
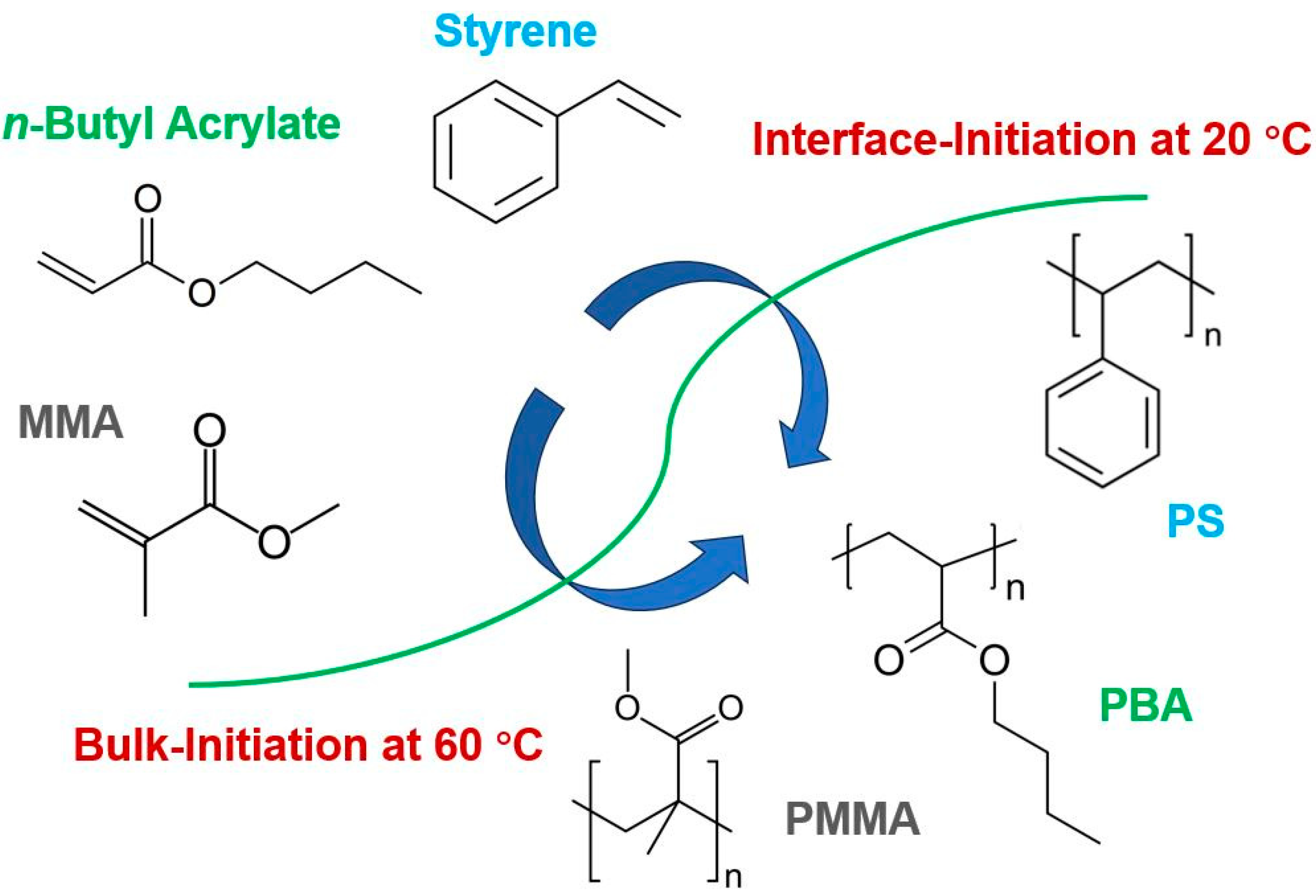
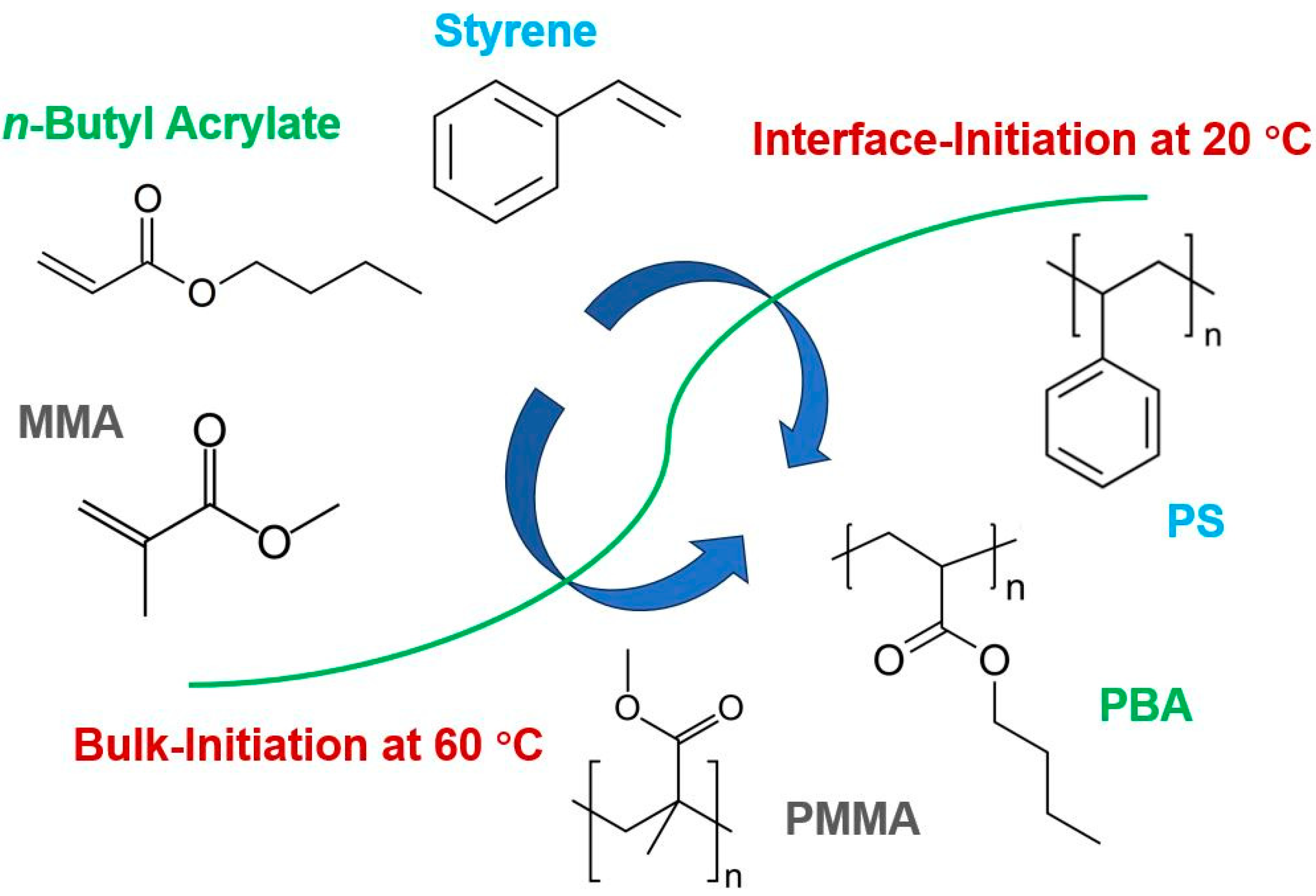
Synthesis of Polymers with Narrow Molecular Mass Distribution through Interface-Initiated Room-Temperature Polymerization in Emulsion Gels


Abstract
:1. Introduction
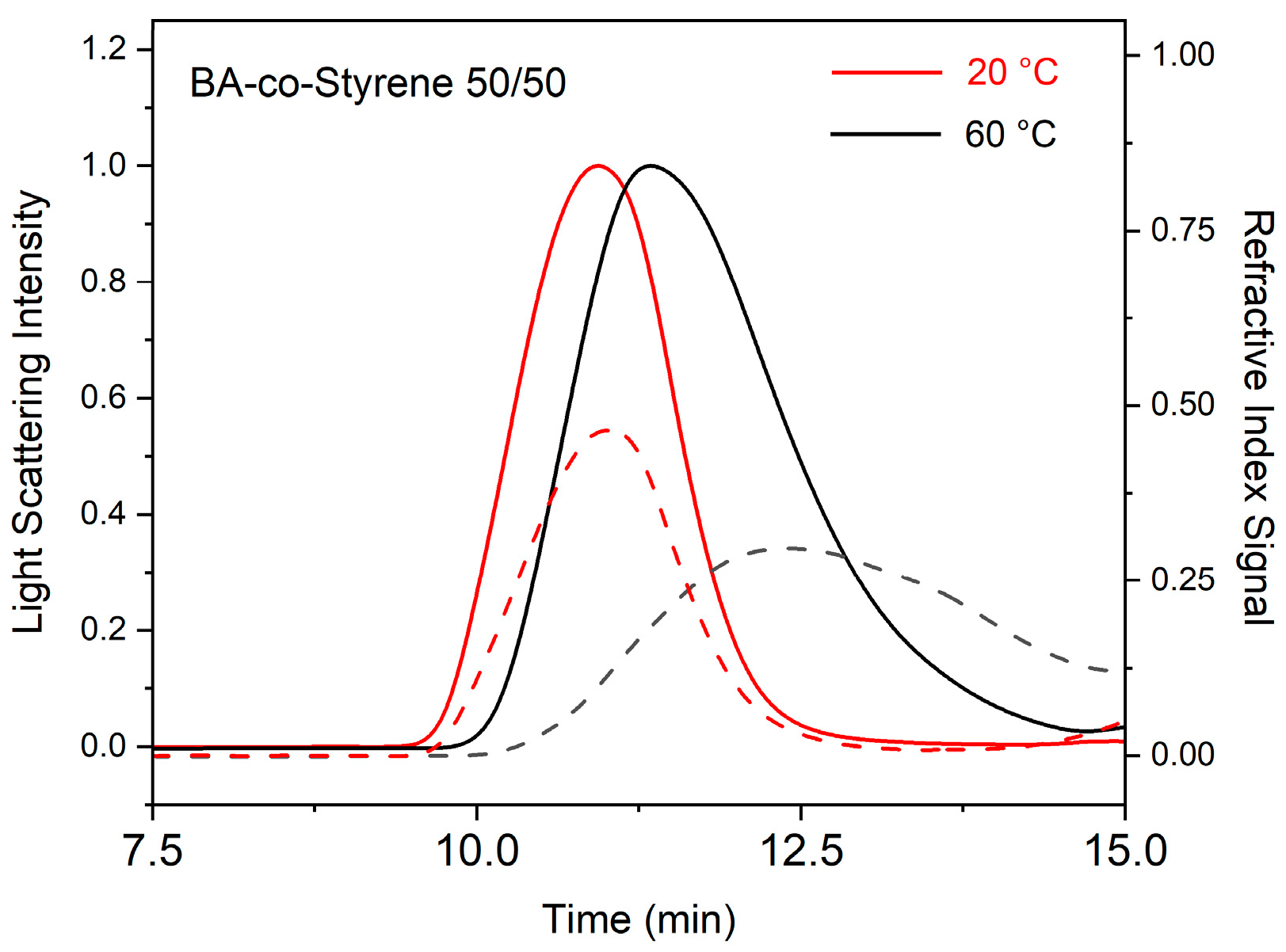
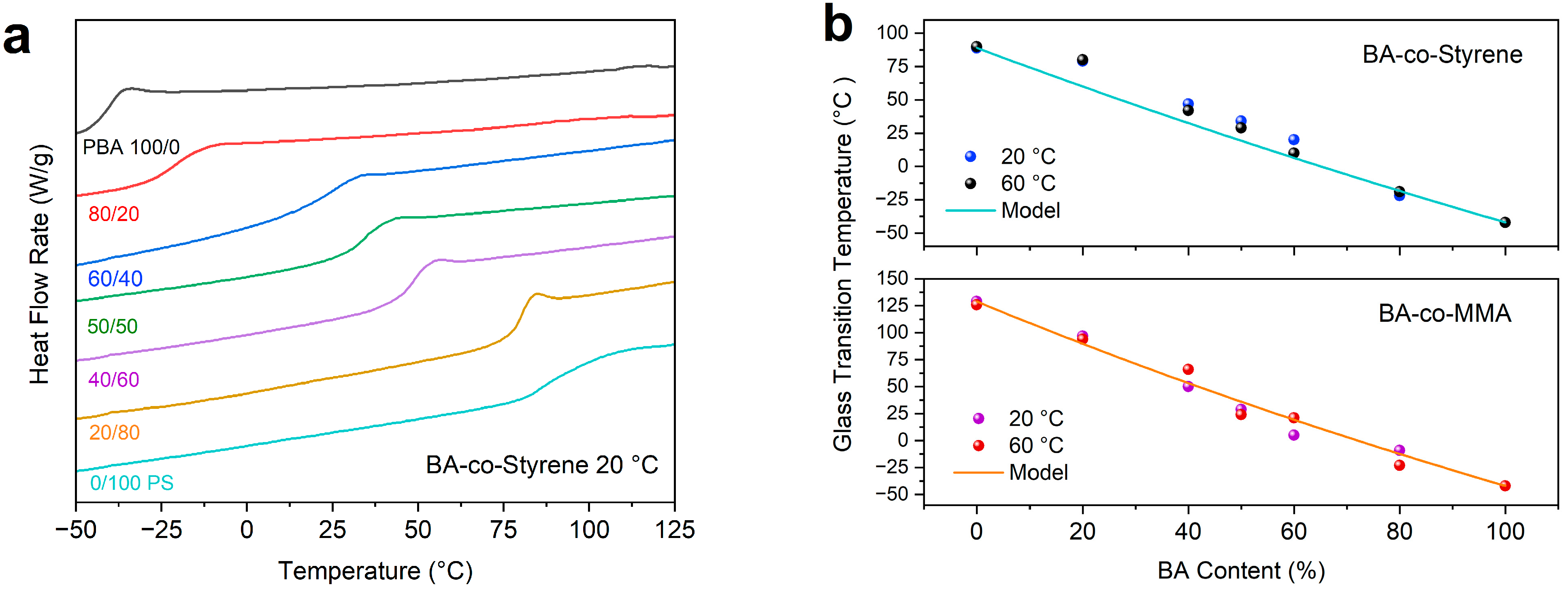
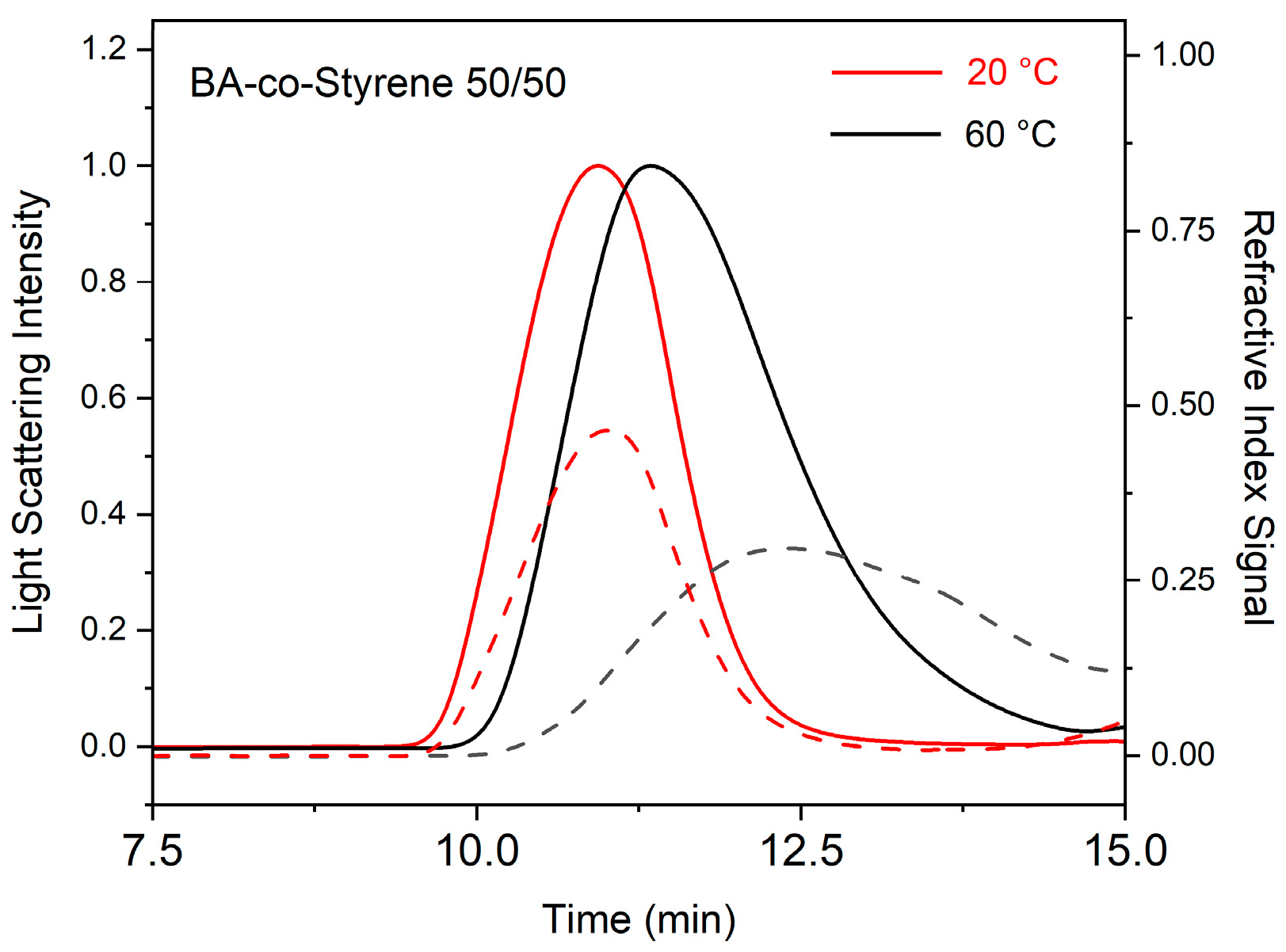
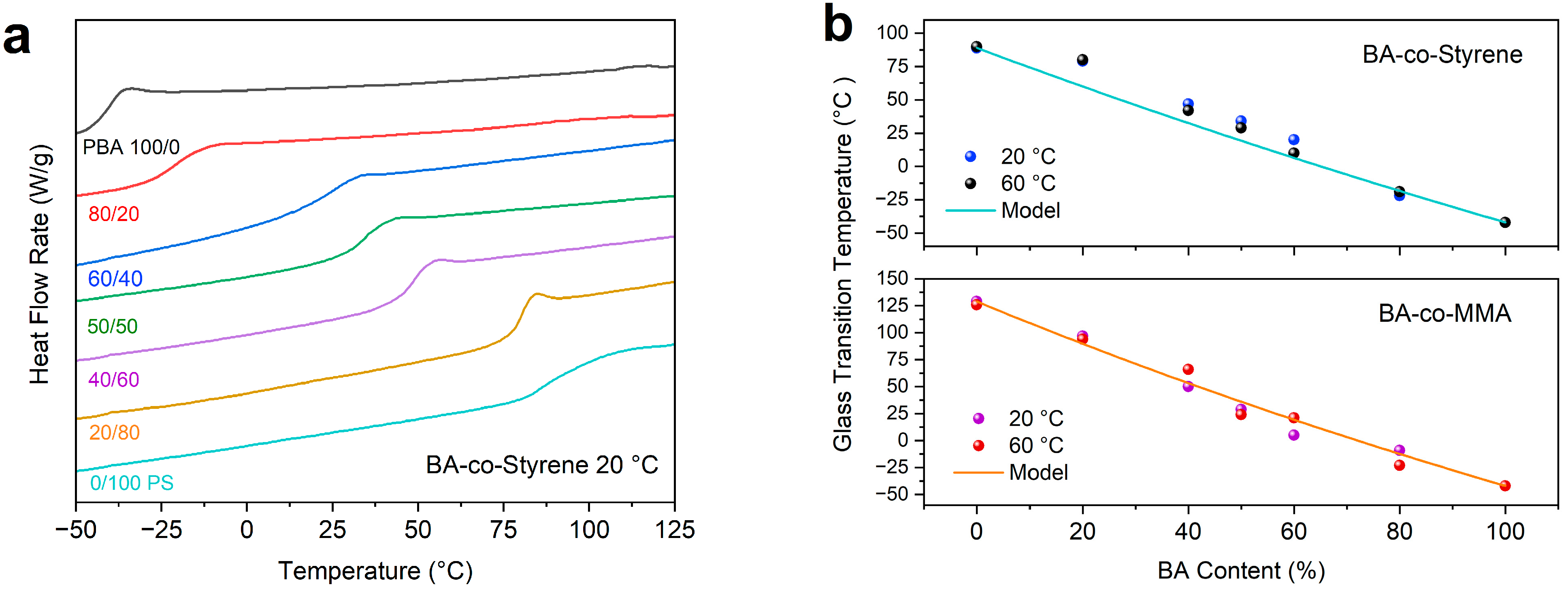
2. Results and Discussion
3. Conclusions
4. Experimental Section
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, T.; Xu, G.; Blum, F.D. Eco-friendly room-temperature polymerization in emulsions and beyond. Polym. Rev. 2023; in press. [Google Scholar] [CrossRef]
- Asua, J.M. Emulsion polymerization: From fundamental mechanisms to process developments. J. Polym. Sci. A Polym. Chem. 2004, 42, 1025. [Google Scholar]
- Zhang, T.; Sanguramath, R.A.; Israel, S.; Silverstein, M.S. Emulsion templating: Porous polymers and beyond. Macromolecules 2019, 52, 5445. [Google Scholar]
- Wang, Y.B.; Sun, B.J.; Hao, Z.W.; Zhang, J.H. Advances in organic-inorganic hybrid latex particles via in situ emulsion polymerization. Polymers 2023, 15, 2995. [Google Scholar]
- Zhang, T.; Xu, G.; Li, Z.F.; Regev, O.; Maddumaarachchi, M.; Blum, F.D. PS/CTAB/silica composites from room temperature polymerization of high internal phase emulsion gels. J. Colloid Interface Sci. 2015, 451, 161. [Google Scholar] [CrossRef]
- Dube, M.A.; Salehpour, S. Applying the principles of green chemistry to polymer production technology. Macromol. React. Eng. 2014, 8, 7. [Google Scholar]
- Anastas, P.T.; Warner, J.C. Green Chemistry: Theory and Practice; Oxford University Press: Oxford, UK, 2000. [Google Scholar]
- Schon, A.; Clarkson, B.R.; Jaime, M.; Freire, E. Temperature stability of proteins: Analysis of irreversible denaturation using isothermal calorimetry. Proteins 2017, 85, 2009. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.Q.; Bulmus, V.; Herlambang, D.L.; Barner-Kowollik, C.; Stenzel, M.H.; Davis, T.P. In situ formation of protein-polymer conjugates through reversible addition fragmentation chain transfer polymerization. Angew. Chem. Int. Ed. 2007, 46, 3099. [Google Scholar] [CrossRef] [PubMed]
- Yamashina, M.; Sei, Y.; Akita, M.; Yoshizawa, M. Safe storage of radical initiators within a polyaromatic nanocapsule. Nat. Commun. 2014, 5, 4662. [Google Scholar] [CrossRef]
- Szekely, A.; Klussmann, M. Molecular radical chain initiators for ambient- to low-temperature applications. Chem. Asian J. 2019, 14, 105. [Google Scholar] [CrossRef]
- Sarac, A.S. Redox polymerization. Prog. Polym. Sci. 1999, 24, 1149. [Google Scholar]
- Kohut-Svelko, N.; Pirri, R.; Asua, J.M.; Leiza, J.R. Redox initiator systems for emulsion polymerization of acrylates. J. Polym. Sci. A Polym. Chem. 2009, 47, 2917. [Google Scholar] [CrossRef]
- Jacob, L.I.; Pauer, W.; Schroeter, B. Influence of redox initiator component ratios on the emulsion copolymerisation of vinyl acetate and neodecanoic acid vinyl ester. RSC Adv. 2022, 12, 14197. [Google Scholar] [CrossRef]
- Schmitt, M.; Garra, P.; Lalevee, J. Bulk polymerization photo-initiator ZnO: Increasing of the benzoyl formic acid concentration and LED illumination. Macromol. Chem. Phys. 2018, 219, 1800208. [Google Scholar] [CrossRef]
- Li, Z.Y.; Lian, Q.J.; Xu, Y.W.; Zhang, Y.C.; Zhang, P.F.; Geng, J. Aggregation-induced emission luminogen catalyzed photocontrolled reversible addition-fragmentation chain transfer polymerization in an aqueous environment. Macromolecules 2022, 55, 2904. [Google Scholar] [CrossRef]
- Watson, M.D.; Fechtenkotter, A.; Mullen, K. Big is beautiful—“Aromaticity” revisited from the viewpoint of macromolecular and supramolecular benzene chemistry. Chem. Rev. 2001, 101, 1267. [Google Scholar]
- Tauer, K.; Oz, N. Interfacial energy promotes radical heterophase polymerization. Macromolecules 2004, 37, 5880. [Google Scholar] [CrossRef]
- Godwin, A.; Hartenstein, M.; Muller, A.H.E.; Brocchini, S. Narrow molecular weight distribution precursors for polymer-drug conjugates. Angew. Chem. Int. Ed. 2001, 40, 594–597. [Google Scholar] [CrossRef]
- Truong, N.P.; Jones, G.R.; Bradford, K.G.E.; Konkolewicz, D.; Anastasaki, A. A comparison of RAFT and ATRP methods for controlled radical polymerization. Nat. Rev. Chem. 2021, 5, 859. [Google Scholar] [CrossRef] [PubMed]
- Rzayev, J.; Penelle, J. HP-RAFT: A free-radical polymerization technique for obtaining living polymers of ultrahigh molecular weights. Angew. Chem. Int. Ed. 2004, 43, 1691. [Google Scholar] [CrossRef] [PubMed]
- Nieswandt, K.; Georgopanos, P.; Held, M.; Sperling, E.; Abetz, V. RAFT emulsion polymerization of styrene using a poly((N,N-dimethyl acrylamide)-co-(N-isopropyl acrylamide)) mCTA: Synthesis and thermosensitivity. Polymers 2022, 14, 62. [Google Scholar] [CrossRef]
- Xu, G.; Nambiar, R.R.; Blum, F.D. Room-temperature decomposition of 2,2’-azobis(isobutyronitrile) in emulsion gels with and without silica. J. Colloid Interface Sci. 2006, 302, 658. [Google Scholar] [CrossRef]
- Zhang, T.; Xu, G.; Regev, O.; Blum, F.D. Low-temperature polymerization of methyl methacrylate emulsion gels through surfactant catalysis. J. Colloid Interface Sci. 2016, 461, 128. [Google Scholar] [CrossRef] [PubMed]
- Chambard, G.; Klumperman, B.; German, A.L. Dependence of chemical composition of styrene/butyl acrylate copolymers on temperature and molecular weight. Polymer 1999, 40, 4459. [Google Scholar] [CrossRef]
- Sosnowski, S.; Szymanski, R.; Lorandi, F.; Olszewski, M.; Sobieski, J.; Yin, R.G.; Bockstaller, M.R.; Matyjaszewski, K. Distribution of alternating sequences in methyl methacrylate/n-butyl acrylate copolymers prepared by atom transfer radical polymerization. Macromolecules 2021, 54, 9837. [Google Scholar] [CrossRef]
- Xu, G.; Blum, F.D. Surfactant-enhanced free radical polymerization of styrene in emulsion gels. Polymer 2008, 49, 3233. [Google Scholar] [CrossRef]
- Buback, M.; Huckestein, B.; Kuchta, F.D.; Russell, G.T.; Schmid, E. Initiator efficiency in 2,2′-azoisobutyronitrile-initiated free-radical polymerizations of styrene. Macromol. Chem. Phys. 1994, 195, 2117. [Google Scholar] [CrossRef]
- Lv, C.N.; Du, Y.X.; Pan, X.C. Alkylboranes in Conventional and Controlled Radical Polymerization. J. Polym. Sci. 2020, 58, 14–19. [Google Scholar] [CrossRef]
- Ballard, N.; Hamzehlou, S.; Ruiperez, F.; Asua, J.M. On the termination mechanism in the radical polymerization of acrylates. Macromol. Rapid Commun. 2016, 37, 1364. [Google Scholar] [CrossRef] [PubMed]
- Kruger, K.; Tauer, K.; Yagci, Y.; Moszner, N. Photoinitiated bulk and emulsion polymerization of styrene—Evidence for photo-controlled radical polymerization. Macromolecules 2011, 44, 9539. [Google Scholar] [CrossRef]
- Zhang, T.; Blum, F.D. Cationic surfactant blocks radical-inhibiting sites on silica. J. Colloid Interface Sci. 2017, 504, 111. [Google Scholar] [CrossRef]
- Charton, N.; Feldermann, A.; Theis, A.; Stenzel, M.H.; Davis, T.P.; Barner-Kowollik, C. Initiator efficiency of 2,2′-azobis(isobutyronitrile) in bulk dodecyl acrylate free-radical polymerizations over a wide conversion and molecular weight range. J. Polym. Sci. A Polym. Chem. 2004, 42, 5170. [Google Scholar] [CrossRef]
- Guan, Z.B.; Combes, J.R.; Menceloglu, Y.Z.; Desimone, J.M. Homogeneous free-radical polymerizations in supercritical carbon-dioxide 2. Thermal-decomposition of 2,2′-azobis(isobutyronitrile). Macromolecules 1993, 26, 2663. [Google Scholar] [CrossRef]
- Burnett, G.M.; Cameron, G.G.; Joiner, S.N. Solvent effects on the free radical polymerization of styrene. J. Chem. Soc. Faraday Trans. 1 Phys. Chem. Condens. Phases 1973, 69, 322. [Google Scholar] [CrossRef]
- Nakamura, Y.; Ogihara, T.; Hatano, S.; Abe, M.; Yamago, S. Control of the termination mechanism in radical polymerization by viscosity: Selective disproportionation in viscous media. Chem. Eur. J. 2017, 23, 1299. [Google Scholar] [CrossRef]
- Luo, Y.; Tsavalas, J.; Schork, F.J. Theoretical aspects of particle swelling in living free radical miniemulsion polymerization. Macromolecules 2001, 34, 5501. [Google Scholar] [CrossRef]
- Zhou, J.H.; Yao, H.T.; Ma, J.Z. Recent advances in RAFT-mediated surfactant-free emulsion polymerization. Polym. Chem. 2018, 9, 2532. [Google Scholar]
- You, Y.Z.; Hong, C.Y.; Bai, R.K.; Pan, C.Y.; Wang, J. Photo-initiated living free radical polymerization in the presence of dibenzyl trithiocarbonate. Macromol. Chem. Phys. 2002, 203, 477. [Google Scholar] [CrossRef]
- Liu, S.; Srinivasan, S.; Grady, M.C.; Soroush, M.; Rappe, A.M. Backbiting and beta-scission reactions in free- radical polymerization of methyl acrylate. Int. J. Quantum Chem. 2014, 114, 34. [Google Scholar] [CrossRef]
- Goikoetxea, M.; Heijungs, R.; Barandiaran, M.J.; Asua, J.M. Energy efficient emulsion polymerization strategies. Macromol. React. Eng. 2008, 2, 90. [Google Scholar] [CrossRef]
- Lin, J.; Yan, D.; Yuan, C. Non-steady-state model for kinetics of free radical polymerization, 3. Direct photo-initiation. Polym. Int. 1992, 28, 123. [Google Scholar] [CrossRef]
- Wu, X.J.; Wang, Y.; Yang, W.; Xie, B.H.; Yang, M.B.; Dan, W. A rheological study on temperature dependent microstructural changes of fumed silica gels in dodecane. Soft Matter 2012, 8, 10457. [Google Scholar] [CrossRef]
- Frelichowska, J.; Bolzinger, M.A.; Chevalier, Y. Effects of solid particle content on properties of o/w Pickering emulsions. J. Colloid Interface Sci. 2010, 351, 348. [Google Scholar] [CrossRef]
- Brown, N.; de la Pena, A.; Razavi, S. Interfacial rheology insights: Particle texture and Pickering foam stability. J. Phys. Condens. Matter 2023, 35, 384002. [Google Scholar] [CrossRef]
- Khezri, K.; Mahdavi, H. Polystyrene-silica aerogel nanocomposites by in situ simultaneous reverse and normal initiation technique for ATRP. Microporous Mesoporous Mater. 2016, 228, 132. [Google Scholar] [CrossRef]
- Khezri, K.; Roghani-Mamaqani, H. Effect of MCM-41 nanoparticles on ARGET ATRP of styrene: Investigating thermal properties. J. Compos. Mater. 2015, 49, 1525. [Google Scholar] [CrossRef]
- Salami-Kalajahi, M.; Haddadi-Asl, V.; Rahimi-Razin, S.; Behboodi-Sadabad, F.; Roghani-Mamaqani, H.; Hemmati, M. Investigating the effect of pristine and modified silica nanoparticles on the kinetics of methyl methacrylate polymerization. Chem. Eng. J. 2011, 174, 368–375. [Google Scholar] [CrossRef]
- Gordon, M.; Taylor, S.J. Ideal copolymers and the second-order transitions of synthetic rubbers. I. non-crystalline copolymers. J. Appl. Chem. 1952, 2, 493. [Google Scholar] [CrossRef]
- Penzel, E.; Rieger, J.; Schneider, H.A. The glass transition temperature of random copolymers: 1. Experimental data and the Gordon-Taylor equation. Polymer 1997, 38, 325. [Google Scholar] [CrossRef]
- Miyake, G.M.; Chen, E.Y.-X. Synthesis of highly syndiotactic polymers by discrete catalysts or initiators. Polym. Chem. 2011, 2, 2462. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymer | 20 °C | 60 °C | ||
|---|---|---|---|---|
| Mw (kg/mol) | PDI | Mw (kg/mol) | PDI | |
| PBA | 757 | 1.26 | 31 | 2.46 |
| PS | 1661 | 1.37 | 172 | 2.04 |
| PMMA | 674 | 1.12 | 108 | 3.86 |
| Butyl acrylate-co-styrene 50/50 | 1807 | 1.17 | 96 | 3.06 |
| Butyl acrylate-co-MMA 50/50 | 888 | 1.25 | 257 | 1.96 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Duan, M.P.; Zhou, Z.; Zhang, T. Synthesis of Polymers with Narrow Molecular Mass Distribution through Interface-Initiated Room-Temperature Polymerization in Emulsion Gels. Polymers 2023, 15, 4081. https://doi.org/10.3390/polym15204081
Duan MP, Zhou Z, Zhang T. Synthesis of Polymers with Narrow Molecular Mass Distribution through Interface-Initiated Room-Temperature Polymerization in Emulsion Gels. Polymers. 2023; 15(20):4081. https://doi.org/10.3390/polym15204081
Chicago/Turabian StyleDuan, Miles Pamueles, Zhirong Zhou, and Tan Zhang. 2023. "Synthesis of Polymers with Narrow Molecular Mass Distribution through Interface-Initiated Room-Temperature Polymerization in Emulsion Gels" Polymers 15, no. 20: 4081. https://doi.org/10.3390/polym15204081
APA StyleDuan, M. P., Zhou, Z., & Zhang, T. (2023). Synthesis of Polymers with Narrow Molecular Mass Distribution through Interface-Initiated Room-Temperature Polymerization in Emulsion Gels. Polymers, 15(20), 4081. https://doi.org/10.3390/polym15204081