open 2022, 5, 74. [Google Scholar] [CrossRef]
- Liu, L.; Yang, S.; Zhang, Z.; Wang, Q.; Xie, Z. Synthesis and characterization of poly (diethylsiloxane) and its copolymers with different diorganosiloxane units. J. Polym. Sci. 2003, 41, 2722. [Google Scholar] [CrossRef]
- Caspari, P.; Nüesch, F.A.; Opris, D.M. Synthesis of solvent-free processable and on-demand cross-linkable dielectric elastomers for actuators. J. Mater. Chem. C 2019, 7, 12139. [Google Scholar] [CrossRef]
- Perju, E.; Ko, Y.S.; Dünki, S.J.; Opris, D.M. Increased electromechanical sensitivity of polysiloxane elastomers by chemical modification with thioacetic groups. Mater. Des. 2020, 186, 108319. [Google Scholar] [CrossRef]
- Sheima, Y.; Caspari, P.; Opris, D.M. Artificial muscles: Dielectric elastomers responsive to low voltages. Macromol. Rapid Commun. 2019, 40, e1900205. [Google Scholar] [CrossRef]
- Sheima, Y.; Yuts, Y.; Frauenrath, H.; Opris, D.M. Polysiloxanes modified with different types and contents of polar groups: Synthesis, structure, and thermal and dielectric properties. Macromolecules 2021, 54, 5737. [Google Scholar] [CrossRef]
- Dünki, S.J.; Nüesch, F.A.; Opris, D.M. Elastomers with tunable dielectric and electromechanical properties. J. Mater. Chem. C 2016, 4, 10545. [Google Scholar] [CrossRef]
- Perju, E.; Cuervo-Reyes, E.; Shova, S.; Opris, D.M. Synthesis of novel cyclosiloxane monomers containing push-pull moieties and their anionic ring opening polymerization. RCS Adv. 2018, 8, 7569. [Google Scholar] [CrossRef]
- Perju, E.; Shova, S.; Opris, D.M. Electrically Driven Artificial Muscles Using Novel Polysiloxane Elastomers Modified Nitroaniline Push-Pull Moieties. ACS Appl. Mater. Interfaces 2020, 12, 23432. [Google Scholar] [CrossRef]
- Mukbaniani, O.; Tatrishvili, T.; Koberidze, K.; Scherf, U. Hydride addition of methylhydridesiloxanes to conjugated cyclohexa-1, 3-diene. J. Appl. Polym. Sci. 2010, 116, 1131. [Google Scholar] [CrossRef]
- Wang, D.; Klein, J.; Mejía, E. Catalytic systems for the cross-linking of organosilicon polymers. Chem. Asian J. 2017, 12, 1180. [Google Scholar] [CrossRef]
- Sunitha, K.; Bhuvaneswari, S.; Mathew, D.; Unnikrishnan, G.; Nair, C.R. Comb polymer network of polydimethylsiloxane with a novolac stem: Synthesis via click coupling and surface morphology architecturing by solvents. Macromolecules 2017, 50, 9656. [Google Scholar] [CrossRef]
- Khatir, B.; Azimi Dijvejin, Z.; Serles, P.; Filleter, T.; Golovin, K. Molecularly Capped Omniphobic Polydimethylsiloxane Brushes with Ultra-Fast Contact Line Dynamics. Small 2023, 19, 2301142. [Google Scholar] [CrossRef]
- Cao, J.; Wang, Y.; Wang, D.; Sun, R.; Guo, M.; Feng, S. A Super-Amphiphilic 3D Silicone Sponge with High Porosity for the Efficient Adsorption of Various Pollutants. Macromol. Rapid Commun. 2021, 42, e2000603. [Google Scholar] [CrossRef]
- Huang, Y.; Yan, J.; Wang, D.; Feng, S.; Zhou, C. Construction of self-healing disulfide-linked silicone elastomers by thiol oxidation coupling reaction. Polymers 2021, 13, 3729. [Google Scholar] [CrossRef]
- Caspari, P.; Dünki, S.J.; Nüesch, F.A.; Opris, D.M. Dielectric elastomer actuators with increased dielectric permittivity and low leakage current capable of suppressing electromechanical instability. J. Mater. Chem. C 2018, 6, 2043. [Google Scholar] [CrossRef]
- Sun, H.; Liu, X.; Liu, S.; Yu, B.; Ning, N.; Tian, M.; Zhang, L. Silicone dielectric elastomer with improved actuated strain at low electric field and high self-healing efficiency by constructing supramolecular network. J. Chem. Eng. 2020, 384, 123242. [Google Scholar] [CrossRef]
- Sun, H.; Liu, X.; Liu, S.; Yu, B.; Ning, N.; Tian, M.; Zhang, L. A supramolecular silicone dielectric elastomer with a high dielectric constant and fast and highly efficient self-healing under mild conditions. J. Mater. Chem. A 2020, 8, 23330. [Google Scholar] [CrossRef]
- Xu, Y.; Lu, S.; Wei, Z.; Feng, S. Supramolecular Elastomers with Excellent Dielectric Properties and High Recyclability Based on the Coordinative Bond. Macromol. Rapid Commun. 2023, 44, e2200766. [Google Scholar] [CrossRef]
- Goff, J.; Sulaiman, S.; Arkles, B. Applications of hybrid polymers generated from living anionic ring opening polymerization. Molecules 2021, 26, 2755. [Google Scholar] [CrossRef]
- Chruściel, J.J. Silicon-Based Polymers and Materials; Walter de Gruyter GmbH & Co KG: Berlin, Boston, 2022. [Google Scholar] [CrossRef]
- Park, Y.; Kang, D.W.; Kang, H.J. Synthesis and Characterization of Vinyl-Terminated Poly (dimethyl-co-methylvinyl) siloxane by Ring Opening Polymerization. Macromol. Res. 2021, 29, 569–575. [Google Scholar] [CrossRef]
- Egorova, E.V.; Vasilenko, N.G.; Demchenko, N.V.; Tatarinova, E.A.; Muzafarov, A.M. Polycondensation of alkoxysilanes in an active medium as a versatile method for the preparation of polyorganosiloxanes. Dokl. Chem. 2009, 424, 15. [Google Scholar] [CrossRef]
- Kalinina, A.; Strizhiver, N.; Vasilenko, N.; Perov, N.; Demchenko, N.; Muzafarov, A. Polycondensation of diethoxydimethylsilane in active medium. Silicon 2014, 7, 95. [Google Scholar] [CrossRef]
- Bychkova, A.A.; Soskov, F.V.; Demchenko, A.I.; Storozhenko, P.A.; Muzafarov, A.M. Condensation of methylphenylalkoxysilanes in an active medium as a selective method for synthesis of cyclic or linear methylphenylsiloxanes. Russ. Chem. Bull. 2011, 60, 2384. [Google Scholar] [CrossRef]
- Kalinina, A.A.; Talalaeva, E.V.; Demchenko, A.I.; Vasilenko, N.G.; Molodtsova, Y.A.; Demchenko, N.V.; Muzafarov, A.M. Synthesis of dimethylcyclosiloxanes in the active medium. Russ. Chem. Bull. 2016, 65, 1013. [Google Scholar] [CrossRef]
- Armaredo, L.F.; Perkin, D.D. Purification of Laboratory Chemicals, 4th ed.; Butterworth Heinemann: Oxford, UK, 1999. [Google Scholar]
- Czerwinski, W.K. Improved increments for characterization of comonomer sequencing in binary copolymers. Polymer 1997, 38, 1381. [Google Scholar] [CrossRef]
- Shragin, D.I.; Kopylov, V.M.; Ivanov, P.V.; Sokol’skaya, I.B.; Savitskii, A.A. Study of anionic copolymerization of α, ω-bis (trimethylsiloxy) polydimethylsiloxane with organocyclosiloxanes. Polym. Sci. Ser. A 2007, 49, 381. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| № | Process Conditions | Characteristics | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PDMS-X-Vin | t a, h | N b (HO), wt. % | Products’ Composition, wt. % | Molecular Weight Characteristics of L, wt. % | MeVinSiO/ Me2SiO for L | ||||||||
| D3–7 | DnVm | V3–7 | L | Mp | Mw | Mn | PDI | Th. | Real | ||||
| 1 | PDMS-100-Vin-Ⅰ | 8 | 7.3 | - | - | 42 | 58 | 1150 | 860 | 670 | 1.3 | - | - |
| 2 | PDMS-25-Vin-Ⅰ | 8 | 4.8 | 10 | 35.5 | - | 55 | 1000 | 1050 | 750 | 1.4 | 3/1 | 3/0.75 |
| 3 | PDMS-50-Vin-Ⅰ | 8 | 6.5 | 1 | 38.5 | 0.5 | 60 | 1000 | 870 | 660 | 1.3 | 1/1 | 1/0.85 |
| 4 | PDMS-75-Vin-Ⅰ | 8 | 6.8 | - | 34.6 | 2.4 | 63 | 1000 | 900 | 700 | 1.3 | 1/3 | 1/2.22 |
| 5 | PDMS-0-Vin-Ⅰ ** | - | - | 55 | - | - | 45 | 1700 | - | - | - | - | - |
| 6 | PDMS-100-Vin-Ⅱ | 6 * | 7.0 | - | - | 27 | 73 | 1600 | 900 | 680 | 1.3 | - | - |
| 7 | PDMS-25-Vin-Ⅱ | 6 * | 4.7 | 7 | 23 | - | 70 | 1600 | 1200 | 900 | 1.3 | 3/1 | 3/0.88 |
| 8 | PDMS-50-Vin-Ⅱ | 6 * | 5.7 | 1 | 19.6 | 0.4 | 79 | 1700 | 1300 | 900 | 1.4 | 1/1 | 1/0.85 |
| 9 | PDMS-75-Vin-Ⅱ | 6 * | 6.5 | 0.2 | 16 | 9.8 | 74 | 1400 | 1100 | 800 | 1.3 | 1/3 | 1/2.36 |
| 10 | PDMS-0-Vin-Ⅱ ** | - | - | 12 | - | - | 88 | 1700 | - | - | - | - | - |
| PDMS-X-Vin | Monomer Introduction Rate | Cyclic/Linear, %/% |
|---|---|---|
| PDMS-75-Vin | 0.1 mL/min | 26/74 |
| 0.3 mL/min | 38/62 | |
| Simultaneous mixing | 45.5/54.5 | |
| PDMS-50-Vin | 0.1 mL/min | 21/79 |
| 0.3 mL/min | 35/65 | |
| Simultaneous mixing | 40/60 | |
| PDMS-25-Vin | 0.1 mL/min | 30/70 |
| 0.3 mL/min | 36/64 | |
| Simultaneous mixing | 37/63 |
| № | Process Conditions | Characteristics | |||||
|---|---|---|---|---|---|---|---|
| t, h | T, °C/AcOK | Before Condensation | After Condensation | ||||
| Mp | Cycles Content, % | Mp | Cycles Content, % | PDI | |||
| initial | - | 25/- | 1100 | 36 | - | - | |
| 1 | 5 | 50/- | 1100 | 36 | 4200 | 26 | 1.49 |
| 2 | 5 | 100/- | 4200 | 26 | 4200 | 9 | 1.55 |
| 3 | 5 | 150/- | 4200 | 9 | 4700 | 0 | 1.73 |
| 4 | 5 | 150/AcOK | 4700 | 0 | gel | - | - |
| 5 | 5 | 150/AcOK | 1100 | 36 | 29,000 | 0 | - |
| 6 | 5 | 150/AcOK | 29,000 | 0 | 124,000 | 0 | - |
| № | Conditions | Characteristics | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PDMS-75-Vin | PDMS-50-Vin | PDMS-25-Vin | ||||||||||||
| t, h | T, °C/ AcOK | Before | After | Before | After | Before | After | |||||||
| Mp | C *, % | Mp | C *, % | Mp | C *, % | Mp | C *, % | Mp | C *, % | Mp | C *, % | |||
| 1 | 5 | 50/- | 1400 | 27 | 7500 | 17 | 1700 | 20 | 7900 | 10 | 1600 | 25 | 8000 | 9 |
| 2 | 5 | 100/- | 7500 | 17 | 18,000 | 5 | 7900 | 10 | 17,500 | 4 | 8000 | 9 | 15,000 | 2 |
| 3 | 5 | 150/- | 18,000 | 5 | 20,000 | 0 | 17,500 | 4 | 18,000 | 0 | 15,000 | 2 | 18,000 | 0 |
| 4 | 5 | 150/AcOK | 1400 | 27 | 25,000 | 0 | 1700 | 20 | 22,000 | 0 | 1600 | 25 | 21,500 | 0 |
| № | PDMS-X-Vin | Molecular Weight Characteristics | |||
|---|---|---|---|---|---|
| Mp | Mw | Mn | PDI | ||
| 1 | PDMS-25-Vin | 18,000 | 31,500 | 16,700 | 1.9 |
| 2 | PDMS-50-Vin | 18,000 | 29,700 | 20,000 | 1.5 |
| 3 | PDMS-75-Vin | 20,000 | 25,500 | 15,200 | 1.7 |
| Triad | δSi, ppm | Integral Intensity of Triad (%) | ||
|---|---|---|---|---|
| PDMS-75-Vin (R = 1.21) | PDMS-50-Vin (R = 1.22) | PDMS-25-Vin (R = 1.02) | ||
| DVD | –(35.7–36.0) | 3 | 6 | 5 |
| DVV | –(35.3–35.6) | 17 | 13 | 3 |
| VVV | –(34.9–35.2) | 26 | 7 | 1 |
| DDD | –(21.7–22.1) | 4 | 12 | 49 |
| VDD | –(21.2–21.6) | 19 | 39 | 35 |
| VDV | –(20.7–21.1) | 30 | 23 | 7 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khmelnitskaia, A.G.; Kalinina, A.A.; Meshkov, I.B.; Tukhvatshin, R.S.; Cherkaev, G.V.; Ponomarenko, S.A.; Muzafarov, A.M. Synthesis of Vinyl-Containing Polydimethylsiloxane in An Active Medium. Polymers 2024, 16, 257. https://doi.org/10.3390/polym16020257
Khmelnitskaia AG, Kalinina AA, Meshkov IB, Tukhvatshin RS, Cherkaev GV, Ponomarenko SA, Muzafarov AM. Synthesis of Vinyl-Containing Polydimethylsiloxane in An Active Medium. Polymers. 2024; 16(2):257. https://doi.org/10.3390/polym16020257
Chicago/Turabian StyleKhmelnitskaia, Alina G., Aleksandra A. Kalinina, Ivan B. Meshkov, Rinat S. Tukhvatshin, Georgii V. Cherkaev, Sergey A. Ponomarenko, and Aziz M. Muzafarov. 2024. "Synthesis of Vinyl-Containing Polydimethylsiloxane in An Active Medium" Polymers 16, no. 2: 257. https://doi.org/10.3390/polym16020257
APA StyleKhmelnitskaia, A. G., Kalinina, A. A., Meshkov, I. B., Tukhvatshin, R. S., Cherkaev, G. V., Ponomarenko, S. A., & Muzafarov, A. M. (2024). Synthesis of Vinyl-Containing Polydimethylsiloxane in An Active Medium. Polymers, 16(2), 257. https://doi.org/10.3390/polym16020257