Polysaccharide-Based In Situ Self-Healing Hydrogels for Tissue Engineering Applications


 ,
,  ,
,  and
and 
Abstract
1. Introduction
2. Self-Healing Mechanisms
2.1. Dynamic Covalent Bonding
2.1.1. Imine Bonds
2.1.2. Acylhydrazone Bonds
2.1.3. Disulfide Bonds
2.1.4. Boronate-Ester Complexation
2.1.5. Diels-Alder “Click Chemistry”
2.2. Physical Bonding
2.2.1. Hydrogen Bonding
2.2.2. Ionic Bonding
2.2.3. Host-Guest Interaction
2.2.4. Hydrophobic Bonding
3. In Situ Self-Healing Polysaccharide-Based Hydrogels
3.1. Hydrophobic Self-Healing Hydrogels
3.2. Ionically Induced Self-Healing Hydrogels
3.3. In Situ Polymerization and Self-Healing Hydrogels
3.4. In Situ Hydrogels Formed via Dynamic Covalent Bonds with Self-Healing
4. Tissue Engineering Applications
4.1. Bone Tissue
4.2. Cartilage
4.3. Muscle
4.4. Skin
4.5. Cardiac
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Pertici, V.; Pin-barre, C.; Rivera, C.; Pellegrino, C.; Gigmes, D.; Trimaille, T. Degradable and Injectable Hydrogel for Drug Delivery in Soft Tissues. Biomacromolecules 2019, 20, 149–163. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H. Injectable hydrogels delivering therapeutic agents for disease treatment and tissue engineering. Biomater. Res. 2018, 22, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Ekerdt, B.L.; Fuentes, C.M.; Lei, Y.; Adil, M.M.; Ramasubramanian, A.; Segalman, R.A.; Schaffer, D.V. Thermoreversible Hyaluronic Acid-PNIPAAm Hydrogel Systems for 3D Stem Cell Culture. Adv. Healthc. Mater. 2018, 18, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Wichterle, O.; Lím, D. Hydrophilic Gels for Biological Use. Nature 1960, 185, 117–118. [Google Scholar] [CrossRef]
- Mohammed, S.; Chouhan, G.; Anuforom, O.; Cooke, M.; Walsh, A.; Morgan-Warren, P.; Jenkins, M.; de Cogan, F. Thermosensitive hydrogel as an in situ gelling antimicrobial ocular dressing. Mater. Sci. Eng. C 2017, 78, 203–209. [Google Scholar] [CrossRef]
- Akhtar, M.F.; Hanif, M.; Ranjha, N.M. Methods of synthesis of hydrogels: A review. Saudi Pharm. J. 2016, 24, 554–559. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Gao, Q.; Lu, X.; Zhou, H. In situ forming hydrogels based on chitosan for drug delivery and tissue regeneration. Asian J. Pharm. Sci. 2016, 11, 673–683. [Google Scholar] [CrossRef]
- Ahmed, E.M. Hydrogel: Preparation, characterization and applications: A review. J. Adv. Res. 2015, 6, 105–121. [Google Scholar] [CrossRef]
- Zhu, J. Bioactive Modification of Poly(ethylenglykol) Hydrogels for Tissue Engineering. Biomaterials 2010, 31, 4639–4656. [Google Scholar] [CrossRef]
- Chae, T.; Yang, H.; Leung, V.; Ko, F.; Troczynski, T. Novel biomimetic hydroxyapatite/alginate nanocomposite fibrous scaffolds for bone tissue regeneration. J. Mater. Sci. Mater. Med. 2013, 24, 1885–1894. [Google Scholar] [CrossRef]
- Loebsack, A.; Greene, K.; Wyatt, S.; Culberson, C.; Austin, C.; Beiler, R.; Roland, W.; Eiselt, P.; Rowley, J.; Burg, K.; et al. In vivo characterization of a porous hydrogel material for use as a tissue engineering bulking agent. J. Biomed. Mater. Res. 2001, 57, 575–581. [Google Scholar] [CrossRef]
- Biondi, M.; Ungaro, F.; Quaglia, F.; Netti, P.A. Controlled drug delivery in tissue engineering. Adv. Drug Deliv. Rev. 2008, 60, 229–242. [Google Scholar] [CrossRef] [PubMed]
- Orienti, I.; Treré, R.; Zecchi, V. Hydrogels formed by cross-linked polyvinylalcohol as colon-specific drug delivery systems. Drug Dev. Ind. Pharm. 2001, 27, 877–884. [Google Scholar] [CrossRef] [PubMed]
- Solorio, L.; Zwolinski, C.; Lund, A.W.; Farrell, M.J.; Jan, P. Gelatin microspheres crosslinked with genipin for local delivery of growth factors. J. Tissue Eng. 2011, 4, 514–523. [Google Scholar] [CrossRef]
- Hoare, T.R.; Kohane, D.S. Hydrogels in drug delivery: Progress and challenges. Polymer 2008, 49, 1993–2007. [Google Scholar] [CrossRef]
- Donnelly, R.; Shaikh, R.; Raj Singh, T.; Garland, M.; Woolfson, A.D. Mucoadhesive drug delivery systems. J. Pharm. Bioallied Sci. 2011, 3, 89. [Google Scholar] [CrossRef] [PubMed]
- Jeong, B.; Kim, S.W.; Bae, Y.H. Thermosensitive sol-gel reversible hydrogels. Adv. Drug Deliv. Rev. 2012, 64, 154–162. [Google Scholar] [CrossRef]
- Leach, J.B.; Bivens, K.A.; Collins, C.N.; Schmidt, C.E. Development of photocrosslinkable hyaluronic acid-polyethylene glycol-peptide composite hydrogels for soft tissue engineering. J. Biomed. Mater. Res. Part A 2004, 70, 74–82. [Google Scholar] [CrossRef]
- Palumbo, F.S.; Fiorica, C.; Di Stefano, M.; Pitarresi, G.; Gulino, A.; Agnello, S.; Giammona, G. In situ forming hydrogels of hyaluronic acid and inulin derivatives for cartilage regeneration. Carbohydr. Polym. 2015, 122, 408–416. [Google Scholar] [CrossRef]
- Xiao Ding, X.; Xian Zheng, Z.; Si Xue, C.; Ren Xi, Z.; Kennedy, J.F. A strategy to introduce the pH sensitivity to temperature sensitive PNIPAAm hydrogels without weakening the thermosensitivity. Carbohydr. Polym. 2007, 68, 416–423. [Google Scholar] [CrossRef]
- Hennink, W.E.; van Nostrum, C.F. Novel crosslinking methods to design hydrogels. Adv. Drug Deliv. Rev. 2012, 64, 223–236. [Google Scholar] [CrossRef]
- Laftah, W.A.; Hashim, S.; Ibrahim, A.N. Polymer hydrogels: A review. Polym. Plast. Technol. Eng. 2011, 50, 1475–1486. [Google Scholar] [CrossRef]
- Maitra, J.; Shukla, V.K. Cross-linking in Hydrogels—A Review. Am. J. Polym. Sci. 2014, 4, 25–31. [Google Scholar] [CrossRef]
- Croisier, F.; Jérôme, C. Chitosan-based biomaterials for tissue engineering. Eur. Polym. J. 2013, 49, 780–792. [Google Scholar] [CrossRef]
- Oliveira, J.T.; Martins, L.; Picciochi, R.; Malafaya, P.B.; Sousa, R.A.; Neves, N.M.; Mano, J.F.; Reis, R.L. Gellan gum: A new biomaterial for cartilage tissue engineering applications. J. Biomed. Mater. Res. Part A 2010, 93, 852–863. [Google Scholar] [CrossRef]
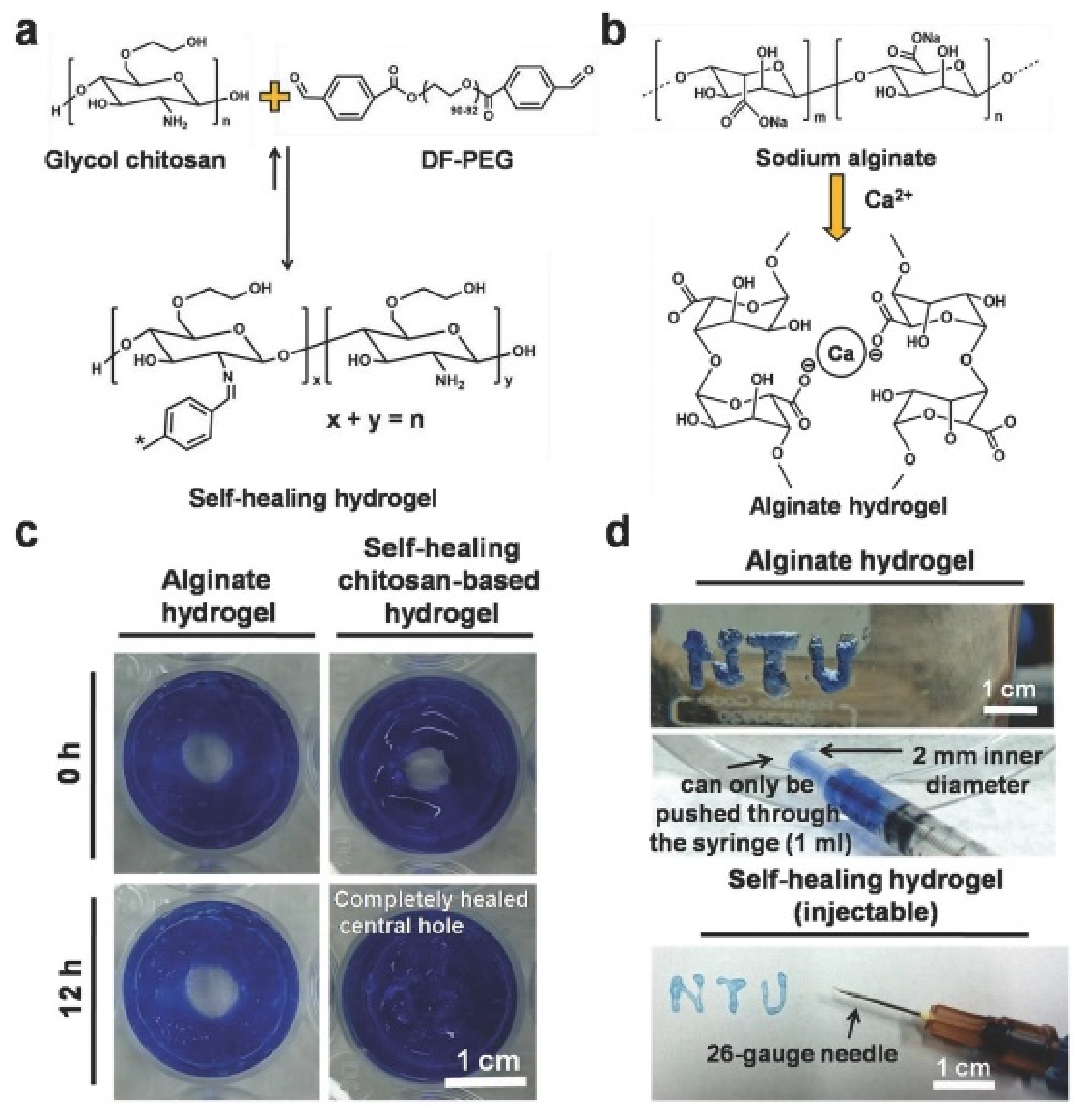
- Venkatesan, J.; Bhatnagar, I.; Manivasagan, P.; Kang, K.-H.; Kim, S.-K. Alginate composites for bone tissue engineering: A review. Int. J. Biol. Macromol. 2015, 72, 269–281. [Google Scholar] [CrossRef]
- Draget, K.I.; Skjak Braek, G.; Smidsod, O. Alginic acid gels: The effect of alginate chemical composition and molecular weight. Carbohydr. Polym. 1994, 25, 31–38. [Google Scholar] [CrossRef]
- Dong Woog, L. Strong adhesion and cohesion of chitosan in aqueous solutions. Langmuir 2013, 29, 14222–14229. [Google Scholar]
- Ozdil, D.; Aydin, H.M. Polymers for medical and tissue engineering applications. J. Chem. Technol. Biotechnol. 2014, 89, 1793–1810. [Google Scholar] [CrossRef]
- Mann, B.K.; Gobin, A.S.; Tsai, A.T.; Schmedlen, R.H.; West, J.L. Smooth muscle cell growth in photopolymerized hydrogels with cell adhesive and proteolytically degradable domains: Synthetic ECM analogs for tissue engineering. Biomaterials 2001, 22, 3045–3051. [Google Scholar] [CrossRef]
- Coviello, T.; Matricardi, P.; Marianecci, C.; Alhaique, F. Polysaccharide hydrogels for modified release formulations. J. Control. Release 2007, 119, 5–24. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Ding, J. Injectable hydrogels as unique biomedical materials. Chem. Soc. Rev. 2008, 37, 1473. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, N.N.; Perez, T.A.; Pedreiro, L.N.; Prezotti, F.G.; Boni, F.I.; Cardoso, V.M.D.O.; Venâncio, T.; Gremião, M.P.D. A novel pH-responsive hydrogel-based on calcium alginate engineered by the previous formation of polyelectrolyte complexes (PECs) intended to vaginal administration. Drug Dev. Ind. Pharm. 2017, 43, 1656–1668. [Google Scholar] [CrossRef] [PubMed]
- Ghanian, M.H.; Mirzadeh, H.; Baharvand, H. In Situ Forming, Cytocompatible, and Self-Recoverable Tough Hydrogels Based on Dual Ionic and Click Cross-Linked Alginate. Biomacromolecules 2018, 19, 1646–1662. [Google Scholar] [CrossRef]
- Han, Y.; Zeng, Q.; Li, H. The calcium silicate/alginate composite: Preparation and evaluation of its behavior as bioactive injectable hydrogels. Acta Biomater. 2013, 9, 9107–9117. [Google Scholar] [CrossRef]
- Qu, J.; Zhao, X.; Ma, P.X.; Guo, B. pH-responsive self-healing injectable hydrogel based on N-carboxyethyl chitosan for hepatocellular carcinoma therapy. Acta Biomater. 2017, 58, 168–180. [Google Scholar] [CrossRef]
- Zhang, L.; Li, Y.; Li, L.; Guo, B.; Ma, P.X. Reactive & Functional Polymers Non-cytotoxic conductive carboxymethyl-chitosan/aniline pentamer hydrogels. React. Funct. Polym. 2014, 82, 81–88. [Google Scholar] [CrossRef]
- Tao, Y.; Ai, L.; Bai, H.; Liu, X. Synthesis of pH-responsive photocrosslinked hyaluronic acid-based hydrogels for drug delivery. J. Polym. Sci. Part A Polym. Chem. 2012, 50, 3507–3516. [Google Scholar] [CrossRef]
- Wieland, J.A.; Houchin-Ray, T.L.; Shea, L.D. Non-viral vector delivery from PEG-hyaluronic acid hydrogels Julie. J. Control. Release 2009, 120, 233–241. [Google Scholar] [CrossRef]
- Segura, T.; Chung, P.H.; Shea, L.D. DNA delivery from hyaluronic acid-collagen hydrogels via a substrate-mediated approach. Biomaterials 2005, 26, 1575–1584. [Google Scholar] [CrossRef]
- Roy, K.; Mao, H.Q.; Huang, S.K.; Leong, K.W. Oral gene delivery with chitosan--DNA nanoparticles generates immunologic protection in a murine model of peanut allergy. Nat. Med. 1999, 5, 387. [Google Scholar] [CrossRef]
- Li, Y.; Wang, X.; Fu, Y.; Wei, Y.; Zhao, L.; Tao, L. Self-Adapting Hydrogel to Improve the Therapeutic Effect in Wound-Healing. ACS Appl. Mater. Interfaces 2018, 10, 26046–26055. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Liu, K.; Li, T.; Zhang, W.; Dong, Y.; Lv, J.; Wang, W.; Sun, J.; Li, M.; Wang, M.; et al. An in situ hydrogel based on carboxymethyl chitosan and sodium alginate dialdehyde for corneal wound healing after alkali burn. J. Biomed. Mater. Res. Part A 2019, 107, 742–754. [Google Scholar] [CrossRef]
- Hu, Y.; Zhang, Z.; Li, Y.; Ding, X.; Li, D.; Shen, C.; Xu, F.J. Dual-Crosslinked Amorphous Polysaccharide Hydrogels Based on Chitosan/Alginate for Wound Healing Applications. Macromol. Rapid Commun. 2018, 39, 1–5. [Google Scholar] [CrossRef]
- Nguyen, Q.V.; Huynh, D.P.; Park, J.H.; Lee, D.S. Injectable polymeric hydrogels for the delivery of therapeutic agents: A review. Eur. Polym. J. 2015, 72, 602–619. [Google Scholar] [CrossRef]
- Jain, A.; Kim, Y.T.; McKeon, R.J.; Bellamkonda, R.V. In situ gelling hydrogels for conformal repair of spinal cord defects, and local delivery of BDNF after spinal cord injury. Biomaterials 2006, 27, 497–504. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.; Yang, J.H.; Liu, Z.Q.; Xu, F.; Zhou, J.X.; Zrínyi, M.; Osada, Y.; Chen, Y.M. Novel biocompatible polysaccharide-based self-healing hydrogel. Adv. Funct. Mater. 2015, 25, 1352–1359. [Google Scholar] [CrossRef]
- Taylor, D.L.; in het Panhuis, M. Self-Healing Hydrogels. Adv. Mater. 2016, 28, 9060–9093. [Google Scholar] [CrossRef]
- Phadke, A.; Zhang, C.; Arman, B.; Hsu, C.C.; Mashelkar, R.A.; Lele, A.K.; Tauber, M.J.; Arya, G.; Varghese, S. Rapid self-healing hydrogels. Proc. Natl. Acad. Sci. USA 2012, 109, 4383–4388. [Google Scholar] [CrossRef] [PubMed]
- Tu, Y.; Chen, N.; Li, C.; Liu, H.; Zhu, R.; Chen, S.; Xiao, Q.; Liu, J.; Ramakrishna, S.; He, L. Advances in injectable self-healing biomedical hydrogels. Acta Biomater. 2019, 90, 1–20. [Google Scholar] [CrossRef]
- Barroso, N.; Guaresti, O.; Pérez-Álvarez, L.; Ruiz-Rubio, L.; Gabilondo, N.; Vilas-Vilela, J.L. Self-healable hyaluronic acid/chitosan polyelectrolyte complex hydrogels and multilayers. Eur. Polym. J. 2019, 120, 109268. [Google Scholar] [CrossRef]
- Li, Q.; Liu, C.; Wen, J.; Wu, Y.; Shan, Y.; Liao, J. The design, mechanism and biomedical application of self-healing hydrogels. Chin. Chem. Lett. 2017, 28, 1857–1874. [Google Scholar] [CrossRef]
- Zou, W.; Dong, J.; Luo, Y.; Zhao, Q.; Xie, T. Dynamic Covalent Polymer Networks: From Old Chemistry to Modern Day Innovations. Adv. Mater. 2017, 29, 1606100. [Google Scholar] [CrossRef]
- García, F.; Smulders, M.M.J. Dynamic covalent polymers. J. Polym. Sci. Part A Polym. Chem. 2016, 54, 3551–3577. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Hsu, S.H. Synthesis and biomedical applications of self-healing hydrogels. Front. Chem. 2018, 6, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Zhang, Y.; Zhang, X.; Tao, L.; Li, S.; Wei, Y. Facilely prepared inexpensive and biocompatible self-healing hydrogel: A new injectable cell therapy carrier. Polym. Chem. 2012, 3, 3235–3238. [Google Scholar] [CrossRef]
- Lü, S.; Gao, C.; Xu, X.; Bai, X.; Duan, H.; Gao, N.; Feng, C.; Xiong, Y.; Liu, M. Injectable and Self-Healing Carbohydrate-Based Hydrogel for Cell Encapsulation. ACS Appl. Mater. Interfaces 2015, 7, 13029–13037. [Google Scholar] [CrossRef]
- Li, S.; Pei, M.; Wan, T.; Yang, H.; Gu, S.; Tao, Y.; Liu, X.; Zhou, Y.; Xu, W.; Xiao, P. Self-healing hyaluronic acid hydrogels based on dynamic Schiff base linkages as biomaterials. Carbohydr. Polym. 2020, 250, 116922. [Google Scholar] [CrossRef]
- Zhang, Y.; Tao, L.; Li, S.; Wei, Y. Synthesis of multiresponsive and dynamic chitosan-based hydrogels for controlled release of bioactive molecules. Biomacromolecules 2011, 12, 2894–2901. [Google Scholar] [CrossRef]
- Liu, F.; Li, F.; Deng, G.; Chen, Y.; Zhang, B.; Zhang, J.; Liu, C.Y. Rheological images of dynamic covalent polymer networks and mechanisms behind mechanical and self-healing properties. Macromolecules 2012, 45, 1636–1645. [Google Scholar] [CrossRef]
- Chang, R.; Wang, X.; Li, X.; An, H.; Qin, J. Self-Activated Healable Hydrogels with Reversible Temperature Responsiveness. ACS Appl. Mater. Interfaces 2016, 8, 25544–25551. [Google Scholar] [CrossRef] [PubMed]
- Deng, G.; Tang, C.; Li, F.; Jiang, H.; Chen, Y. Covalent cross-linked polymer gels with reversible sol-gel transition and self-healing properties. Macromolecules 2010, 43, 1191–1194. [Google Scholar] [CrossRef]
- Fairbanks, B.D.; Singh, S.P.; Bowman, C.N.; Anseth, K.S. Photodegradable, photoadaptable hydrogels via radical-mediated disulfide fragmentation reaction. Macromolecules 2011, 44, 2444–2450. [Google Scholar] [CrossRef] [PubMed]
- Guan, Y.; Zhang, Y. Boronic acid-containing hydrogels: Synthesis and their applications. Chem. Soc. Rev. 2013, 42, 8106–8121. [Google Scholar] [CrossRef]
- Yesilyurt, V.; Webber, M.J.; Appel, E.A.; Godwin, C.; Langer, R.; Anderson, D.G. Injectable Self-Healing Glucose-Responsive Hydrogels with pH-Regulated Mechanical Properties. Adv. Mater. 2016, 28, 86–91. [Google Scholar] [CrossRef]
- Yan, J.; Springsteen, G.; Deeter, S.; Wang, B. The relationship among pK a, pH, and binding constants in the interactions between boronic acids and diols—It is not as simple as it appears. Tetrahedron 2004, 60, 11205–11209. [Google Scholar] [CrossRef]
- Axthelm, J.; Askes, S.H.C.; Elstner, M.; Upendar Reddy, G.; Görls, H.; Bellstedt, P.; Schiller, A. Fluorinated Boronic Acid-Appended Pyridinium Salts and 19F NMR Spectroscopy for Diol Sensing. J. Am. Chem. Soc. 2017, 139, 11413–11420. [Google Scholar] [CrossRef]
- Cai, B.; Luo, Y.; Guo, Q.; Zhang, X.; Wu, Z. A glucose-sensitive block glycopolymer hydrogel based on dynamic boronic ester bonds for insulin delivery. Carbohydr. Res. 2017, 445, 32–39. [Google Scholar] [CrossRef]
- Liu, Y.L.; Chuo, T.W. Self-healing polymers based on thermally reversible Diels-Alder chemistry. Polym. Chem. 2013, 4, 2194–2205. [Google Scholar] [CrossRef]
- Zhang, H.; Xia, H.; Zhao, Y. Poly(vinyl alcohol) hydrogel can autonomously self-heal. ACS Macro Lett. 2012, 1, 1233–1236. [Google Scholar] [CrossRef]
- Mcqueen-Mason, S.; Cosgrove, D.J. Disruption of hydrogen bonding between plant cell wall polymers by proteins that induce wall extension. Proc. Natl. Acad. Sci. USA 1994, 91, 6574–6578. [Google Scholar] [CrossRef] [PubMed]
- Shi, D.; Liu, R.; Dong, W.; Li, X.; Zhang, H.; Chen, M.; Akashi, M. PH-dependent and self-healing properties of mussel modified poly(vinyl alcohol) hydrogels in a metal-free environment. RSC Adv. 2015, 5, 82252–82258. [Google Scholar] [CrossRef]
- Guo, Y.; Zhou, X.; Tang, Q.; Bao, H.; Wang, G.; Saha, P. A self-healable and easily recyclable supramolecular hydrogel electrolyte for flexible supercapacitors. J. Mater. Chem. A 2016, 4, 8769–8776. [Google Scholar] [CrossRef]
- Yavvari, P.S.; Srivastava, A. Robust, self-healing hydrogels synthesised from catechol rich polymers. J. Mater. Chem. B 2015, 3, 899–910. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Narain, R.; Zeng, H. Rational design of self-healing tough hydrogels: A mini review. Front. Chem. 2018, 6, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Cai, L.; Jia, Y.G.; Liu, S.; Chen, Y.; Ren, L. Progress in self-healing hydrogels assembled by host-guest interactions: Preparation and biomedical applications. J. Mater. Chem. B 2019, 7, 1637–1651. [Google Scholar] [CrossRef] [PubMed]
- Nakahata, M.; Takashima, Y.; Yamaguchi, H.; Harada, A. Redox-responsive self-healing materials formed from hostĝ€" guest polymers. Nat. Commun. 2011, 2, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.G.; Zhang, M.; Zhu, X.X. CO2-Switchable Self-Healing Host-Guest Hydrogels. Macromolecules 2017, 50, 9696–9701. [Google Scholar] [CrossRef]
- Xiong, C.; Zhang, L.; Xie, M.; Sun, R. Photoregulating of Stretchability and Toughness of a Self-Healable Polymer Hydrogel. Macromol. Rapid Commun. 2018, 39, 2–7. [Google Scholar] [CrossRef]
- Loh, X.J. Supramolecular host-guest polymeric materials for biomedical applications. Mater. Horiz. 2014, 1, 185–195. [Google Scholar] [CrossRef]
- Zhang, J.; Guo, D.S.; Wang, L.H.; Wang, Z.; Liu, Y. Supramolecular binary hydrogels from calixarenes and amino acids and their entrapment-release of model dye molecules. Soft Matter 2011, 7, 1756–1762. [Google Scholar] [CrossRef]
- Tuncaboylu, D.C.; Sari, M.; Oppermann, W.; Okay, O. Tough and self-healing hydrogels formed via hydrophobic interactions. Macromolecules 2011, 44, 4997–5005. [Google Scholar] [CrossRef]
- Can, V.; Kochovski, Z.; Reiter, V.; Severin, N.; Siebenbürger, M.; Kent, B.; Just, J.; Rabe, J.P.; Ballauff, M.; Okay, O. Nanostructural Evolution and Self-Healing Mechanism of Micellar Hydrogels. Macromolecules 2016, 49, 2281–2287. [Google Scholar] [CrossRef]
- Klouda, L.; Mikos, A.G. Thermoresponsive hydrogels in biomedical applications—A review. Eur. J. Pharm. Biopharm. 2011, 68, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Tai, H.; Howard, D.; Takae, S.; Wang, W.; Vermonden, T.; Hennink, W.E.; Stayton, P.S.; Hoffman, A.S.; Endruweit, A.; Alexander, C.; et al. Photo-cross-linked hydrogels from thermoresponsive PEGMEMA-PPGMA-EGDMA copolymers containing multiple methacrylate groups: Mechanical property, swelling, protein release, and cytotoxicity. Biomacromolecules 2009, 10, 2895–2903. [Google Scholar] [CrossRef] [PubMed]
- Ren, Z.; Wang, Y.; Ma, S.; Duan, S.; Yang, X.; Gao, P.; Zhang, X.; Cai, Q. Effective Bone Regeneration Using Thermosensitive Poly(N-Isopropylacrylamide) Grafted Gelatin as Injectable Carrier for Bone Mesenchymal Stem Cells. ACS Appl. Mater. Interfaces 2015, 7, 19006–19015. [Google Scholar] [CrossRef] [PubMed]
- Gioffredi, E.; Boffito, M.; Calzone, S.; Giannitelli, S.M.; Rainer, A.; Trombetta, M.; Mozetic, P.; Chiono, V. Pluronic F127 Hydrogel Characterization and Biofabrication in Cellularized Constructs for Tissue Engineering Applications. Procedia CIRP 2016, 49, 125–132. [Google Scholar] [CrossRef]
- Chung, H.J.; Lee, Y.; Park, T.G. Thermo-sensitive and biodegradable hydrogels based on stereocomplexed Pluronic multi-block copolymers for controlled protein delivery. J. Control. Release 2008, 127, 22–30. [Google Scholar] [CrossRef]
- Xu, J.; Strandman, S.; Zhu, J.X.X.; Barralet, J.; Cerruti, M. Biomaterials Genipin-crosslinked catechol-chitosan mucoadhesive hydrogels for buccal drug delivery. Biomaterials 2015, 37, 395–404. [Google Scholar] [CrossRef]
- Shi, A.; Dai, X.; Jing, Z. Tough and Self-Healing Chitosan/Poly(acrylamide-co-acrylic acid) Double Network Hydrogels. Polym. Sci. Ser. A 2020, 62, 228–239. [Google Scholar] [CrossRef]
- Matsuno, T.; Hashimoto, Y.; Adachi, S. Preparation of injectable 3D-formed β-tricalcium phosphate bead/alginate composite for bone tissue engineering. Dent. Mater. 2008, 27, 827–834. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.H.; Lim, S.M.; Han, D.K.; Yuk, S.H.; Im, G.I.; Lee, J.H. Time-dependent alginate/polyvinyl alcohol hydrogels as injectable cell carriers. J. Biomater. Sci. Polym. Ed. 2009, 20, 863–876. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, G.S.; Lal, H. Novel grafted cellulose-based hydrogels for water technologies. Desalination 2003, 159, 131–138. [Google Scholar] [CrossRef]
- Jaipan, P.; Nguyen, A.; Narayan, R.J. Gelatin-based hydrogels for biomedical applications. MRS Commun. 2017, 7, 416–426. [Google Scholar] [CrossRef]
- Seidlits, S.K.; Khaing, Z.Z.; Petersen, R.R.; Nickels, J.D.; Vanscoy, J.E.; Shear, J.B.; Schmidt, C.E. The effects of hyaluronic acid hydrogels with tunable mechanical properties on neural progenitor cell differentiation. Biomaterials 2010, 31, 3930–3940. [Google Scholar] [CrossRef]
- Luan, S.; Zhu, Y.; Wu, X.; Wang, Y.; Liang, F.; Song, S. Hyaluronic-Acid-Based pH-Sensitive Nanogels for Tumor-Targeted Drug Delivery. ACS Biomater. Sci. Eng. 2017, 3, 2410–2419. [Google Scholar] [CrossRef]
- Miao, T.; Fenn, S.L.; Charron, P.N.; Oldinski, R.A. Self-Healing and Thermoresponsive Dual-Cross-Linked Alginate Hydrogels Based on Supramolecular Inclusion Complexes. Biomacromolecules 2015, 16, 3740–3750. [Google Scholar] [CrossRef]
- Hao, X.; Liu, H.; Xie, Y.; Fang, C.; Yang, H. Thermal-responsive self-healing hydrogel based on hydrophobically modified chitosan and vesicle. Colloid Polym. Sci. 2013, 291, 1749–1758. [Google Scholar] [CrossRef]
- Schmaljohann, D. Thermo- and pH-responsive polymers in drug delivery. Adv. Drug Deliv. Rev. 2006, 58, 1655–1670. [Google Scholar] [CrossRef]
- Du, H.; Liu, M.; Yang, X.; Zhai, G. The design of pH-sensitive chitosan-based formulations for gastrointestinal delivery. Drug Discov. Today 2015, 20, 1004–1011. [Google Scholar] [CrossRef]
- Li, M.; Mondrinos, M.J.; Chen, X.; Gandhi, M.R.; Ko, F.K.; Lelkes, P.I. Elastin Blends for Tissue Engineering Scaffolds. J. Biomed. Mater. Res. Part A 2006, 79, 963–973. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Lou, R.; Liu, X.; Gao, M.; Zheng, H.; Yang, T.; Xie, H.; Yu, W.; Ma, X. A self-healing hydrogel formation strategy: Via exploiting endothermic interactions between polyelectrolytes. Chem. Commun. 2016, 52, 6273–6276. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.; Datta, L.P.; Roy, S. A Stimuli-Responsive Supramolecular Hydrogel for Controlled Release of Drug. J. Mol. Eng. Mater. 2017, 5, 1750011. [Google Scholar] [CrossRef]
- Alegre-Requena, J.V.; Häring, M.; Herrera, R.P.; Díaz Díaz, D. Regulatory parameters of self-healing alginate hydrogel networks prepared via mussel-inspired dynamic chemistry. New J. Chem. 2016, 40, 8493–8501. [Google Scholar] [CrossRef]
- Dai, X.; Zhang, Y.; Gao, L.; Bai, T.; Wang, W.; Cui, Y.; Liu, W. A mechanically strong, highly stable, thermoplastic, and self-healable supramolecular polymer hydrogel. Adv. Mater. 2015, 27, 3566–3571. [Google Scholar] [CrossRef]
- Fu, B.; Cheng, B.; Bao, X.; Wang, Z.; Shangguan, Y.; Hu, Q. Self-healing and conductivity of chitosan-based hydrogels formed by the migration of ferric ions. J. Appl. Polym. Sci. 2019, 136, 1–8. [Google Scholar] [CrossRef]
- You, J.; Xie, S.; Cao, J.; Ge, H.; Xu, M.; Zhang, L.; Zhou, J. Quaternized Chitosan/Poly(acrylic acid) Polyelectrolyte Complex Hydrogels with Tough, Self-Recovery, and Tunable Mechanical Properties. Macromolecules 2016, 49, 1049–1059. [Google Scholar] [CrossRef]
- Sun, J.-Y.; Zhao, X.; Illeperuma, W.R.K.; Chaudhuri, O.; Oh, K.H.; Mooney, D.J.; Vlassak, J.J.; Suo, Z. Highly stretchable and tough hydrogels. Nature 2012, 489, 133–136. [Google Scholar] [CrossRef]
- Haque, M.A.; Kurokawa, T.; Kamita, G.; Gong, J.P. Lamellar bilayers as reversible sacrificial bonds to toughen hydrogel: Hysteresis, self-recovery, fatigue resistance, and crack blunting. Macromolecules 2011, 44, 8916–8924. [Google Scholar] [CrossRef]
- Seitz, M.E.; Martina, D.; Baumberger, T.; Krishnan, V.R.; Hui, C.Y.; Shull, K.R. Fracture and large strain behavior of self-assembled triblock copolymer gels. Soft Matter 2009, 5, 447–456. [Google Scholar] [CrossRef]
- Gong, J.P.; Katsuyama, Y.; Kurokawa, T.; Osada, Y. Double-network hydrogels with extremely high mechanical strength. Adv. Mater. 2003, 15, 1155–1158. [Google Scholar] [CrossRef]
- Vahedi, M.; Barzin, J.; Shokrolahi, F.; Shokrollahi, P. Self-Healing, Injectable Gelatin Hydrogels Cross-Linked by Dynamic Schiff Base Linkages Support Cell Adhesion and Sustained Release of Antibacterial Drugs. Macromol. Mater. Eng. 2018, 303, 1–10. [Google Scholar] [CrossRef]
- Tseng, T.C.; Tao, L.; Hsieh, F.Y.; Wei, Y.; Chiu, I.M.; Hsu, S.H. An injectable, self-healing hydrogel to repair the central nervous system. Adv. Mater. 2015, 27, 3518–3524. [Google Scholar] [CrossRef]
- Liu, Y.; Hsu, Y.-H.; Huang, A.P.-H.; Hsu, S. Semi-interpenetrating polymer network of hyaluronan and chitosan self-healing hydrogels for central nervous system repair. ACS Appl. Mater. Interfaces 2020, 12, 40108–40120. [Google Scholar] [CrossRef] [PubMed]
- Ye, B.; Zhang, S.; Li, R.; Li, L.; Lu, L.; Zhou, C. An in-situ formable and fibrils-reinforced polysaccharide composite hydrogel by self-crosslinking with dual healing ability. Compos. Sci. Technol. 2018, 156, 238–246. [Google Scholar] [CrossRef]
- Malda, J.; Visser, J.; Melchels, F.P.; Jüngst, T.; Hennink, W.E.; Dhert, W.J.A.; Groll, J.; Hutmacher, D.W. 25th Anniversary Article: Engineering Hydrogels for Biofabrication. Adv. Mater. 2013, 25, 5011–5028. [Google Scholar] [CrossRef]
- Ensrud, K.E. Epidemiology of Fracture Risk With Advancing Age. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2013, 68, 1236–1242. [Google Scholar] [CrossRef]
- Zhang, K.; Feng, Q.; Xu, J.; Xu, X.; Tian, F.; Yeung, K.W.K.; Bian, L. Self-Assembled Injectable Nanocomposite Hydrogels Stabilized by Bisphosphonate-Magnesium (Mg2+) Coordination Regulates the Differentiation of Encapsulated Stem Cells via Dual Crosslinking. Adv. Funct. Mater. 2017, 27, 1–11. [Google Scholar] [CrossRef]
- Shi, L.; Wang, F.; Zhu, W.; Xu, Z.; Fuchs, S.; Hilborn, J.; Zhu, L.; Ma, Q.; Wang, Y.; Weng, X.; et al. Self-Healing Silk Fibroin-Based Hydrogel for Bone Regeneration: Dynamic Metal-Ligand Self-Assembly Approach. Adv. Funct. Mater. 2017, 27, 1700591. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, M.; Dai, Z.; Cao, H.; Li, J.; Zhang, W. Sustained protein therapeutics enabled by self-healing nanocomposite hydrogels for non-invasive bone regeneration. Biomater. Sci. 2020, 8, 682–693. [Google Scholar] [CrossRef]
- Vahedi, M.; Shokrolahi, F.; Barzin, J.; Shokrollahi, P.; Taghiyar, L.; Ashtiani, M.K. Amylopectin Multiple Aldehyde Crosslinked Hydrogel as an Injectable and Self-Healing Cell Carrier for Bone Tissue Engineering. Macromol. Mater. Eng. 2020, 305, 1–13. [Google Scholar] [CrossRef]
- Hou, S.; Wang, X.; Park, S.; Jin, X.; Ma, P.X. Rapid Self-Integrating, Injectable Hydrogel for Tissue Complex Regeneration. Adv. Healthc. Mater. 2015, 4, 1491–1495. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Cao, X.; Du, J.; Wang, G.; Chen, X. Multifunctional Hydrogel with Good Structure Integrity, Self-Healing, and Tissue-Adhesive Property Formed by Combining Diels-Alder Click Reaction and Acylhydrazone Bond. ACS Appl. Mater. Interfaces 2015, 7, 24023–24031. [Google Scholar] [CrossRef] [PubMed]
- Guo, B.; Qu, J.; Zhao, X.; Zhang, M. Degradable conductive self-healing hydrogels based on dextran-graft-tetraaniline and N-carboxyethyl chitosan as injectable carriers for myoblast cell therapy and muscle regeneration. Acta Biomater. 2019, 84, 180–193. [Google Scholar] [CrossRef] [PubMed]
- Qu, J.; Zhao, X.; Liang, Y.; Zhang, T.; Ma, P.X.; Guo, B. Antibacterial adhesive injectable hydrogels with rapid self-healing, extensibility and compressibility as wound dressing for joints skin wound healing. Biomaterials 2018, 183, 185–199. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Zhao, X.; Hu, T.; Han, Y.; Guo, B. Mussel-inspired, antibacterial, conductive, antioxidant, injectable composite hydrogel wound dressing to promote the regeneration of infected skin. J. Colloid Interface Sci. 2019, 556, 514–528. [Google Scholar] [CrossRef]
- Zhao, X.; Wu, H.; Guo, B.; Dong, R.; Qiu, Y.; Ma, P.X. Antibacterial anti-oxidant electroactive injectable hydrogel as self-healing wound dressing with hemostasis and adhesiveness for cutaneous wound healing. Biomaterials 2017, 122, 34–47. [Google Scholar] [CrossRef]
- Wang, C.; Liang, C.; Wang, R.; Yao, X.; Guo, P.; Yuan, W.; Liu, Y.; Song, Y.; Li, Z.; Xie, X. The fabrication of a highly efficient self-healing hydrogel from natural biopolymers loaded with exosomes for the synergistic promotion of severe wound healing. Biomater. Sci. 2020, 8, 313–324. [Google Scholar] [CrossRef]
- Miyahara, Y.; Nagaya, N.; Kataoka, M.; Yanagawa, B.; Tanaka, K.; Hao, H.; Ishino, K.; Ishida, H.; Shimizu, T.; Kangawa, K.; et al. Monolayered mesenchymal stem cells repair scarred myocardium after myocardial infarction. Nat. Med. 2006, 12, 459–465. [Google Scholar] [CrossRef]
- Koudstaal, S.; Bastings, M.M.C.; Feyen, D.A.M.; Waring, C.D.; Van Slochteren, F.J.; Dankers, P.Y.W.; Torella, D.; Sluijter, J.P.G.; Nadal-Ginard, B.; Doevendans, P.A.; et al. Sustained delivery of insulin-like growth factor-1/hepatocyte growth factor stimulates endogenous cardiac repair in the chronic infarcted pig heart. J. Cardiovasc. Transl. Res. 2014, 7, 232–241. [Google Scholar] [CrossRef]
- Dong, R.; Zhao, X.; Guo, B.; Ma, P.X. Self-Healing Conductive Injectable Hydrogels with Antibacterial Activity as Cell Delivery Carrier for Cardiac Cell Therapy. ACS Appl. Mater. Interfaces 2016, 8, 17138–17150. [Google Scholar] [CrossRef] [PubMed]
- Bastings, M.M.C.; Koudstaal, S.; Kieltyka, R.E.; Nakano, Y.; Pape, A.C.H.; Feyen, D.A.M.; van Slochteren, F.J.; Doevendans, P.A.; Sluijter, J.P.G.; Meijer, E.W.; et al. A fast pH-switchable and self-healing supramolecular hydrogel carrier for guided, local catheter injection in the infarcted myocardium. Adv. Healthc. Mater. 2014, 3, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Yavvari, P.S.; Pal, S.; Kumar, S.; Kar, A.; Awasthi, A.K.; Naaz, A.; Srivastava, A.; Bajaj, A. Injectable, Self-Healing Chimeric Catechol-Fe(III) Hydrogel for Localized Combination Cancer Therapy. ACS Biomater. Sci. Eng. 2017, 3, 3404–3413. [Google Scholar] [CrossRef]



















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
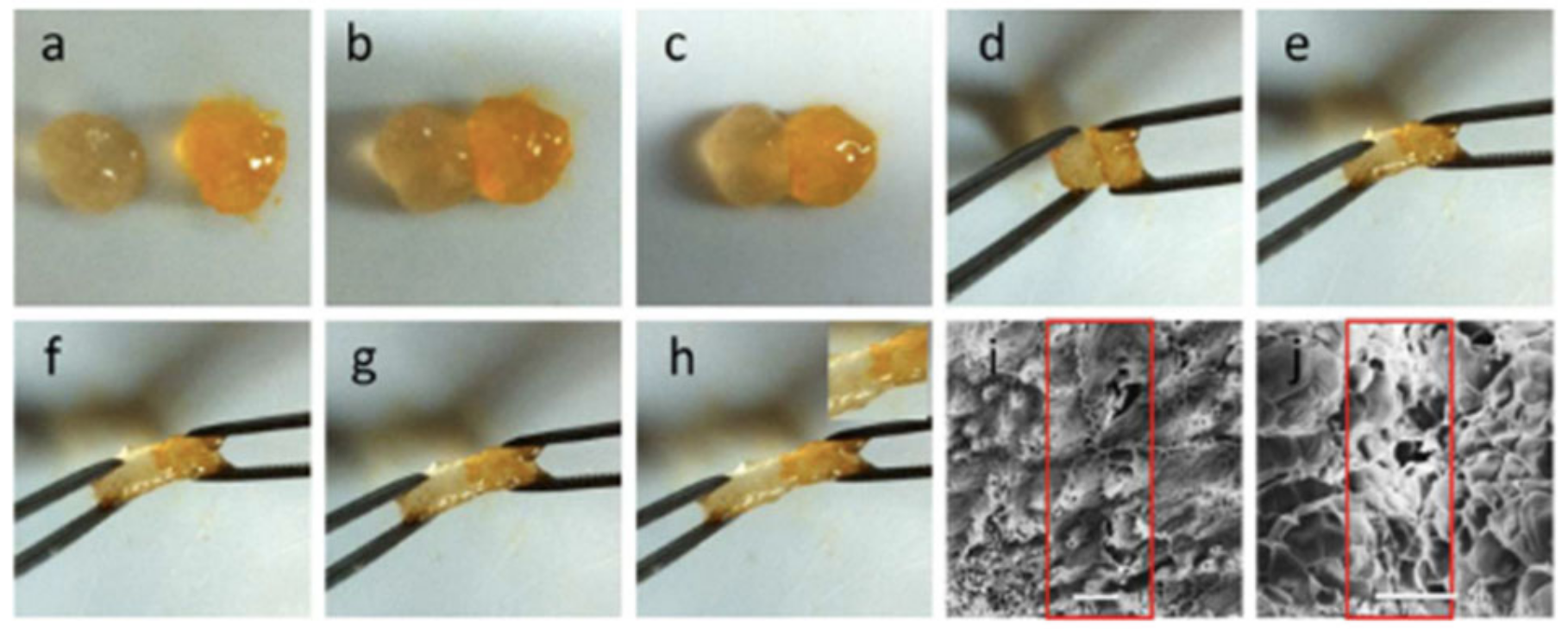{kind=link}
{kind=link}
{kind=link}
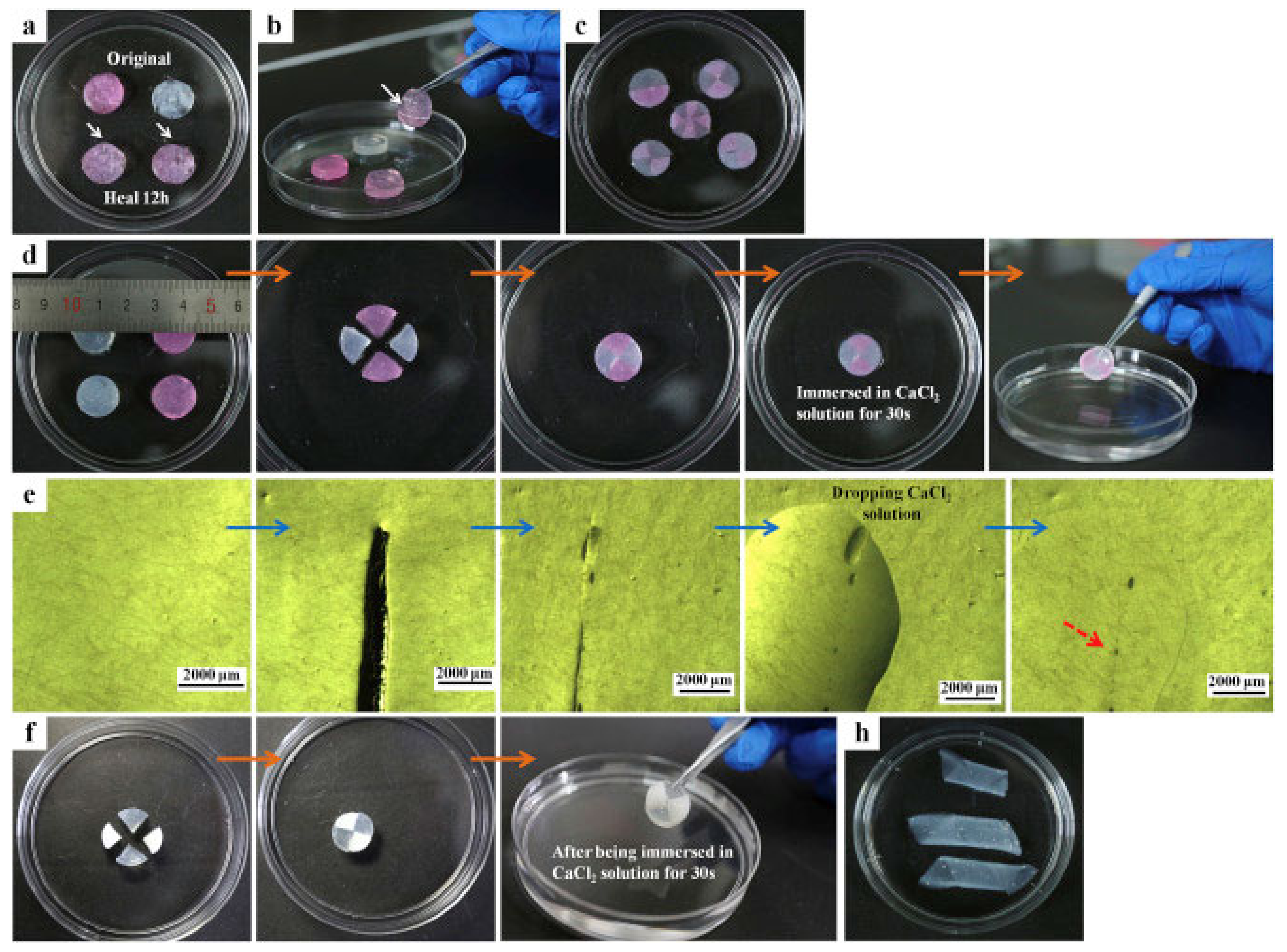{kind=link}
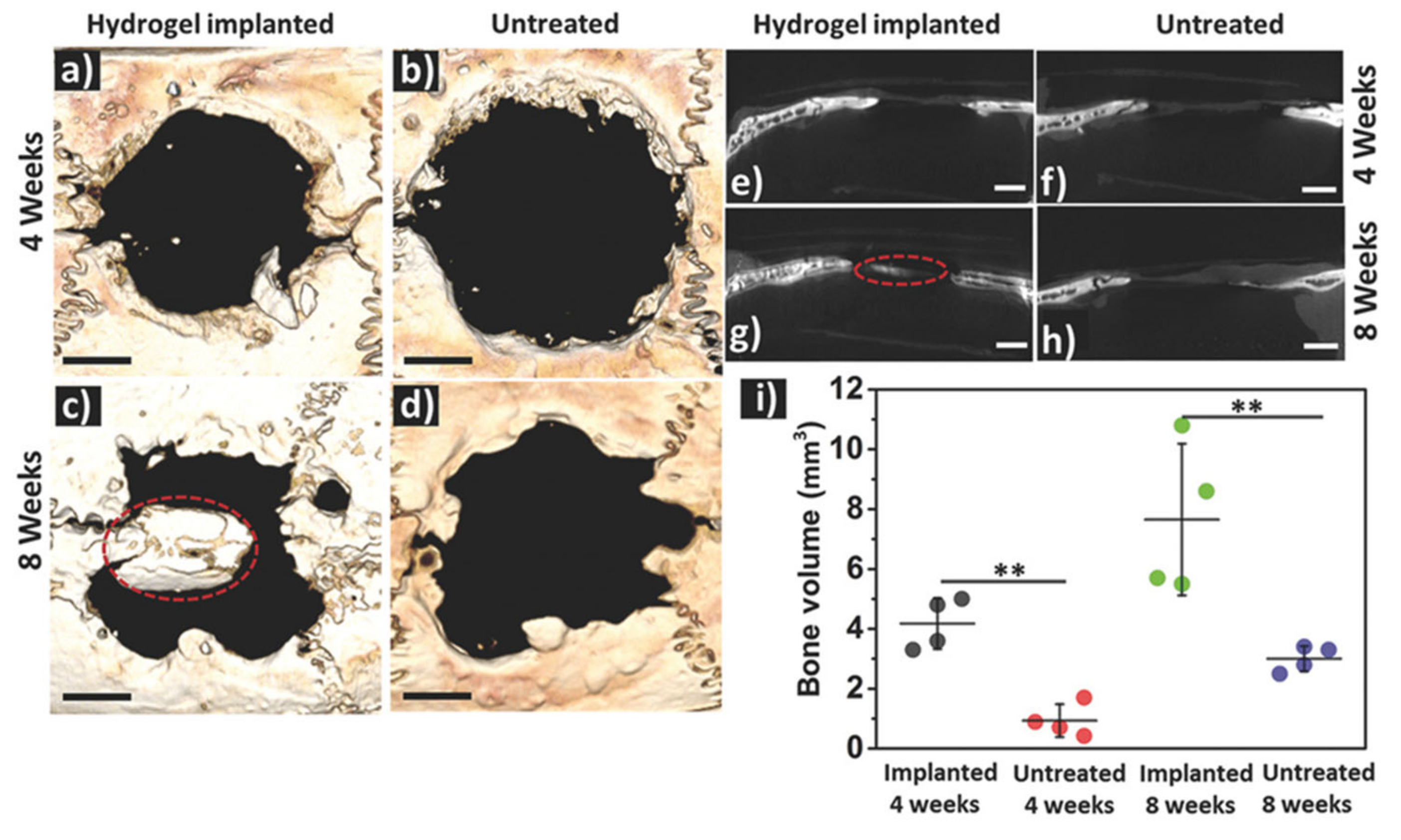{kind=link}
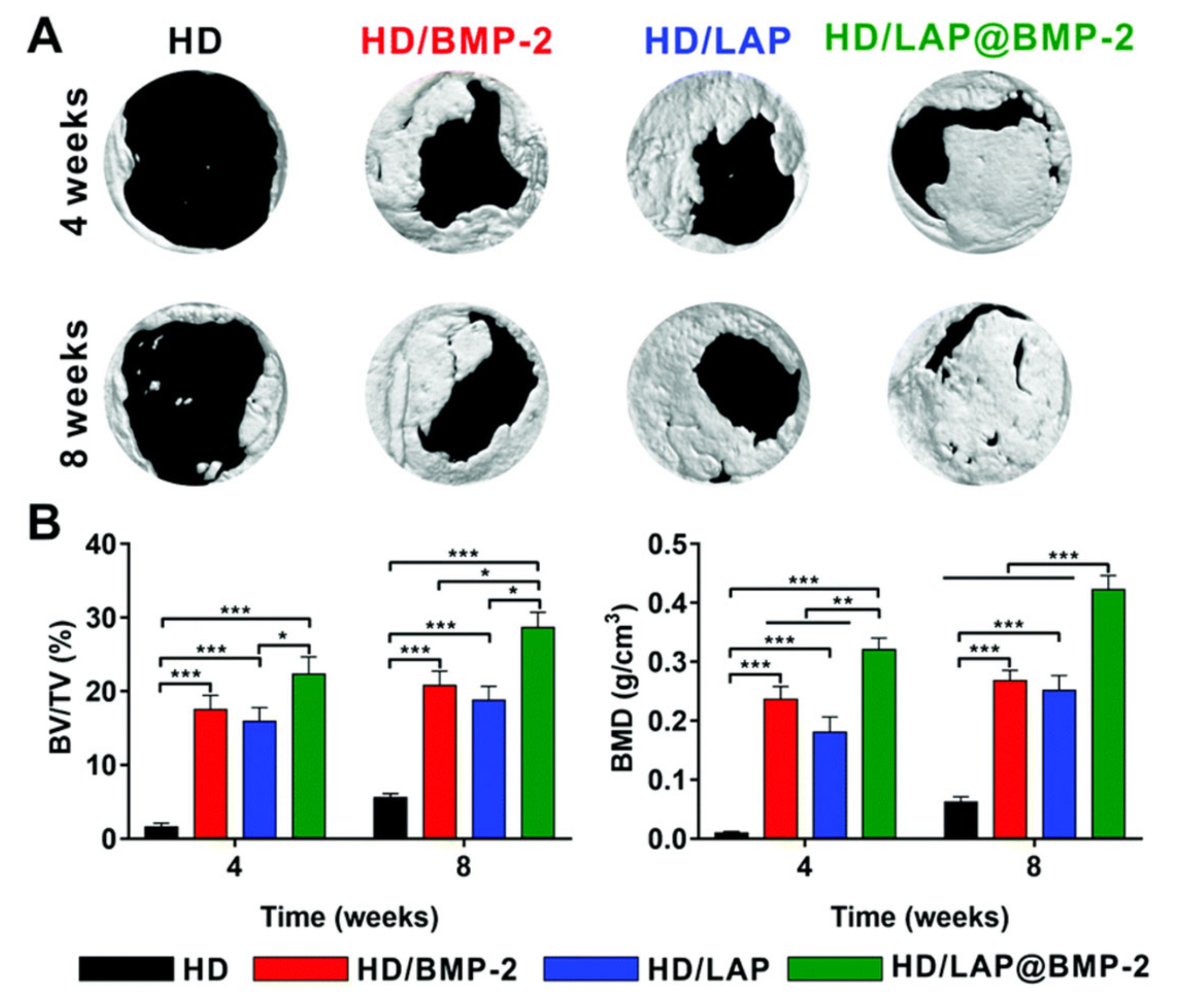{kind=link}
{kind=link}
| Hydrogel | In Situ Gelling Mechanism | Self-Healing Mechanism | Applications | References |
|---|---|---|---|---|
| Alginate grafted onto β-CD and Pluronic® F108 | Hydrophobic interactions | Host-guest interaction | Drug delivery and cell transplantantion | [97] |
| Hydrophobically modified chitosan with thermal-responsive vesicle composed of 5 mS and DTAB | Hydrophobic interactions | Host-guest interaction | Scaffold for tissue engineering | [98] |
| Polyethylene glycol derivated functionalized with/ureido-pyrimidinone | Hydrophobic interactions | Hydrophobic interactions | Cardiac tissue regeneration | [132] |
| 2-Hydroxypropyltrimethylammonium chitosan chloride/Alginate polycomplex | Electrostatic interactions | Ionically induced mechanism | Surface coating and 3D bioprinting | [102] |
| Chitosan/Hyaluronic acid polycomplex | Electrostatic interactions | Ionically induced mechanism | Functional biomedical supports and coatings | [51] |
| Polyacrylic acid (PAA)—chitosan copolymeric hydrogels crosslinked with guanidine | Electrostatic interactions | Ionically induced mechanism | Drug delivery and tissue engineering applications | [103] |
| Chitosan/poly(acrylamide-co-acrylic acid) in presence of Fe3+ | Electrostatic interactions | Ionically induced mechanism | Drug delivery and tissue engineering applications | [90] |
| Dextran functionalized with Ureido-pyrimidinone | Hydrogen bonds | Hydrogen bonds | Encapsulation of chondrocytes for cartilage tissue engineering | [122] |
| Chitosan/Catechol assembled through Fe3+ | Metal-ligand interactions | Ionically induced mechanism | Localized delivery of chemotherapeutic drugs | [133] |
| Alginate/Catechol assembled through Fe3+ | Metal-ligand coordinative interactions | Ionically induced mechanism | Drug delivery and tissue differentiation | [104] |
| N-carboxyethyl chitosan/Polyacrylic acid coordinated with Fe3+ | Metal-ligand coordinative interactions | Ionically induced mechanism | Drug delivery and cartilage tissue differentiation | [106] |
| Hyaluronic acid modified with bisphosphonate and acrylated bisphosphonate and Mg2+ | Metal-ligand coordinative interactions | Metal-ligand coordinative interactions | Bone tissue engineering | [118] |
| Hyaluronic acid hydrogel with silk fibroin nanofibers coated with calcium phosphate | Dynamic metal-bisphosphonate coordination bonds | Dynamic metal-bisphosphonate coordination bonds | Bone tissue engineering | [119] |
| Quaternized chitosan/Polyacrylic acid | In situ polymerization of monomer and electrostatic interactions | Ionically induced mechanism | Load-bearing artificial soft tissues. | [107] |
| Alginate/Ca2+/Polyacrylamide | In situ polymerization and electrostatic interactions | Ionically induced mechanism | 3D cell culture | [108] |
| Gelatin/polyethylene glycol dibenzaldehyde | Dynamic imine bonds | Dynamic imine bonds | Drug delivery systems, as tissue regeneration scaffolds and also as wound dressing for skin injuries | [112] |
| Sodium alginate dialdehyde/Carboxymethyl chitosan | Dynamic Schiff-base covalent bonds | Hydrogen bonds and dynamic Schiff-base covalent bonds | Corneal epithelial reconstruction based on tissue engineering | [43] |
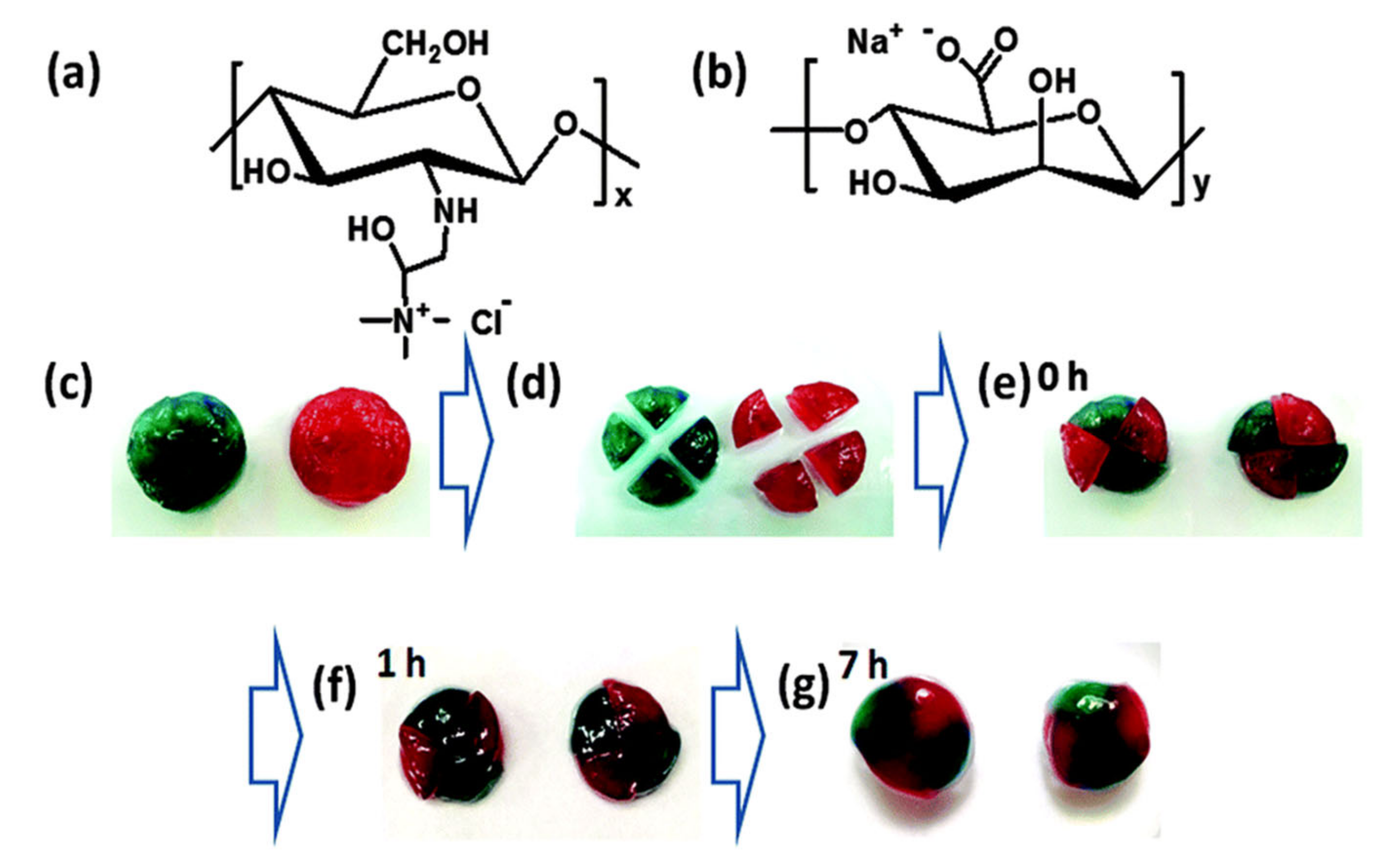
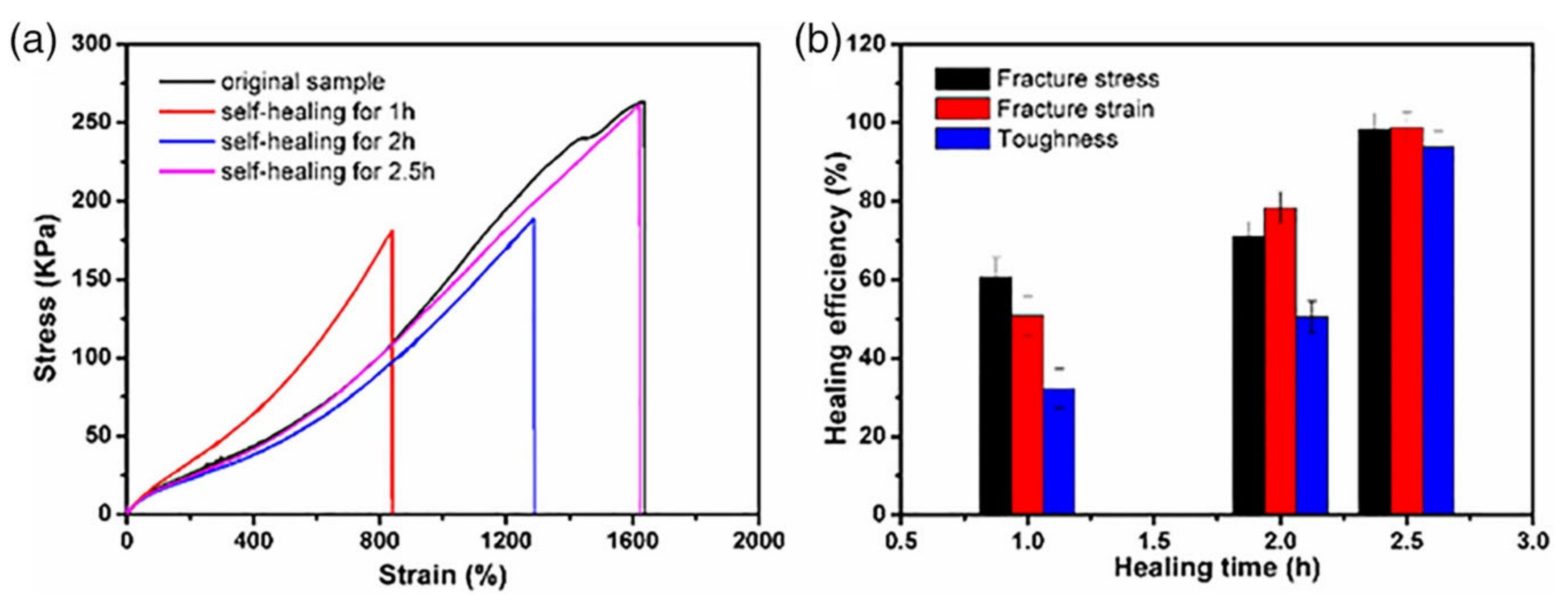
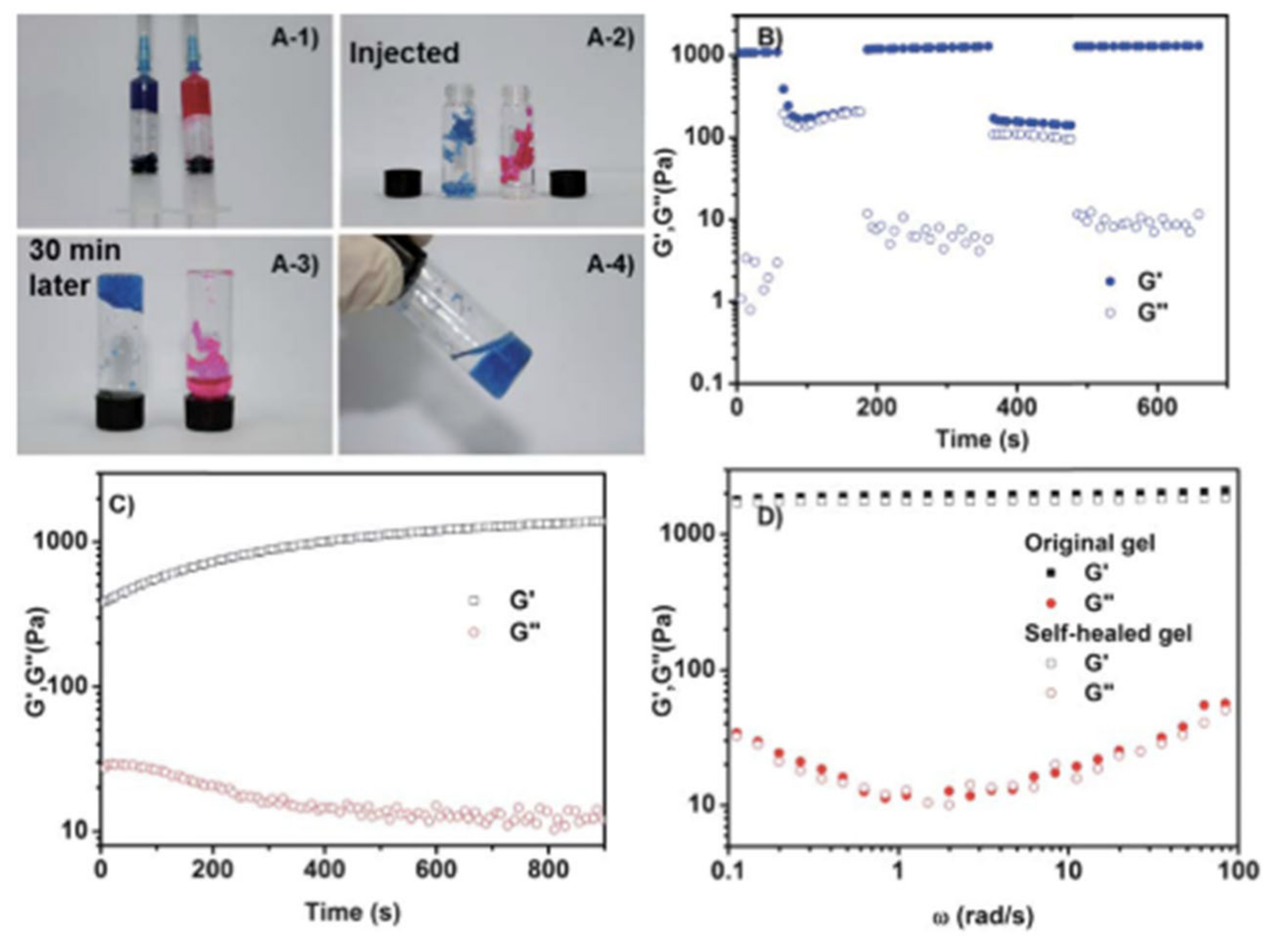
| Glycol chitosan/telechelic difunctional poly(ethylene glycol) | Dynamic Schiff-base covalent bonds | Dynamic Schiff-base covalent bonds | 3D cell culture and injection cell therapy | [56] |
| N-carboxyethyl chitosan/dibenzaldehyde-terminated poly(ethylene glycol) | Dynamic Schiff-base covalent bonds | Dynamic Schiff-base covalent bonds | Anticancer drug delivery for hepatocellular cancer therapy | [36] |
| Hyaluronic acid/Dextran/LAPONITE® | Dynamic Schiff-base covalent bonds and hydrogen bonds | Dynamic Schiff-base covalent bonds and hydrogen bonds | Bone tissue engineering | [120] |
| Chitosan/dextran-graft-aniline tetramer-graft-4-formylbenzoic acid | Dynamic Schiff-base covalent bonds | Dynamic Schiff-base covalent bonds | Encapsulation of C2C12 myoblasts for muscle tissue engineering | [124] |
| Quaternized chitosan-g-polyaniline and poly(ethylene glycol)-co-poly(glycerol sebacate) funtionalized with benzaldehyde | Dynamic Schiff-base covalent bonds | Dynamic Schiff-base covalent bonds | Wound repair | [127] |
| Chitosan-graft-aniline tetramer and dibenzaldehyde-terminated poly(ethylene glycol) | Dynamic Schiff-base covalent bonds | Dynamic Schiff-base covalent bonds | Regeneration of cardiac tissue for myocardial infarction | [131] |
| Hyaluronic acid/furan/adipic dihydrazide | Diels alder click reaction and dynamic acylhydrazone bonds | Dynamic acylhydrazone bond | Cartilage tissue engineering | [123] |
| Maleilated chitosan/Thiol derivatized sodium alginate | Michael addition reaction and ionic interactions | Dynamic disulfide exchange | 3D cell culture | [115] |
| Polyethylene glycol derivatives functionalized with/ureido-pyrimidinone | Hydrophobic interactions | Hydrophobic interactions | Cardiac tissue regeneration | [132] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maiz-Fernández, S.; Pérez-Álvarez, L.; Ruiz-Rubio, L.; Vilas-Vilela, J.L.; Lanceros-Mendez, S. Polysaccharide-Based In Situ Self-Healing Hydrogels for Tissue Engineering Applications. Polymers 2020, 12, 2261. https://doi.org/10.3390/polym12102261
Maiz-Fernández S, Pérez-Álvarez L, Ruiz-Rubio L, Vilas-Vilela JL, Lanceros-Mendez S. Polysaccharide-Based In Situ Self-Healing Hydrogels for Tissue Engineering Applications. Polymers. 2020; 12(10):2261. https://doi.org/10.3390/polym12102261
Chicago/Turabian StyleMaiz-Fernández, Sheila, Leyre Pérez-Álvarez, Leire Ruiz-Rubio, Jose Luis Vilas-Vilela, and Senentxu Lanceros-Mendez. 2020. "Polysaccharide-Based In Situ Self-Healing Hydrogels for Tissue Engineering Applications" Polymers 12, no. 10: 2261. https://doi.org/10.3390/polym12102261
APA StyleMaiz-Fernández, S., Pérez-Álvarez, L., Ruiz-Rubio, L., Vilas-Vilela, J. L., & Lanceros-Mendez, S. (2020). Polysaccharide-Based In Situ Self-Healing Hydrogels for Tissue Engineering Applications. Polymers, 12(10), 2261. https://doi.org/10.3390/polym12102261