Preparation of Half- and Post-Metallocene Hafnium Complexes with Tetrahydroquinoline and Tetrahydrophenanthroline Frameworks for Olefin Polymerization

 ,
, 

Abstract
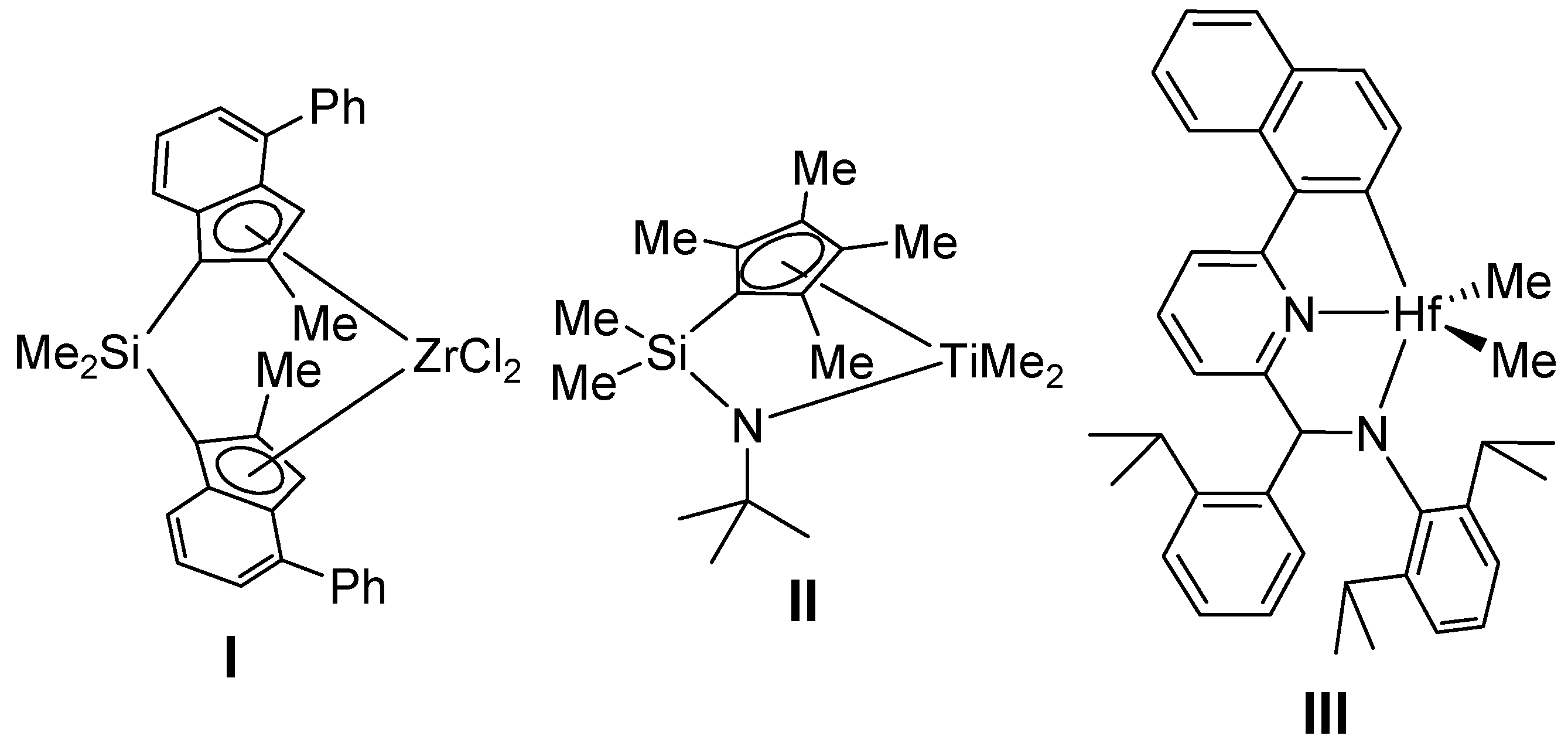
1. Introduction
2. Materials and Methods
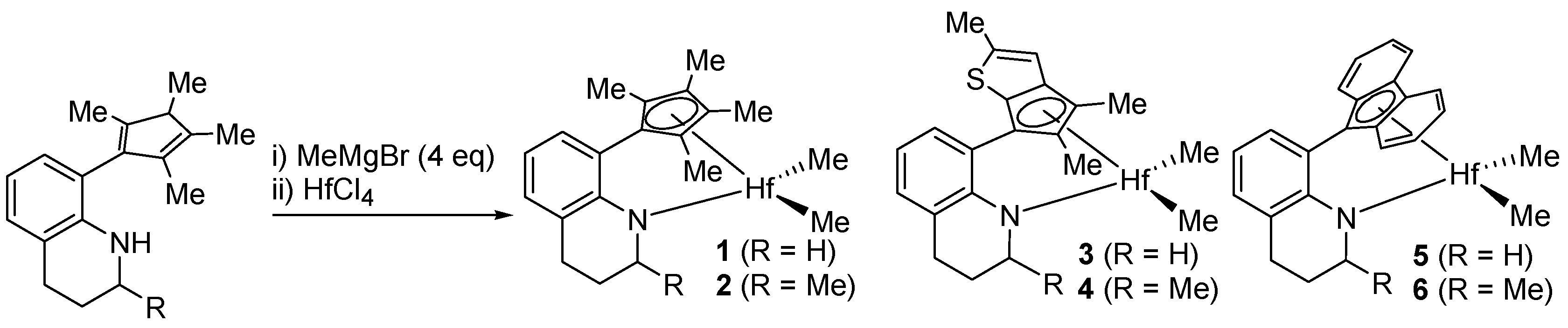
2.1. Preparation of 1
2.2. Preparation of 2
2.3. Preparation of 3
2.4. Preparation of 4
2.5. Preparation of 5
2.6. Preparation of 6
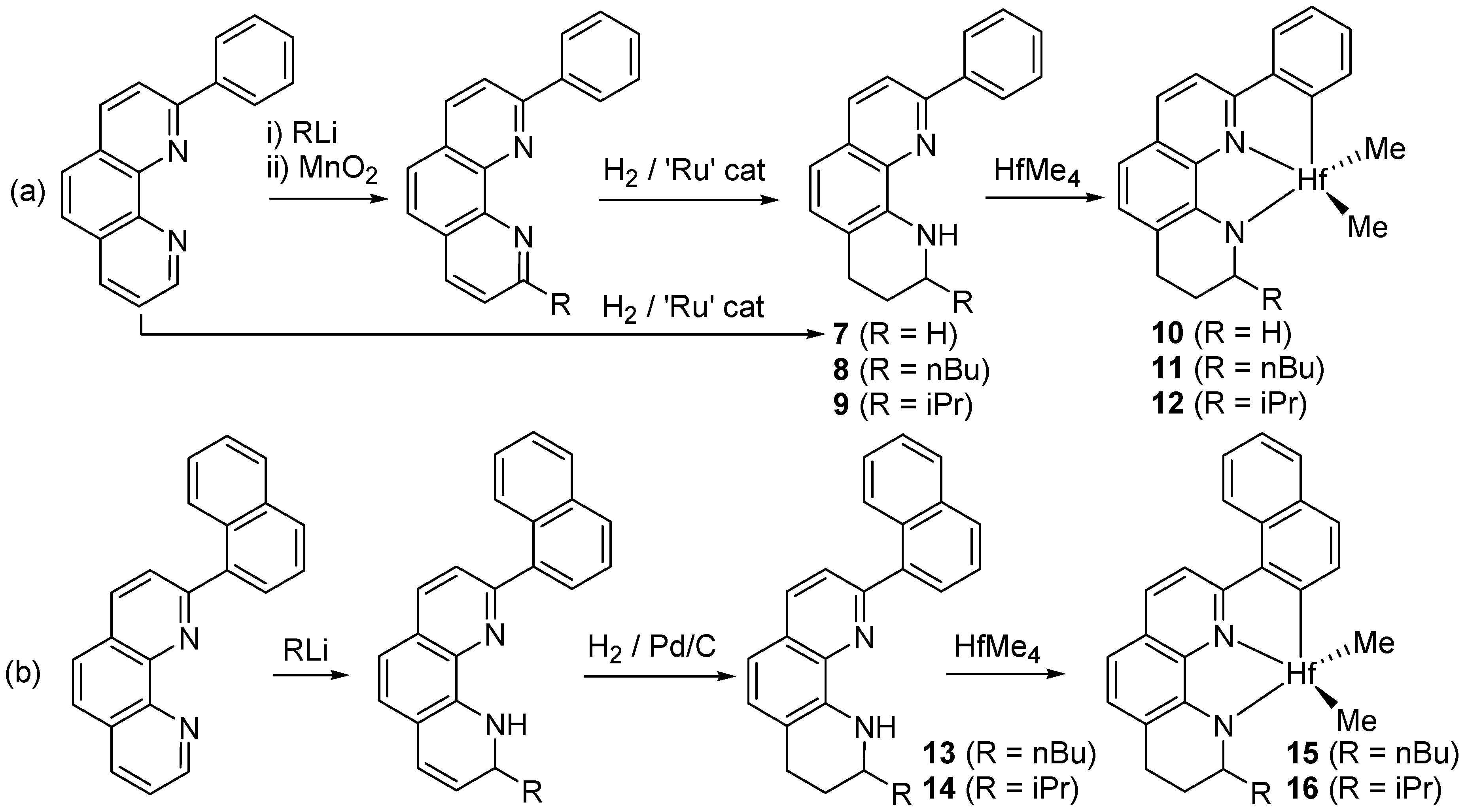
2.7. Preparation of 9
2.8. Preparation of 10
2.9. Preparation of 11
2.10. Preparation of 12
2.11. Preparation of 13
2.12. Preparation of 14
2.13. Preparation of 15
2.14. Preparation of 16
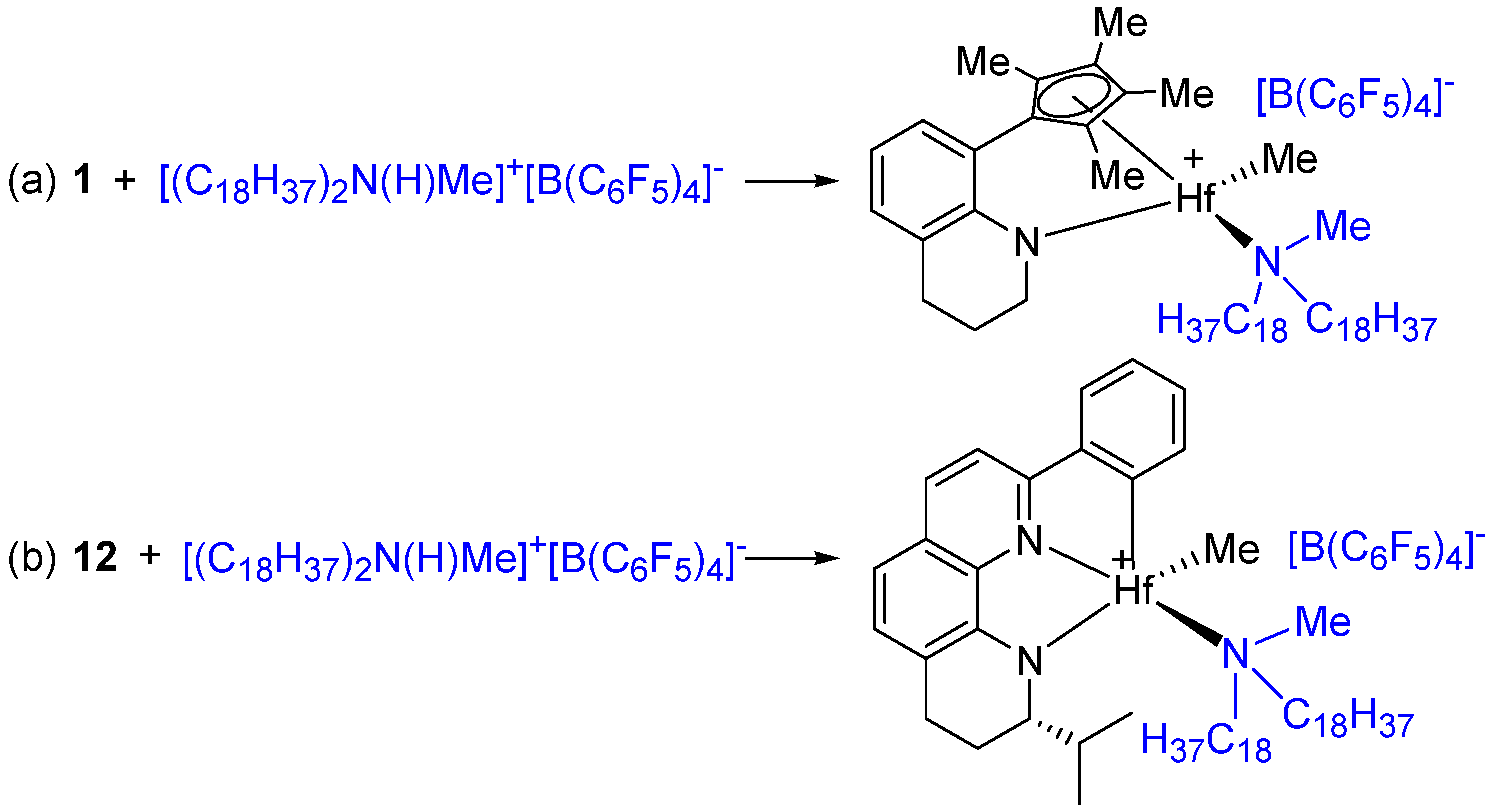
2.15. Preparation of Anhydrous [(C18H37)2N(H)Me]+[B(C6F5)4]−
2.16. A Typical Polymerization (Entry 8 in Table 1)
2.17. X-ray Crystallography
3. Results and Discussion
3.1. Preparation of Hf Complexes
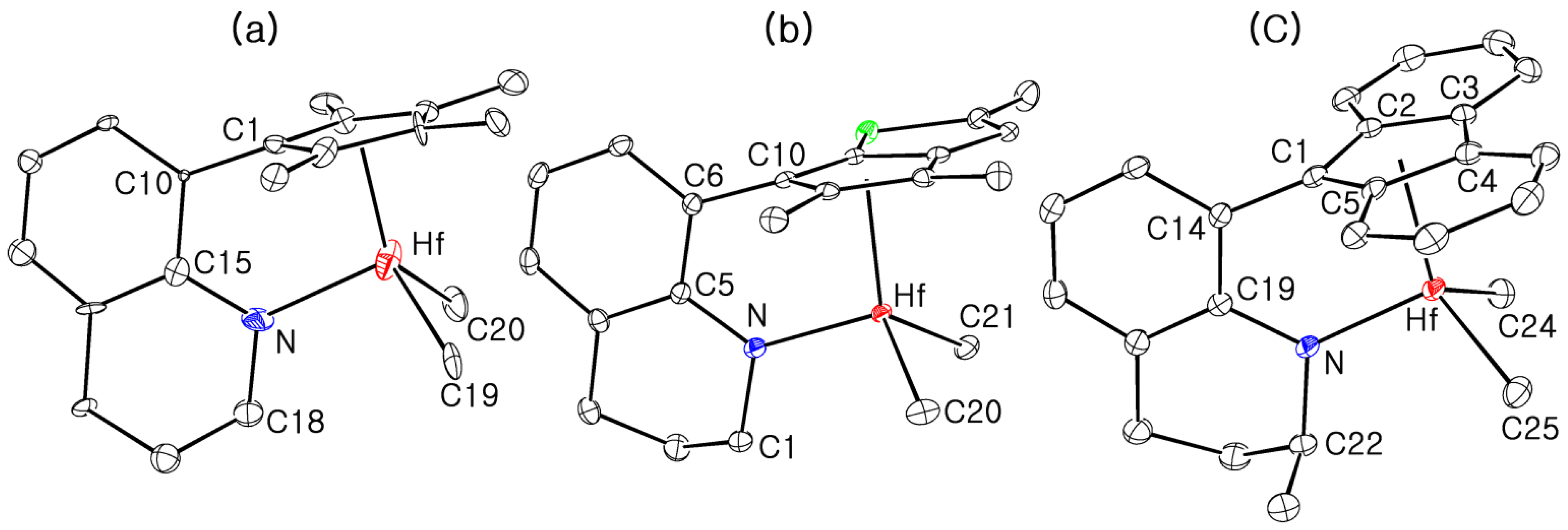
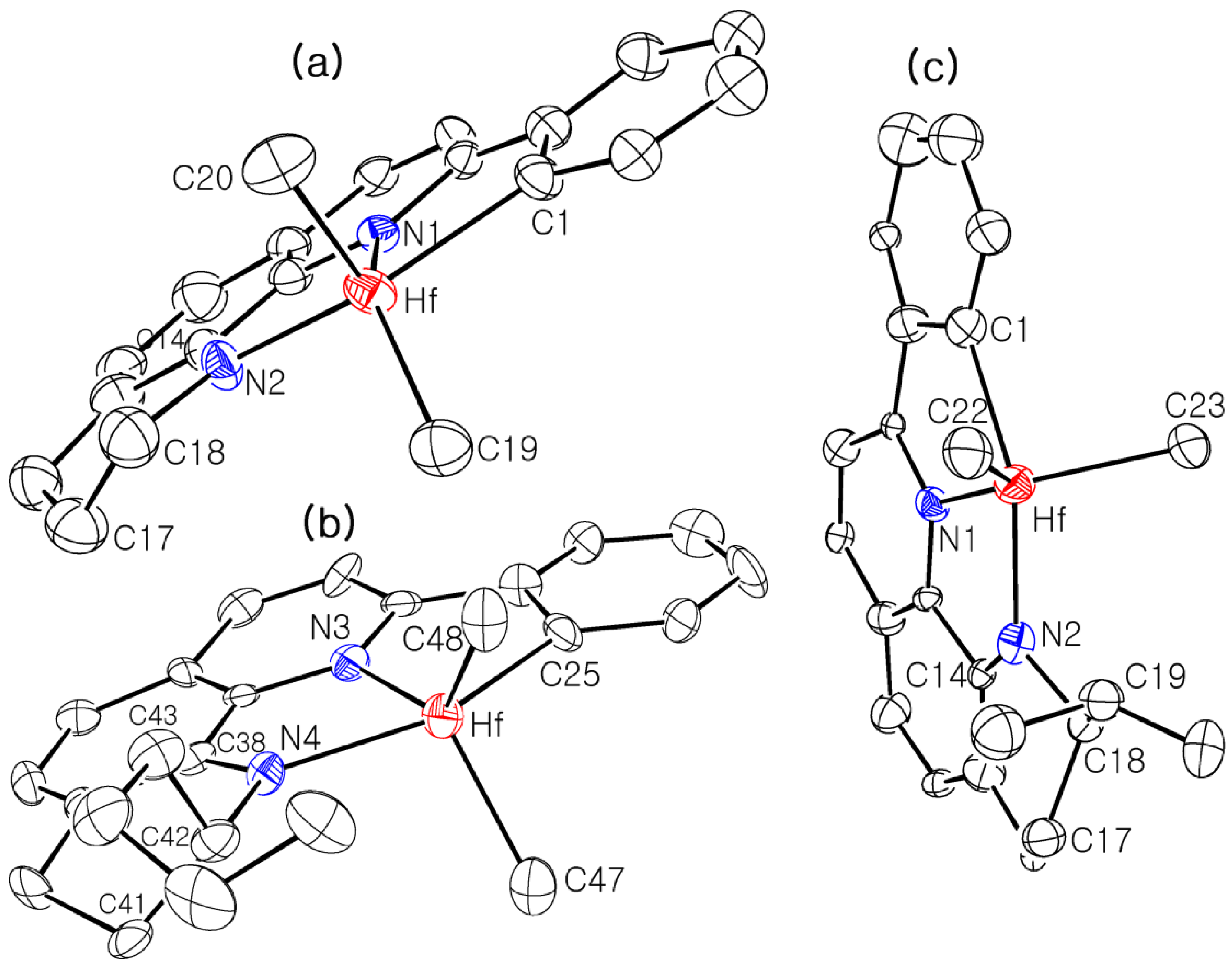
3.2. X-ray Crystallographic Studies
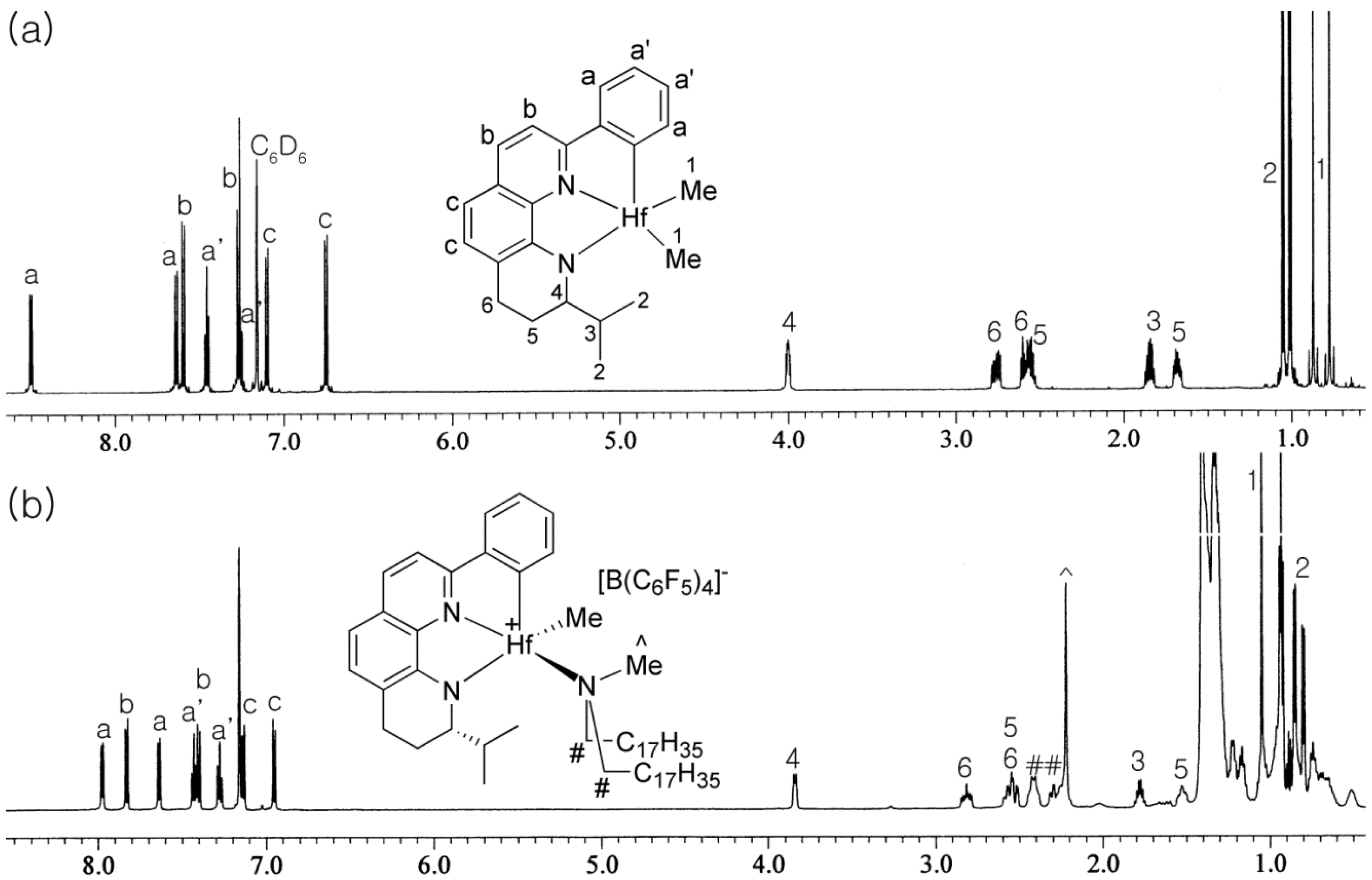
3.3. Activation Reactions
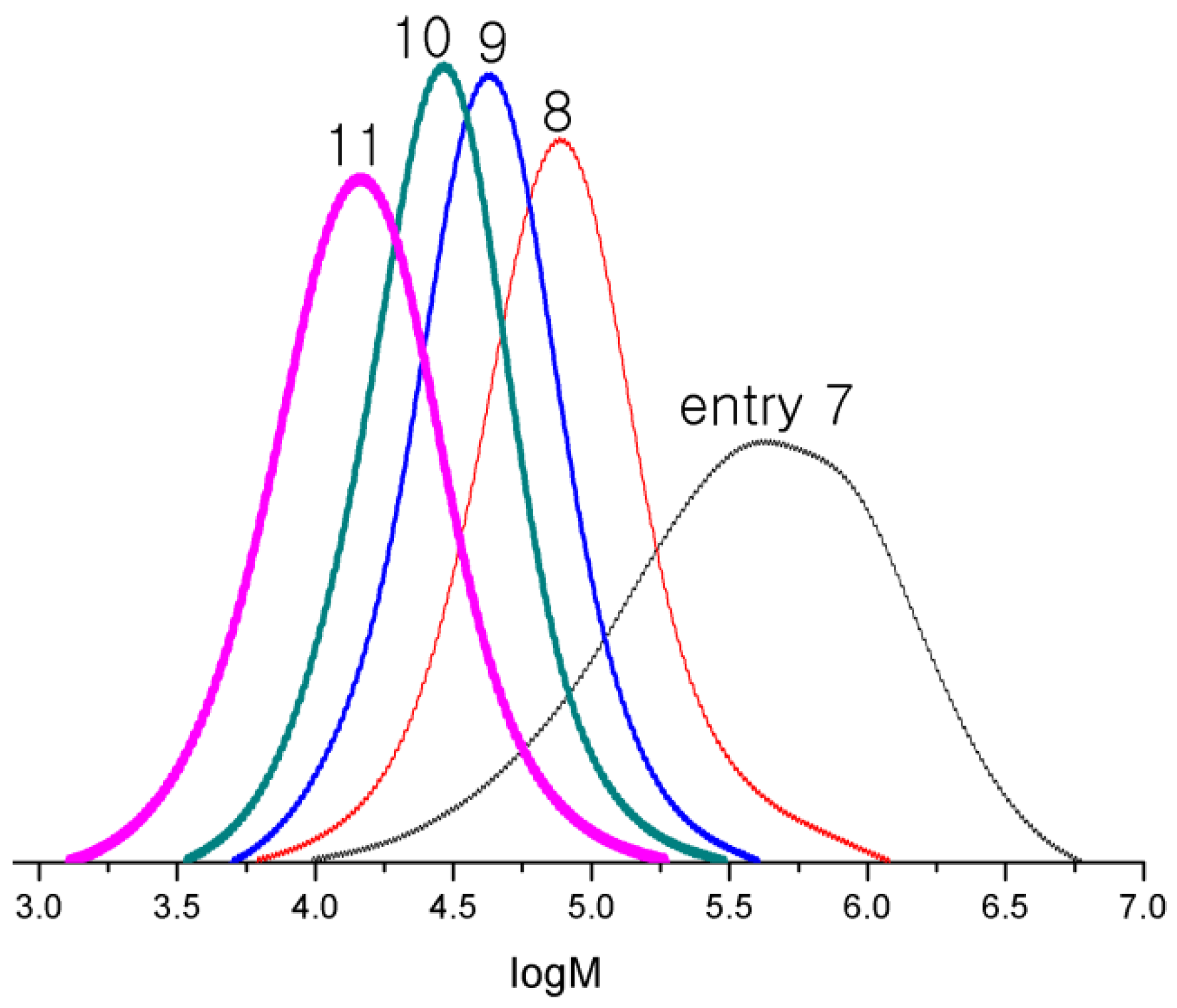
3.4. Polymerization Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Kaminsky, W. Discovery of Methylaluminoxane as Cocatalyst for Olefin Polymerization. Macromolecules 2012, 45, 3289–3297. [Google Scholar] [CrossRef]
- Lee, S.; Park, S.S.; Kim, J.G.; Kim, C.S.; Lee, B.Y. Preparation of "Constrained geometry" titanium complexes of [1,2]azasilinane framework for ethylene/1-octene copolymerization. Molecules 2017, 22, 258. [Google Scholar] [CrossRef] [PubMed]
- Makio, H.; Terao, H.; Iwashita, A.; Fujita, T. FI catalysts for olefin polymerization—A comprehensive treatment. Chem. Rev. 2011, 111, 2363–2449. [Google Scholar] [CrossRef] [PubMed]
- Cano, J.; Kunz, K. How to synthesize a constrained geometry catalyst (CGC)—A survey. J. Organomet. Chem. 2007, 692, 4411–4423. [Google Scholar] [CrossRef]
- Arriola, D.J.; Carnahan, E.M.; Hustad, P.D.; Kuhlman, R.L.; Wenzel, T.T. Catalytic production of olefin block copolymers via chain shuttling polymerization. Science 2006, 312, 714–719. [Google Scholar] [CrossRef]
- Boussie, T.R.; Diamond, G.M.; Goh, C.; Hall, K.A.; LaPointe, A.M.; Leclerc, M.K.; Murphy, V.; Shoemaker, J.A.W.; Turner, H.; Rosen, R.K.; et al. Nonconventional Catalysts for Isotactic Propene Polymerization in Solution Developed by Using High-Throughput-Screening Technologies. Angew. Chem. Int. Ed. 2006, 45, 3278–3283. [Google Scholar] [CrossRef] [PubMed]
- Frazier, K.A.; Froese, R.D.; He, Y.; Klosin, J.; Theriault, C.N.; Vosejpka, P.C.; Zhou, Z.; Abboud, K.A. Pyridylamido hafnium and zirconium complexes: Synthesis, dynamic behavior, and ethylene/1-octene and propylene polymerization reactions. Organometallics 2011, 30, 3318–3329. [Google Scholar] [CrossRef]
- Alfano, F.; Boone, H.W.; Busico, V.; Cipullo, R.; Stevens, J.C. Polypropylene “Chain Shuttling” at Enantiomorphous and Enantiopure Catalytic Species: Direct and Quantitative Evidence from Polymer Microstructure. Macromolecules 2007, 40, 7736–7738. [Google Scholar] [CrossRef]
- Domski, G.J.; Eagan, J.M.; De Rosa, C.; Di Girolamo, R.; LaPointe, A.M.; Lobkovsky, E.B.; Talarico, G.; Coates, G.W. Combined Experimental and Theoretical Approach for Living and Isoselective Propylene Polymerization. ACS Catal. 2017, 7, 6930–6937. [Google Scholar] [CrossRef]
- De Rosa, C.; Di Girolamo, R.; Talarico, G. Expanding the Origin of Stereocontrol in Propene Polymerization Catalysis. ACS Catal. 2016, 6, 3767–3770. [Google Scholar] [CrossRef]
- Park, S.S.; Kim, C.S.; Kim, S.D.; Kwon, S.J.; Lee, H.M.; Kim, T.H.; Jeon, J.Y.; Lee, B.Y. Biaxial Chain Growth of Polyolefin and Polystyrene from 1,6-Hexanediylzinc Species for Triblock Copolymers. Macromolecules 2017, 50, 6606–6616. [Google Scholar] [CrossRef]
- Leclerc, M.K.; Brintzinger, H.H. Origins of stereoselectivity and stereoerror formation in ansa-zirconocene-catalyzed isotactic propene polymerization. A deuterium labeling study. J. Am. Chem. Soc. 1995, 117, 1651–1652. [Google Scholar] [CrossRef]
- Eagan, J.M.; Xu, J.; Di Girolamo, R.; Thurber, C.M.; Macosko, C.W.; La Pointe, A.M.; Bates, F.S.; Coates, G.W. Combining polyethylene and polypropylene: Enhanced performance with PE/iPP multiblock polymers. Science 2017, 355, 814–816. [Google Scholar] [CrossRef] [PubMed]
- Valente, A.; Mortreux, A.; Visseaux, M.; Zinck, P. Coordinative chain transfer polymerization. Chem. Rev. 2013, 113, 3836–3857. [Google Scholar] [CrossRef] [PubMed]
- van Meurs, M.; Britovsek, G.J.P.; Gibson, V.C.; Cohen, S.A. Polyethylene Chain Growth on Zinc Catalyzed by Olefin Polymerization Catalysts: A Comparative Investigation of Highly Active Catalyst Systems across the Transition Series. J. Am. Chem. Soc. 2005, 127, 9913–9923. [Google Scholar] [CrossRef] [PubMed]
- Rocchigiani, L.; Busico, V.; Pastore, A.; Macchioni, A. Comparative NMR Study on the Reactions of Hf(IV) Organometallic Complexes with Al/Zn Alkyls. Organometallics 2016, 35, 1241–1250. [Google Scholar] [CrossRef]
- Hustad, P.O.; Kuhlman, R.L.; Arriola, D.J.; Carnahan, E.M.; Wenzel, T.T. Continuous production of ethylene-based diblock copolymers using coordinative chain transfer polymerization. Macromolecules 2007, 40, 7061–7064. [Google Scholar] [CrossRef]
- Kim, S.D.; Kim, T.J.; Kwon, S.J.; Kim, T.H.; Baek, J.W.; Park, H.S.; Lee, H.J.; Lee, B.Y. Peroxide-Mediated Alkyl–Alkyl Coupling of Dialkylzinc: A Useful Tool for Synthesis of ABA-Type Olefin Triblock Copolymers. Macromolecules 2018, 51, 4821–4828. [Google Scholar] [CrossRef]
- Vittoria, A.; Busico, V.; Cannavacciuolo, F.D.; Cipullo, R. Molecular Kinetic Study of “Chain Shuttling” Olefin Copolymerization. ACS Catal. 2018, 8, 5051–5061. [Google Scholar] [CrossRef]
- Jeon, J.Y.; Park, S.H.; Kim, D.H.; Park, S.S.; Park, G.H.; Lee, B.Y. Synthesis of polyolefin-block-polystyrene through sequential coordination and anionic polymerizations. J. Polym. Sci. Part A Polym. Chem. 2016, 54, 3110–3118. [Google Scholar] [CrossRef]
- Kim, C.S.; Park, S.S.; Kim, S.D.; Kwon, S.J.; Baek, J.W.; Lee, B.Y. Polystyrene chain growth from di-end-functional polyolefins for polystyrene-polyolefin-polystyrene block copolymers. Polymers 2017, 9, 481. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Park, S.S.; Park, S.H.; Jeon, J.Y.; Kim, H.B.; Lee, B.Y. Preparation of polystyrene-polyolefin multiblock copolymers by sequential coordination and anionic polymerization. RSC Adv. 2017, 7, 5948–5956. [Google Scholar] [CrossRef]
- Liu, C.-C.; Liu, Q.; Lo, P.-K.; Lau, K.-C.; Yiu, S.-M.; Chan, M.C.W. Olefin Polymerization Reactivity of Group 4 Post-Metallocene Catalysts Bearing a Four-Membered C(sp3)-Donor Chelate Ring. ChemCatChem 2019, 11, 628–635. [Google Scholar] [CrossRef]
- Kwon, S.J.; Baek, J.W.; Lee, H.J.; Kim, T.J.; Ryu, J.Y.; Lee, J.; Shin, E.J.; Lee, K.S.; Lee, B.Y. Preparation of Pincer Hafnium Complexes for Olefin Polymerization. Molecules 2019, 24, 1676. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, K.; Takayanagi, M.; Suzuki, Y.; Koga, N.; Nagaoka, M. Atomistic chemical computation of Olefin polymerization reaction catalyzed by (pyridylamido)hafnium(IV) complex: Application of Red Moon simulation. J. Comput. Chem. 2019, 40, 421–429. [Google Scholar] [CrossRef] [PubMed]
- Schnee, G.; Farenc, M.; Bitard, L.; Vantomme, A.; Welle, A.; Brusson, J.M.; Afonso, C.; Giusti, P.; Carpentier, J.F.; Kirillov, E. Synthesis, APPI mass-spectrometric characterization, and polymerization studies of group 4 dinuclear bis(Ansa-metallocene) complexes. Catalysts 2018, 8, 558. [Google Scholar] [CrossRef]
- Xu, J.; Eagan, J.M.; Kim, S.-S.; Pan, S.; Lee, B.; Klimovica, K.; Jin, K.; Lin, T.-W.; Howard, M.J.; Ellison, C.J.; et al. Compatibilization of Isotactic Polypropylene (iPP) and High-Density Polyethylene (HDPE) with iPP–PE Multiblock Copolymers. Macromolecules 2018, 51, 8585–8596. [Google Scholar] [CrossRef]
- Zhang, J.; Motta, A.; Gao, Y.; Stalzer, M.M.; Delferro, M.; Liu, B.; Lohr, T.L.; Marks, T.J. Cationic Pyridylamido Adsorbate on Brønsted Acidic Sulfated Zirconia: A Molecular Supported Organohafnium Catalyst for Olefin Homo- and Co-Polymerization. ACS Catal. 2018, 8, 4893–4901. [Google Scholar] [CrossRef]
- Cueny, E.S.; Johnson, H.C.; Landis, C.R. Selective Quench-Labeling of the Hafnium-Pyridyl Amido-Catalyzed Polymerization of 1-Octene in the Presence of Trialkyl-Aluminum Chain-Transfer Reagents. ACS Catal. 2018, 8, 11605–11614. [Google Scholar] [CrossRef]
- Johnson, H.C.; Cueny, E.S.; Landis, C.R. Chain Transfer with Dialkyl Zinc during Hafnium-Pyridyl Amido-Catalyzed Polymerization of 1-Octene: Relative Rates, Reversibility, and Kinetic Models. ACS Catal. 2018, 8, 4178–4188. [Google Scholar] [CrossRef]
- Gao, Y.; Chen, X.; Zhang, J.; Chen, J.; Lohr, T.L.; Marks, T.J. Catalyst Nuclearity Effects on Stereo- and Regioinduction in Pyridylamidohafnium-Catalyzed Propylene and 1-Octene Polymerizations. Macromolecules 2018, 51, 2401–2410. [Google Scholar] [CrossRef]
- Matsumoto, K.; Takayanagi, M.; Sankaran, S.K.; Koga, N.; Nagaoka, M. Role of the Counteranion in the Reaction Mechanism of Propylene Polymerization Catalyzed by a (Pyridylamido)hafnium(IV) Complex. Organometallics 2018, 37, 343–349. [Google Scholar] [CrossRef]
- Wu, C.J.; Lee, S.H.; Yun, H.; Lee, B.Y. Ortho lithiation of tetrahydroquinoline derivatives and its use for the facile construction of polymerization catalysts. Organometallics 2007, 26, 6685–6687. [Google Scholar] [CrossRef]
- Park, J.H.; Do, S.H.; Cyriac, A.; Yun, H.; Lee, B.Y. Preparation of half-metallocenes of thiophene-fused and tetrahydroquinoline-linked cyclopentadienyl ligands for ethylene/α-olefin copolymerization. Dalton Trans. 2010, 39, 9994–10002. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Chen, F.; Qin, J.; He, Y.-M.; Fan, Q.-H. Asymmetric Ruthenium-Catalyzed Hydrogenation of 2- and 2,9-Substituted 1,10-Phenanthrolines. Angew. Chem. Int. Ed. 2013, 52, 7172–7176. [Google Scholar] [CrossRef] [PubMed]
- Robert, K.R.; VanderLende, D.D. Highly soluble olefin polymerization catalyst activator. U.S. Patent 5,919,983, 6 July 1999. [Google Scholar]
- Cho, D.J.; Wu, C.J.; Sujith, S.; Han, W.S.; Kang, S.O.; Lee, B.Y. o-phenylene-bridged Cp/amido titanium complexes for ethylene/1-hexene copolymerizations. Organometallics 2006, 25, 2133–2134. [Google Scholar] [CrossRef]
- Wu, C.J.; Lee, S.H.; Yu, S.T.; Na, S.J.; Yun, H.; Lee, B.Y. CO2-mediated ortho-lithiatium of N-alkylanilines and its use for the construction of polymerization catalysts. Organometallics 2008, 27, 3907–3917. [Google Scholar] [CrossRef]
- Kim, S.; Park, J.; Song, B.; Yoon, S.-W.; Go, M.; Lee, J.; Lee, B. Preparation of Thiophene-Fused and Tetrahydroquinoline-Linked Cyclopentadienyl Titanium Complexes for Ethylene/α-Olefin Copolymerization. Catalysts 2013, 3, 104–124. [Google Scholar] [CrossRef]
- Resconi, L.; Camurati, I.; Grandini, C.; Rinaldi, M.; Mascellani, N.; Traverso, O. Indenyl-amido titanium and zirconium dimethyl complexes: improved synthesis and use in propylene polymerization. J. Organomet. Chem. 2002, 664, 5–26. [Google Scholar] [CrossRef]
- Okuda, J.; Amor, F.; du Plooy, K.E.; Eberle, T.; Hultzsch, K.C.; Spaniol, T.P. Zirconium, hafnium and yttrium complexes containing two linked amido—tetramethylcyclopentadienyl ligands: Synthesis, reactivity and molecular structure of Hf(η5:η1−C5Me4SiMe2NiPr)2. Polyhedron 1998, 17, 1073–1080. [Google Scholar] [CrossRef]
- Carpenetti, D.W.; Kloppenburg, L.; Kupec, J.T.; Petersen, J.L. Application of Amine Elimination for the Efficient Preparation of Electrophilic ansa-Monocyclopentadienyl Group 4 Complexes Containing an Appended Amido Functionality. Structural Characterization of [(C5H4)SiMe2(N-t-Bu)]ZrCl2(NMe2H). Organometallics 1996, 15, 1572–1581. [Google Scholar] [CrossRef]
- Sun, Y.; Xu, B.; Shiono, T.; Cai, Z. Highly Active ansa-(Fluorenyl)(amido)titanium-Based Catalysts with Low Load of Methylaluminoxane for Syndiotactic-Specific Living Polymerization of Propylene. Organometallics 2017, 36, 3009–3012. [Google Scholar] [CrossRef]
- Liu, C.-C.; Chan, M.C.W. Chelating σ-Aryl Post-Metallocenes: Probing Intramolecular [C–H···F–C] Interactions and Unusual Reaction Pathways. Acc. Chem. Res. 2015, 48, 1580–1590. [Google Scholar] [CrossRef] [PubMed]
- Froese, R.D.J.; Hustad, P.D.; Kuhlman, R.L.; Wenzel, T.T. Mechanism of Activation of a Hafnium Pyridyl-Amide Olefin Polymerization Catalyst: Ligand Modification by Monomer. J. Am. Chem. Soc. 2007, 129, 7831–7840. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Pan, H.; Klosin, J.; Tu, S.; Jaganathan, A.; Fontaine, P.P. Synthetic Optimization and Scale-Up of Imino-Amido Hafnium and Zirconium Olefin Polymerization Catalysts. Org. Proc. Res. Dev. 2015, 19, 1383–1391. [Google Scholar] [CrossRef]
- Cueny, E.S.; Johnson, H.C.; Anding, B.J.; Landis, C.R. Mechanistic Studies of Hafnium-Pyridyl Amido-Catalyzed 1-Octene Polymerization and Chain Transfer Using Quench-Labeling Methods. J. Am. Chem. Soc. 2017, 139, 11903–11912. [Google Scholar] [CrossRef] [PubMed]
- Zuccaccia, C.; Macchioni, A.; Busico, V.; Cipullo, R.; Talarico, G.; Alfano, F.; Boone, H.W.; Frazier, K.A.; Hustad, P.D.; Stevens, J.C.; et al. Intra- and Intermolecular NMR Studies on the Activation of Arylcyclometallated Hafnium Pyridyl-Amido Olefin Polymerization Precatalysts. J. Am. Chem. Soc. 2008, 130, 10354–10368. [Google Scholar] [CrossRef] [PubMed]
- Machat, M.R.; Fischer, A.; Schmitz, D.; Vöst, M.; Drees, M.; Jandl, C.; Pöthig, A.; Casati, N.P.M.; Scherer, W.; Rieger, B. Behind the Scenes of Group 4 Metallocene Catalysis: Examination of the Metal-Carbon Bond. Organometallics 2018, 37, 2690–2705. [Google Scholar] [CrossRef]
- Lee, C.S.; Park, J.H.; Hwang, E.Y.; Park, G.H.; Go, M.J.; Lee, J.; Lee, B.Y. Preparation of [Bis(amido)-phosphine] and [Amido-Phosphine Sulfide or Oxide] Hafnium and Zirconium Complexes for Olefin Polymerization. J. Organom. Chem. 2014, 772, 172–181. [Google Scholar] [CrossRef]
- Hwang, E.Y.; Park, G.H.; Lee, C.S.; Kang, Y.Y.; Lee, J.; Lee, B.Y. Preparation of octahydro- and tetrahydro-[1,10]phenanthroline zirconium and hafnium complexes for olefin polymerization. Dalton Trans. 2015, 44, 3845–3855. [Google Scholar] [CrossRef] [PubMed]
- Jun, S.H.; Park, J.H.; Lee, C.S.; Park, S.Y.; Go, M.J.; Lee, J.; Lee, B.Y. Preparation of phosphine-amido hafnium and zirconium complexes for olefin polymerization. Organometallics 2013, 32, 7357–7365. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Catalyst | Al b (50 µmol) | Zn c (μmol) | Yield (g) | [C3H6] d (mol%) | Tm (°C) | Mne (kDa) | Mw/Mn |
|---|---|---|---|---|---|---|---|---|
| 1 | 10 | TOA | 0 | 1.2 | 10 | 86–129 | 16 | 11.0 |
| 2 | 11 | TOA | 0 | 5.8 | 12 | 101–120 | 87 | 1.5 |
| 3 | 12 | TOA | 0 | 7.5 | 13 | 102–120 | 124 | 3.2 |
| 4 | 16 | TOA | 0 | 3.5 | 11 | 99–115 | 26 | 2.0 |
| 5 | III | TOA | 0 | 16 | 56 | not-detected | 61 | 2.6 |
| 6 | 11 | MMAO | 0 | 5.5 | 10 | 105–120 | 266 | 2.1 |
| 7 | 12 | MMAO | 0 | 7.8 | 11 | 102–117 | 190 | 3.4 |
| 8 | 12 | MMAO | 100 | 6.3 | 9.0 | 100–117 | 58 (30; 32) f | 1.8 |
| 9 | 12 | MMAO | 200 | 6.6 | 11 | 100–112 | 33 (17; 17) f | 1.6 |
| 10 | 12 | MMAO | 300 | 5.4 | 13 | 97–110 | 22 (12; 9.0) f | 1.6 |
| 11 | 12 | MMAO | 400 | 3.6 | 15 | 94–108 | 11 (5.9; 4.5) f | 1.8 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baek, J.W.; Kwon, S.J.; Lee, H.J.; Kim, T.J.; Ryu, J.Y.; Lee, J.; Shin, E.J.; Lee, K.S.; Lee, B.Y. Preparation of Half- and Post-Metallocene Hafnium Complexes with Tetrahydroquinoline and Tetrahydrophenanthroline Frameworks for Olefin Polymerization. Polymers 2019, 11, 1093. https://doi.org/10.3390/polym11071093
Baek JW, Kwon SJ, Lee HJ, Kim TJ, Ryu JY, Lee J, Shin EJ, Lee KS, Lee BY. Preparation of Half- and Post-Metallocene Hafnium Complexes with Tetrahydroquinoline and Tetrahydrophenanthroline Frameworks for Olefin Polymerization. Polymers. 2019; 11(7):1093. https://doi.org/10.3390/polym11071093
Chicago/Turabian StyleBaek, Jun Won, Su Jin Kwon, Hyun Ju Lee, Tae Jin Kim, Ji Yeon Ryu, Junseong Lee, Eun Ji Shin, Ki Soo Lee, and Bun Yeoul Lee. 2019. "Preparation of Half- and Post-Metallocene Hafnium Complexes with Tetrahydroquinoline and Tetrahydrophenanthroline Frameworks for Olefin Polymerization" Polymers 11, no. 7: 1093. https://doi.org/10.3390/polym11071093
APA StyleBaek, J. W., Kwon, S. J., Lee, H. J., Kim, T. J., Ryu, J. Y., Lee, J., Shin, E. J., Lee, K. S., & Lee, B. Y. (2019). Preparation of Half- and Post-Metallocene Hafnium Complexes with Tetrahydroquinoline and Tetrahydrophenanthroline Frameworks for Olefin Polymerization. Polymers, 11(7), 1093. https://doi.org/10.3390/polym11071093