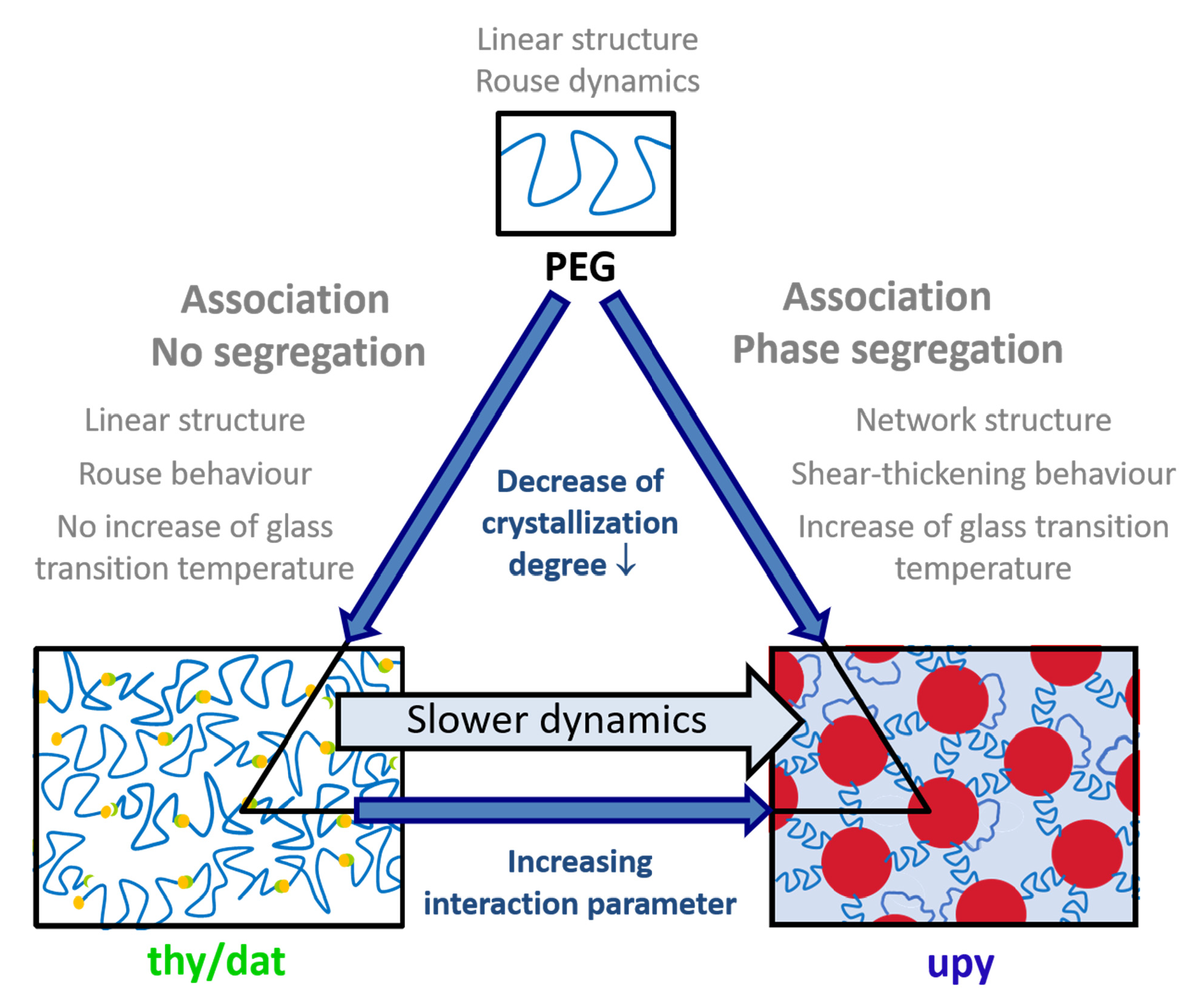
Besides the structure and dynamics of the heterocomplementary supramolecular PEG-thy/dat polymer, the homocomplementary supramolecular PEG-upy in the bulk is here in detail studied. As such the influence of different associative H-bonding functional end-groups on bifunctional PEG polymer properties can be identified. In the following the structure and dynamics of PEG-upy in the bulk is presented.
3.3.1. Structure
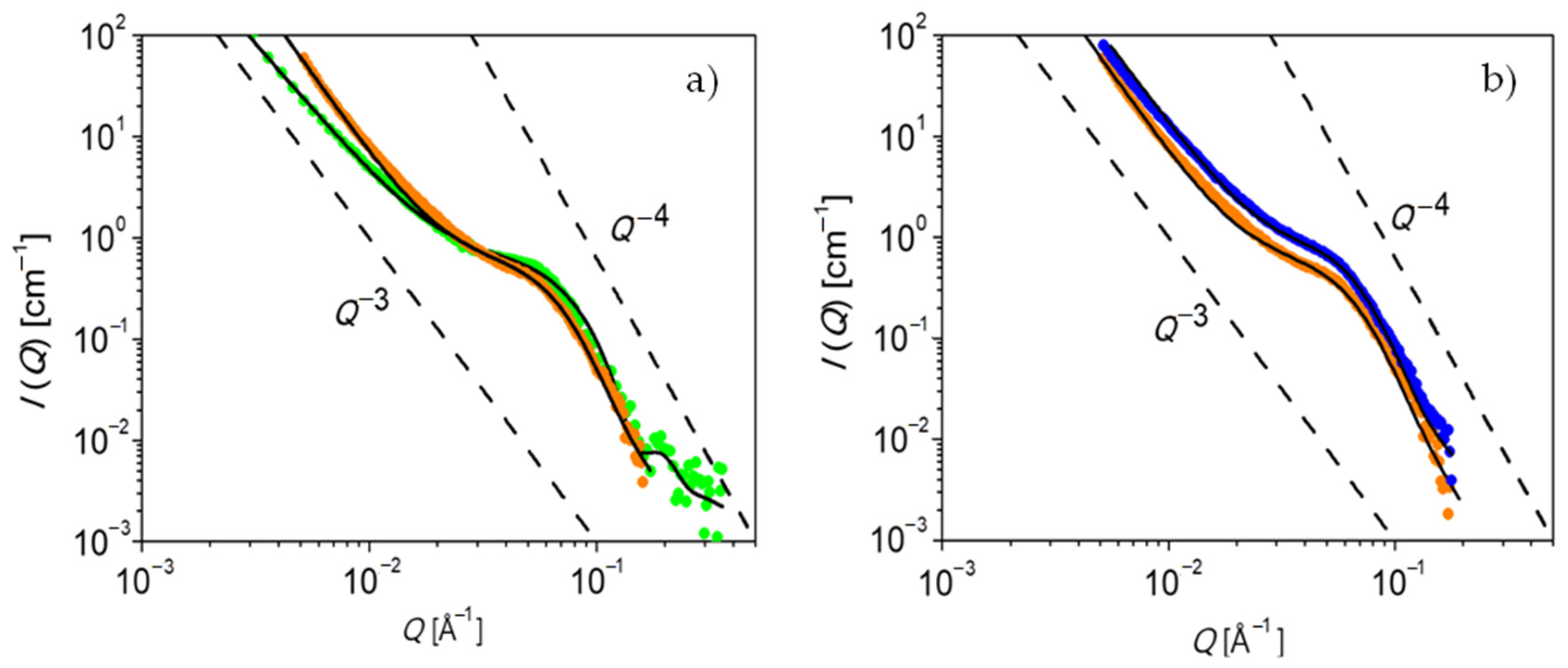
The structure of PEG-upy is studied in the melt by both SAXS and SANS.
Figure 6 shows the obtained data for fully hydrogenated PEG-upy at
T = 333 K and
T = 348 K. The contrast is given by the electron density difference or by the scattering length density difference between the associate upy end groups and the hydrogenous PEG polymer component, depending on the experiment source, either X-rays or neutrons, respectively. The PEG-upy scattering data is corrected for background scattering by subtraction of the contribution of the PEG polymer data at the same temperature. Qualitative observations from the as such corrected small angle scattering pattern show that on one hand the intermediate
Q-range follows a power law,
I(
Q)∝
Q−P, with
and on the other hand an interaction peak can be spotted eventhough toward lower
Q values the scattering pattern is filled with high intensity. Both observations suggest that the morphology of this system consists of interacting compact structures due to phase segregation. Indeed the strong association between upy end-groups is largely favored as the rather high value of
points out [
20,
78]. Therefore, these compact structures are composed by the upy end-groups segregated in clusters separated by the PEG polymer chains, similarly to what is observed in colloidal and micellar systems [
27,
79]. Normally these cluster-like structures are approximated to hard spherical aggregates and are thus treated with the Percus–Yevick model [
64,
80,
81,
82]. The model assumes a suspension of hard spheres with repulsive interaction. The potential is infinite when two spheres touch each other. In the lowest
Q-range a strong decay of the intensity following approximately a
Q−3 power law is also evident; however, no relevant information can be obtained at this
Q-range regarding the morphology.
The scattering intensity per unit of volume of a spherically symmetric particles as in the Percus–Yevick hard sphere model is well known and generally is written as [
65,
83]:
where
is the volume fraction of particles,
the contrast factor between polymer and upy groups forming the spherical particles and
the volume.
is the form factor and
the structure factor due to the contribution of the interactions between particles. Basically, the scattering intensity of the disordered particles can be attributed to the form factor of spheres with radius
(the spherical clusters) and the structure factor of hard spheres (the Percus–Yevick correlation radius due to the interactions between spherical clusters) with a radius of
. We assume that the spherical particles are polydisperse. The detailed description of this model can be found in the
Supporting Information and also in [
64,
80,
83].
The black lines in
Figure 6 correspond to the fit to the data using the Percus–Yevick model. The fit parameters, including the volume fraction of spherical clusters (
ϕ), the mean sphere cluster radius (
) and the Percus–Yevick correlation radius of clusters, (
) are summarized in
Table 5. As displayed in
Figure 6, a reasonable adjustment is obtained and, besides a slight difference at low
Q values, both SAXS and SANS data are very similar for both measured temperatures. In fact, the values from
and
as
and
for both SAXS and SANS data, respectively at
T = 333 K and
for both SAXS and SAXS at
T = 348 K correspond approximately to the end-to-end distance of the PEG polymer (
. Moreover, the volume fractions in PEG-upy are relatively low and roughly agree with the molar fraction of upy groups in the supramolecular polymer (
0.066). This is in accordance with the fact that highly attractive interactions decrease the critical volume concentration that depends sensitively on changes in the interaction between the aggregates or polydispersity, which here is relatively low, ranging from ~11% to ~30%. Actually, this parameter is difficult to characterize as depends greatly on the high
Q scattering, whose range is relatively limited, especially for the SAXS data [
84]. Added to this, the aggregation number,
, defined as the number of end groups per spherical cluster, is an important parameter to characterize the aggregates. From the fit parameter
,
can be calculated as
. Taking the density of the supramolecular polymer melt
= 1.2 g·cm
−3 then
is calculated to be between 25 (SAXS) to 35 (SANS) at
T = 333 K and 31 (SAXS) to 35 (SANS) at
T = 348 K. These results are reasonable, taking into account the high interaction parameter between upy groups and PEG in accordance with literature [
46]. Also concerning the temperature dependence, it is only observed on the SAXS data and therefore the SANS data at
T = 348 K is not shown. Actually, the influence of temperature seems to be mostly seen by an increase of the hard-sphere cluster volume fraction (
Table 5). However, taking into account the error bars of the parameters, this difference cannot be considered, in agreement to what is obtained by SANS. In this context, an increase of temperature from
T = 333 K to
T = 348 K has a very little effect on the PEG-upy structure.
It is demonstrated that PEG-upy forms disordered spherical cluster aggregates as shown by the good agreement of the Percus–Yevick model to the data analysis.
3.3.2. Dynamics
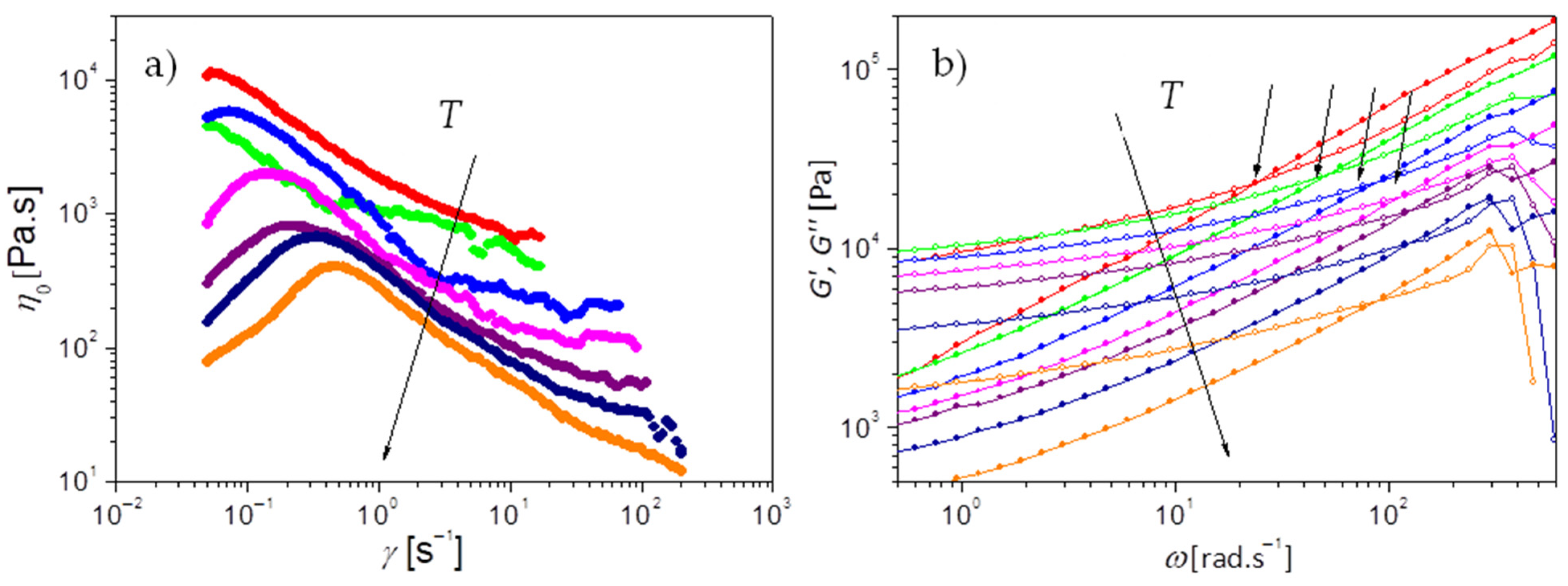
The macroscopic dynamics of PEG-upy is studied by the characterization of the viscoelastic properties measured by rheology in the temperature range from 333 K to 393 K with interval of 10 K.
Figure 7a,b present the viscosity measurements and the complex shear modulus, i.e., the storage (
G′) and loss (
G″) moduli at different angular frequencies, respectively.
Under steady shear, PEG-upy exhibits a shear thickening behavior characterized by an increase in effective viscosity when the shear rate increases past a certain critical value as seen in
Figure 7a. Although rarely observed in common polymer melts or solutions, shear thickening effects have been observed in complex fluids including dense suspensions, wormlike micelles, and associating polymer solutions [
85,
86,
87]. The shear thickening seems to be caused by shear-induced structural changes in all of these systems. Several theoretical models have been proposed to describe the shear thickening behavior, especially in associating polymer solutions [
87]. Marrucci et al. [
88] explored the possibility of shear thickening as arising due to a non-Gaussian chain stretching effect. On the basis of the Tanaka and Edward transient network model, they argued that under flow conditions polymer chains may elongate considerably, well into the non-Gaussian regime, as a result of chain stretching. In an extension, the free path model by Marrucci et al. further assumed that when the chain end dissociates from a network junction, it can only partially relax its extended conformation since it is soon recaptured by the network again. As a consequence, the maximum in the viscosity occurs at a critical shear rate when the ratio of the detachment frequency to the shear rate (which decreases monotonically with increasing shear rate) has not dropped to its asymptotic lower bound, and yet the polymer chains are already stretched close to their maximum extension. In the free path model, the critical shear rate (shear rate at the maximum viscosity)
is then estimated assuming that the elastically active chains reach full extension at the onset of shear thickening. The mean-square distance of the polymer chain is then proportional to the number of Kuhn segments,
and the critical shear rate is approximately:
where
is the network relaxation time. The upy groups that aggregate as spherical clusters are the chain junctions and act as the network crosslinks, eventually forcing the chain extension in case of detachment. The relaxation time of the PEG-upy network
is determined using
taken from
Figure 7a. According to this picture,
is correlated with the detachment relaxation time of the PEG chains from the network junctions, i.e., from the upy clusters.
Moreover, from the oscillatory shear measurements in
Figure 7b it is shown that the storage modulus
G′ dominates over the loss modulus
G″ almost over all the studied frequency range. The slope of around 0.6 (
), is lower than 1 (
), found for Newtonian systems or polymer melts at flow and typical of network-like systems. The elastic behavior dominates as
G’ is relatively constant pointing up to a constant plateau at around 10
4 Pa lower than the plateau module
Pa for PEG polymer [
60]. According to simple rubber elasticity theory, the plateau modulus (
) for a supramolecular network is given by [
89]:
where
is the number density of elastically active strands,
is the Boltzmann constant,
T is the absolute temperature,
is the density of the supramolecular polymer melt (1.2 g·cm
−3), N
A is Avogadro constant,
is the polymer molar mass and
is the fraction of bridging or elastically effective molecules. Considering a perfect network where all PEG polymer chains would adopt a bridging conformation between the upy clusters and thus all would be elastically effective,
= 1 and
mol·m
−3. In this case, the estimated plateau module
Pa for PEG polymer is retrieved [
60]. While simple rubber elasticity theory overestimates
of supramolecular PEG-upy, the overestimation is not surprising. In fact, a lower value and an overestimation of the plateau modulus is found in literature [
89,
90], for systems that also form cluster-like structures due to H-bonding interactions [
90]. Even though H-bonding can create crosslinks that built the network it also introduces defects on the polymeric strands that is formed too due to loops existence, lowering
. Indeed, the possibility of PEG chains to form loops within the same cluster instead of bridges to different clusters has to be considered and can explain the lower
(see further in the text). Besides that, probable contributions from PEG-upy functionalized at only one chain end, which stem from incomplete reaction steps during the functionalization of the polymeric product with subsequent different propensities, inevitably contribute to a lower fraction of effective elastically molecules between the upy crosslinks. In this context, the lower value of
found for PEG-upy can be rationalized to a bridging fraction
close to 1%. Definitely the observation of a plateau modulus clearly shows that the liquid-like viscoelastic properties of PEG at the studied molar mass have changed into a network-like due to the presence of the upy groups, in accordance with what is observed previously by small angle scattering.
Additionally, a crossover () between G′ and G″ is visible at higher frequencies, corresponding to the relaxation time () related to the segmental friction of the polymeric network strands, . It can be defined as the equilibration time or the longest relaxation time of the chains trapped in the network due to the clusters that act as crosslinks.
Figure 8a plots the corresponding temperature dependence of both the network detachment relaxation time
as taken from the viscosity measurements analysis, and the network longest relaxation time
, as obtained from the crossover between storage modulus
G′ and loss modulus
G″.
The data for the network relaxation time
is described using the Arrhenius law [
77]. According to the fit,
kJ/mol and the prefactor for the chain breaking time is 21 μs, which is well in the order of what would be expected for an attempt angular frequency (2 × 10
−5 rad·s
−1). The value of
is also as expected within the typical bond energies for H-bonding (10–65 kJ·mol
−1) [
30,
38] known in literature. According to the free path model, the activation energy of the association is related to the viscosity maximum by
, where
is the viscosity at
,
is the molar density of elastically active strands as defined in Equation (10) and
is the zero-shear viscosity. According to the free path model assumptions, the zero-shear viscosity,
can be calculated, as well as the molar density of elastically active strands
, since from the analysis above,
,
are known and
can be directly extracted from
Figure 7a.
Table 6 summarizes the viscosity maximum
, the plateau modulus
, the molar density of elastically active chains
from both theories and the zero-shear viscosity
at the measured temperatures. The obtained zero-shear viscosity
values seem to agree to the experimental data values at very low shear rates
(
Figure 7a). The molar density of elastically active strands
, is compared to
calculated using Equation (10), and the values are quite compatible. The molar density of elastically active strands
can also be obtained by small angle scattering. According to the structural picture it can be assumed that the molar density related to the Percus–Yevick correlation radius
, between clusters is given by
, and the molar density of elastically active PEG strands correspond to a value between 3.1 mol·m
−3 (SANS) and
mol·m
−3 (SAXS) at
and 3.1 mol·m
−3 (SANS) and
mol·m
−3 (SAXS) at
Undeniably, these values are in very good agreement to the values calculated using the free path model assumptions on the rheology data. Moreover, from
Table 6 we can also get the Weissenberg number
(shear rate multiplied with the relaxation time), at the shear rate at which the viscosity reaches its maximum point (
). The Weissenberg number indicates the degree of orientation generated by the deformation. The Weissenberg number for our system is
, corresponding to a very high orientation of the polymer chains, which is also in conformity with the free path model.
All the above-mentioned arguments show the applicability of the free path model to understand the dynamics of PEG-upy and the good compatibility to the structural analysis.
Finally, information on the segmental dynamics of the network strands is being discussed. The temperature dependence of the longest relaxation time
, related to the segmental friction of the polymeric network strands for PEG-upy as known by polymer theory [
91] is described using the VFTH equation as displayed in
Figure 8b. The glass transition temperature
, for PEG-upy is obtained by doing an extrapolation to
τ = 100 s with the parameters obtained from the fit to the data of
using Equation (7). The glass transition temperature
of PEG-upy polymeric network strands can be also obtained by DSC measurements.
Figure 9 displays the calorimetric curve obtained for PEG-upy. As observed before for PEG polymer and for PEG-thy/dat, here also two-phase transitions are observed, i.e., the glass transition
, and melting temperature
, with increasing temperatures, respectively. Though the value of the glass transition temperature here is also influenced by the crystallinity degree, it gives a clear signal as depicted in
Figure 9. Indeed, the crystallization degree is lowest of all the three studied polymers of around 30%. The analysis of the thermogram thus revealed
≈ 234.2 K and
= 306.5 K and it is summarized further in Table 9. The extrapolation of the temperature dependence of network longest relaxation time
for the low temperature dynamic regime gives a rheological value of
= 245.6 K, which as expected, is closer to the results from the DSC (
≈ 234.2 K) than from the estimation, even though the
from viscoelastic measurements is well above the glass region. Interestingly, an estimation done using the known empirical relationship
, correspond to
≈ 197.7 K of the equivalent linear polymer [
71]. This fact seems to point that the network-like structure and the phase segregation interfere with what is theoretically expected for the segmental dynamics of PEG-upy chains, taking only the
and the lower crystallinity degree into account.


 ,
, 












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}