The Impact of Lignin Structural Diversity on Performance of Cellulose Nanofiber (CNF)-Starch Composite Films




Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials and Reagents
2.2. Successive Solvent Fractionation of Lignin
2.3. Preparation of Tunicate CNF-Starch-Lignin Films
2.4. Lignin Characterization
2.4.1. Size-Exclusion Chromatography (SEC)
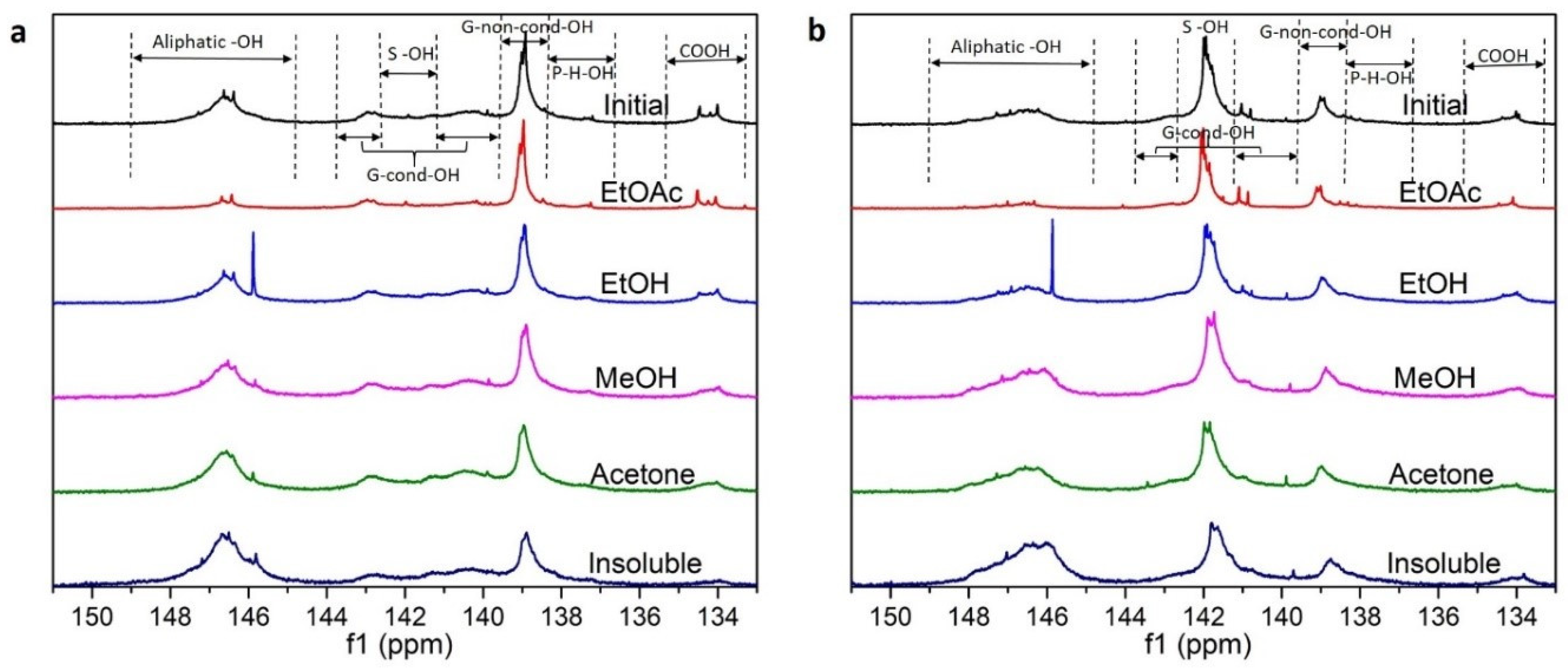
2.4.2. 31P Nuclear Magnetic Resonance (31P NMR)
2.4.3. Pyrolysis-Gas Chromatography/Mass Spectrometry/Fire Ionization Detector (Py-GC/MS/FID)
2.5. Tunicate CNF-Starch-Lignin Films Characterization
2.5.1. Density Measurement
2.5.2. Scanning Electron Microscope (SEM)
2.5.3. Transmittance Measurement
2.5.4. Tensile Test
2.5.5. Thermal Gravimetric Analysis (TGA)
3. Results
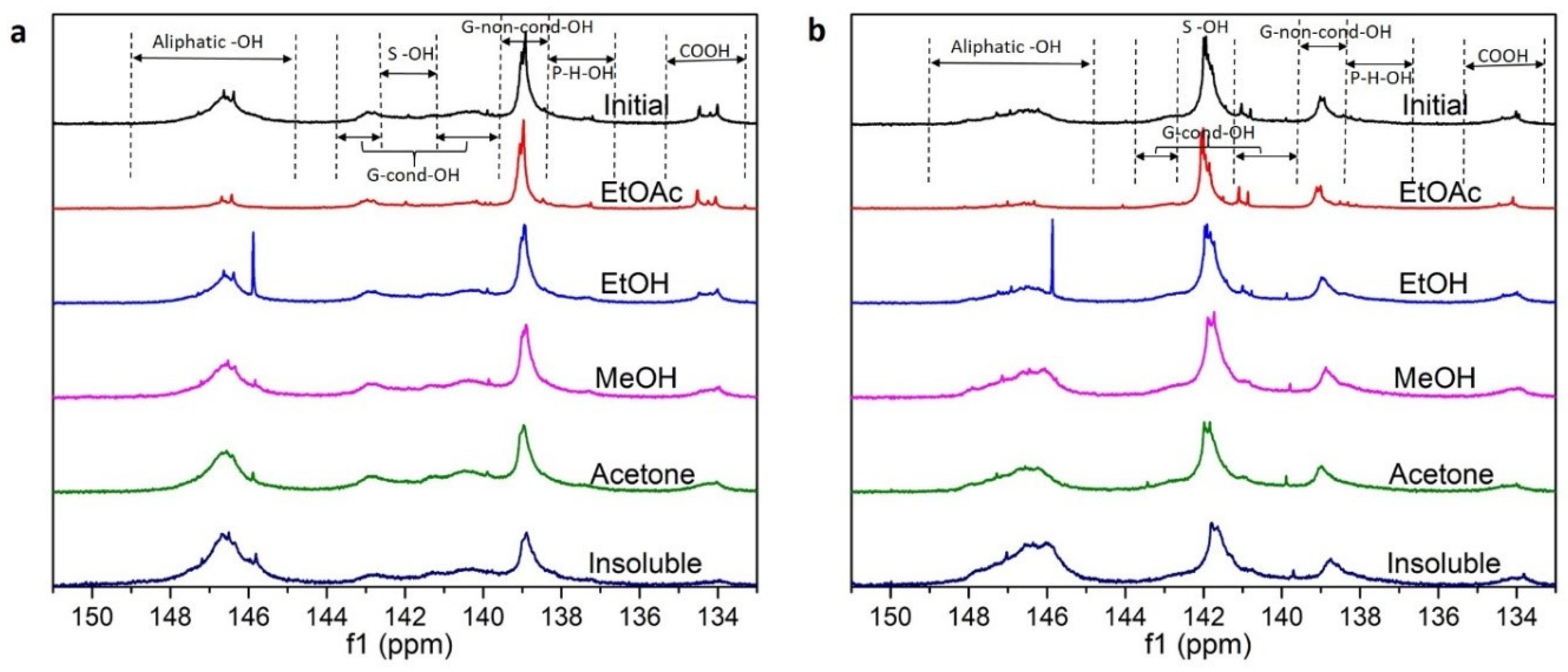
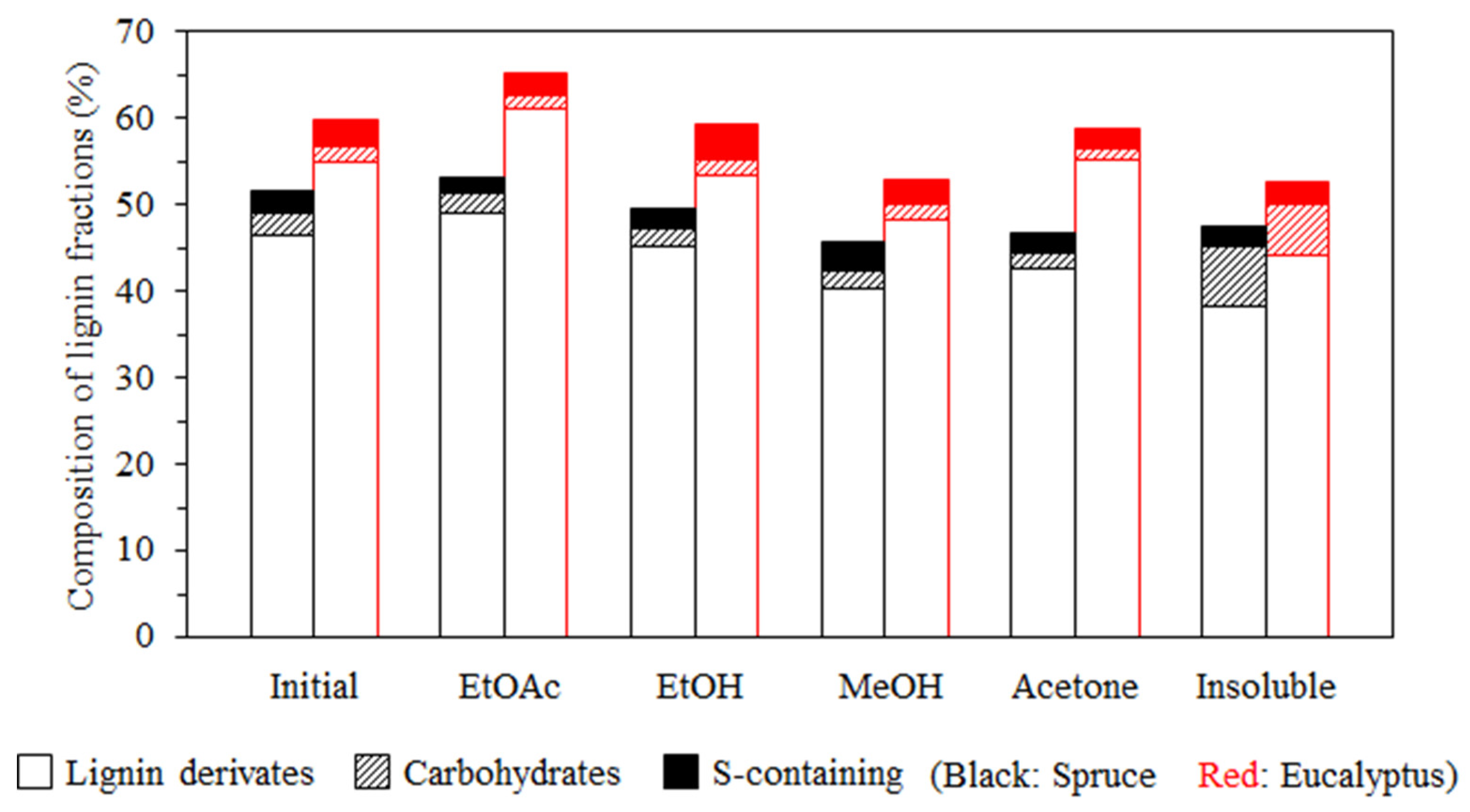
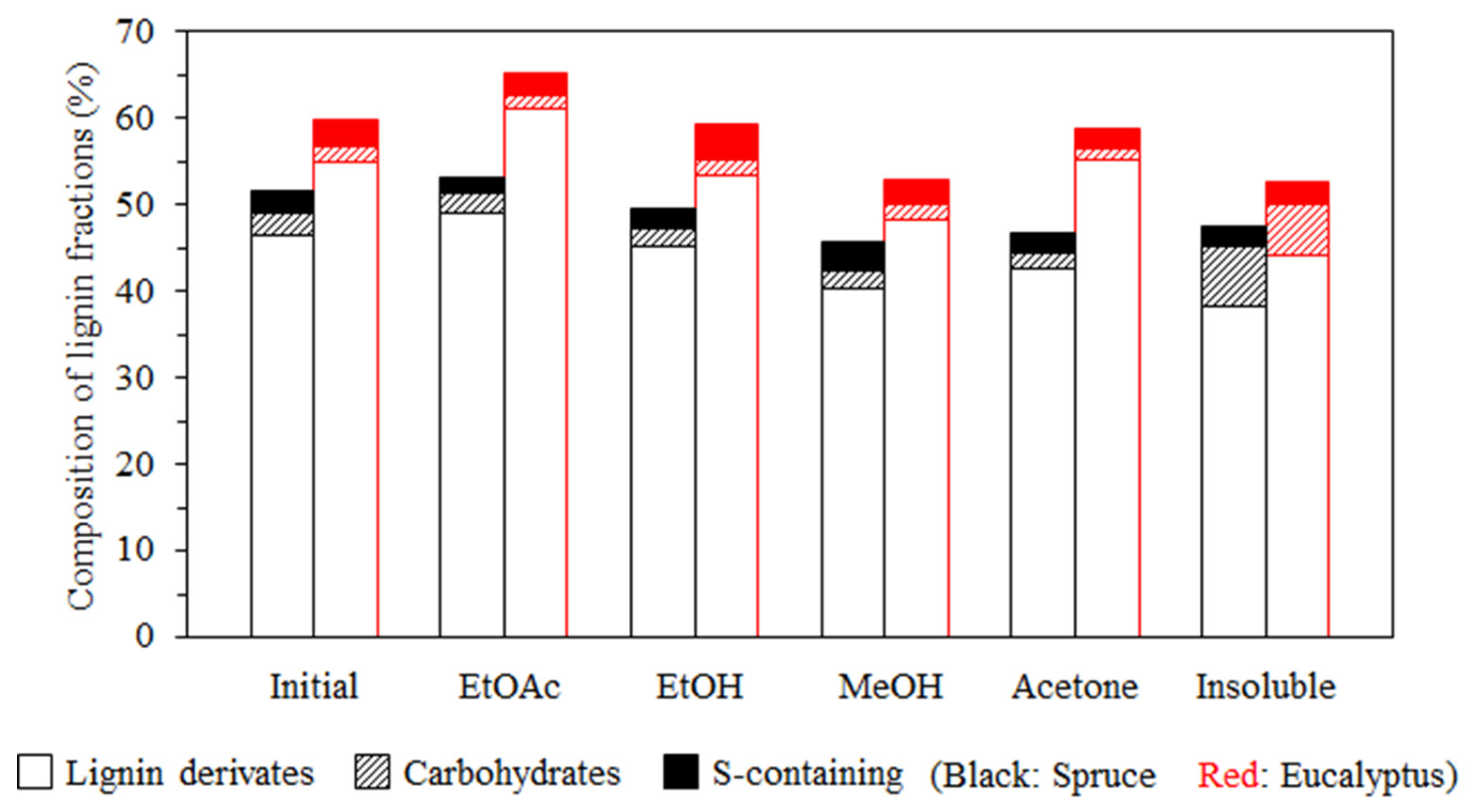
3.1. Lignin Diversities after Successive Solvent Extraction
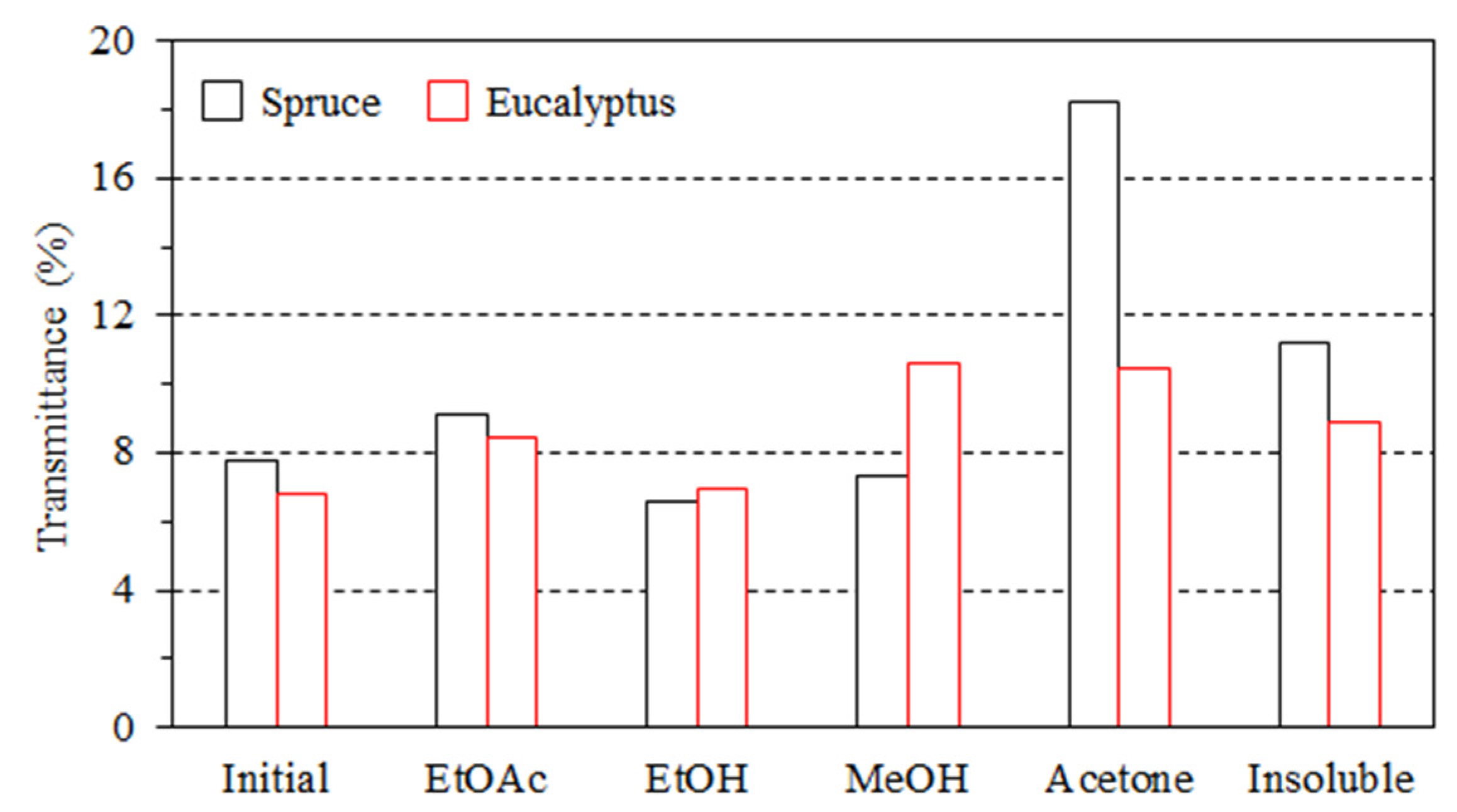
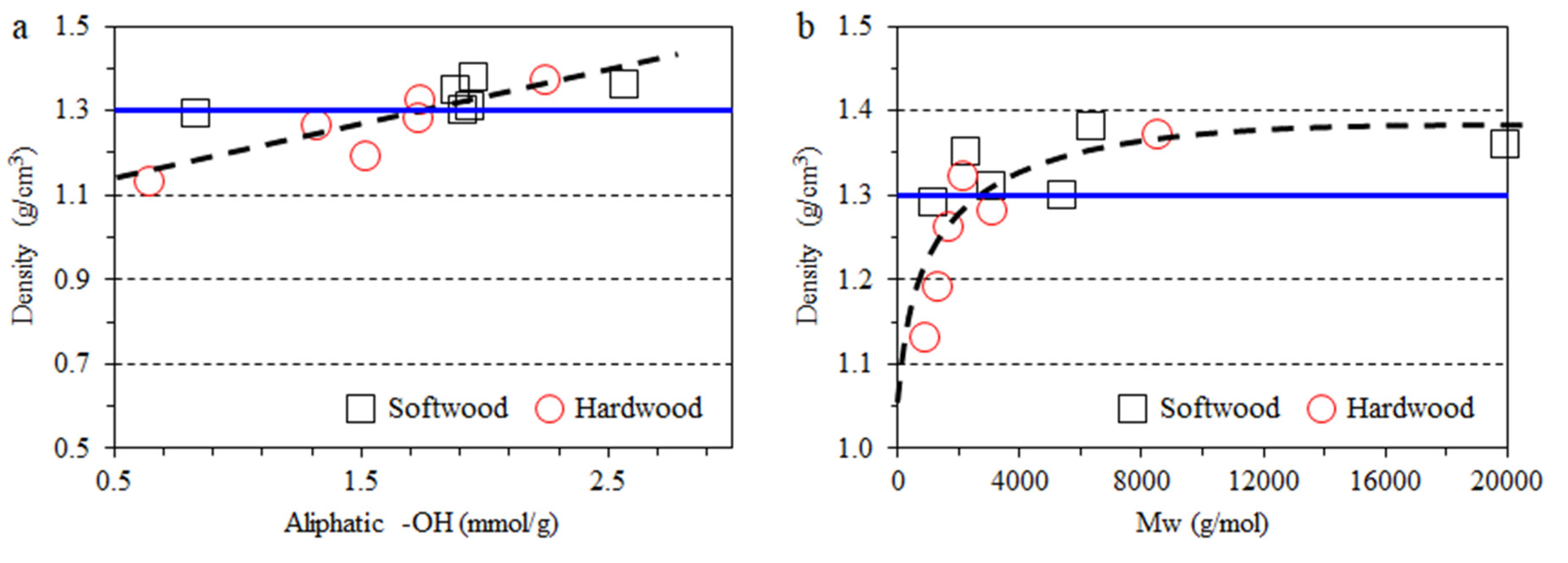
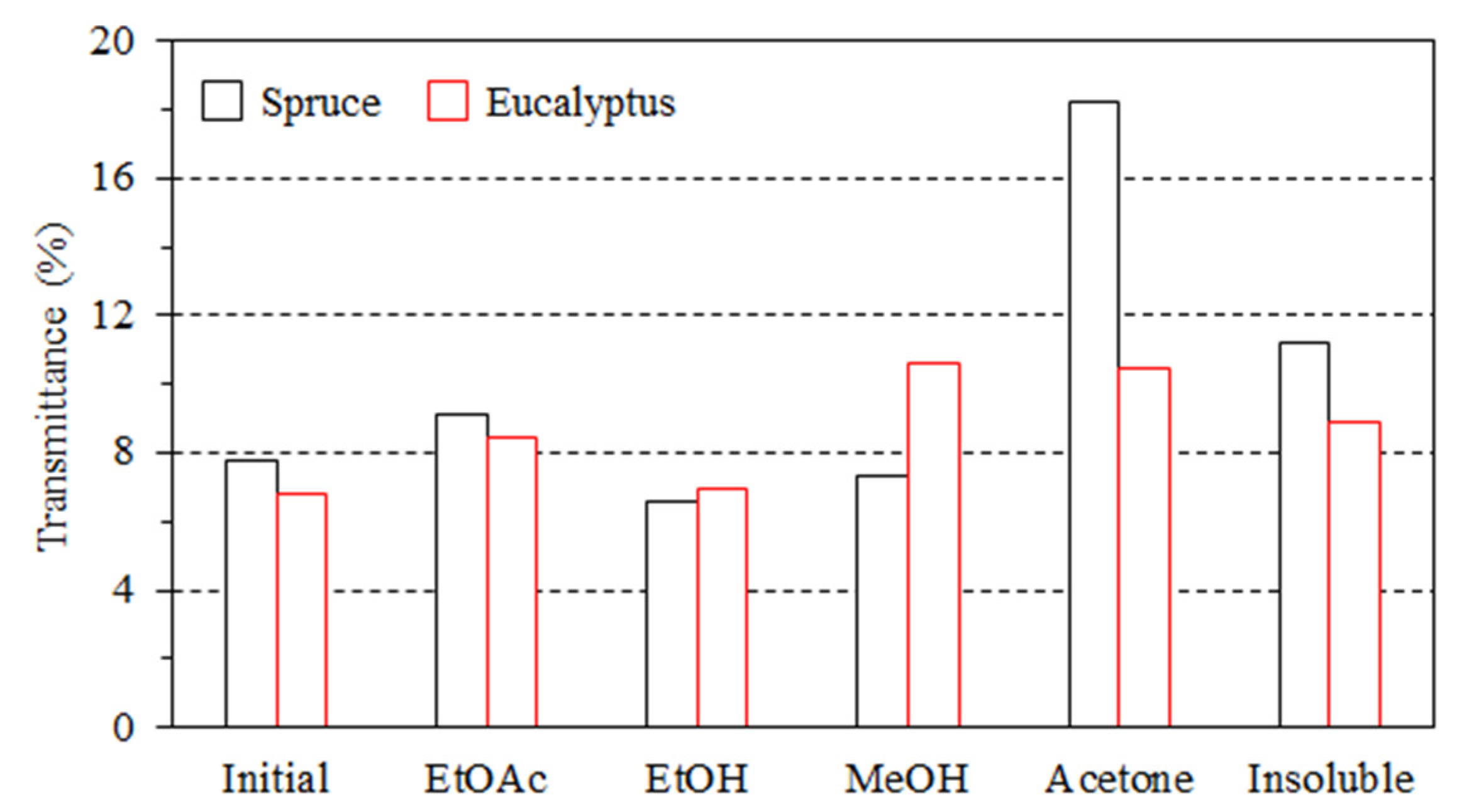
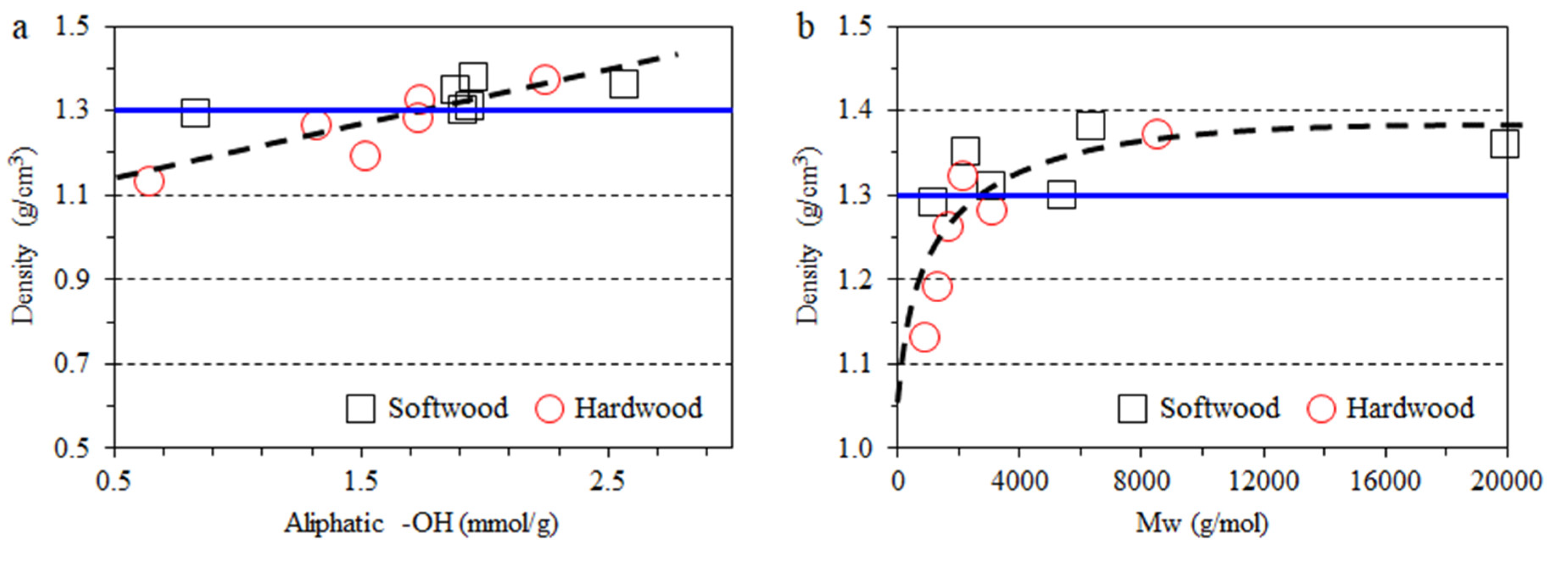
3.2. Impact of Lignin on Film Appearance and Morphology
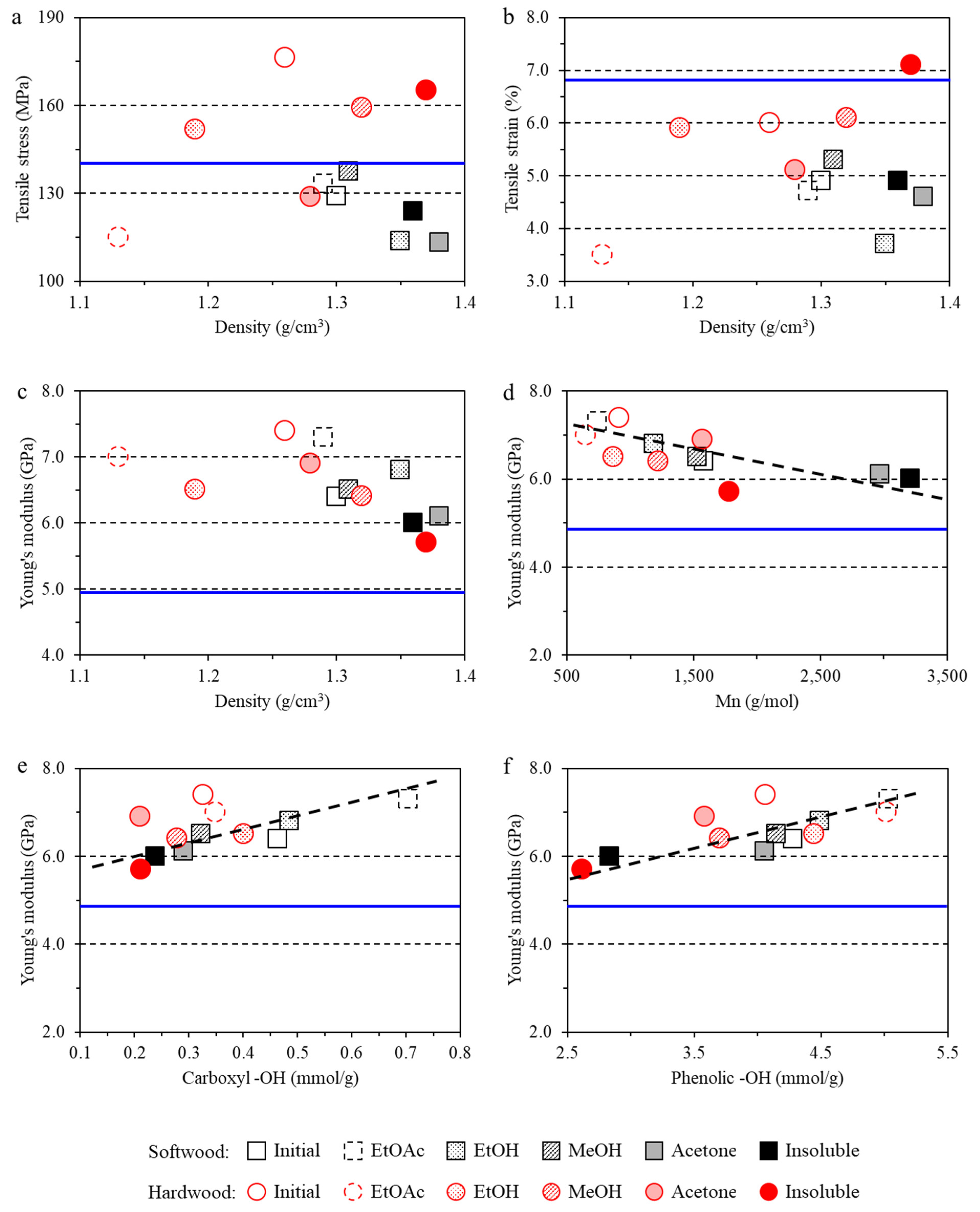
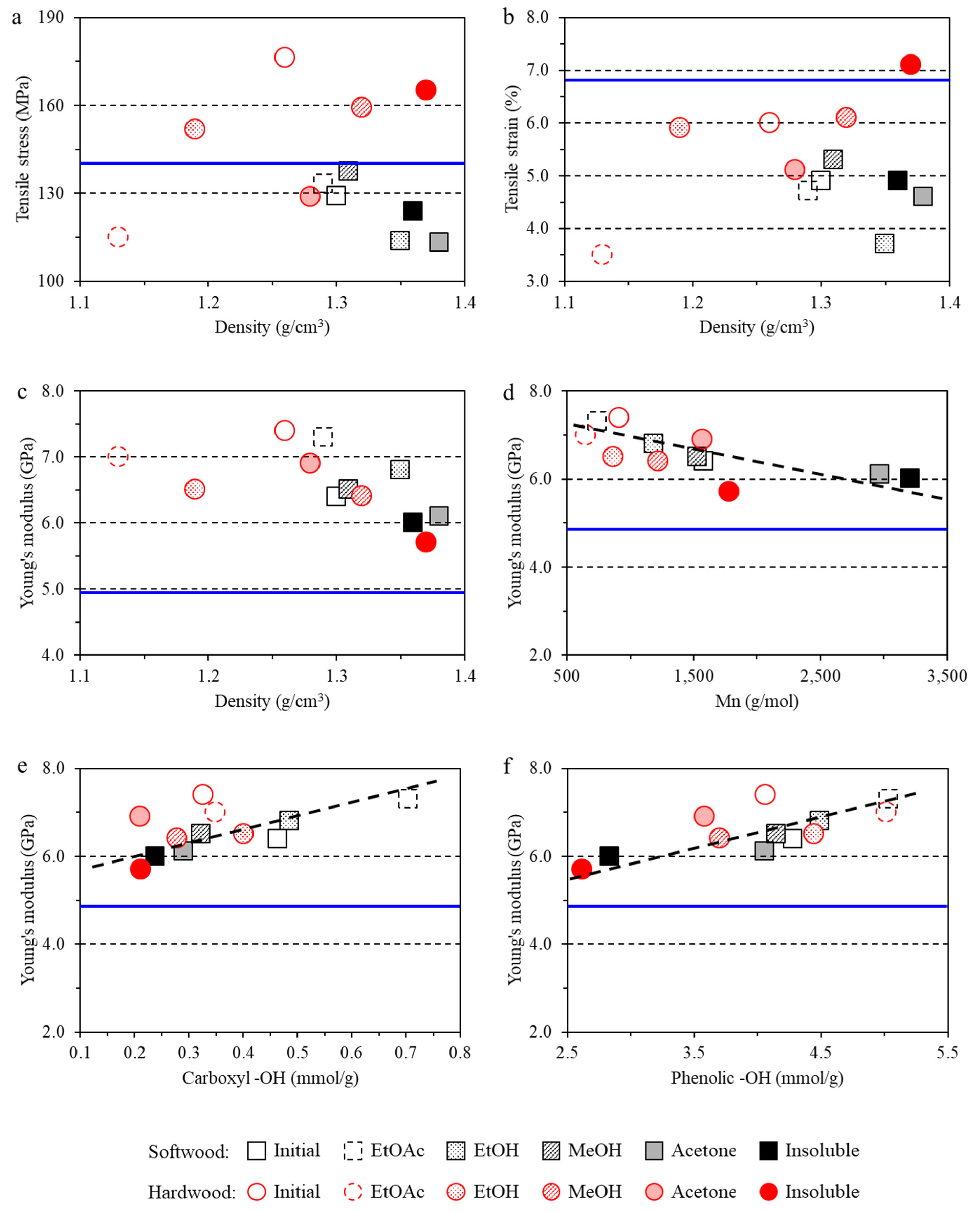
3.3. Impact of Lignin on Film Mechanical Properties
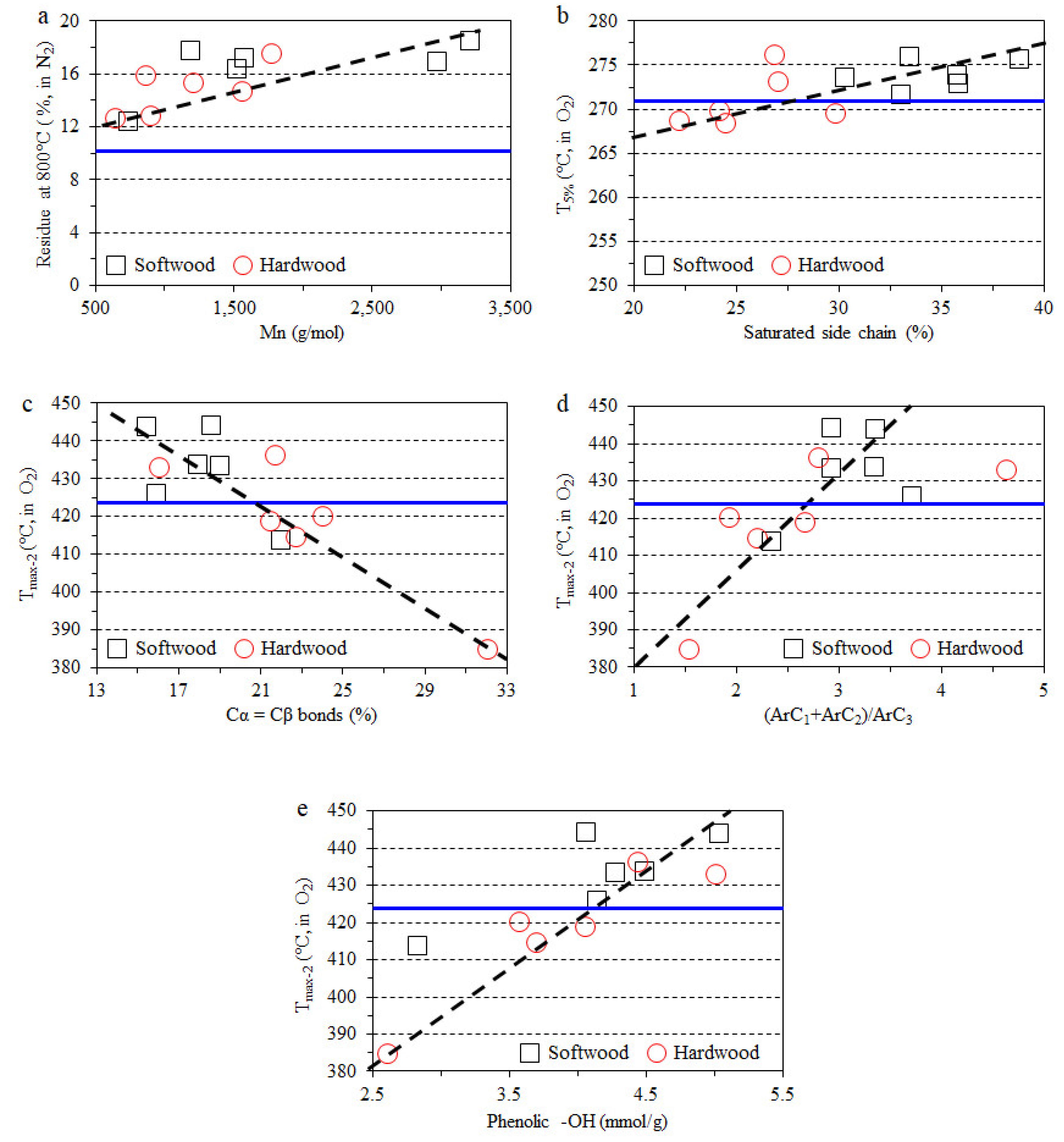
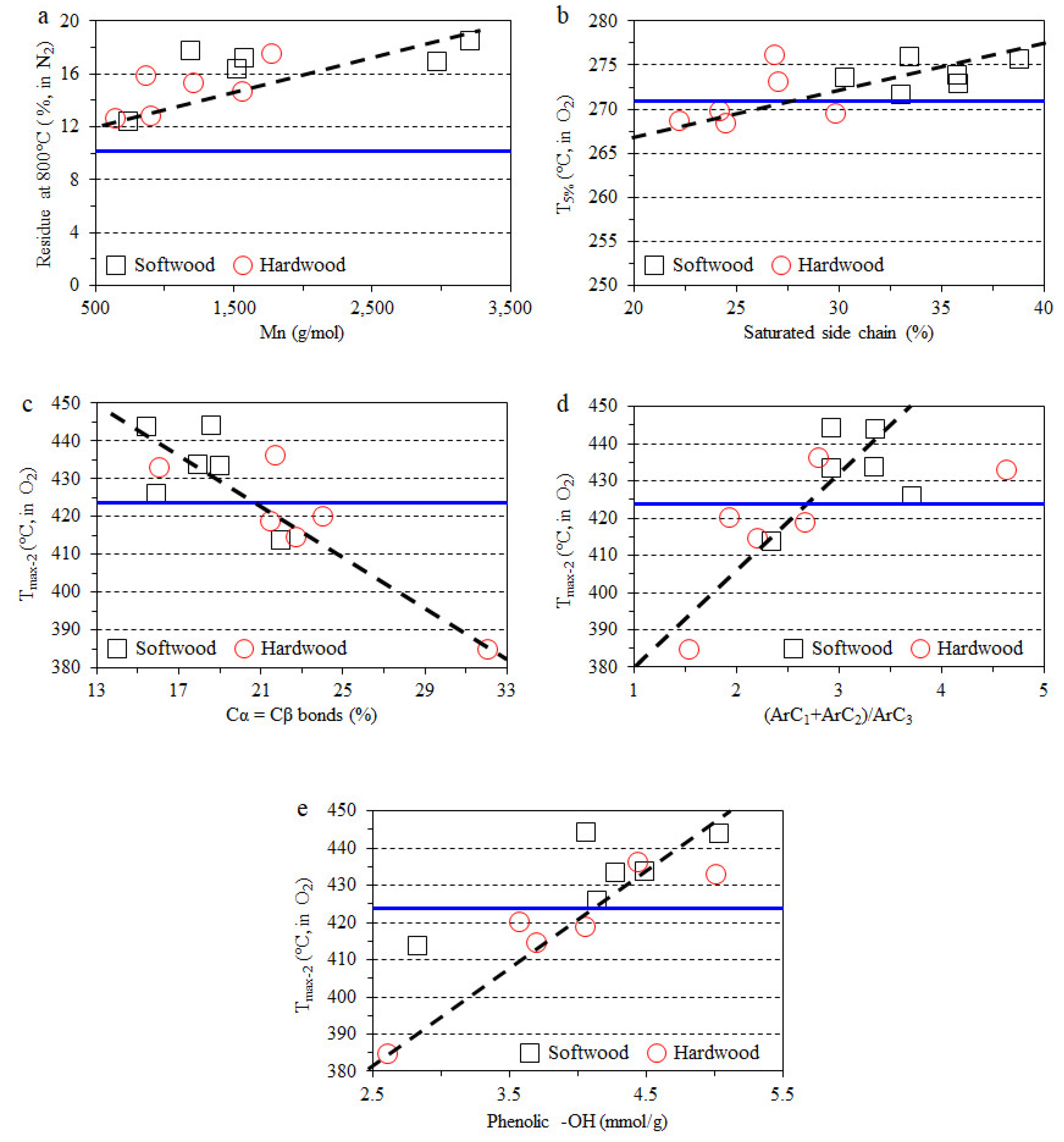
3.4. Impact of Lignin on Film Thermal Stability
4. Conclusions
- In this study, the lignin fractions with great diversities have been found to affect the properties of the tunicate CNF-starch composite films significantly:
- -
- The lignin aggregated to form particles in the composites, thus affecting the color and transmittance value.
- -
- The lignin imparted to the films a lower tensile strain and higher Young’s modulus due to the inherent rigidity of lignin. However, hardwood lignin with lower molecular weight and polydispersity reinforced the film structure, while softwood lignin with a higher molecular weight and lower G+S unit content negatively affected the tensile stress of the films.
- The thermal stability of composite films in a nitrogen atmosphere have a clear positive correlation with the molecular weight of lignin, and the char formation was improved with the addition of the lignin fraction.
- The direct relationship between the certain lignin structural features and thermo-oxidative stability of lignin-containing composite films was also observed. The same structural features positively influence the antioxidant properties of lignin.
- The structure–performance relationships found here will help to tune the properties of lignin to fit in specific application in the materials field. For example, as found in this study, the acetone lignin fraction is the best candidate to prepare uniform and highly transparent composite films; insoluble lignin fraction with high molecular weight could render the composite film dense structure; the ethyl acetate lignin fraction with low molecular weight, low PDI, and high content of phenolic –OH group are the best for composite films to have high Young’s modulus and good thermal stability.
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Geyer, R.; Jambeck, J.R.; Law, K.L. Production, use, and fate of all plastics ever made. Sci. Adv. 2017, 3, e1700782. [Google Scholar] [CrossRef] [PubMed]
- Venkata Mohan, S.; Nikhil, G.N.; Chiranjeevi, P.; Nagendranatha Reddy, C.; Rohit, M.V.; Kumar, A.N.; Sarkar, O. Waste biorefinery models towards sustainable circular bioeconomy: Critical review and future perspectives. Bioresour. Technol. 2016, 215, 2–12. [Google Scholar] [CrossRef]
- Sharma, P.R.; Chattopadhyay, A.; Zhan, C.; Sharma, S.K.; Geng, L.; Hsiao, B.S. Lead removal from water using carboxycellulose nanofibers prepared by nitro-oxidation method. Cellulose 2018, 25, 1961–1973. [Google Scholar] [CrossRef]
- Sharma, P.R.; Sharma, S.K.; Antoine, R.; Hsiao, B.S. Efficient removal of arsenic using zinc oxide nanocrystal-decorated regenerated microfibrillated cellulose scaffolds. ACS Sustain. Chem. Eng. 2019, 7, 6140–6151. [Google Scholar] [CrossRef]
- Sharma, P.R.; Chattopadhyay, A.; Sharma, S.K.; Geng, L.; Amiralian, N.; Martin, D.; Hsiao, B.S. Nanocellulose from spinifex as an effective adsorbent to remove cadmium(ii) from water. ACS Sustain. Chem. Eng. 2018, 6, 3279–3290. [Google Scholar] [CrossRef]
- Wang, Z.; Ma, H.; Chu, B.; Hsiao, B.S. Fabrication of cellulose nanofiber-based ultrafiltration membranes by spray coating approach. J. Appl. Polym. Sci. 2017. [Google Scholar] [CrossRef]
- Isogai, A.; Saito, T.; Fukuzumi, H. TEMPO-oxidized cellulose nanofibers. Nanoscale 2011, 3, 71–85. [Google Scholar] [CrossRef]
- Sharma, P.R.; Joshi, R.; Sharma, S.K.; Hsiao, B.S. A simple approach to prepare carboxycellulose nanofibers from untreated biomass. Biomacromolecules 2017, 18, 2333–2342. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.R.; Varma, A.J. Functional nanoparticles obtained from cellulose: Engineering the shape and size of 6-carboxycellulose. Chem. Commun. 2013, 49, 8818–8820. [Google Scholar] [CrossRef]
- Mohanty, A.K.; Misra, M.; Drzal, L.T. Sustainable bio-composites from renewable resources: Opportunities and challenges in the green materials world. J. Polym. Environ. 2002, 10, 19–26. [Google Scholar] [CrossRef]
- Crouvisier-Urion, K.; Bodart, P.R.; Winckler, P.; Raya, J.; Gougeon, R.D.; Cayot, P.; Domenek, S.; Debeaufort, F.; Karbowiak, T. Biobased composite films from chitosan and lignin: Antioxidant activity related to structure and moisture. ACS Sustain. Chem. Eng. 2016, 4, 6371–6381. [Google Scholar] [CrossRef]
- Väisänen, T.; Haapala, A.; Lappalainen, R.; Tomppo, L. Utilization of agricultural and forest industry waste and residues in natural fiber-polymer composites: A review. Waste Manag. 2016, 54, 62–73. [Google Scholar] [CrossRef] [PubMed]
- Sixta, H.; Potthast, A.; Krotschek, A.W. Chemical pulping processe: Sections 4.1–4.2. In Handbook of Pulp; Wiley-VCH Verlag GmbH: Hoboken, NJ, USA, 2008; pp. 109–229. [Google Scholar]
- Lora, J. Chapter 10—Industrial Commercial Lignins: Sources, Properties and Applications A2—Belgacem, Mohamed Naceur. In Monomers, Polymers and Composites from Renewable Resources; Gandini, A., Ed.; Elsevier: Amsterdam, The Netherlands, 2008; pp. 225–241. [Google Scholar]
- Zakzeski, J.; Bruijnincx, P.C.A.; Jongerius, A.L.; Weckhuysen, B.M. The catalytic valorization of lignin for the production of renewable chemicals. Chem. Rev. 2010, 110, 3552–3599. [Google Scholar] [CrossRef] [PubMed]
- Li, M.-F.; Sun, S.-N.; Xu, F.; Sun, R.-C. Sequential solvent fractionation of heterogeneous bamboo organosolv lignin for value-added application. Sep. Purif. Technol. 2012, 101, 18–25. [Google Scholar] [CrossRef]
- Rials, T.G.; Glasser, W.G. Multiphase materials with lignin. Iv. Blends of hydroxypropyl cellulose with lignin. J. Appl. Polym. Sci. 1989, 37, 2399–2415. [Google Scholar] [CrossRef]
- Sadeghifar, H.; Venditti, R.; Jur, J.; Gorga, R.E.; Pawlak, J.J. Cellulose-lignin biodegradable and flexible uv protection film. ACS Sustain. Chem. Eng. 2017, 5, 625–631. [Google Scholar] [CrossRef]
- Košíková, B.; Demianová, V.; Kačuráková, M. Sulfur-free lignins as composites of polypropylene films. J. Appl. Polym. Sci. 1993, 47, 1065–1073. [Google Scholar] [CrossRef]
- Çalgeris, İ.; Çakmakçı, E.; Ogan, A.; Kahraman, M.V.; Kayaman-Apohan, N. Preparation and drug release properties of lignin-starch biodegradable films. Starch Stärke 2012, 64, 399–407. [Google Scholar] [CrossRef]
- Cazacu, G.; Mihaies, M.; Pascu, M.C.; Profire, L.; Kowarskik, A.L.; Vasile, C. Polyolefin/lignosulfonate blends, 9. Macromol. Mater. Eng. 2004, 289, 880–889. [Google Scholar] [CrossRef]
- Farooq, M.; Zou, T.; Riviere, G.; Sipponen, M.H.; Österberg, M. Strong, ductile, and waterproof cellulose nanofibril composite films with colloidal lignin particles. Biomacromolecules 2019, 20, 693–704. [Google Scholar] [CrossRef]
- Aadil, K.R.; Barapatre, A.; Jha, H. Synthesis and characterization of Acacia lignin-gelatin film for its possible application in food packaging. Bioresour. Bioprocess. 2016, 3, 27. [Google Scholar] [CrossRef]
- Kim, Y.; Suhr, J.; Seo, H.-W.; Sun, H.; Kim, S.; Park, I.-K.; Kim, S.-H.; Lee, Y.; Kim, K.-J.; Nam, J.-D. All biomass and uv protective composite composed of compatibilized lignin and poly(lactic-acid). Sci. Rep. 2017, 7, 43596. [Google Scholar] [CrossRef]
- Cui, C.; Sun, R.; Argyropoulos, D.S. Fractional precipitation of softwood kraft lignin: Isolation of narrow fractions common to a variety of lignins. ACS Sustain. Chem. Eng. 2014, 2, 959–968. [Google Scholar] [CrossRef]
- Gordobil, O.; Moriana, R.; Zhang, L.; Labidi, J.; Sevastyanova, O. Assesment of technical lignins for uses in biofuels and biomaterials: Structure-related properties, proximate analysis and chemical modification. Ind. Crops Prod. 2016, 83, 155–165. [Google Scholar] [CrossRef]
- Watkins, D.; Nuruddin, M.; Hosur, M.; Tcherbi-Narteh, A.; Jeelani, S. Extraction and characterization of lignin from different biomass resources. J. Mater. Res. Technol. 2015, 4, 26–32. [Google Scholar] [CrossRef]
- Gierer, J. Chemical aspects of kraft pulping. Wood Sci. Technol. 1980, 14, 241–266. [Google Scholar] [CrossRef]
- Kirk, T.K.; Brown, W.; Cowling, E.B. Preparative fractionation of lignin by gel-permeation chromatography. Biopolymers 1969, 7, 135–153. [Google Scholar] [CrossRef]
- Toledano, A.; García, A.; Mondragon, I.; Labidi, J. Lignin separation and fractionation by ultrafiltration. Sep. Purif. Technol. 2010, 71, 38–43. [Google Scholar] [CrossRef]
- Sevastyanova, O.; Helander, M.; Chowdhury, S.; Lange, H.; Wedin, H.; Zhang, L.; Ek, M.; Kadla, J.F.; Crestini, C.; Lindström, M.E. Tailoring the molecular and thermo–mechanical properties of kraft lignin by ultrafiltration. J. Appl. Polym. Sci. 2014, 131, 9505–9515. [Google Scholar] [CrossRef]
- García, A.; Toledano, A.; Serrano, L.; Egüés, I.; González, M.; Marín, F.; Labidi, J. Characterization of lignins obtained by selective precipitation. Sep. Purif. Technol. 2009, 68, 193–198. [Google Scholar] [CrossRef]
- Helander, M.; Theliander, H.; Lawoko, M.; Henriksson, G.; Zhang, L.; Lindström, M.E. Fractionation of technical lignin: Molecular mass and ph effects. BioResources 2013, 8, 2270–2282. [Google Scholar] [CrossRef]
- Jääskeläinen, A.S.; Liitiä, T.; Mikkelson, A.; Tamminen, T. Aqueous organic solvent fractionation as means to improve lignin homogeneity and purity. Ind. Crops Prod. 2017, 103, 51–58. [Google Scholar] [CrossRef]
- Duval, A.; Vilaplana, F.; Crestini, C.; Lawoko, M. Solvent screening for the fractionation of industrial kraft lignin. Holzforschung 2016, 70, 11–20. [Google Scholar] [CrossRef]
- Sun, R.; Tomkinson, J.; Griffiths, S. Fractional and physico-chemical analysis of soda-aq lignin by successive extraction with organic solvents from oil palm efb fiber. Int. J. Polym. Anal. Charact. 2000, 5, 531–547. [Google Scholar] [CrossRef]
- Wang, K.; Xu, F.; Sun, R. Molecular characteristics of kraft-aq pulping lignin fractionated by sequential organic solvent extraction. Int. J. Mol. Sci. 2010, 11, 2988–3001. [Google Scholar] [CrossRef] [PubMed]
- Gioia, C.; Lo Re, G.; Lawoko, M.; Berglund, L. Tunable thermosetting epoxies based on fractionated and well-characterized lignins. J. Am. Chem. Soc. 2018, 140, 4054–4061. [Google Scholar] [CrossRef]
- Arshanitsa, A.; Ponomarenko, J.; Dizhbite, T.; Andersone, A.; Gosselink, R.J.A.; van der Putten, J.; Lauberts, M.; Telysheva, G. Fractionation of technical lignins as a tool for improvement of their antioxidant properties. J. Anal. Appl. Pyrolysis 2013, 103, 78–85. [Google Scholar] [CrossRef]
- Baumberger, S.; Lapierre, C.; Monties, B. Utilization of pine kraft lignin in starch composites: Impact of structural heterogeneity. J. Agric. Food Chem. 1998, 46, 2234–2240. [Google Scholar] [CrossRef]
- Baumberger, S.; Lapierre, C.; Monties, B.; Valle, G.D. Use of kraft lignin as filler for starch films. Polym. Degrad. Stab. 1998, 59, 273–277. [Google Scholar] [CrossRef]
- Tomani, P. The lignoboost process. Cell. Chem. Technol. 2010, 44, 53–58. [Google Scholar]
- Tagami, A.; Gioia, C.; Lauberts, M.; Budnyak, T.; Moriana, R.; Lindström, M.E.; Sevastyanova, O. Solvent fractionation of softwood and hardwood kraft lignins for more efficient uses: Compositional, structural, thermal, antioxidant and adsorption properties. Ind. Crops Prod. 2019, 129, 123–134. [Google Scholar] [CrossRef]
- Zhao, Y.; Li, J. Excellent chemical and material cellulose from tunicates: Diversity in cellulose production yield and chemical and morphological structures from different tunicate species. Cellulose 2014, 21, 3427–3441. [Google Scholar] [CrossRef]
- Zhao, Y.; Moser, C.; Lindström, M.E.; Henriksson, G.; Li, J. Cellulose nanofibers from softwood, hardwood, and tunicate: Preparation–structure–film performance interrelation. ACS Appl. Mater. Interfaces 2017, 9, 13508–13519. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zhang, Y.; Lindström, M.E.; Li, J. Tunicate cellulose nanocrystals: Preparation, neat films and nanocomposite films with glucomannans. Carbohydr. Polym. 2015, 117, 286–296. [Google Scholar] [CrossRef]
- Guerra, A.; Lucia Lucian, A.; Argyropoulos Dimitris, S. Isolation and characterization of lignins from eucalyptus grandis hill ex maiden and eucalyptus globulus labill. By enzymatic mild acidolysis (Emal). Holzforschung 2008, 62, 24–30. [Google Scholar] [CrossRef]
- Asikkala, J.; Tamminen, T.; Argyropoulos, D.S. Accurate and reproducible determination of lignin molar mass by acetobromination. J. Agric. Food Chem. 2012, 60, 8968–8973. [Google Scholar] [CrossRef]
- Guerra, A.; Filpponen, I.; Lucia, L.A.; Argyropoulos, D.S. Comparative evaluation of three lignin isolation protocols for various wood species. J. Agric. Food Chem. 2006, 54, 9696–9705. [Google Scholar] [CrossRef]
- Alves, A.; Gierlinger, N.; Schwanninger, M.; Rodrigues, J. Analytical pyrolysis as a direct method to determine the lignin content in wood: Part 3. Evaluation of species-specific and tissue-specific differences in softwood lignin composition using principal component analysis. J. Anal. Appl. Pyrolysis 2009, 85, 30–37. [Google Scholar] [CrossRef]
- Ponomarenko, J.; Dizhbite, T.; Lauberts, M.; Viksna, A.; Dobele, G.; Bikovens, O.; Telysheva, G. Characterization of softwood and hardwood lignoboost kraft lignins with emphasis on their antioxidant activity. BioResources 2014, 9, 2051–2068. [Google Scholar] [CrossRef]
- Gosselink, R.J.A.; Snijder, M.H.B.; Kranenbarg, A.; Keijsers, E.R.P.; de Jong, E.; Stigsson, L.L. Characterisation and application of novafiber lignin. Ind. Crops Prod. 2004, 20, 191–203. [Google Scholar] [CrossRef]
- Faix, O.; Bremer, J.; Meier, D.; Fortmann, I.; Scheijen, M.A.; Boon, J.J. Characterization of tobacco lignin by analytical pyrolysis and fourier transform-infrared spectroscopy. J. Anal. Appl. Pyrolysis 1992, 22, 239–259. [Google Scholar] [CrossRef]
- Meier, D.; Faix, O. Pyrolysis-gas chromatography-mass spectrometry. In Methods in Lignin Chemistry; Lin, S.Y., Dence, C.W., Eds.; Springer: Berlin/Heidelberg, Germany, 1992; pp. 177–199. [Google Scholar]
- Ponomarenko, J.; Dizhbite, T.; Lauberts, M.; Volperts, A.; Dobele, G.; Telysheva, G. Analytical Pyrolysis—A Tool for Revealing of Lignin Structure-Antioxidant Activity Relationship. J. Anal. Appl. Pyrolysis 2015, 113, 360–369. [Google Scholar] [CrossRef]
- Ponomarenko, J.; Lauberts, M.; Dizhbite, T.; Lauberte, L.; Jurkjane, V.; Telysheva, G. Antioxidant Activity of Various Lignins and Lignin-Related Phenylpropanoid Units with High and Low Molecular Weight. Holzforschung 2015, 69, 795–805. [Google Scholar] [CrossRef]
- Wu, R.-L.; Wang, X.-L.; Li, F.; Li, H.-Z.; Wang, Y.-Z. Green composite films prepared from cellulose, starch and lignin in room-temperature ionic liquid. Bioresour. Technol. 2009, 100, 2569–2574. [Google Scholar] [CrossRef]
- Lupoi, J.S.; Gjersing, E.; Davis, M.F. Evaluating lignocellulosic biomass, its derivatives, and downstream products with Raman spectroscopy. Front. Bioeng. Biotechnol. 2015, 3, 50. [Google Scholar] [CrossRef]
- Prakobna, K.; Galland, S.; Berglund, L.A. High-performance and moisture-stable cellulose-starch nanocomposites based on bioinspired core-shell nanofibers. Biomacromolecules 2015, 16, 904–912. [Google Scholar] [CrossRef]
- Dufresne, A.; Vignon, M.R. Improvement of starch film performances using cellulose microfibrils. Macromolecules 1998, 31, 2693–2696. [Google Scholar] [CrossRef]
- Avelino, F.; Almeida, S.L.; Duarte, E.B.; Sousa, J.R.; Mazzetto, S.E.; de Souza Filho, M.D.S.M. Thermal and mechanical properties of coconut shell lignin-based polyurethanes synthesized by solvent-free polymerization. J. Mater. Sci. 2018, 53, 1470–1486. [Google Scholar] [CrossRef]
- Chen, K.; Ye, D.; Gu, S.; Zhou, Y. Thermal-oxidative effect of kraft lignin antioxidant in polypropylene: Uncovering the key factor using correlation analysis model. Int. J. Biol. Macromol. 2018, 107, 478–485. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type | Fraction | Yield (%) | Mn (g/mol) | Mw (g/mol) | PDI | Aliph –OH (mmol/g) | –COOH (mmol/g) | Ph –OH (mmol/g) |
|---|---|---|---|---|---|---|---|---|
| Spruce | Initial | - | 1490 | 6650 | 4.48 | 1.93 | 0.46 | 4.27 |
| EtOAc | 24.5 | 740 | 1200 | 1.63 | 0.85 | 0.67 | 5.10 | |
| EtOH | 32.5 | 1190 | 2280 | 1.93 | 1.91 | 0.48 | 4.60 | |
| MeOH | 8.2 | 1530 | 3080 | 2.01 | 1.96 | 0.31 | 4.15 | |
| Acetone | 14.7 | 2970 | 6360 | 2.14 | 1.95 | 0.27 | 4.02 | |
| Insoluble | 20.0 | 3210 | 19,930 | 6.20 | 2.76 | 0.21 | 3.04 | |
| Eucalyptus | Initial | - | 900 | 1980 | 2.20 | 1.33 | 0.32 | 4.23 |
| EtOAc | 35.2 | 650 | 950 | 1.47 | 0.65 | 0.35 | 5.02 | |
| EtOH | 32.8 | 870 | 1390 | 1.60 | 1.52 | 0.40 | 4.45 | |
| MeOH | 15.4 | 1220 | 2170 | 1.78 | 1.74 | 0.28 | 3.71 | |
| Acetone | 6.1 | 1570 | 3150 | 2.00 | 1.74 | 0.21 | 3.58 | |
| Insoluble | 10.5 | 1780 | 8570 | 4.82 | 2.25 | 0.21 | 2.62 |
| Type | Fraction | G + S Units % | Saturated Side Chain % | Cα = Cβ Bonds % | O-Atoms in Side Chain % | (ArC1 + ArC2)/ArC3 * |
|---|---|---|---|---|---|---|
| Spruce | Initial | 83.8 | 33.5 | 19.0 | 10.9 | 2.9 |
| EtOAc | 84.6 | 30.3 | 15.4 | 12.5 | 3.4 | |
| EtOH | 84.6 | 35.8 | 18.0 | 9.6 | 3.4 | |
| MeOH | 82.2 | 38.8 | 15.9 | 9.8 | 3.7 | |
| Acetone | 82.7 | 35.8 | 18.6 | 11.7 | 2.9 | |
| Insoluble | 85.3 | 33.1 | 22.0 | 13.3 | 2.3 | |
| Eucalyptus | Initial | 93.6 | 24.3 | 21.5 | 15.7 | 2.7 |
| EtOAc | 94.0 | 24.5 | 16.1 | 20.5 | 4.6 | |
| EtOH | 92.6 | 29.9 | 21.7 | 10.0 | 2.8 | |
| MeOH | 92.7 | 27.1 | 22.8 | 9.2 | 2.2 | |
| Acetone | 92.3 | 26.9 | 24.1 | 11.5 | 1.9 | |
| Insoluble | 92.9 | 22.3 | 32.1 | 12.0 | 1.5 |
| Type | Fraction | TGA (N2) | TGA (O2) | |||||
|---|---|---|---|---|---|---|---|---|
| T5% | Tmax | Residue at 800 °C (char) | T5% | Tmax-1 | Tmax-2 | Residue at 800 °C (ash) | ||
| °C | °C | % | °C | °C | °C | % | ||
| Blank | 283.3 | 354.7 | 10.3 | 270.8 | 325.2 | 423.6 | 0.8 | |
| Spruce | Initial | 270.8 | 354.8 | 17.2 | 275.8 | 319.0 | 433.2 | 0.5 |
| EtOAc | 266.7 | 359.9 | 12.4 | 273.5 | 320.1 | 443.8 | 0.6 | |
| EtOH | 274.2 | 355.2 | 17.7 | 272.8 | 321.3 | 433.5 | 0.5 | |
| MeOH | 272.8 | 354.2 | 16.4 | 275.7 | 321.5 | 425.9 | 0.9 | |
| Acetone | 277.5 | 353.7 | 16.9 | 273.8 | 321.2 | 444.1 | 0.6 | |
| Insoluble | 271.2 | 355.3 | 18.5 | 271.7 | 326.2 | 413.5 | 0.8 | |
| Eucalyptus | Initial | 268.7 | 358.7 | 12.7 | 269.7 | 319.2 | 418.6 | 0.7 |
| EtOAc | 265.2 | 359.7 | 12.6 | 268.3 | 318.9 | 432.8 | 0.5 | |
| EtOH | 265.7 | 355.9 | 15.8 | 270.5 | 311.4 | 439.4 | 0.4 | |
| MeOH | 270.7 | 354.8 | 15.2 | 273.0 | 326.7 | 414.4 | 0.8 | |
| Acetone | 275.7 | 356.2 | 14.6 | 276.0 | 326.1 | 419.9 | 0.8 | |
| Insoluble | 268.8 | 357.6 | 17.4 | 268.5 | 331.3 | 384.4 | 1.3 | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, Y.; Tagami, A.; Dobele, G.; Lindström, M.E.; Sevastyanova, O. The Impact of Lignin Structural Diversity on Performance of Cellulose Nanofiber (CNF)-Starch Composite Films. Polymers 2019, 11, 538. https://doi.org/10.3390/polym11030538
Zhao Y, Tagami A, Dobele G, Lindström ME, Sevastyanova O. The Impact of Lignin Structural Diversity on Performance of Cellulose Nanofiber (CNF)-Starch Composite Films. Polymers. 2019; 11(3):538. https://doi.org/10.3390/polym11030538
Chicago/Turabian StyleZhao, Yadong, Ayumu Tagami, Galina Dobele, Mikael E. Lindström, and Olena Sevastyanova. 2019. "The Impact of Lignin Structural Diversity on Performance of Cellulose Nanofiber (CNF)-Starch Composite Films" Polymers 11, no. 3: 538. https://doi.org/10.3390/polym11030538
APA StyleZhao, Y., Tagami, A., Dobele, G., Lindström, M. E., & Sevastyanova, O. (2019). The Impact of Lignin Structural Diversity on Performance of Cellulose Nanofiber (CNF)-Starch Composite Films. Polymers, 11(3), 538. https://doi.org/10.3390/polym11030538