Self-Assembled Structures of Diblock Copolymer/Homopolymer Blends through Multiple Complementary Hydrogen Bonds

1
Department of Materials and Optoelectronic Science, Center for Functional Polymers and Supramolecular Materials, National Sun Yat-Sen University, Kaohsiung 80424, Taiwan
2
Graduate Institute of Applied Science and Technology, National Taiwan University of Science and Technology, Taipei 106, Taiwan
3
Department of Medicinal and Applied Chemistry, Kaohsiung Medical University, Kaohsiung 80424, Taiwan
*
Author to whom correspondence should be addressed.
Crystals 2018, 8(8), 330; https://doi.org/10.3390/cryst8080330
Submission received: 13 August 2018
/
Revised: 17 August 2018
/
Accepted: 17 August 2018
/
Published: 19 August 2018
(This article belongs to the Special Issue Self-Assembled Supramolecular Polymers via Strong H Bonding)
Abstract
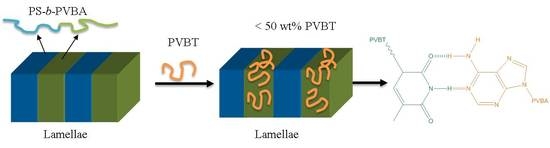
:A poly(styrene-b-vinylbenzyl triazolylmethyl methyladenine) (PS-b-PVBA) diblock copolymer and a poly(vinylbenzyl triazolylmethyl methylthymine) (PVBT) homopolymer were prepared through a combination of nitroxide-mediated radical polymerizations and click reactions. Strong multiple hydrogen bonding interactions of the A···T binary pairs occurred in the PVBA/PVBT miscible domain of the PS-b-PVBA/PVPT diblock copolymer/homopolymer blend, as evidenced in Fourier transform infrared and 1H nuclear magnetic resonance spectra. The self-assembled lamellar structure of the pure PS-b-PVBA diblock copolymer after thermal annealing was transformed to a cylinder structure after blending with PVBT at lower concentrations and then to a disordered micelle or macrophase structure at higher PVBT concentrations, as revealed by small-angle X-ray scattering and transmission electron microscopy.
1. Introduction
The self-assembly of diblock copolymers (A-b-B) has received much attention for possible applications in, for example, drug delivery and the preparation of mesoporous materials, photonic crystals, and photovoltaic devices [1,2,3,4,5]. In the bulk state, diblock copolymers form several typical self-assembled nanostructures including lamellae, cylinders, gyroids, and spherical structures depending on the volume fractions and overall molecular weights of their block copolymer segments and their Flory-Huggins interaction parameters [6,7,8]. Although block copolymers with various types (and volume fractions) of segments can be synthesized using several variations of living polymerizations, these approaches can be expensive and time-consuming [9,10,11]. As a result, blending of a homopolymer C into a diblock copolymer (A-b-B) segment, with stabilization through strong hydrogen bonding, and varying the volume fractions of the diblock copolymer, has become the preferred method for the preparation of a range of well-defined self-assembled structures [10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35]. Examples of such A-b-B/C blends stabilized through hydrogen bonding have been reported for four different systems, both experimentally and theoretically: poly(styrene-b-vinylphenol) (PS-b-PVPh) blended with poly(4-vinylpyridine) (P4VP), poly(methyl methacrylate) (PMMA), polycaprolactone (PCL), or poly(ethylene oxide) (PEO) homopolymers; PVPh-b-PMMA and PVPh-b-PCL blended with PVP homopolymers; and PS-b-P4VP and PCL-b-P4VP blending with phenolic or PVPh homopolymers [10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35]. In all of these cases, however, the block segments and homopolymers feature units capable of forming only a single hydrogen bond (e.g., phenolic hydroxyl, pyridine, ether, and carbonyl groups). For example, PVPh/P4VP miscible blends have been considered as examples of systems featuring strong hydrogen bonds, with an inter-/self-association equilibrium constant ratio (KA/KB) of 1200/66 = 18, and have been investigated widely [36,37,38]. Nevertheless, their single-site hydrogen bonding interactions are considerably weaker than those in systems featuring multiple hydrogen bonding interactions [39,40,41,42,43].
The applications of complementary multiple hydrogen bonding in supramolecular structures, formed from synthetic polymers featuring nucleotide bases on their side chains, have received much recent interest. The values of KA can be tuned from several hundreds to greater than 107 for polymer chains capable of forming multiple hydrogen bonding interactions [44,45,46,47,48,49]. The most important multiple hydrogen bonds are those found in complex biological systems, in particular, in DNA, a near-perfect example of a macromolecular structure in polymer science. It remains a challenge to develop synthetic polymers capable of mimicking the behavior of DNA. The self-assembly of DNA strands is mediated through complementary multiple hydrogen bonds formed between guanine (G) and cytosine (C) and between adenine (A) and thymine (T) nucleobases attached to the phosphate sugar backbone [50,51].
We have also studied the application of multiple hydrogen bonds in modifying the miscibility behavior and supramolecular structures of several nucleobase-functionalized polymers prepared through free radical polymerization [52,53,54,55,56,57,58,59,60,61,62,63,64,65,66]. We found that the preparation of highly A- or T-functionalized vinylbenzyl monomers can be difficult, due to the polarities of these hetero-nucleobase monomers being significantly different from those of typical commercial monomers (e.g., methacrylate or styrene monomers) [52,53,54]. In general, highly nucleobase-functionalized polymers are prepared through chemical modification of the side chains and not directly through free radical copolymerization. Yang et al. summarized the general methods for the preparation of highly nucleobase-functionalized polymers using various living polymerizations, including nitroxide-mediated radical (NMRP), reversible addition-fragmentation chain transfer (RAFT), and atom transfer radical (ATRP) polymerization [67]. For example, Long et al. prepared A- and T-functionalized triblock copolymers through NMRP and observed dramatic increases in the viscosity and thermal properties as a result of complementary polymer blending of binary A···T pairs [47,68]. Haddleton et al. synthesized polymers containing uridine (U) and A nucleobases through ATRP [69]. Cheng et al. synthesized various nucleobase homopolymers through ATRP, including vinylbenzyl adenine (VBA) and vinylbenzyl thymine (VBT) monomers, but they only obtained low and inconsistent molecular weights for these two homopolymers [70]. Therefore, we suspected that a combination of NMRP and click reactions might allow us to prepare highly A- and T-functionalized polymers of the same molecular weights [71].
Our approach to mimicking DNA-like interactions through the preparation of an A-containing diblock copolymer, as well as a T-containing homopolymer, was synthesized using a combination of NMRP and click reactions. First, we used NMRP to prepare a poly(styrene-b-vinyl chloride) diblock copolymer (PS-b-PVBC) and a PVBC homopolymer, which we then converted to the corresponding poly(styrene–b–vinylbenzyl azide) (PS-b-PVBN3) diblock copolymer and the homopolymer PVBN3, respectively, through reactions with NaN3. Finally, we obtained the PS-b-PVBA diblock copolymer and PVBT homopolymer through click reactions of the PS-b-PVBN3 diblock copolymer and the PVBN3 homopolymer with propargyl adenine (PA) and propargyl thymine (PT), respectively. This approach allowed us to investigate the behavior of PS-b-PVBA/PVBT diblock copolymer/homopolymer blends. To the best of our knowledge, this study is the first to prepare diblock copolymer/homopolymer blend systems stabilized through strong complementary multiple hydrogen bonds. We have used Fourier transform infrared (FTIR) and 1H nuclear magnetic resonance (NMR) spectroscopy, differential scanning calorimetry (DSC), small-angle X-ray scattering (SAXS), and transmission electron microscopy (TEM) to determine the chemical structures, specific interactions, and self-assembled structures of these PS-b-PVBA/PVBT blends stabilized through strong complementary multiple hydrogen bonding interactions.
2. Experimental Section
2.1. Materials
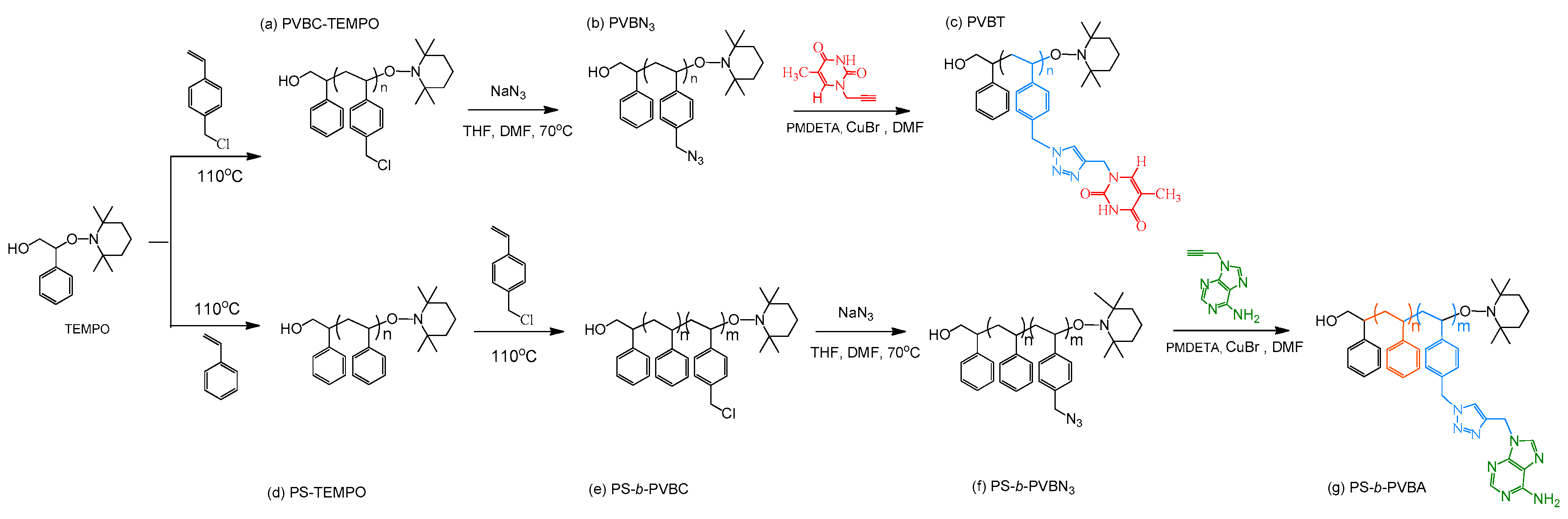
Styrene, benzoyl peroxide, vinylbenzyl chloride (VBC), pentamethyldiethylenetriamine (PMDETA), copper(I) bromide (CuBr), and sodium azide (NaN3) were purchased from Aldrich (St. Louis, MO, USA). Adenine, thymine, propargyl bromide, and K2CO3 were obtained from Showa Chemical (Tokyo, Japan). 1-Hydroxy-2-phenyl-2-(2′,2′,6′,6′-tetramethyl-1-piperidinyloxy)ethane (TEMPO-OH), PA, PT, poly(vinylbenzyl chloride) (PVBC) (Scheme 1a), poly(vinylbenzyl azide) (PVBN3) (Scheme 1b), and poly(vinylbenzyl triazolylmethyl methylthymine) (PVBT) (Scheme 1c) were prepared according to procedures described previously [21,29,56,58,60]. All solvents and other chemicals were used as received.
2.2. PS-b-PVBA
TEMPO-OH (95.4 mg) and purified styrene (60 mL) were placed in a 100 mL flask. The solution was stirred at 95 °C for 3 h and then heated at 125 °C for 4 h to provide the PS-TEMPO homopolymer (Scheme 1d), which was purified through three repeated precipitations from CH2Cl2 into MeOH. The PS-b-PVBC copolymer was synthesized through bulk polymerization of the VBC monomer (20 mL), using PS-TEMPO (200 mg) as the macroinitiator, at 125 °C for 24 h. The product was dissolved in THF and then poured into excess MeOH to obtain the PS-b-PVBC copolymer (Scheme 1e). NaN3 (1.56 g, 23.8 mmol) was added to a solution of PS-b-PVBC (0.820 g, 4.86 mmol of PVBC) in dimethylformamide (DMF) (100 mL) and the mixture was then stirred at room temperature for 24 h before being precipitated into H2O. PS-b-PVBN3 (Scheme 1f) was obtained after dissolving the precipitate in CH2Cl2 and reprecipitating from MeOH. PS-b-PVBN3 (0.84 g, 4.8 mmol), CuBr (4.8 mg, 0.32 mmol), and PA (16 mg, 0.98 mmol) were dissolved in DMF (100 mL). After three freeze/thaw/pump cycles, PMDETA (8.4 mg, 0.32 mmol) was added and the mixture was then heated at 60 °C for 24 h. After passing the solution through a neutral alumina column, the eluate was poured into excess EtOAc to give PS-b-PVBA (Scheme 1g) as a precipitate.
2.3. PS-b-PVBA/PVBT Blend Complexes
Various weight fractions of PS-b-PVBA and PVBT were dissolved in DMF. The solutions were stirred at room temperature for 24 h and then concentrated slowly at 90 °C over two days and finally in a vacuum oven for three days at 130 °C.
2.4. Characterization
1H NMR spectra were recorded using a Bruker ARX500 spectrometer (McKinley Scientific, Sparta, NJ, USA); samples were dissolved in d6-DMSO or CDCl3. Molecular weights and polydispersities were determined using gel permeation chromatography (GPC; Waters, Taipei, Taiwan, 510 HPLC), with a UV detector, a 410 differential refractometer, and DMF as the eluent (flow rate: 0.8 mL min−1). FTIR spectra were recorded using a Bruker Tensor 27 FTIR spectrophotometer under an N2 atmosphere at room temperature at a spectral resolution of 1 cm−1; the KBr disk method was employed, and the sample thickness was sufficiently thin to obey the Beer-Lambert law. The thermal behavior of the PS-b-PVBA/PVBT blend samples was measured through differential scanning calorimetry (DSC, TA Q-20, TA Instrument, New Castle, DE, USA) over the temperature range from −90 to +260 °C under an N2 atmosphere (scan rate: 20 °C min−1). SAXS analyses were performed at the National Synchrotron Radiation Research Center (NSRRC, Taiwan), using the BL17B3 beamline (λ = 1.1273 Å). The blend samples were sealed between two Kapton windows and the various temperature-resolved measurements were made on a hot-stage under an N2 atmosphere. TEM images were recorded using a JEOL 2100 microscope (Tokyo, Japan). The samples were prepared using a Leica microtome equipped with a diamond knife; the thickness was controlled at approximately 700–1000 Å. The PS-b-PVBA/PVBT blend samples were placed onto Cu grids coated with carbon films. The samples were measured after staining with I2 to reveal the PVBA and PVBT domains.
3. Results and Discussion
3.1. Synthesis of PS-b-PVBA
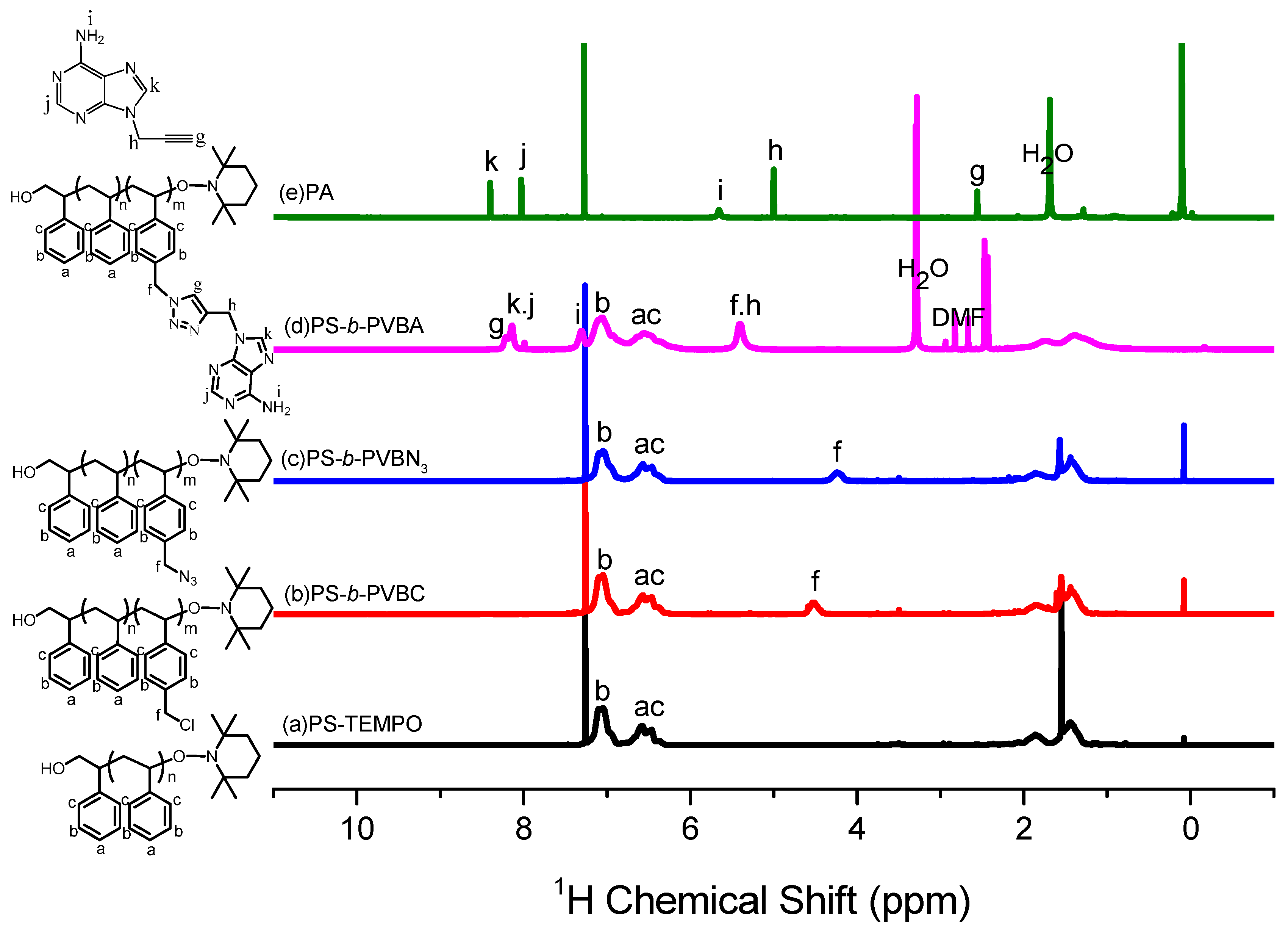
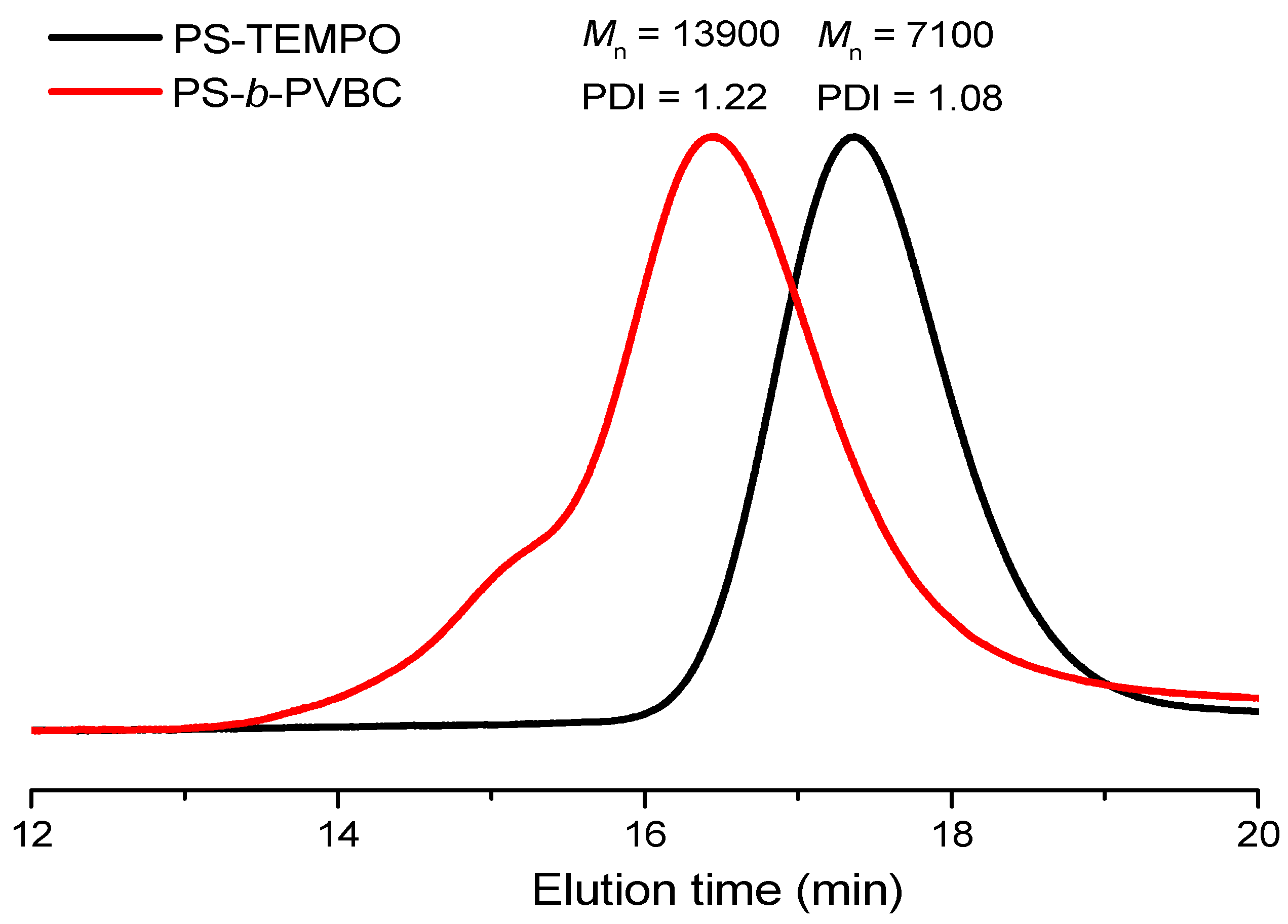
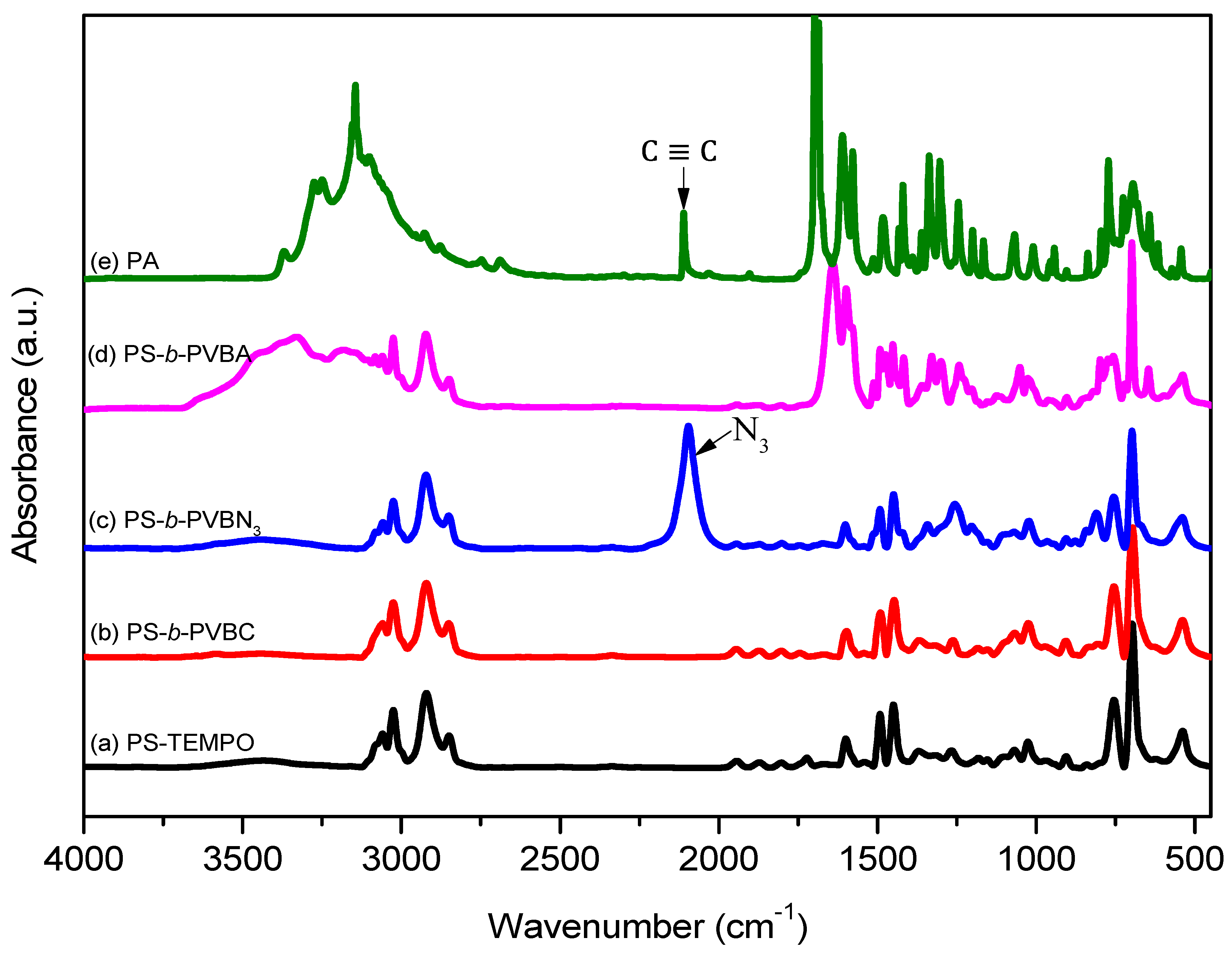
The thermal bulk polymerization of styrene and the VBC monomer at high temperature was performed using TEMPO-OH as the initiator. Figure 1a,b display 1H NMR spectra of PS-TEMPO and PS-b-PVBC, respectively. The spectrum of PS-TEMPO (Figure 1a) features signals for the aromatic protons at 6.2–6.8 and 7.2 ppm and for the tetramethyl protons near 0.9 ppm. The molecular weight of PS-TEMPO was calculated based on the integrated area ratios of the aromatic protons of PS and the tetramethyl protons of TEMPO-OH. The PS-b-PVBC copolymer was synthesized through the polymerization of VBC, using PS-TEMPO as the macroinitiator. Figure 1b reveals the presence of a signal for the CH2Cl unit of the PVBC segment near 4.51 ppm, suggesting the formation of the PS-b-PVBC diblock copolymer. The molecular weight of the PS-b-PVBC diblock copolymer was calculated based on the 1H NMR spectrum, considering the integrated area ratios of the signals for the CH2Cl and aromatic units. GPC analysis of PS-TEMPO and PS-b-PVBC confirmed the formation of the PS-b-PVBC diblock copolymer (Figure 2). The PS-b-PVBC diblock copolymer had narrow polydispersity; the absence of any signal for the PS-TEMPO macroinitiator and the shift to a lower retention time upon increasing the molecular weight of PS-b-PVBC were also consistent with the formation of PS67-b-PVBC27 diblock copolymer (Table 1). The 1H NMR spectroscopic signal of the benzylic methylene unit significantly shifted upfield from 4.51 ppm for the PVBC segment (CH2Cl) to 4.24 ppm for the PVBN3 segment (CH2N3), as displayed in Figure 1c; the disappearance of the signal at 4.51 ppm indicated the complete substitution of the chloride units to form the PS-b-PVBN3 block copolymer. The corresponding FTIR spectrum (Figure 3c) featured a signal at 2094 cm−1 for the azido units of the PVBN3 block segment, confirming the successful synthesis of the PS-b-PVBN3 diblock copolymer.
The PS-b-PVBA block copolymer was synthesized through a click reaction between PS-b-PVBN3 and PT. After this click reaction, 1H NMR spectroscopy revealed that the signal of the propargylic CH2 unit of PA (5.0 ppm, Figure 1e) had shifted downfield to 5.41 ppm for the PS-b-PVBA block copolymer (Figure 1d). Furthermore, the signal of the benzylic methylene unit of PS-b-PVBN3 (CH2N3, 4.24 ppm, Figure 1c) also significantly shifted downfield to 5.45 ppm for the PS-b-PVBA block copolymer, with a signal appearing for the CH units of the triazole groups at 8.22 ppm, indicating the successful synthesis of the PS-b-PVBA diblock copolymer. FTIR spectra provided further evidence for the completeness of the click reaction (Figure 3): the signal at 2094 cm−1, due to the azido units of the PVBN3 segment (Figure 3c), and the signal at 2110 cm−1, due to the acetylene unit of PA (Figure 3e), were both absent, while a signal for the NH groups from the PA units appeared in the range 3200–3400 cm−1 in the spectrum of PS-b-PVBA (Figure 3d). Thus, the 1H NMR and FTIR spectra confirmed the successful preparation of the PS-b-PVBA diblock copolymer; Table 1 summarizes the pertinent information regarding the PS-b-PVBA block copolymer used in this study.
3.2. Thermal Analyses of PS-b-PVBA/PVBT
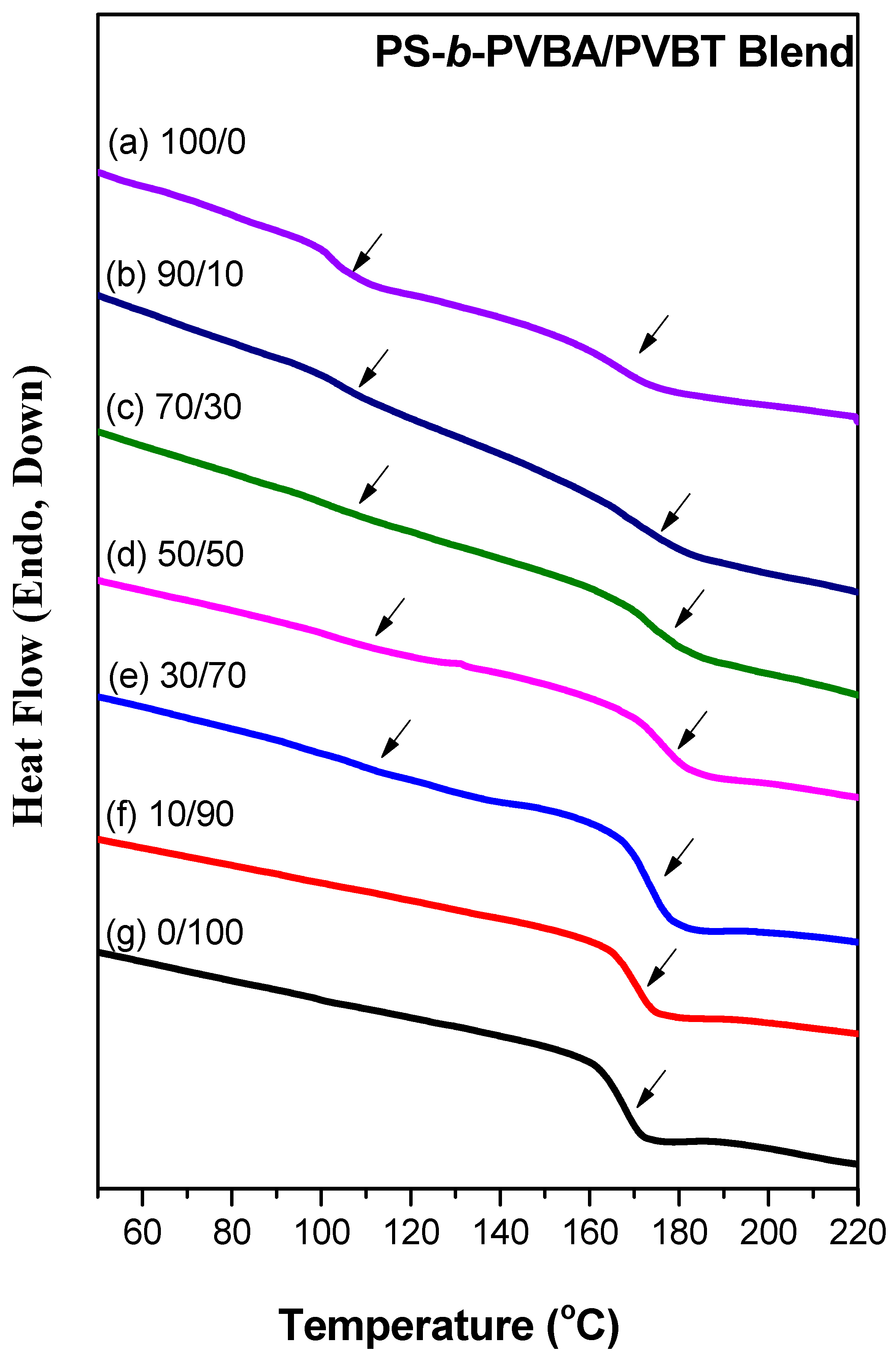
DSC is a general method for examining the miscibility behavior of polymer blends. Figure 4 presents the DSC thermograms of the PS-b-PVBA/PVBT blends of various compositions. The PS-b-PVBA block copolymer exhibited two glass transition temperatures (Tg), at 104 and 166 °C, respectively; the lower value corresponds to that of the PS block segment, while the higher value represents the PVBA block segment. The pure PVBT homopolymer exhibited a single value of Tg of 167 °C. Significantly, the values of Tg of PVBA and PVBT were greater than that of the pure PS segment because the A and T units of their side chains (in the PVBA and PVBT segments) were capable of strong self-complementary multiple hydrogen bonding. For the PS-b-PVBA/PVBT blends, we observed two values of Tg at all blend compositions, suggesting that microphase separation occurred in these blends; the higher values of Tg (167–177 °C) corresponded to the inter-complementary multiple hydrogen bonds of A···T pairs in the PVBA/PVBT miscible domain, whilst the lower values (104–112 °C) represented the PS domains. Interestingly, the values of Tg of the PVBA/PVBT miscible domain were higher than those of the individual PVBA and PVBT homopolymers for all blend compositions, due to the strong inter-complementary multiple hydrogen bonding interactions of the A···T pairs. The Kwei equation has been used widely to predict the glass transition temperatures of strongly hydrogen-bonded polymer blend systems [72]:
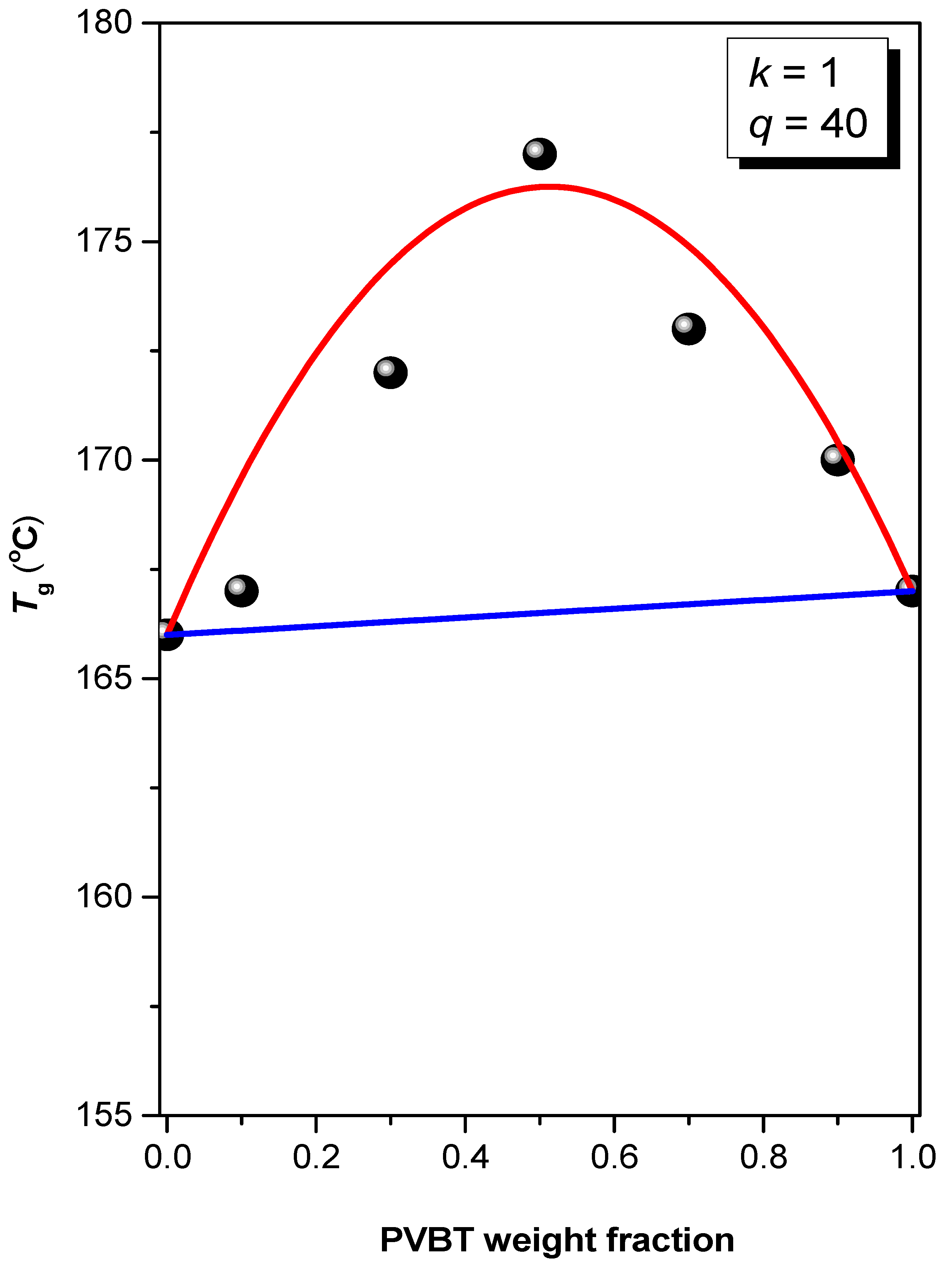
where Wi is the weight fraction of each segment; Tgi represents the corresponding value of Tg; and k and q are fitting constants. Figure 5 displays the dependence on the value of Tg of the miscible PVBA/PVBT domains on the blend composition; the highest value of Tg was obtained at the PVBA/PVBT = 50/50 blend composition, presumably because multiple hydrogen bonding induced physical crosslinking. Furthermore, values of k and q of 1 and 40, respectively, were determined through the nonlinear least-squares analyses. The parameter q is related to the strength of the multiple hydrogen bonding in the PVBA/PVBT domain, suggesting a balance between inter-association A···T multiple hydrogen bonding and the breaking of self-association (A···A, T···T) multiple hydrogen bonding. The positive value of q of 40 suggests strong inter-complementary multiple hydrogen bonding in the PVBA/PVBT domain, consistent with an inter-association equilibrium constant (Ka) for A···T binary pairs of approximately 102–103 M−1 and self-association equilibrium constants (Kb) for A···A and T···T units of approximately 2–3 M−1 [52]. In addition, the value of q for the PS-b-PVBA/PVBT diblock copolymer/homopolymer blends (q = 40) was also greater than that for the binary PVBA/PVBT homopolymer blend (q = 35) [70], presumably due to the nano-confinement effect arising from microphase separation of the diblock copolymer. As a result, the chain mobility in the PVBA/PVBT miscible domains of this PS-b-PVBA/PVBT blend was relatively restricted, resulting in a smaller free volume arising from the ordered nanostructure with compact packing and a higher value of Tg.
3.3. Hydrogen Bonding in PS-b-PVBA/PVBT Blends
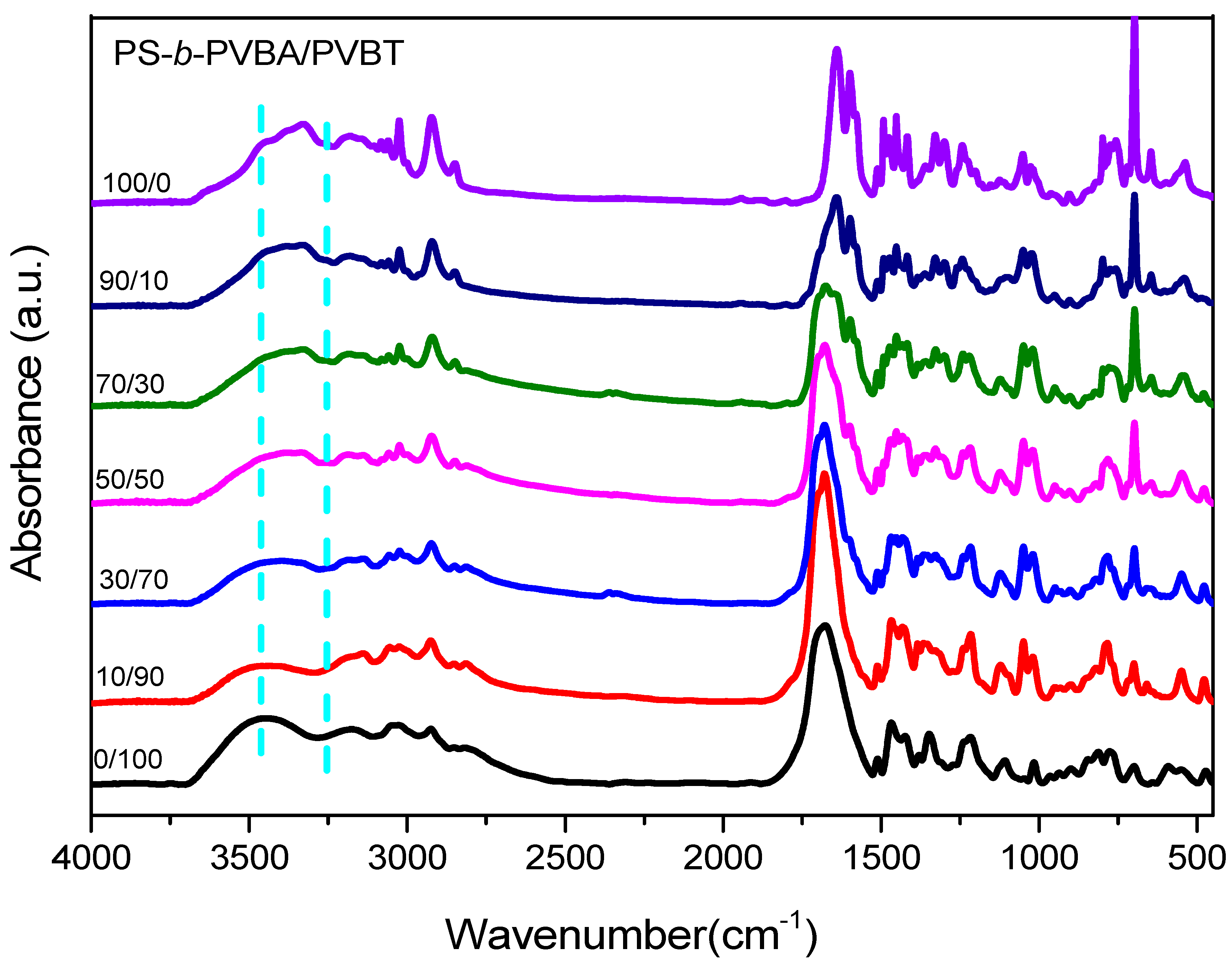
We used FTIR spectroscopy—a general technique for examining hydrogen bonding interactions in supramolecular systems—to obtain evidence for the multiple hydrogen bonding interactions between the A and T nucleobases of the PVBA and PVBT segments, respectively. Figure 6 displays the FTIR spectra of the PS-b-PVBA/PVBT blend, recorded at room temperature. The broad absorption peaks in the range from 3200 to 3600 cm−1 were due to NH and NH2 stretching vibrations. The band at 3469 cm−1 corresponded to the free NH units; this signal shifted to 3331 cm−1 for the interacting A···T units. Furthermore, the peak at 3252 cm−1 was due to the NH units of the T groups interacting with the A units in the PVBA/PVBT miscible domain [72]. Figure S1 presents expanded views of the C=O and C=N regions of the FTIR spectra of the PS-b-PVBA/PVBT blend, revealing several characteristic peaks, including those for (i) the multiple-hydrogen-bonded C4=O units of the T groups at 1662 cm−1; (ii) the free C4=O units of the T groups at 1680 cm−1, and (iii) the free C2=O units of the T groups at 1662 cm−1 for the PVBT homopolymer [69]. The characteristic signals for the A units of the PVBA block segment appeared at 1600 cm−1, due to the ring-stretching plus bonded NH2 scissoring, and at 1657 cm−1, due to the bonded NH2 scissoring plus ring stretching. The signals for the multiple hydrogen-bonded A···T pairs appeared at 1665–1655 cm−1. The fraction of free C2=O groups of the T units decreased upon increasing the concentration of PS-b-PVBA, consistent with the presence of multiple hydrogen-bonded A···T pairs in the PVBA/PVBT miscible domain. The 1H NMR spectra of model compounds, the VBA and VBT monomers in CDCl3 solution, were recorded to confirm the formation of multiple hydrogen bonds between A···T pairs (Figure S2) [72]. The addition of VBA into a solution of VBT led to a significant downfield shift of the signal for the NH protons of the T unit, from 8.9 ppm initially to 11.9 ppm at a VBA content of 70 wt%—the result of multiple hydrogen bonds in A···T pairs. In a previous study, we calculated a value of Ka for A···T pairs of 534 M−1 based on 1H NMR spectroscopic titration experiments and the Benesi–Hildebrand approach [72]. In this present study, however, our PVBA and PVBT segments only dissolved in highly polar solvents (e.g., DMSO, DMF) and, therefore, the values of Ka would presumably be lower than those in low-polarity solvents (e.g., chloroform).
3.4. Self-Assembled Supramolecular Structures of PS-b-PVBA/PVBT Blends
Figure 7 presents SAXS patterns of the pure PS67-b-PVBA27 diblock copolymer recorded at various temperatures. At room temperature, the SAXS pattern featured a broad peak at a value of q of 0.052 Å−1 (d = 12.0 nm) with no other resolvable high-order peaks, suggesting a nanostructure with a short-range order. This broad peak shifted to a lower value of q and become sharper upon increasing the temperature, forming a lamellar nanostructure of a long-range order, as evidenced by highly ordered peak ratios of 1:2:3 at 180 °C. The first peak at a value of q of 0.033 Å−1 (d = 19.0 nm) suggests a lamellar structure having a long period of 19.0 nm with a volume fraction of the PS segment of approximately 0.57 in this case, consistent with a lamellar phase diagram. As a result, this PS-b-PVBA diblock copolymer required thermal annealing at a temperature higher than the corresponding value of Tg (166 °C for the PVBA segment), such that the chain mobility would be enhanced to form the long-range order of a lamellar structure. The TEM image in Figure 8a of the pure PS-b-PVBA diblock copolymer after thermal annealing at 180 °C also indicates the presence of alternative lamellar structures and a lamellar period of approximately 20 nm, consistent with the SAXS data.
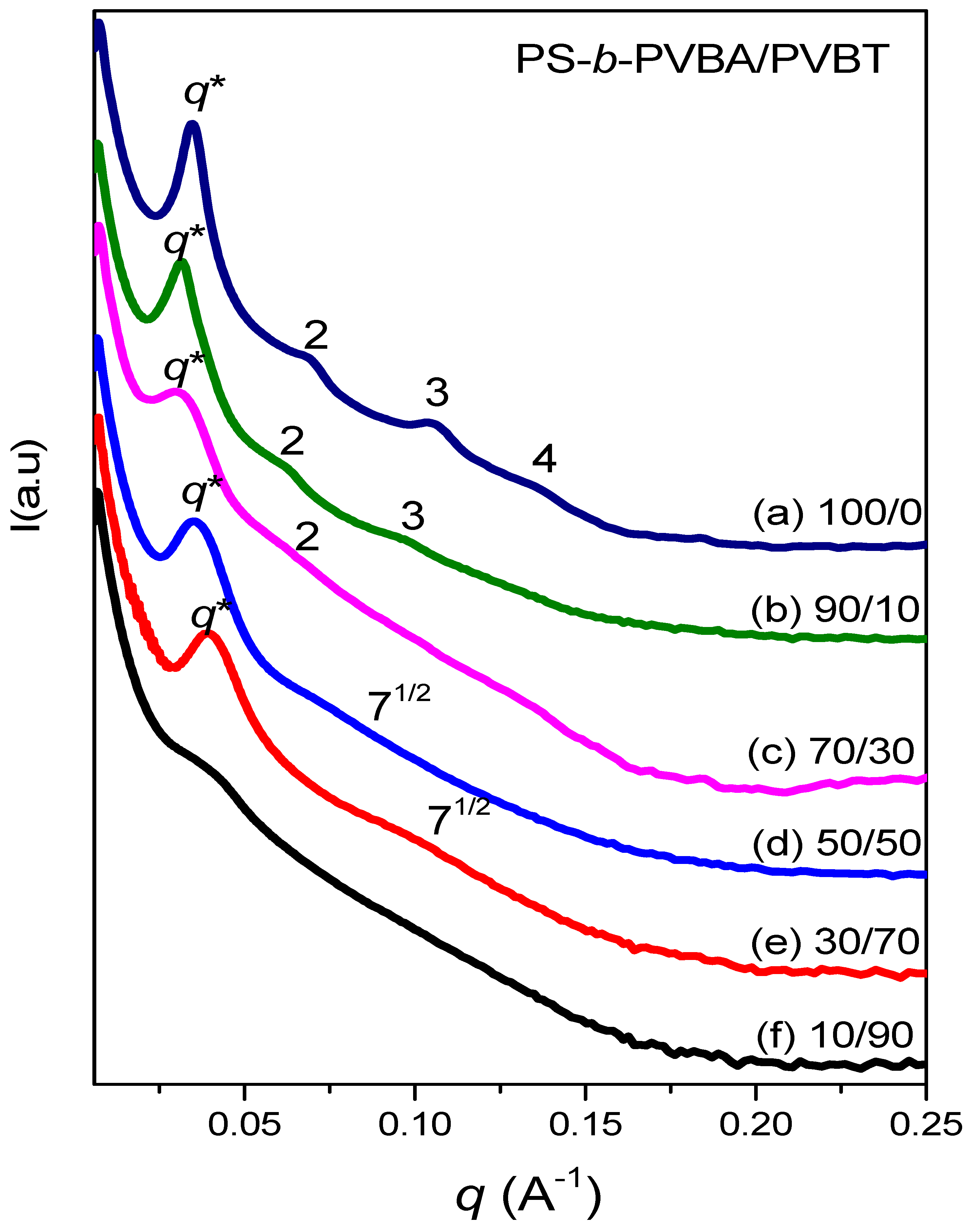
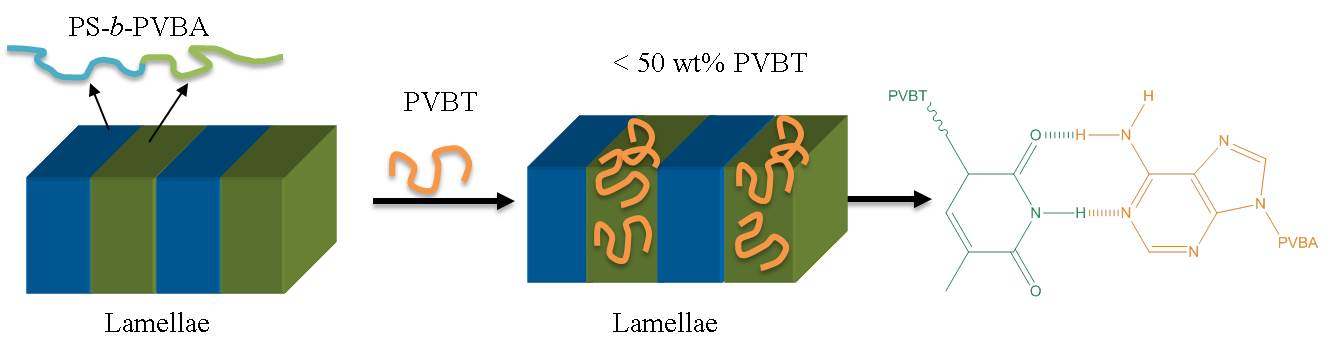
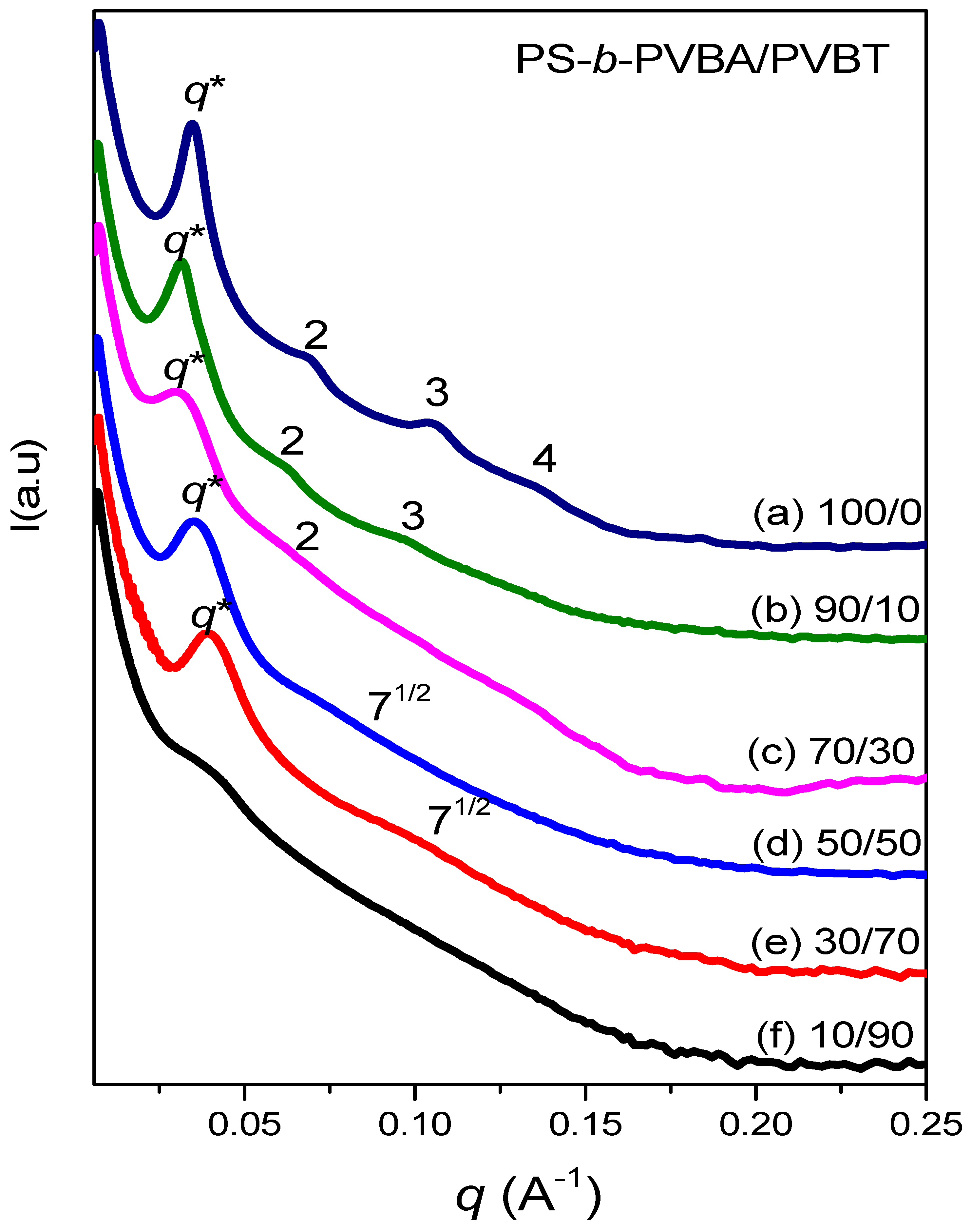
Figure 9 displays SAXS patterns of PS-b-PVBA/PVBT blends at various compositions, recorded at room temperature after thermal annealing at 180 °C. The SAXS pattern in Figure 9a indicates that the pure PS-b-PVBA diblock copolymer formed a highly ordered lamellar nanostructure, as evidenced by a scattering peak ratio of 1:2:3:4, with the long period of the lamellar structure being 18.4 nm, determined from the first peak with a value of q of 0.034 Å−1, which is slightly smaller than the value obtained in Figure 7 after heating at 180 °C. We also observed a highly ordered lamellar nanostructure, from the scattering peaks at a ratio of 1:2:3, after blending with PVBT at a concentration of 10 wt% (Figure 9b), consistent with the TEM data in Figure 8b. Furthermore, the first-order scattering peak shifted to a slightly lower value of q (0.0316 Å−1; d = 19.87 nm) at a PVBT concentration of 10 wt%, indicating an increase in the inter-lamellar spacing. Increasing the content of PVBT to 30 wt% also maintained the lamellar structure, but the peak ratio was only 1:2, indicating a transformation to a short-range-order lamellar structure, which is also consistent with the TEM data in Figure 8c. The first scattering peak again shifted to a lower value of q (0.0306 Å−1; d = 20.52 nm), suggesting an increase in the block copolymer spacing. This phenomenon could be explained by considering an increase in the size of the PVBA/PVBT domain upon increasing the content of PVBT. Increasing the concentration of PVBT to 50 wt% led to a short-range-ordered cylinder nanostructure with a peak ratio of 1:√4:√7—again consistent with the TEM data in Figure 8d In this case, however, the first scattering peak was shifted slightly to a higher value of q (0.0348 Å−1; d = 18.04 nm), suggesting a decrease in the spacing of the block copolymer. Further increasing the concentration of PVBT to 70 wt%, the system retained its short-range-ordered cylinder nanostructure with a peak ratio of 1:√4:√7, once again consistent with the TEM image in Figure 8e. This first scattering peak was shifted to a higher value of q (0.039 Å−1; d = 16.1 nm), implying a further decrease in the spacing of the block copolymer segment. The thickness of the PS domain presumably decreased in the spacing of the block copolymer segment, because of the relatively higher cross-sectional area among the PS and PVBA block segments, upon blending with PVBT, which was the result of an increase in the distances between the chemical junction points along the interface, thereby inducing the different self-assembled structures (from lamellar to cylindrical) observed in this study. When the concentration of PVBT reached 90 wt%, the SAXS pattern only featured the first broad scattering signal corresponding to disordered micelles or macrophase separation. This result is consistent with the behavior of many other block copolymer/homopolymer blend systems, observed either experimentally or theoretically, having disordered micelles or macrophase-separated structures. The addition of the homopolymer PVBT could induce a change in the self-assembly of the PS-b-PVBT diblock copolymer from a lamellar to a cylindrical structure, due to the increase in the effective interaction parameter resulting from the multiple hydrogen bonding interactions of the miscible PVBA/PVBT domains and the change in the overall volume fraction of the block copolymer segment; this phenomenon corresponds to the wet-brush behavior of diblock copolymer/homopolymer blends. The system did, however, display a disordered micelle structure at higher PVBT concentrations, presumably arising from the strength of the hydrogen bonding decreasing in the highly polar solvent (DMF), which disrupted the complementary multiple hydrogen bonding in the PVBA/PVBT domains.
4. Conclusions
We have synthesized a PS-b-PVBA diblock copolymer and a PVBT homopolymer through a combination of NMRP and click reactions. A positive deviation in the value of Tg of the miscible PVBA/PVBT domain was observed for the PS-b-PVBA/PVPT diblock copolymer/homopolymer blends, which was the result of strong multiple hydrogen bonds between their A and T units, as determined through FTIR and NMR spectroscopic analyses. The self-assembled lamellar structure of the pure PS-b-PVBA diblock copolymer transformed into cylinder structures, disordered micelles, and macrophase structures upon increasing the concentration of PVBT, as determined using SAXS and TEM; this phenomenon also arose as a result of the multiple hydrogen bonding of the binary A···T pairs.
Supplementary Materials
The following are available online at https://www.mdpi.com/2073-4352/8/8/330/s1, Figure S1: Scale expanded FTIR spectra of PS-b-PVBA/PVBT diblock copolymer/homopolymer blends with various compositions based on Figure 6, Figure S2: 1H NMR spectra displaying the chemical shift of the NH groups of VBT/VBA mixtures of various compositions.
Author Contributions
W.-C.S. and Y.-S.W. contributed to the synthesis of block copolymers; and C.-F.W. and S.-W.K. coordinated the study, interpreted the results, and wrote the paper.
Funding
This study was supported financially by the Ministry of Science and Technology, Taiwan, under contracts MOST 106-2221-E-110-067-MY3 and 105-2221-E-110-092-MY3.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Jenekhe, S.A.; Chen, X.L. Self-assembly of ordered microporous materials from rod-coil block copolymers. Science 1999, 283, 372–375. [Google Scholar] [CrossRef] [PubMed]
- Guiod, A.R.; Vandermeulen, W.M.; Klok, V.H. Advanced drug delivery devices via self-assembly of amphiphilic block copolymers. Adv. Drug Deliv. Rev. 2012, 64, 270–279. [Google Scholar]
- Lin, E.L.; Hsu, W.L.; Chiang, Y.W. Trapping structural coloration by a bioinspired gyroid microstructure in solid state. ACS Nano 2018, 12, 485–493. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Liu, J.; Li, C.; Li, Y.; Tade, M.O.; Dai, S.; Yamauchi, Y. Synthesis of nitrogen-doped mesoporous carbon spheres with extra-large pores through assembly of diblock copolymer micelles. Angew. Chem. Int. Ed. 2015, 54, 588–593. [Google Scholar] [CrossRef]
- Kuo, S.W. Hydrogen Bonding in Polymeric Materials; John Wiley & Sons: Hoboken, NJ, USA, 2018. [Google Scholar]
- Benoit, H.; Hadiioannou, G. Scattering theory and properties of block copolymers with various architectures in the homogeneous bulk state. Macromolecules 1988, 21, 1449–1464. [Google Scholar] [CrossRef]
- Shull, K.R. Mean-field theory of block copolymers: Bulk melts, surfaces, and thin films. Macromolecules 1992, 25, 2122–2133. [Google Scholar] [CrossRef]
- Heng, Z.G.; Li, R.; Chen, Y.; Zou, H.W.; Liang, M. Preparation of damping structural integration materials via the formation of nanostructure in triblock copolymer modified epoxy resins. J. Polym. Res. 2016, 23, 128. [Google Scholar] [CrossRef]
- Han, Y.K.; Pearce, E.M.; Kwei, T.K. Poly(styrene-b-vinylphenyldimethylsilanol) and its blends with homopolymers. Macromolecules 2000, 33, 1321–1329. [Google Scholar] [CrossRef]
- Jiang, M.; Xie, H. Miscibility and morphology in block copolymer/homopolymer blends. Prog. Polym. Sci. 1991, 16, 977–1026. [Google Scholar] [CrossRef]
- Zhao, J.Q.; Pearce, E.M.; Kwei, T.K. Binary and ternary blends of polystyrene-block-poly(p-hydroxystyrene). Macromolecules 1997, 30, 7119–7126. [Google Scholar] [CrossRef]
- Kosoneen, H.; Ruokolainen, J.; Nyholm, P.; Ikkala, O. Self-organized cross-linked phenolic thermosets: Thermal and dynamic mechanical properties of novolac/block copolymer blends. Polymer 2001, 42, 9481–9486. [Google Scholar] [CrossRef]
- Dobrosielska, K.; Wakao, S.; Takano, A.; Matsushita, Y. Nanophase-separated structures of AB block copolymer/C homopolymer blends with complementary hydrogen-bonding interactions. Macromolecules 2008, 41, 7695–7698. [Google Scholar] [CrossRef]
- Dobrosielska, K.; Wakao, S.; Suzuki, J.; Noda, K.; Takano, A.; Matsushita, Y. Effect of homopolymer molecular weight on nanophase-separated structures of AB block copolymer/C homopolymer blends with hydrogen-bonding interactions. Macromolecules 2009, 42, 7098–7102. [Google Scholar] [CrossRef]
- Chen, S.C.; Kuo, S.W.; Jeng, U.S.; Su, C.J.; Chang, F.C. On modulating the phase behavior of block copolymer/homopolymer blends via hydrogen bonding. Macromolecules 2010, 43, 1083–1092. [Google Scholar] [CrossRef]
- Dehghan, A.; Shi, A.C. Modeling hydrogen bonding in diblock copolymer/homopolymer blends. Macromolecules 2013, 46, 5796–5805. [Google Scholar] [CrossRef]
- Tsai, S.C.; Lin, Y.C.; Lin, E.L.; Chiang, Y.W.; Kuo, S.W. Hydrogen bonding strength effect on self-assembly supramolecular structures of diblock copolymer/homopolymer blends. Polym. Chem. 2016, 7, 2395–2409. [Google Scholar] [CrossRef]
- Hameed, N.; Guo, Q. Nanostructure and hydrogen bonding in interpolyelectrolyte complexes of poly(ε-caprolactone)-block-poly(2-vinyl pyridine) and poly(acrylic acid). Polymer 2008, 49, 5268–5275. [Google Scholar] [CrossRef]
- Hameed, N.; Liu, J.; Guo, Q. Self-assembled complexes of poly(4-vinylphenol) and poly(ε-caprolactone)-block-poly (2-vinylpyridine) via competitive hydrogen bonding. Macromolecules 2008, 41, 7596–7605. [Google Scholar] [CrossRef]
- Hameed, N.; Guo, Q. Selective hydrogen bonding and hierarchical nanostructures in poly(hydroxyether of bisphenol A)/poly(ε-caprolactone)-block-poly(2-vinyl pyridine) blends. Polymer 2008, 49, 922–933. [Google Scholar] [CrossRef]
- Chen, W.C.; Kuo, S.W.; Lu, C.H.; Jeng, U.S.; Chang, F.C. Self-assembly structures through competitive interactions of crystalline−amorphous diblock copolymer/homopolymer blends: Poly(ε-caprolactone-b-4-vinyl pyridine)/poly(vinyl phenol). Macromolecules 2009, 42, 3580–3590. [Google Scholar] [CrossRef]
- Salim, N.V.; Hanley, T.; Guo, Q. Microphase separation through competitive hydrogen bonding in double crystalline diblock copolymer/homopolymer blends. Macromolecules 2010, 43, 7695–7704. [Google Scholar] [CrossRef]
- Li, J.G.; Lin, Y.D.; Kuo, S.W. From microphase separation to self-organized mesoporous phenolic resin through competitive hydrogen bonding with double-crystalline diblock copolymers of poly(ethylene oxide-b-ε-caprolactone). Macromolecules 2011, 44, 9295–9309. [Google Scholar] [CrossRef]
- Salim, N.V.; Hameed, N.; Guo, Q. Competitive hydrogen bonding and self-assembly in poly(2-vinyl pyridine)-block-poly(methyl methacrylate)/poly(hydroxyether of bisphenol A) blends. J. Polym. Sci. Part B Polym. Phys. 2009, 47, 1894–1905. [Google Scholar] [CrossRef]
- Hameed, N.; Salim, N.V.; Guo, Q. Microphase separation through competitive hydrogen bonding in self-assembled diblock copolymer/homopolymer complexes. J. Chem. Phys. 2009, 131, 214905. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.F.; Kuo, S.W.; Huang, C.F.; Lu, J.S.; Chan, S.C.; Wang, C.F.; Chang, F.C. Hydrogen-bonding interactions mediate the phase behavior of an AB/C block copolymer/homopolymer blend comprising poly (methyl methacrylate-b-vinylpyrrolidone) and poly (vinylphenol). Macromolecules 2006, 39, 5458–5465. [Google Scholar] [CrossRef]
- Chen, W.C.; Kuo, S.W.; Jeng, U.S.; Chang, F.C. Self-assembly through competitive interactions of miscible diblock copolymer/homopolymer blends: Poly(vinylphenol-b-methyl methacrylate)/poly (vinylpyrrolidone) blend. Macromolecules 2008, 41, 1401–1410. [Google Scholar] [CrossRef]
- Zhou, J.; Shi, A.C. Microphase separation induced by differential interactions in diblock copolymer/homopolymer blends. J. Chem. Phys. 2009, 130, 234904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, I.; Kuo, S.W.; Chang, F.C. Self-Assembly structures through competitive interactions of miscible crystalline–amorphous diblock copolymer/homopolymer blends. Polymer 2009, 50, 5276–5287. [Google Scholar] [CrossRef]
- Matsushita, Y. Creation of hierarchically ordered nanophase structures in block polymers having various competing interactions. Macromolecules 2007, 40, 771–776. [Google Scholar] [CrossRef]
- Miyase, H.; Asai, Y.; Takano, A.; Matsushita, Y. Kaleidoscopic tiling patterns with large unit cells from ABC star-shaped terpolymer/diblock copolymer blends with hydrogen bonding interaction. Macromolecules 2017, 50, 979–986. [Google Scholar] [CrossRef]
- Asari, T.; Matsuo, S.; Takano, A.; Matsushita, Y. Three-phase hierarchical structures from AB/CD diblock copolymer blends with complemental hydrogen bonding interaction. Macromolecules 2005, 38, 8811–8815. [Google Scholar] [CrossRef]
- Asari, T.; Arai, S.; Takano, A.; Matsushita, Y. Archimedean tiling structures from ABA/CD block copolymer blends having intermolecular association with hydrogen bonding. Macromolecules 2006, 39, 2232–2237. [Google Scholar] [CrossRef]
- Kuo, S.W. Hydrogen bond-mediated self-assembly and supramolecular structures of diblock copolymer mixtures. Polym. Inter. 2009, 58, 455–464. [Google Scholar] [CrossRef]
- Chen, W.C.; Kuo, S.W.; Chang, F.C. Self-assembly of an A–B diblock copolymer blended with a C homopolymer and a C–D diblock copolymer through hydrogen bonding interaction. Polymer 2010, 51, 4176–4184. [Google Scholar] [CrossRef]
- Kuo, S.W.; Lin, C.L.; Chang, F.C. The Study of Hydrogen Bonding and Miscibility in Poly(vinyl pyridines) with Phenolic Resin. Polymer 2002, 43, 3943–3949. [Google Scholar] [CrossRef]
- Kuo, S.W.; Tung, P.H.; Chang, F.C. Syntheses and the Study of Strong Hydrogen-Bonded Poly(vinyl phenol-b-vinyl pyridine) Diblock Copolymer through Anionic Polymerization. Macromolecules 2006, 39, 9388–9395. [Google Scholar] [CrossRef]
- Wang, M.; Jiang, M.; Ning, F.L.; Chen, D.Y.; Liu, S.Y.; Duan, H.W. Block-copolymer-free strategy for preparing micelles and hollow spheres: Self-assembly of poly(4-vinylpyridine) and modified polystyrene. Macromolecules 2002, 35, 5980–5989. [Google Scholar] [CrossRef]
- IIhan, F.; Gray, M.; Rotello, V.M. Reversible side chain modification through noncovalent interactions. “Plug and play” polymers. Macromolecules 2001, 34, 2597–2601. [Google Scholar]
- Sherrington, D.C.; Taskinen, K.A. Self-assembly in synthetic macromolecular systems via multiple hydrogen bonding interactions. Chem. Soc. Rev. 2001, 30, 83–93. [Google Scholar] [CrossRef]
- Feldman, K.E.; Kade, M.J.; de Greef, T.F.A.; Meijer, E.W.; Kramer, E.J.; Hawker, C.J. Polymers with multiple hydrogen-bonded end groups and their blends. Macromolecules 2008, 41, 4694. [Google Scholar] [CrossRef]
- Sivakava, S.; Wu, J.; Campo, C.J.; Mather, P.T.; Rowan, S.J. Liquid-crystalline supramolecular polymers formed through complementary nucleobase-pair interactions. Chem. A Eur. J. 2005, 12, 446–456. [Google Scholar] [CrossRef] [PubMed]
- Altintas, O.; Schulze-Suenninghausen, D.; Luy, B.; Barner-Kowollik, C. Facile preparation of supramolecular H-shaped (Ter)polymers via multiple hydrogen bonding. ACS Macro Lett. 2013, 2, 211–216. [Google Scholar] [CrossRef]
- Park, T.; Zimmerman, S.C. Formation of a miscible supramolecular polymer blend through self-assembly mediated by a quadruply hydrogen-bonded heterocomplex. J. Am. Chem. Soc. 2006, 128, 11582–11590. [Google Scholar] [CrossRef] [PubMed]
- Mather, B.D.; Baker, M.B.; Beyer, F.L.; Berg, M.A.G.; Green, M.D.; Long, T.E. Supramolecular triblock copolymers containing complementary nucleobase molecular recognition. Macromolecules 2007, 40, 6834–6845. [Google Scholar] [CrossRef]
- Nair, K.P.; Breedveld, V.; Weck, M. Complementary hydrogen-bonded thermoreversible polymer networks with tunable properties. Macromolecules 2008, 41, 3429–3438. [Google Scholar] [CrossRef]
- Mather, B.D.; Lizotte, J.R.; Long, T.E. Synthesis of chain end functionalized multiple hydrogen bonded polystyrenes and poly(alkyl acrylates) using controlled radical polymerization. Macromolecules 2004, 37, 9331–9337. [Google Scholar] [CrossRef]
- Satoh, K.; Yokoyama, T.; Shimasaki, T.; Teramoto, N.; Shibata, M. Bio-based epoxy networks incorporating covalent and melamine cyanurate-type multiple hydrogen-bonding crosslinkages. J. Polym. Res. 2016, 23, 176. [Google Scholar] [CrossRef]
- Park, T.; Zimmerman, S.C. Interplay of fidelity, binding strength, and structure in supramolecular polymers. J. Am. Chem. Soc. 2006, 128, 14236–14237. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.R.; Walter, T. Nucleic acid models. Prog. Polym. Sci. 1996, 21, 209–253. [Google Scholar] [CrossRef]
- Watson, J.D. The Secret of Life; Knopf: New York, NY, USA, 2003. [Google Scholar]
- Kuo, S.W.; Cheng, R.S. DNA-Like interactions enhance the miscibility of supramolecular polymer blends. Polymer 2009, 50, 177–188. [Google Scholar] [CrossRef]
- Kuo, S.W.; Tsai, H.T. Complementary multiple hydrogen-bonding interactions increase the glass transition temperatures to PMMA copolymer mixtures. Macromolecules 2009, 42, 4701–4711. [Google Scholar] [CrossRef]
- Kuo, S.W.; Hsu, C.H. Miscibility enhancement of supramolecular polymer blends through complementary multiple hydrogen bonding interactions. Polym. Inter. 2010, 59, 998–1005. [Google Scholar] [CrossRef]
- Hu, W.H.; Huang, K.W.; Kuo, S.W. Heteronucleobase-functionalized benzoxazine: Synthesis, thermal property and multiple hydrogen bonding interactions. Polym. Chem. 2012, 3, 1546–1554. [Google Scholar] [CrossRef]
- Wu, Y.C.; Kuo, S.W. Complementary multiple hydrogen bonding interactions mediate the self-assembly of supramolecular structures from thymine-containing block copolymers and hexadecyladenine. Polym. Chem. 2012, 3, 3100–3111. [Google Scholar] [CrossRef]
- Wang, J.H.; Altukhov, O.; Cheng, C.C.; Chang, F.C.; Kuo, S.W. Supramolecular structures of uracil-functionalized PEG with multi-diamidopyridine POSS through complementary hydrogen bonding interactions. Soft Matter 2013, 9, 5196–5206. [Google Scholar] [CrossRef]
- Huang, K.W.; Wu, Y.R.; Kuo, S.W. From random coil to helical structure induced by carbon nanotube through supramolecular interactions. Macromol. Rapid Commun. 2013, 34, 1530–1536. [Google Scholar] [CrossRef] [PubMed]
- Shih, H.K.; Chen, Y.H.; Chu, Y.L.; Cheng, C.C.; Chang, F.C.; Zhu, C.Y.; Kuo, S.W. Photo-crosslinking of pendent uracil units provides supramolecular hole injection/transport conducting polymers for highly efficient light-emitting diodes. Polymers 2015, 7, 804–818. [Google Scholar] [CrossRef]
- Wu, Y.C.; Bastakoti, B.P.; Pramanik, M.; Yamauchi, Y.; Kuo, S.W. Multiple hydrogen bonding mediates the formation of multicompartment micelles and hierarchical self-assembled structures from pseudo A-block-(B-graft-C) terpolymers. Polym. Chem. 2015, 6, 5110–5124. [Google Scholar] [CrossRef]
- Huang, C.W.; Wu, P.W.; Su, W.H.; Zhu, C.Y.; Kuo, S.W. Stimuli-Responsive Supramolecular Materials: Photo-Tunable and Molecular Recognition Behavior. Polym. Chem. 2016, 7, 795–806. [Google Scholar] [CrossRef]
- Huang, C.W.; Mohamed, M.G.; Zhu, C.Y.; Kuo, S.W. Functional supramolecular polypeptides involving π-π stacking and strong hydrogen-bonding interactions: A conformation study toward carbon nanotubes (CNTs) dispersion. Macromolecules 2016, 49, 5374–5385. [Google Scholar] [CrossRef]
- Huang, C.W.; Ji, W.Y.; Kuo, S.W. Water-soluble fluorescent nanoparticles from supramolecular amphiphiles featuring heterocomplementary multiple hydrogen bondings. Macromolecules 2017, 50, 7091–7101. [Google Scholar] [CrossRef]
- Huang, C.W.; Ji, W.Y.; Kuo, S.W. Stimuli-responsive supramolecular conjugated polymer with phototunable surface relief grating. Polym. Chem. 2018, 9, 2813–2820. [Google Scholar] [CrossRef]
- Jain, A.; George, S.J. New directions in supramolecular electronics. Mater. Today 2015, 18, 206–214. [Google Scholar] [CrossRef]
- Aliprandi, A.; Mauro, M.; Cola, L.D. Controlling and imaging biomimetic self-assembly. Nat. Chem. 2016, 8, 10–15. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Xi, W. Nucleobase-Containing Polymers: Structure, Synthesis, and Applications. Polymers 2017, 9, 666. [Google Scholar] [CrossRef]
- Yamauchi, K.; Kanomata, A.; Inoue, T.; Long, T.E. Thermoreversible polyesters consisting of multiple hydrogen bonding (MHB). Macromolecules 2004, 37, 3519–3522. [Google Scholar] [CrossRef]
- Garcia, M.; Beecham, M.P.; Kempe, K.; Haddleton, D.M.; Khan, A.; Marsh, A. Water soluble triblock and pentablock poly(methacryloyl nucleosides) from copper-mediated living radical polymerisation using PEG macroinitiators. Eur. Polym. J. 2015, 66, 444–451. [Google Scholar] [CrossRef]
- Cheng, C.C.; Huang, C.F.; Yen, Y.C.; Chang, F.C. A “plug and play” polymer through biocomplementary hydrogen bonding. J. Polym. Sci. A Polym. Chem. 2008, 46, 6416–6424. [Google Scholar] [CrossRef]
- Wu, Y.S.; Wu, Y.C.; Kuo, S.W. Thymine- and Adenine-Functionalized Polystyrene Form Self-Assembled Structures through Multiple Complementary Hydrogen Bonds. Polymers 2014, 6, 1827–1845. [Google Scholar] [CrossRef] [Green Version]
- Kwei, T. The effect of hydrogen bonding on the glass transition temperatures of polymer mixtures. J. Polym. Sci. Polym. Lett. Ed. 1984, 22, 307–313. [Google Scholar] [CrossRef]
Scheme 1.
Synthesis of (c) PVBT from (a) PVBC-TEMPO and (b) PVBN3 and of (g) PS-b-PVBA from (d) PS-TEMPO; (e) PS-b-PVBC and (f) PS-b-PVBN3.
Scheme 1.
Synthesis of (c) PVBT from (a) PVBC-TEMPO and (b) PVBN3 and of (g) PS-b-PVBA from (d) PS-TEMPO; (e) PS-b-PVBC and (f) PS-b-PVBN3.
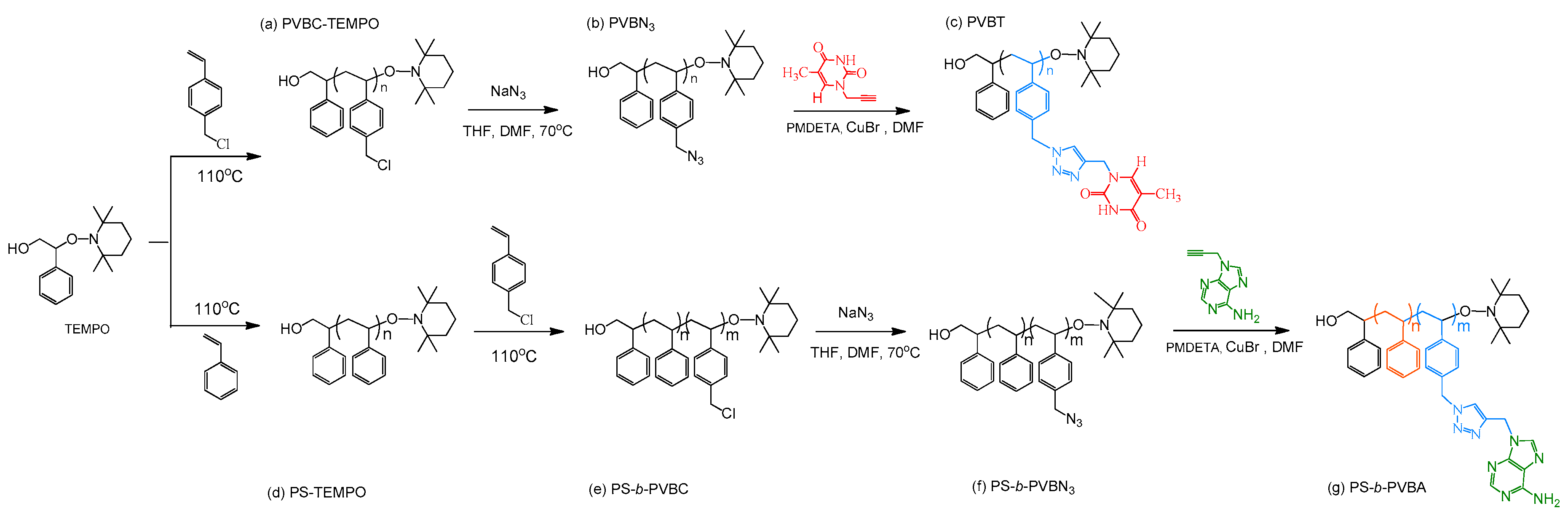
Figure 1.
1H NMR spectra of (a) PS-TEMPO; (b) PS-b-PVBC; (c) PS-b-PVBN3; (d) PS-b-PVBA; and (e) PA.
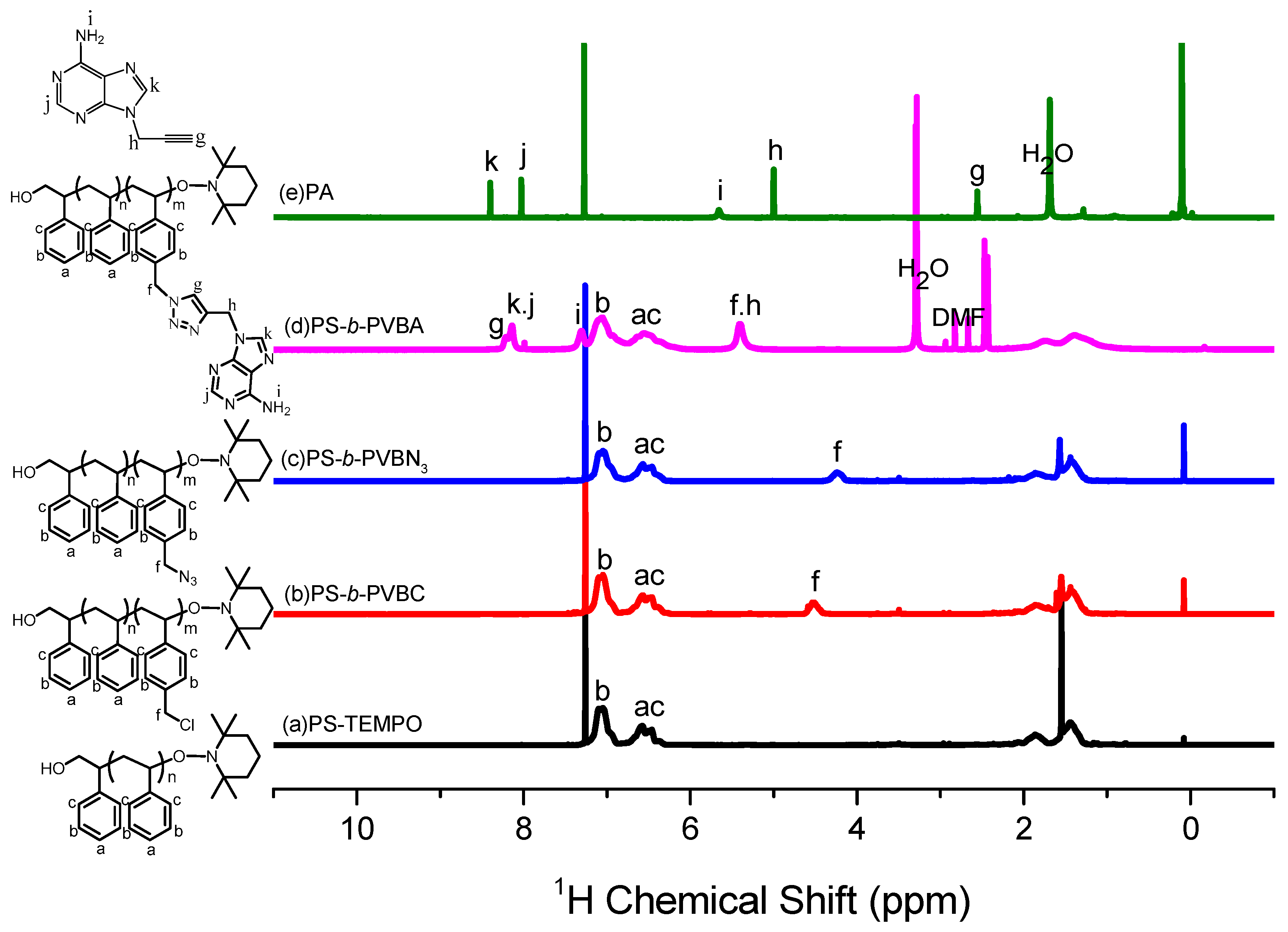
Figure 2.
GPC analyses of PS-TEMPO and the PS-b-PVBC diblock copolymer.
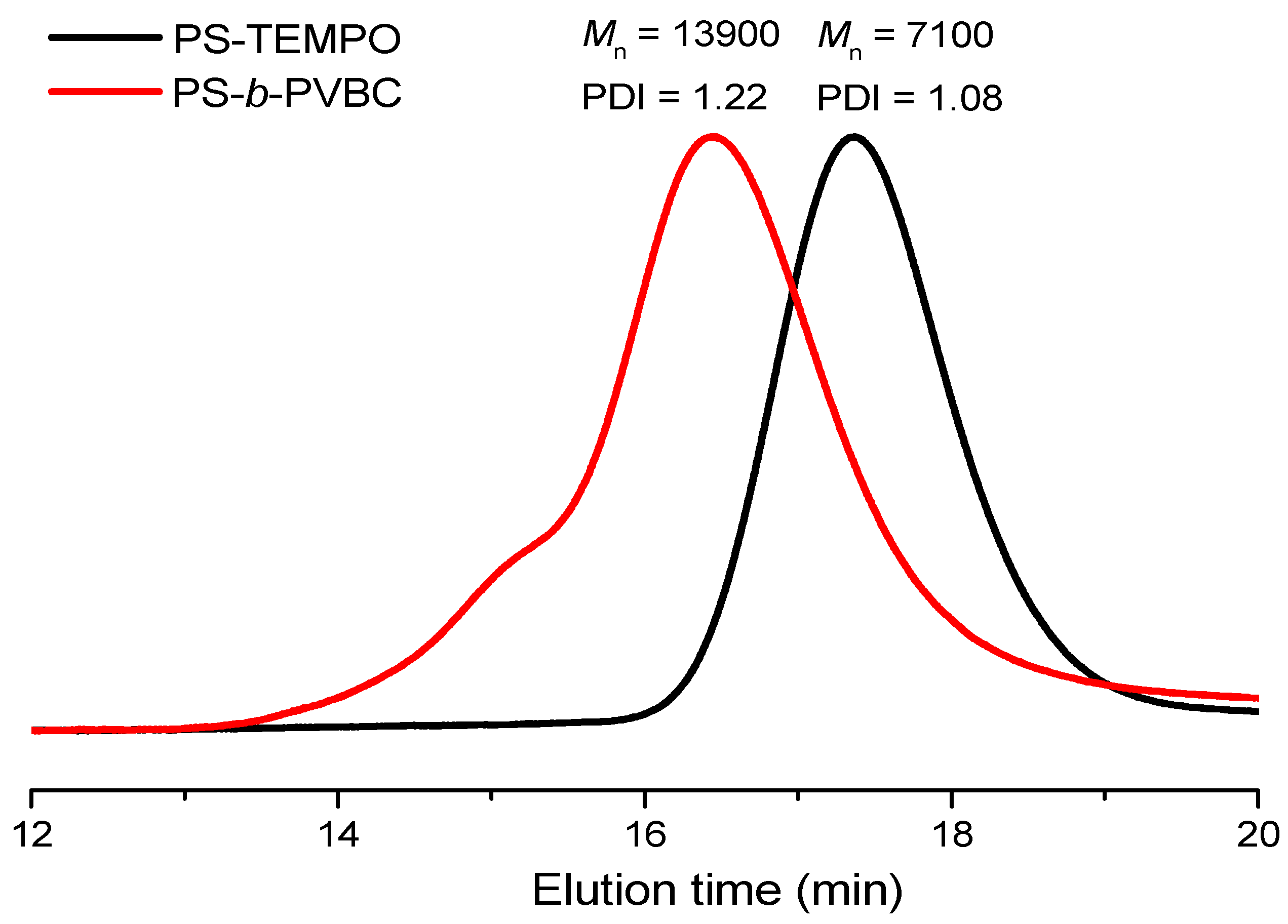
Figure 3.
FTIR spectra of (a) PS-TEMPO; (b) PS-b-PVBC; (c) PS-b-PVBN3; (d) PS-b-PVBA and (e) PA.
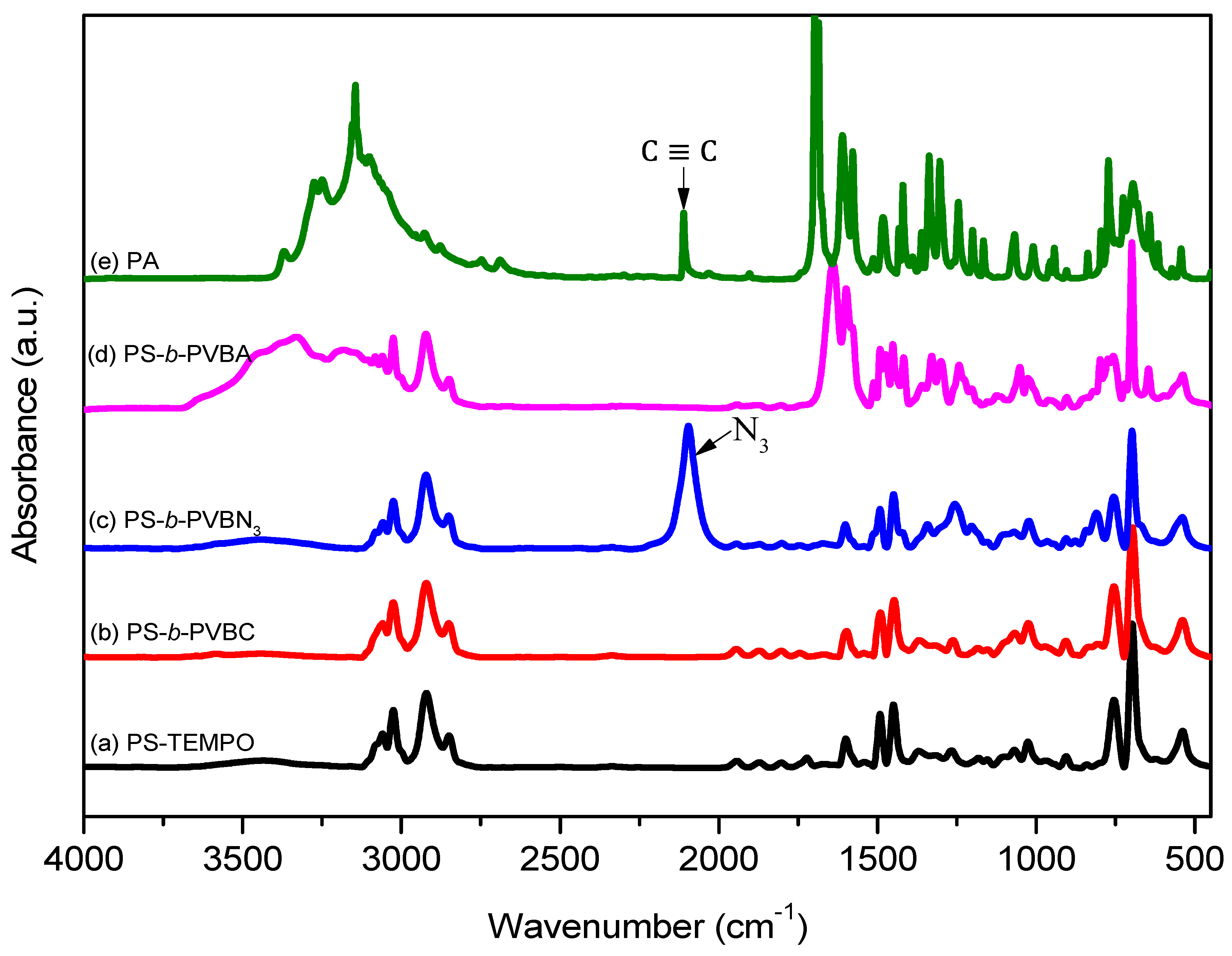
Figure 4.
DSC thermograms of PS-b-PVBA/PVBT diblock copolymer/homopolymer blends of various compositions.
Figure 4.
DSC thermograms of PS-b-PVBA/PVBT diblock copolymer/homopolymer blends of various compositions.
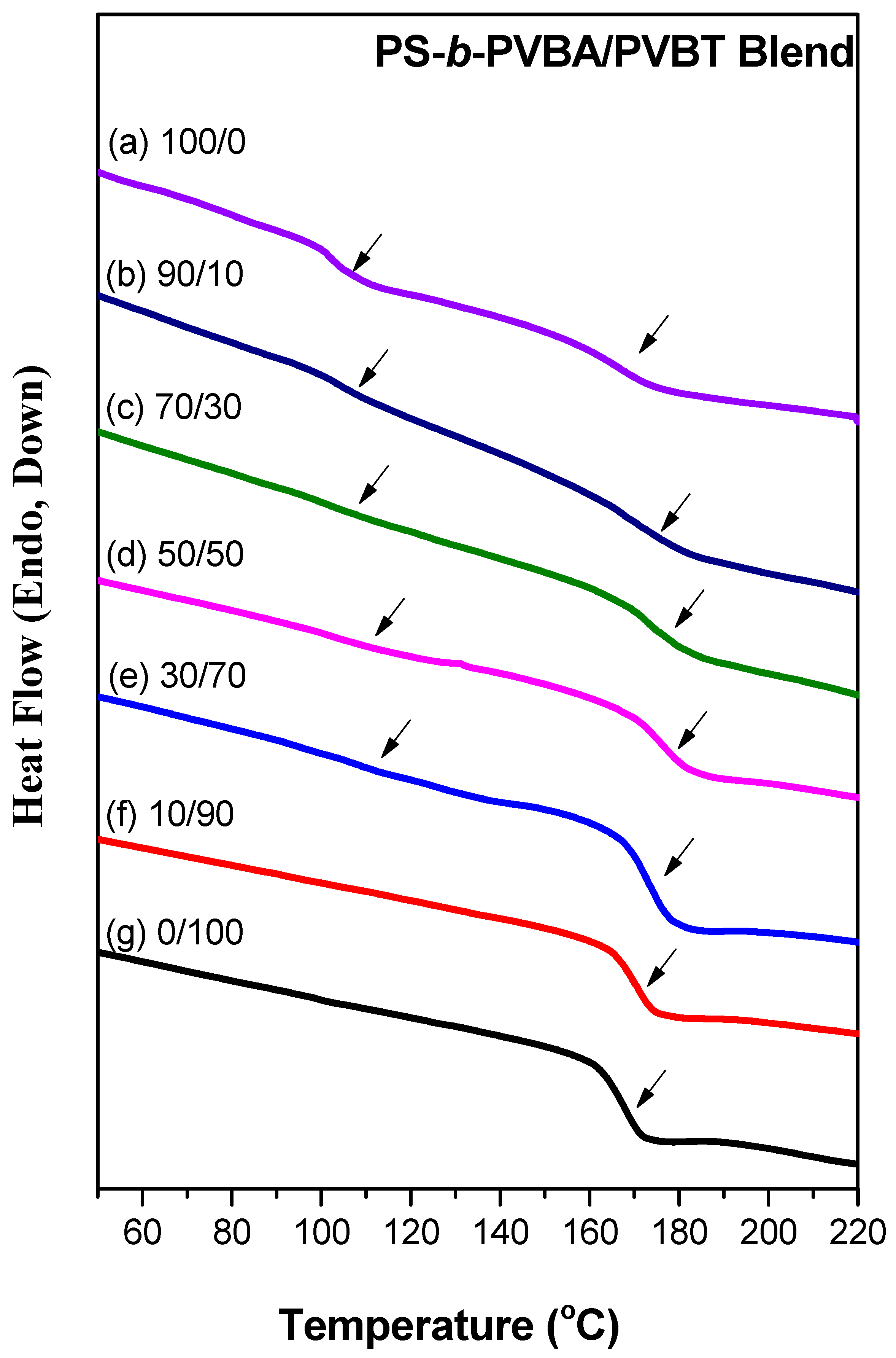
Figure 5.
Glass transition temperature composition curve for PS-b-PVBA/PVBT blends, based on the Kwei equation.
Figure 5.
Glass transition temperature composition curve for PS-b-PVBA/PVBT blends, based on the Kwei equation.
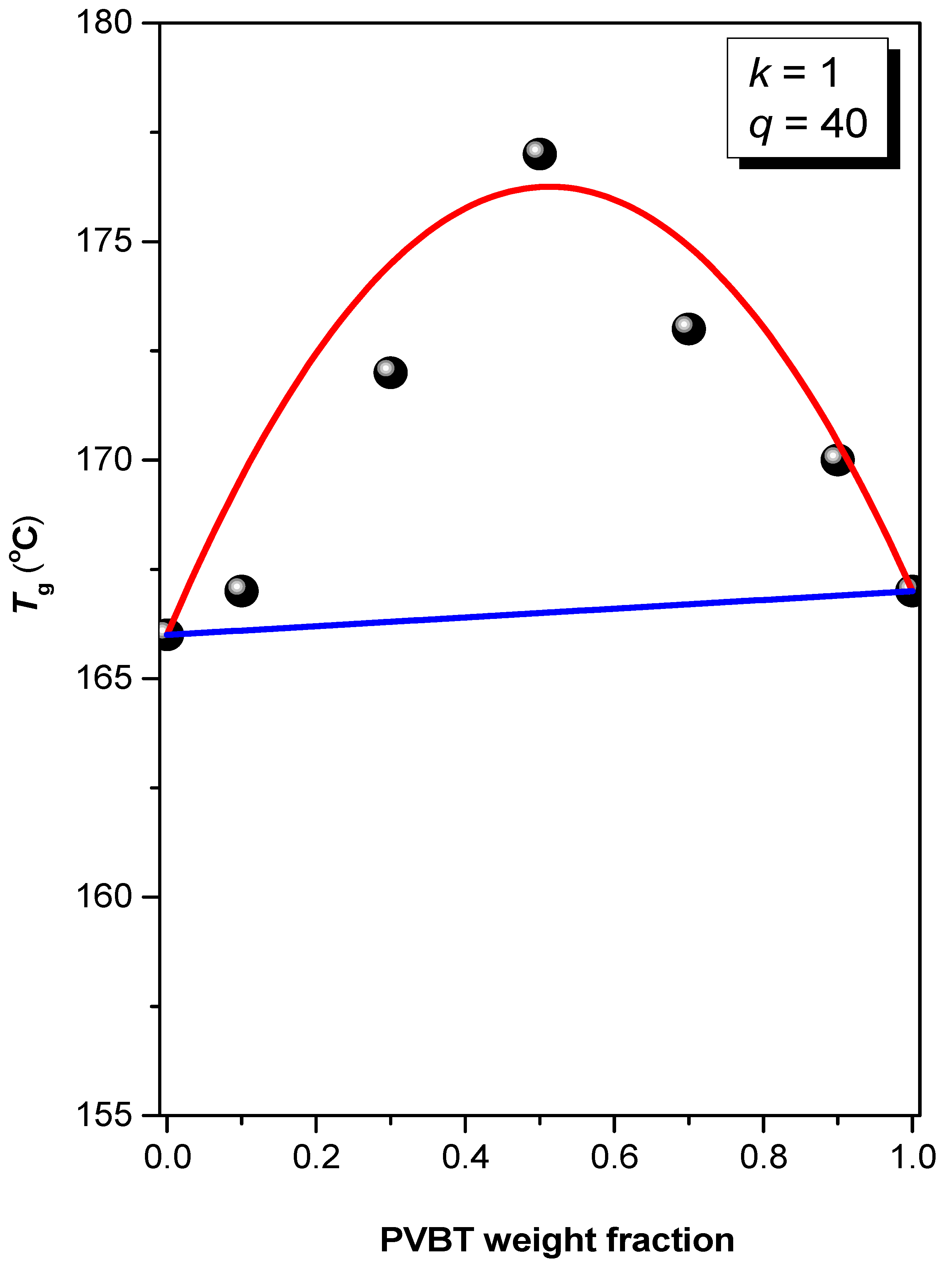
Figure 6.
FTIR spectra of PS-b-PVBA/PVBT diblock copolymer/homopolymer blends of various compositions.
Figure 6.
FTIR spectra of PS-b-PVBA/PVBT diblock copolymer/homopolymer blends of various compositions.
Figure 7.
SAXS analyses of the pure PS-b-PVBA diblock copolymer, recorded at various temperatures.
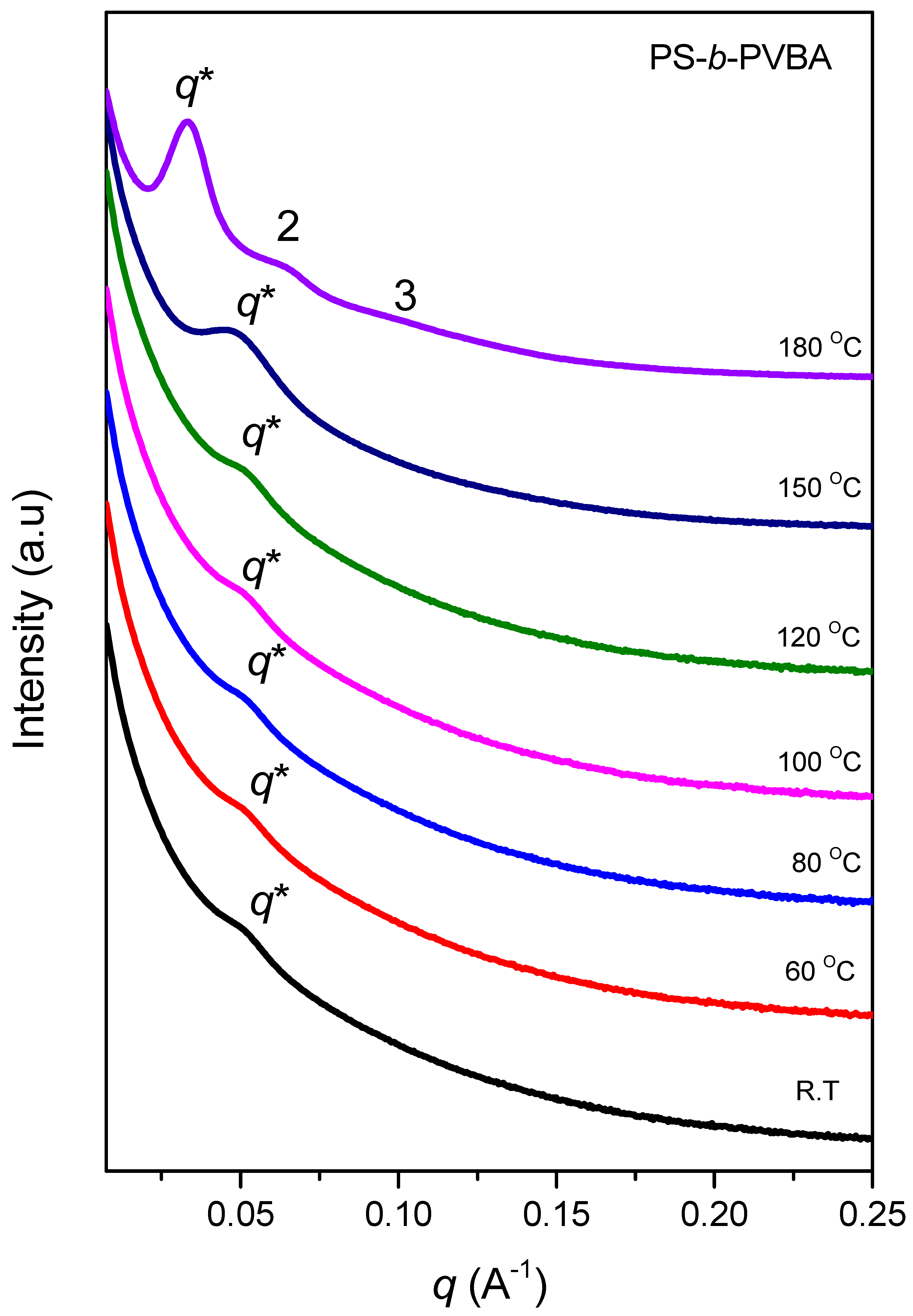
Figure 8.
TEM images of PS-b-PVBA/PVBT blends, after thermal annealing at 180 °C, at compositions of (a) 100/0; (b) 90/10; (c) 70/30; (d) 50/50 and (e) 30/70.
Figure 8.
TEM images of PS-b-PVBA/PVBT blends, after thermal annealing at 180 °C, at compositions of (a) 100/0; (b) 90/10; (c) 70/30; (d) 50/50 and (e) 30/70.

Figure 9.
SAXS analyses of PS-b-PVBA/PVBT blends, after thermal annealing at 180 °C, at compositions of (a) 100/0; (b) 90/10; (c) 70/30; (d) 50/50; (e) 30/70 and (f) 10/90.
Figure 9.
SAXS analyses of PS-b-PVBA/PVBT blends, after thermal annealing at 180 °C, at compositions of (a) 100/0; (b) 90/10; (c) 70/30; (d) 50/50; (e) 30/70 and (f) 10/90.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Characteristics of the diblock copolymer PS-b-PVBA prepared in this study.
| Sample | PS (Mn) a | PVBC (Mn) a | PVBA (Mn) a | PS Tg (°C) b | PVBA Tg (°C) b | PDI c |
|---|---|---|---|---|---|---|
| PS67-b-PVBA27 | 6900 | 4100 | 8900 | 104 | 166 | 1.22 |
a Determined from 1H NMR spectrum; b Obtained through DSC analysis at 20 °C min−1; c Measured through GPC with PS-standard calibration.
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Su, W.-C.; Wu, Y.-S.; Wang, C.-F.; Kuo, S.-W. Self-Assembled Structures of Diblock Copolymer/Homopolymer Blends through Multiple Complementary Hydrogen Bonds. Crystals 2018, 8, 330. https://doi.org/10.3390/cryst8080330
AMA Style
Su W-C, Wu Y-S, Wang C-F, Kuo S-W. Self-Assembled Structures of Diblock Copolymer/Homopolymer Blends through Multiple Complementary Hydrogen Bonds. Crystals. 2018; 8(8):330. https://doi.org/10.3390/cryst8080330
Chicago/Turabian StyleSu, Wei-Chen, Yu-Shian Wu, Chih-Feng Wang, and Shiao-Wei Kuo. 2018. "Self-Assembled Structures of Diblock Copolymer/Homopolymer Blends through Multiple Complementary Hydrogen Bonds" Crystals 8, no. 8: 330. https://doi.org/10.3390/cryst8080330
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.