

Origin of Optoelectronic Contradictions in 3,4-Cycloalkyl[c]-chalcogenophenes: A Computational Study


Abstract
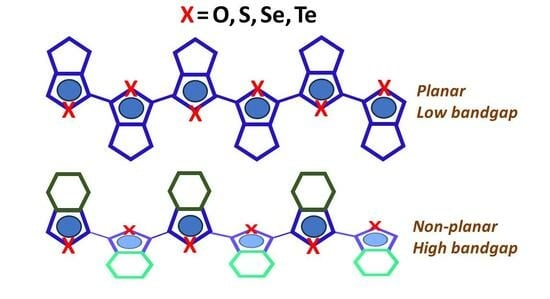
:
1. Introduction
2. Materials and Methods
2.1. Single Crystal X-ray Diffraction (SCXRD)
2.2. Computational Methodology
3. Results
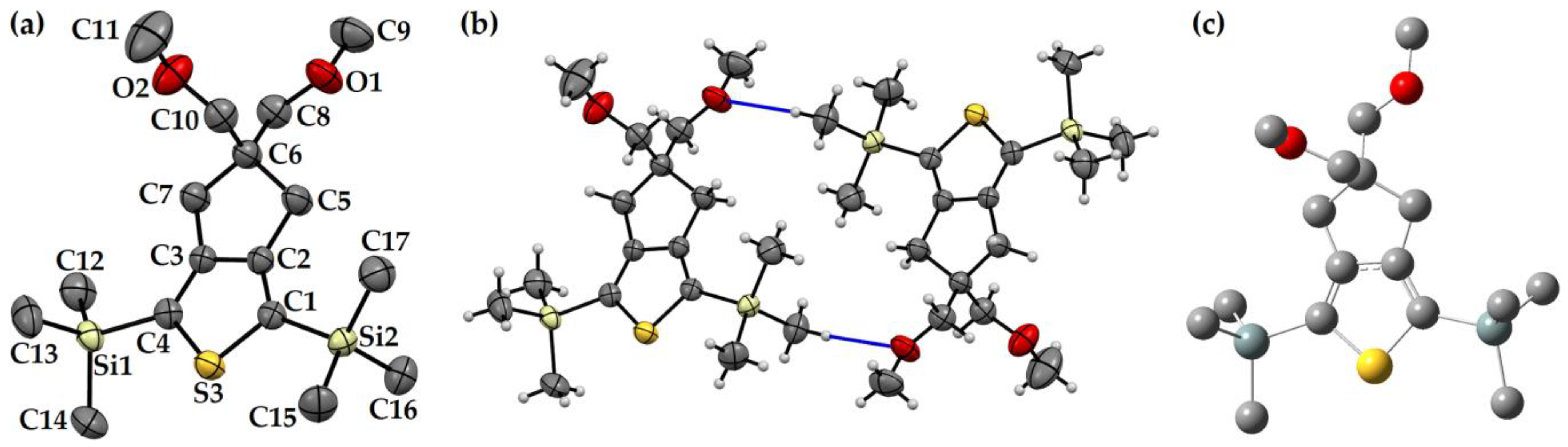
3.1. Crystal Structure
3.2. Computational Analysis
3.2.1. Gas-Phase Morphology
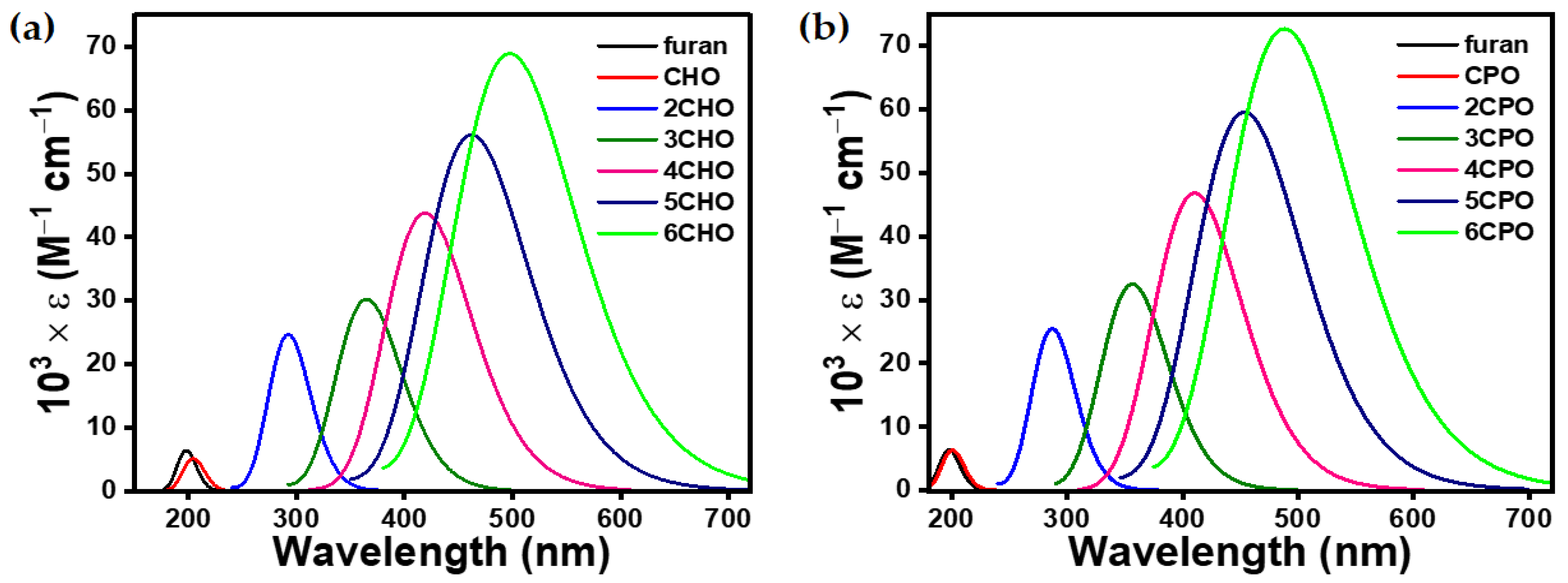
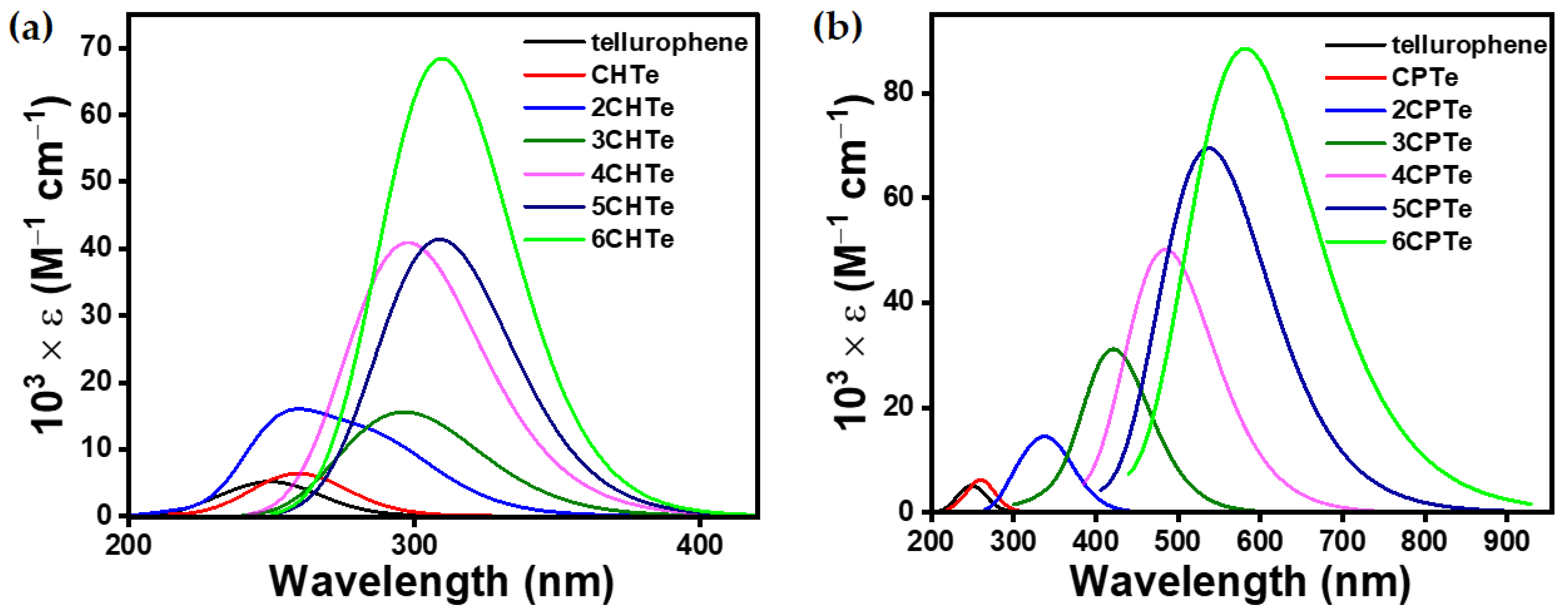
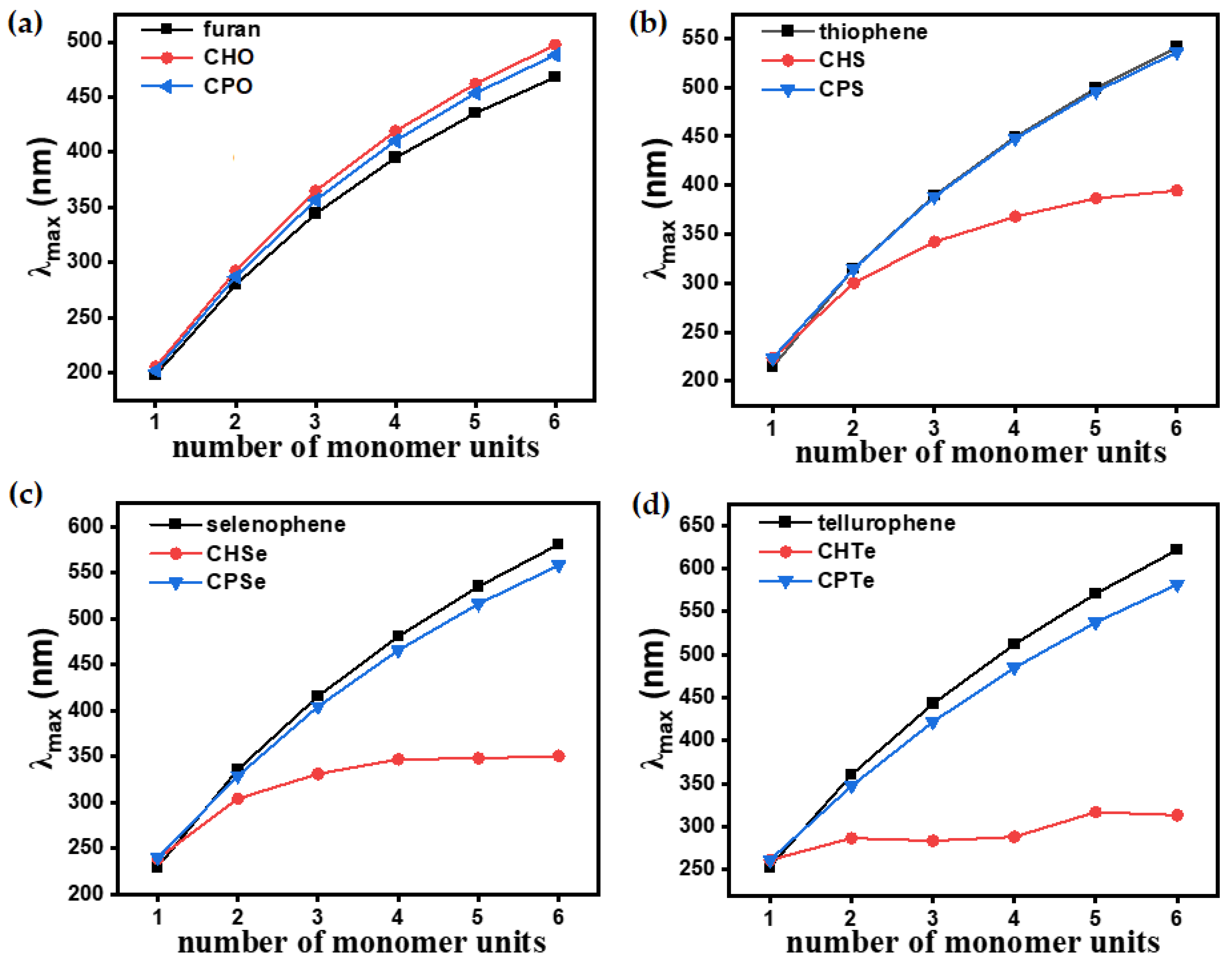
3.2.2. Optoelectronic Properties
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ye, S.Y.; Lotocki, V.; Xu, H.; Seferos, D.S. Group 16 conjugated polymers based on furan, thiophene, selenophene, and tellurophene. Chem. Soc. Rev. 2022, 51, 6442–6474. [Google Scholar] [CrossRef] [PubMed]
- Lunzer, M.; Beckwith, J.S.; Chalupa-Gantner, F.; Rosspeintner, A.; Licari, G.; Steiger, W.; Hametner, C.; Liska, R.; Fröhlich, J.; Vauthey, E.; et al. Beyond the Threshold: A Study of Chalcogenophene-Based Two-Photon Initiators. Chem. Mater. 2022, 34, 3042–3052. [Google Scholar] [CrossRef] [PubMed]
- Pan, Q.; Wu, Q.; Sun, Q.; Zhou, X.; Cheng, L.; Zhang, S.; Yuan, Y.; Zhang, Z.; Ma, J.; Zhang, Y.; et al. Biomolecule-friendly conducting PEDOT interface for long-term bioelectronic devices. Sens. Actuators B Chem. 2022, 373, 132703–132713. [Google Scholar] [CrossRef]
- Li, Y.; Qi, R.; Wang, X.; Yuan, H. Recent Strategies to Develop Conjugated Polymers for Detection and Therapeutics. Polymers 2023, 15, 3570. [Google Scholar] [CrossRef] [PubMed]
- Sung, M.; Yoo, H.; Yoo, D.; Ahn, H.; Park, G.; Park, K.H.; Song, S.H.; Lee, B.H.; Lee, J. Fluorinated Benzothiadiazole-based polymers with chalcogenophenes for organic field-effect transistors. Org. Electron. 2022, 111, 106649–106657. [Google Scholar] [CrossRef]
- Lee, S.M.; Lee, H.R.; Dutta, G.K.; Lee, J.; Oh, J.H.; Yang, C. Furan-flanked diketopyrrolopyrrole-based chalcogenophene copolymers with siloxane hybrid side chains for organic field-effect transistors. Polym. Chem. 2019, 10, 2854–2862. [Google Scholar] [CrossRef]
- Fei, Z.; Han, Y.; Gann, E.; Hodsden, T.; Chesman, A.S.R.; McNeill, C.R.; Anthopoulos, T.D.; Heeney, M. Alkylated Selenophene-Based Ladder-Type Monomers via a Facile Route for High-Performance Thin-Film Transistor Applications. J. Am. Chem. Soc. 2017, 139, 8552–8561. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, E.A.; Yasa, M.; Cirpan, A.; Toppare, L. A follow-up investigation: Organic solar cells based on chalcogenophene-Thieno[3,4-c]pyrrole-4,6-dione-chalcogenophene containing random conjugated polymers. J. Electroanal. Chem. 2023, 932, 117213–117220. [Google Scholar] [CrossRef]
- Chai, G.; Zhang, J.; Pan, M.; Wang, Z.; Yu, J.; Liang, J.; Yu, H.; Chen, Y.; Shang, A.; Liu, X.; et al. Deciphering the Role of Chalcogen-Containing Heterocycles in Nonfullerene Acceptors for Organic Solar Cells. ACS Energy Lett. 2020, 5, 3415–3425. [Google Scholar] [CrossRef]
- Park, Y.S.; Kale, T.S.; Nam, C.Y.; Choi, D.; Grubbs, R.B. Effects of heteroatom substitution in conjugated heterocyclic compounds on photovoltaic performance: From sulfur to tellurium. Chem. Commun. 2014, 50, 7964–7967. [Google Scholar] [CrossRef]
- Zampetti, A.; Minotto, A.; Squeo, B.M.; Gregoriou, V.G.; Allard, S.; Scherf, U.; Chochos, C.L.; Cacialli, F. Highly Efficient Solid-State Near-infrared Organic Light-Emitting Diodes incorporating A-D-A Dyes based on α,β-unsubstituted “BODIPY” Moieties. Sci. Rep. 2017, 7, 1611–1617. [Google Scholar] [CrossRef]
- Tao, Y.; Yang, C.; Qin, J. Organic host materials for phosphorescent organic light-emitting diodes. Chem. Soc. Rev. 2011, 40, 2943–2970. [Google Scholar] [CrossRef] [PubMed]
- Shah, K.W.; Wang, S.-X.; Soo, D.X.Y.; Xu, J. Viologen-Based Electrochromic Materials: From Small Molecules, Polymers and Composites to Their Applications. Polymers 2019, 11, 1839. [Google Scholar] [CrossRef] [PubMed]
- Planells, M.; Schroeder, B.C.; McCulloch, I. Effect of Chalcogen Atom Substitution on the Optoelectronic Properties in Cyclopentadithiophene Polymers. Macromolecules 2014, 47, 5889–5894. [Google Scholar] [CrossRef]
- Han, Y.; Xiao, J.; Wu, X.; Zhou, W.; Shen, L.; Zhang, J.; Wang, Y.; Zhang, X.; Song, Y. Tuning Nonlinear Optical Behavior by Incorporation of the Chalcogenophene into Twistacenes. J. Phys. Chem. B 2020, 124, 10766–10775. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, J.M.; Jagadamma, L.K.; Cameron, J.; Wiles, A.A.; Wilson, C.; Skabara, P.J.; Samuel, I.D.W.; Cooke, G. New thiophene-based conjugated macrocycles for optoelectronic applications. J. Mater. Chem. C 2021, 9, 16257–16271. [Google Scholar] [CrossRef]
- Ren, S.; Ding, Y.; Zhang, W.; Wang, Z.; Wang, S.; Yi, Z. Rational Design of Novel Conjugated Terpolymers Based on Diketopyrrolopyrrole and Their Applications to Organic Thin-Film Transistors. Polymers 2023, 15, 3803. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Hu, W.; Russell, T.P. Measuring the Degree of Crystallinity in Semicrystalline Regioregular Poly(3-hexylthiophene). Macromolecules 2016, 49, 4501–4509. [Google Scholar] [CrossRef]
- Hai, T.A.P.; Sugimoto, R. Synthesis and characterization of copolymers composed of 3-hexylthiophene and fluorene via chemical oxidation with FeCl3. Polym. J. 2016, 48, 1115–1121. [Google Scholar] [CrossRef]
- Timmermans, B.; Koeckelberghs, G. Chiral expression of co-crystallizing poly(thiophene)-block-poly(selenophene) copolymers. Polym. Chem. 2020, 11, 2715–2723. [Google Scholar] [CrossRef]
- Nakao, K.; Nishimura, M.; Tamachi, T.; Kuwatani, Y.; Miyasaka, H.; Nishinaga, T.; Iyoda, M. Giant Macrocycles Composed of Thiophene, Acetylene, and Ethylene Building Blocks. J. Am. Chem. Soc. 2006, 128, 16740–16747. [Google Scholar] [CrossRef]
- Weiland, K.J.; Brandl, T.; Atz, K.; Prescimone, A.; Häussinger, D.; Šolomek, T.; Mayor, M. Mechanical Stabilization of Helical Chirality in a Macrocyclic Oligothiophene. J. Am. Chem. Soc. 2019, 141, 2104–2110. [Google Scholar] [CrossRef]
- Topolskaia, V.; Pollit, A.A.; Cheng, S.; Seferos, D.S. Trends in Conjugated Chalcogenophenes: A Theoretical Study. Chem. Eur. J. 2021, 27, 9038–9043. [Google Scholar] [CrossRef] [PubMed]
- Ren, S.; Wang, Z.; Zhang, W.; Ding, Y.; Yi, Z. Donor-Acceptor-Based Organic Polymer Semiconductor Materials to Achieve High Hole Mobility in Organic Field-Effect Transistors. Polymers 2023, 15, 3713. [Google Scholar] [CrossRef] [PubMed]
- Hellwig, P.S.; Peglow, T.J.; Penteado, F.; Bagnoli, L.; Perin, G.; Lenardão, E.J. Recent Advances in the Synthesis of Selenophenes and Their Derivatives. Molecules 2020, 25, 5907. [Google Scholar] [CrossRef] [PubMed]
- Kumar, G.J.; Bogoslavsky, B.; Debnath, S.; Bedi, A. Effect of Chalcogenophenes on Chiroptical Activity of Twisted Tetracenes: Computational Analysis, Synthesis and Crystal Structure Thereof. Molecules 2023, 28, 5074. [Google Scholar] [CrossRef] [PubMed]
- Yaghoobi Nia, N.; Bonomo, M.; Zendehdel, M.; Lamanna, E.; Desoky, M.M.H.; Paci, B.; Zurlo, F.; Generosi, A.; Barolo, C.; Viscardi, G.; et al. Impact of P3HT Regioregularity and Molecular Weight on the Efficiency and Stability of Perovskite Solar Cells. ACS Sustain. Chem. Eng. 2021, 9, 5061–5073. [Google Scholar] [CrossRef]
- Patra, A.; Agrawal, V.; Bhargav, R.; Shahjad; Bhardwaj, D.; Chand, S.; Sheynin, Y.; Bendikov, M. Metal Free Conducting PEDOS, PEDOT, and Their Analogues via an Unusual Bromine-Catalyzed Polymerization. Macromolecules 2015, 48, 8760–8764. [Google Scholar] [CrossRef]
- Das, S.; Bedi, A.; Krishna, G.R.; Reddy, C.M.; Zade, S.S. Cyclopenta[c]selenophene based cooligomers and their polymers: Comparative study with thiophene analogues. Org. Biomol. Chem. 2011, 9, 6963–6972. [Google Scholar] [CrossRef]
- Patra, A.; Wijsboom, Y.H.; Zade, S.S.; Li, M.; Sheynin, Y.; Leitus, G.; Bendikov, M. Poly(3,4-ethylenedioxyselenophene). J. Am. Chem. Soc. 2008, 130, 6734–6736. [Google Scholar] [CrossRef]
- Das, S.; Zade, S.S. Poly(cyclopenta[c]selenophene): A new polyselenophene. Chem. Commun. 2010, 46, 1168–1170. [Google Scholar] [CrossRef] [PubMed]
- Zade, S.S.; Zamoshchik, N.; Bendikov, M. From Short Conjugated Oligomers to Conjugated Polymers. Lessons from Studies on Long Conjugated Oligomers. Acc. Chem. Res. 2011, 44, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Fagan, P.J.; Nugent, W.A. Synthesis of main group heterocycles by metallacycle transfer from zirconium. J. Am. Chem. Soc. 1988, 110, 2310–2312. [Google Scholar] [CrossRef]
- Lucht, B.L.; Mao, S.S.H.; Tilley, T.D. A Zirconocene-Coupling Route to Substituted Poly(p-phenylenedienylene)s: Band Gap Tuning via Conformational Control. J. Am. Chem. Soc. 1998, 120, 4354–4365. [Google Scholar] [CrossRef]
- Luppi, B.T.; McDonald, R.; Ferguson, M.J.; Sang, L.; Rivard, E. Rapid access to (cycloalkyl)tellurophene oligomer mixtures and the first poly(3-aryltellurophene). Chem. Commun. 2019, 55, 14218–14221. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Shimo, S.; Hupf, E.; Ochiai, J.; Braun, C.A.; Torres Delgado, W.; Xu, L.; He, G.; Rivard, E.; Iwasawa, N. Self-Assembly of Macrocyclic Boronic Esters Bearing Tellurophene Moieties and Their Guest-Responsive Phosphorescence. Chem. Eur. J. 2019, 25, 8479–8483. [Google Scholar] [CrossRef] [PubMed]
- Bedi, A.; Senanayak, S.P.; Narayan, K.S.; Zade, S.S. Synthesis and characterization of copolymers based on cyclopenta[c]thiophene and bithiazole and their transistor properties. J. Polym. Sci. Part A Polym. Chem. 2013, 51, 4481–4488. [Google Scholar] [CrossRef]
- Debnath, S.; Singh, S.; Bedi, A.; Krishnamoorthy, K.; Zade, S.S. Synthesis, Optoelectronic, and Transistor Properties of BODIPY- and Cyclopenta[c]thiophene-Containing π-Conjugated Copolymers. J. Phys. Chem. C 2015, 119, 15859–15867. [Google Scholar] [CrossRef]
- Debnath, S.; Masilamani, G.; Agrawal, A.; Kumar, N.R.; Kumar, C.; Zade, S.S.; Bedi, A. Cyclopenta[c]thiophene- and Diketopyrrolopyrrole-Based Red-Green-Blue Electrochromic Polymers. Org. Mater. 2022, 4, 268–276. [Google Scholar] [CrossRef]
- He, G.; Kang, L.; Torres Delgado, W.; Shynkaruk, O.; Ferguson, M.J.; McDonald, R.; Rivard, E. The Marriage of Metallacycle Transfer Chemistry with Suzuki–Miyaura Cross-Coupling To Give Main Group Element-Containing Conjugated Polymers. J. Am. Chem. Soc. 2013, 135, 5360–5363. [Google Scholar] [CrossRef]
- Bedi, A.; Debnath, S.; Chandak, H.S.; Zade, S.S. Phenyl-capped cyclopenta[c]chalcogenophenes: Synthesis, crystal structures, electrochemistry and theoretical insights. RSC Adv. 2014, 4, 35653–35658. [Google Scholar] [CrossRef]
- Bedi, A.; Debnath, S.; Zade, S.S. Diselenolodiselenole: A selenium containing fused heterocycle for conjugated systems. Chem. Commun. 2014, 50, 13454–13456. [Google Scholar] [CrossRef] [PubMed]
- Parke, S.M.; Boone, M.P.; Rivard, E. Marriage of heavy main group elements with π-conjugated materials for optoelectronic applications. Chem. Commun. 2016, 52, 9485–9505. [Google Scholar] [CrossRef] [PubMed]
- Al-Hashimi, M.; Han, Y.; Smith, J.; Bazzi, H.S.; Alqaradawi, S.Y.A.; Watkins, S.E.; Anthopoulos, T.D.; Heeney, M. Influence of the heteroatom on the optoelectronic properties and transistor performance of soluble thiophene-, selenophene- and tellurophene–vinylene copolymers. Chem. Sci. 2016, 7, 1093–1099. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.; Zhang, T.; Liu, R.; Tian, W.; Wu, K.; Zhu, J.; Wang, Z.; He, C.; Feng, J.; Guo, X.; et al. Chalcogen-bridged coordination polymer for the photocatalytic activation of aryl halides. Nat. Commun. 2023, 14, 4002. [Google Scholar] [CrossRef] [PubMed]
- Kawashima, K.; Osaka, I.; Takimiya, K. Effect of Chalcogen Atom on the Properties of Naphthobischalcogenadiazole-Based π-Conjugated Polymers. Chem. Mater. 2015, 27, 6558–6570. [Google Scholar] [CrossRef]
- Biot, N.; Romito, D.; Bonifazi, D. Substituent-Controlled Tailoring of Chalcogen-Bonded Supramolecular Nanoribbons in the Solid State. Cryst. Growth Des. 2021, 21, 536–543. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Song, L.; Borup, K.A.; Jørgensen, M.R.V.; Iversen, B.B. New Insight on Tuning Electrical Transport Properties via Chalcogen Doping in n-type Mg3Sb2-Based Thermoelectric Materials. Adv. Energy Mater. 2018, 8, 1702776–1702782. [Google Scholar] [CrossRef]
- Kumar, S.S.; Thakuria, R.; Nangia, A. Pharmaceutical cocrystals and a nitrate salt of voriconazole. CrystEngComm 2014, 16, 4722–4731. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Rev. A.03; Gaussian, Inc.: Wallingford, UK, 2016. [Google Scholar]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Becke, A.D. Density—Functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Bergner, A.; Dolg, M.; Küchle, W.; Stoll, H.; Preuß, H. Ab initio energy-adjusted pseudopotentials for elements of groups 13–17. Mol. Phys. 1993, 80, 1431–1441. [Google Scholar] [CrossRef]
- Kaupp, M.; Schleyer, P.V.R.; Stoll, H.; Preuss, H. Pseudopotential approaches to Ca, Sr, and Ba hydrides. Why are some alkaline earth MX2 compounds bent? J. Chem. Phys. 1991, 94, 1360–1366. [Google Scholar] [CrossRef]
- Dolg, M.; Stoll, H.; Preuss, H.; Pitzer, R.M. Relativistic and correlation effects for element 105 (hahnium, Ha): A comparative study of M and MO (M = Nb, Ta, Ha) using energy-adjusted ab initio pseudopotentials. J. Phys. Chem. 1993, 97, 5852–5859. [Google Scholar] [CrossRef]
- Shahjad; Bhargav, R.; Bhardwaj, D.; Mishra, A.; Patra, A. Synthesis and Characterization of Benzodithiophene–Chalcogenophene Based Copolymers: A Comparative Study of Optoelectronic Properties and Photovoltaic Applications. Macromol. Chem. Phys. 2017, 218, 1700038–1700046. [Google Scholar] [CrossRef]
- Gidron, O.; Diskin-Posner, Y.; Bendikov, M. α-Oligofurans. J. Am. Chem. Soc. 2010, 132, 2148–2150. [Google Scholar] [CrossRef]
- Mulay, S.V.; Bogoslavky, B.; Galanti, I.; Galun, E.; Gidron, O. Bifuran-imide: A stable furan building unit for organic electronics. J. Mater. Chem. C 2018, 6, 11951–11955. [Google Scholar] [CrossRef]
- Shida, N.; Nishiyama, H.; Zheng, F.; Ye, S.; Seferos, D.S.; Tomita, I.; Inagi, S. Redox chemistry of π-extended tellurophenes. Commun. Chem. 2019, 2, 124–132. [Google Scholar] [CrossRef]
- Dishi, O.; Gidron, O. Macrocyclic Oligofurans: A Computational Study. J. Org. Chem. 2018, 83, 3119–3125. [Google Scholar] [CrossRef]
- Wu, B.; Melvina; Wu, X.; Lee Yeow, E.K.; Yoshikai, N. Versatile telluracycle synthesis via the sequential electrophilic telluration of C(sp2)-Zn and C(sp2)-H bonds. Chem. Sci. 2017, 8, 4527–4532. [Google Scholar] [CrossRef]
- Kosar, N.; Ayub, K.; Gilani, M.A.; Muhammad, S.; Mahmood, T. Benchmark Density Functional Theory Approach for the Calculation of Bond Dissociation Energies of the M–O2 Bond: A Key Step in Water Splitting Reactions. ACS Omega 2022, 7, 20800–20808. [Google Scholar] [CrossRef] [PubMed]
- Roncali, J. Conjugated poly(thiophenes): Synthesis, functionalization, and applications. Chem. Rev. 1992, 92, 711–738. [Google Scholar] [CrossRef]
- Patra, A.; Bendikov, M. Polyselenophenes. J. Mater. Chem. 2010, 20, 422–433. [Google Scholar] [CrossRef]
- Sun, H.; Autschbach, J. Electronic Energy Gaps for π-Conjugated Oligomers and Polymers Calculated with Density Functional Theory. J. Chem. Theory Comput. 2014, 10, 1035–1047. [Google Scholar] [CrossRef] [PubMed]
- Nikoo, S.; Meister, P.J.; Hayward, J.J.; Gauld, J.W. An Assessment of Computational Methods for Calculating Accurate Structures and Energies of Bio-Relevant Polysulfur/Selenium-Containing Compounds. Molecules 2018, 23, 3323. [Google Scholar] [CrossRef] [PubMed]
- Zander, M.; Kirsch, G. On the Phosphorescence of Benzologues of Furan, Thiophene, Selenophene, and Tellurophene. A Systematic Study of the Intra -annular Internal Heavy-atom Effect. Z. Naturforsch. A 1989, 44, 205–209. [Google Scholar] [CrossRef]
- Chen, H.; Deng, Y.; Zhu, X.; Wang, L.; Lv, L.; Wu, X.; Li, Z.; Shi, Q.; Peng, A.; Peng, Q.; et al. Toward Achieving Single-Molecule White Electroluminescence from Dual Emission of Fluorescence and Phosphorescence. Chem. Mater. 2020, 32, 4038–4044. [Google Scholar] [CrossRef]
- He, G.; Torres Delgado, W.; Schatz, D.J.; Merten, C.; Mohammadpour, A.; Mayr, L.; Ferguson, M.J.; McDonald, R.; Brown, A.; Shankar, K.; et al. Coaxing Solid-State Phosphorescence from Tellurophenes. Angew. Chem. Int. Ed. 2014, 53, 4587–4591. [Google Scholar] [CrossRef]
- Pander, P.; Motyka, R.; Zassowski, P.; Lapkowski, M.; Swist, A.; Data, P. Electrochromic Properties of Novel Selenophene and Tellurophene Derivatives Based on Carbazole and Triphenylamine Core. J. Phys. Chem. C 2017, 121, 11027–11036. [Google Scholar] [CrossRef]
- Carrera, E.I.; Seferos, D.S. Semiconducting Polymers Containing Tellurium: Perspectives Toward Obtaining High-Performance Materials. Macromolecules 2015, 48, 297–308. [Google Scholar] [CrossRef]
- Chivers, T.; Laitinen, R.S. Tellurium: A maverick among the chalcogens. Chem. Soc. Rev. 2015, 44, 1725–1739. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| nCHO | Side View | nCPO | Side View |
|---|---|---|---|
| CHO |  | CPO |  |
| 2CHO |  | 2CPO |  |
| 3CHO |  | 3CPO |  |
| 4CHO |  | 4CPO |  |
| 5CHO |  | 5CPO |  |
| 6CHO |  | 6CPO |  |
| (CHO)n |  | (CPO)n |  |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Masilamani, G.; Krishna, G.R.; Debnath, S.; Bedi, A. Origin of Optoelectronic Contradictions in 3,4-Cycloalkyl[c]-chalcogenophenes: A Computational Study. Polymers 2023, 15, 4240. https://doi.org/10.3390/polym15214240
Masilamani G, Krishna GR, Debnath S, Bedi A. Origin of Optoelectronic Contradictions in 3,4-Cycloalkyl[c]-chalcogenophenes: A Computational Study. Polymers. 2023; 15(21):4240. https://doi.org/10.3390/polym15214240
Chicago/Turabian StyleMasilamani, Ganesh, Gamidi Rama Krishna, Sashi Debnath, and Anjan Bedi. 2023. "Origin of Optoelectronic Contradictions in 3,4-Cycloalkyl[c]-chalcogenophenes: A Computational Study" Polymers 15, no. 21: 4240. https://doi.org/10.3390/polym15214240
APA StyleMasilamani, G., Krishna, G. R., Debnath, S., & Bedi, A. (2023). Origin of Optoelectronic Contradictions in 3,4-Cycloalkyl[c]-chalcogenophenes: A Computational Study. Polymers, 15(21), 4240. https://doi.org/10.3390/polym15214240