Resolution Dependence of an Ab Initio Phasing Method in Protein X-ray Crystallography




Abstract
:1. Introduction
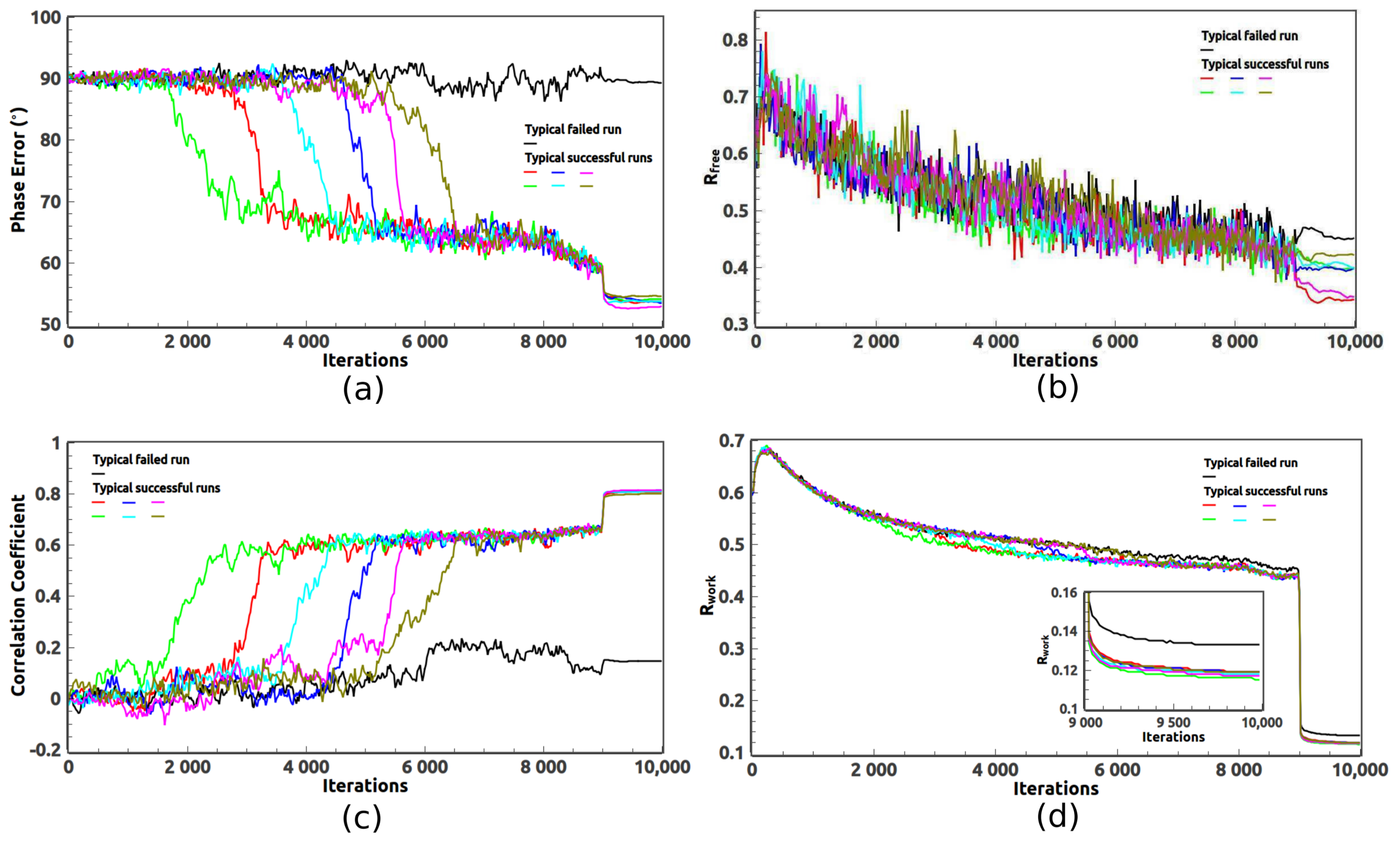
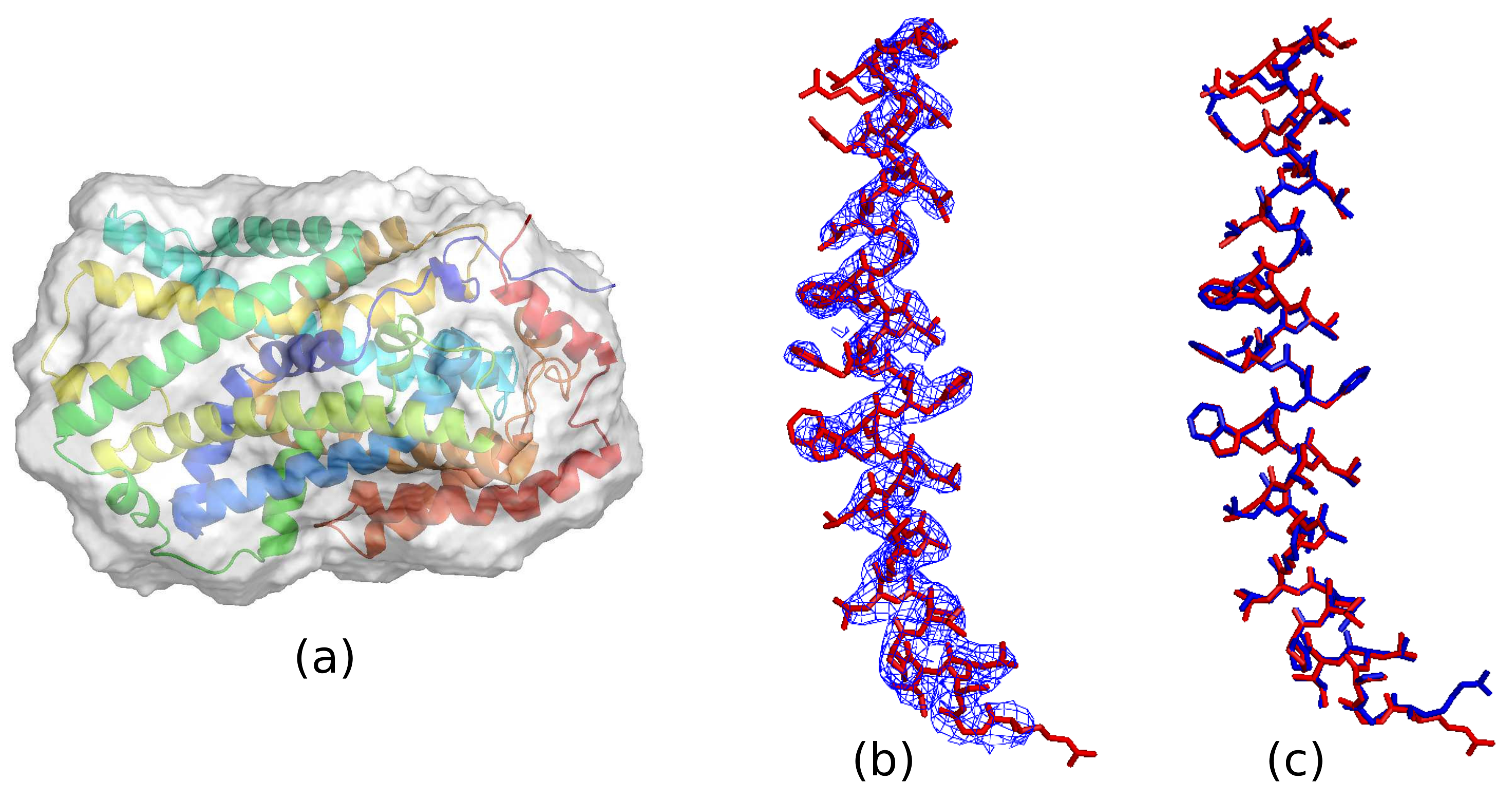
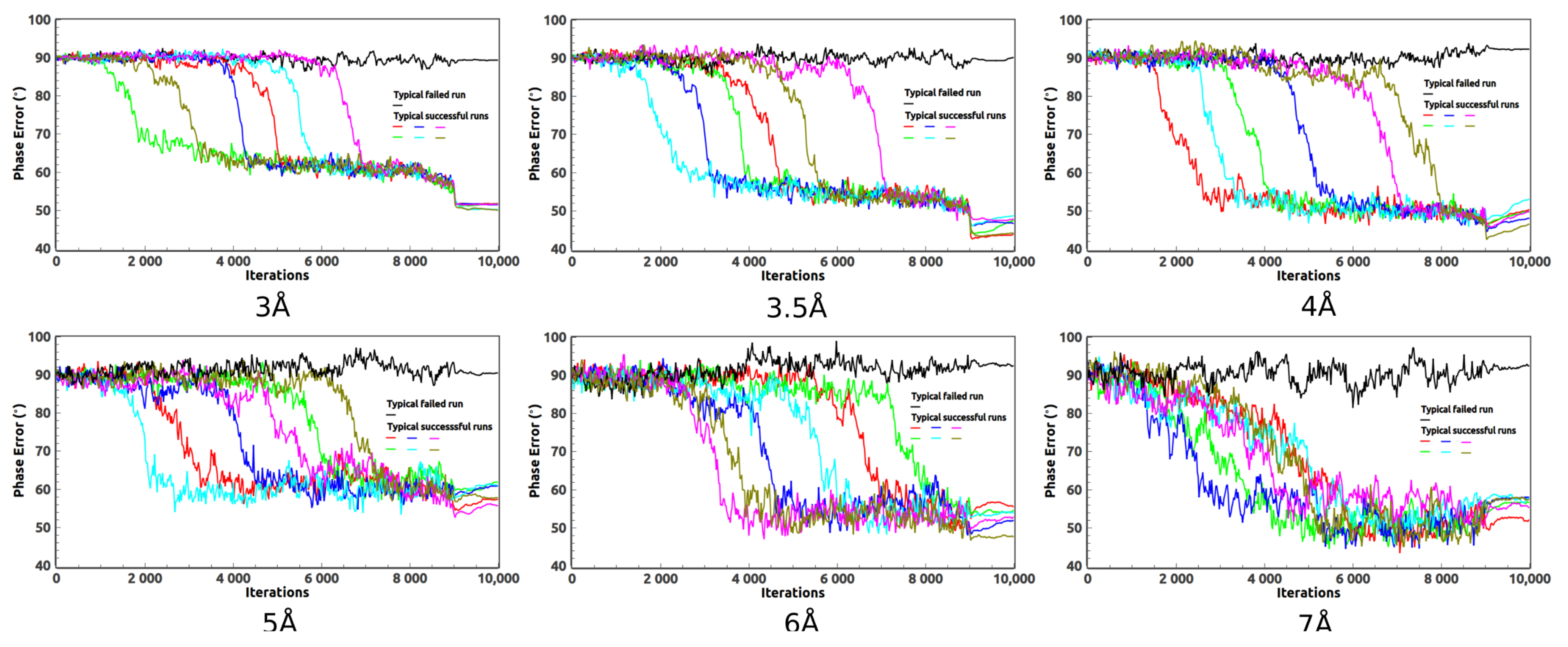
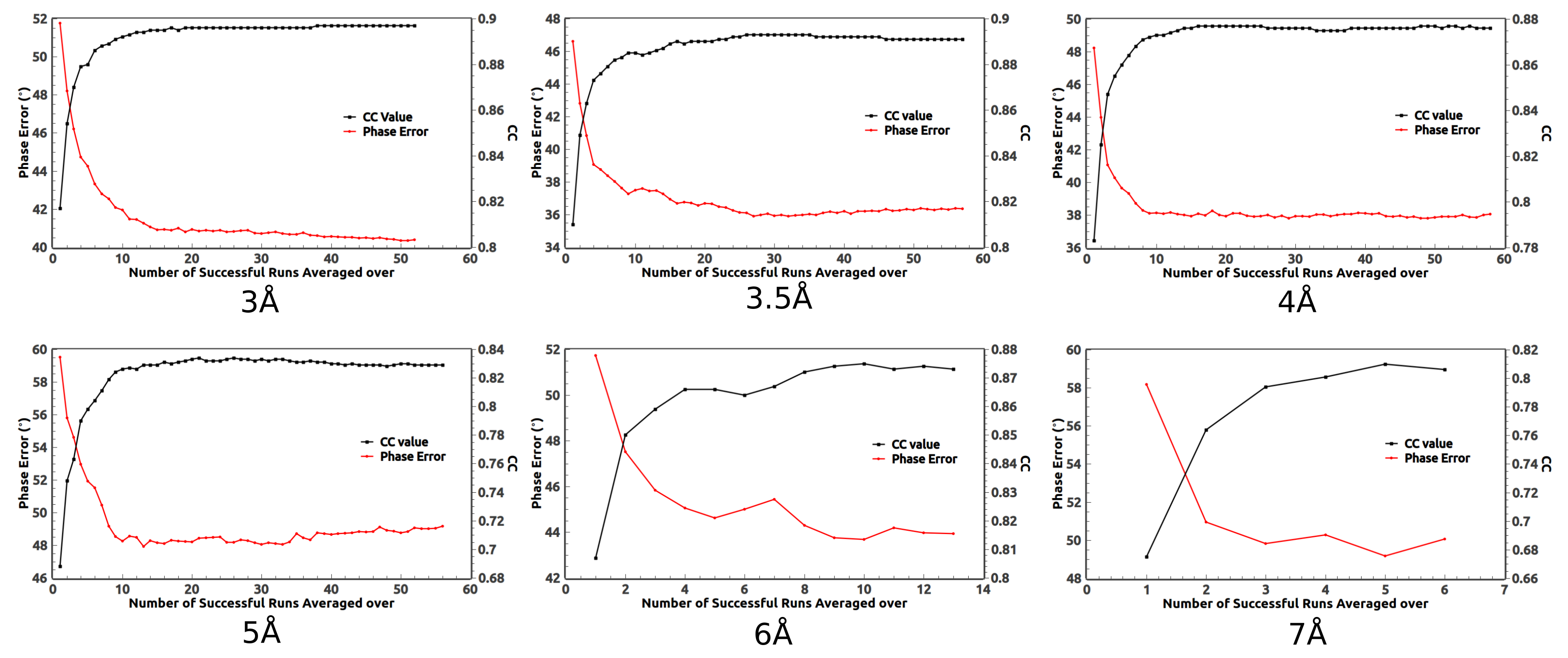
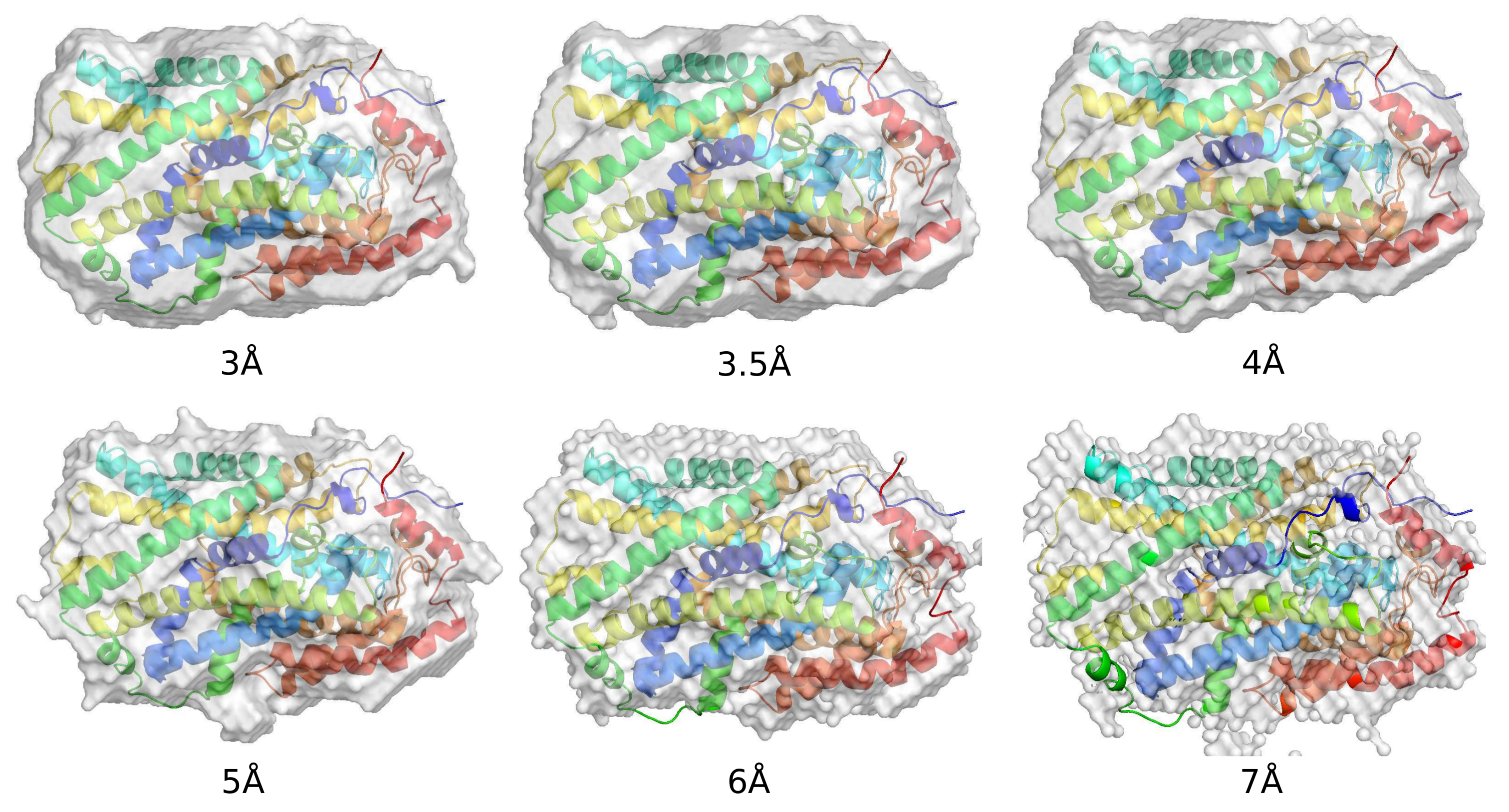
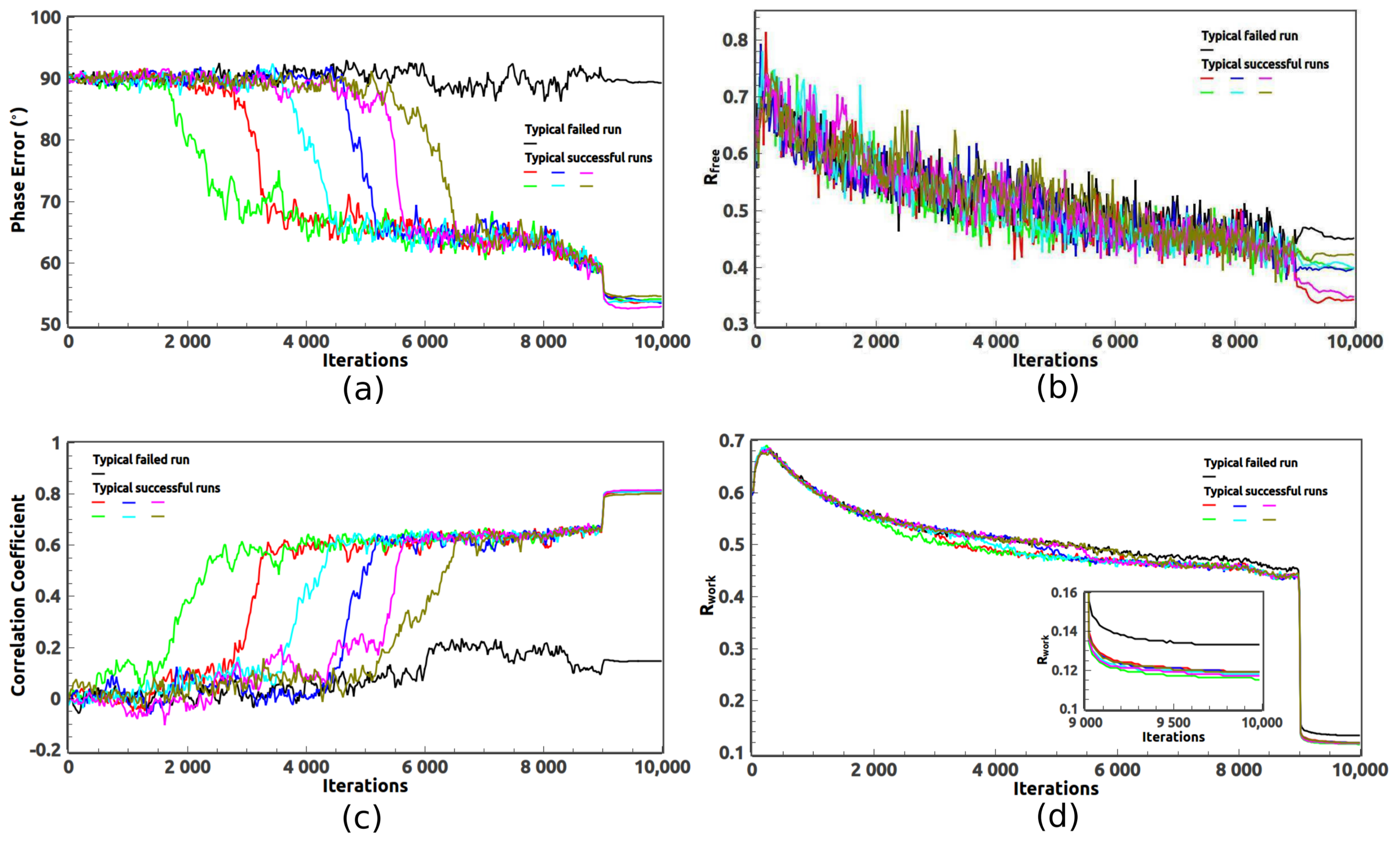
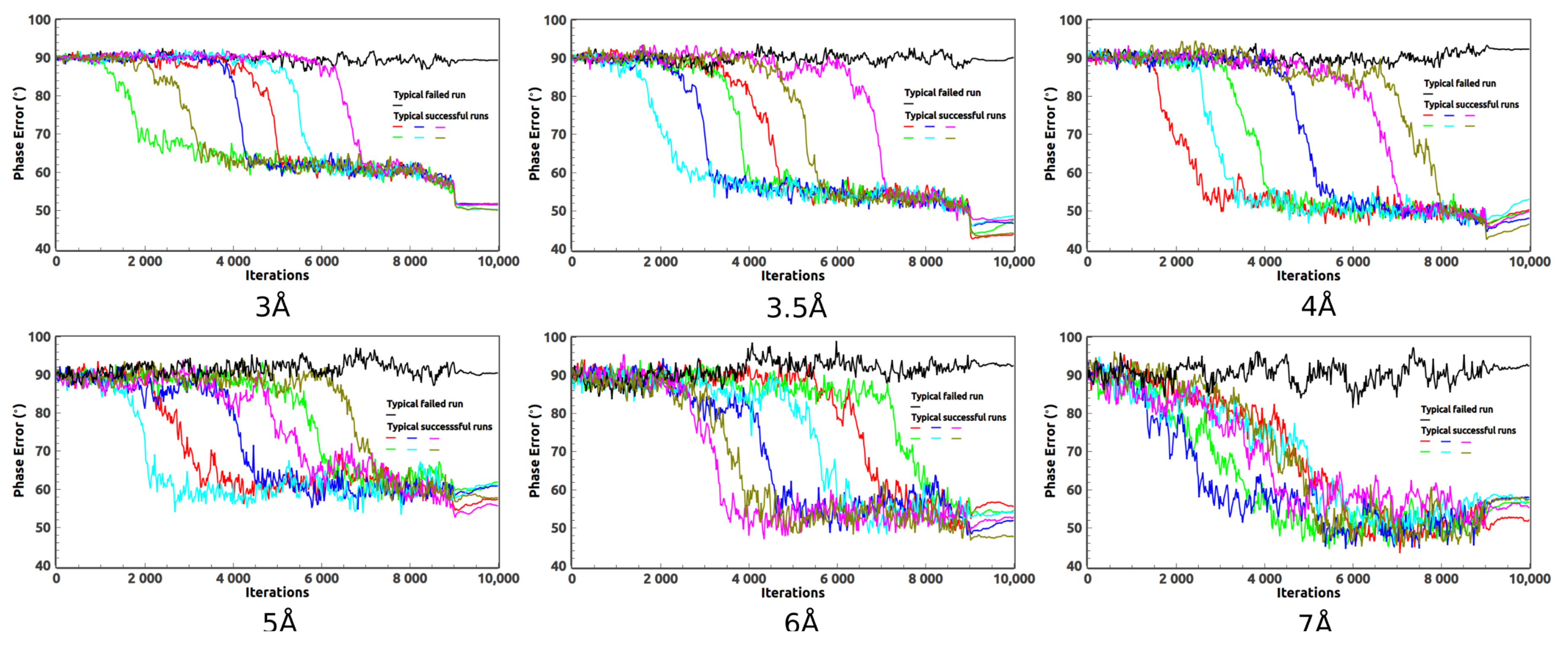
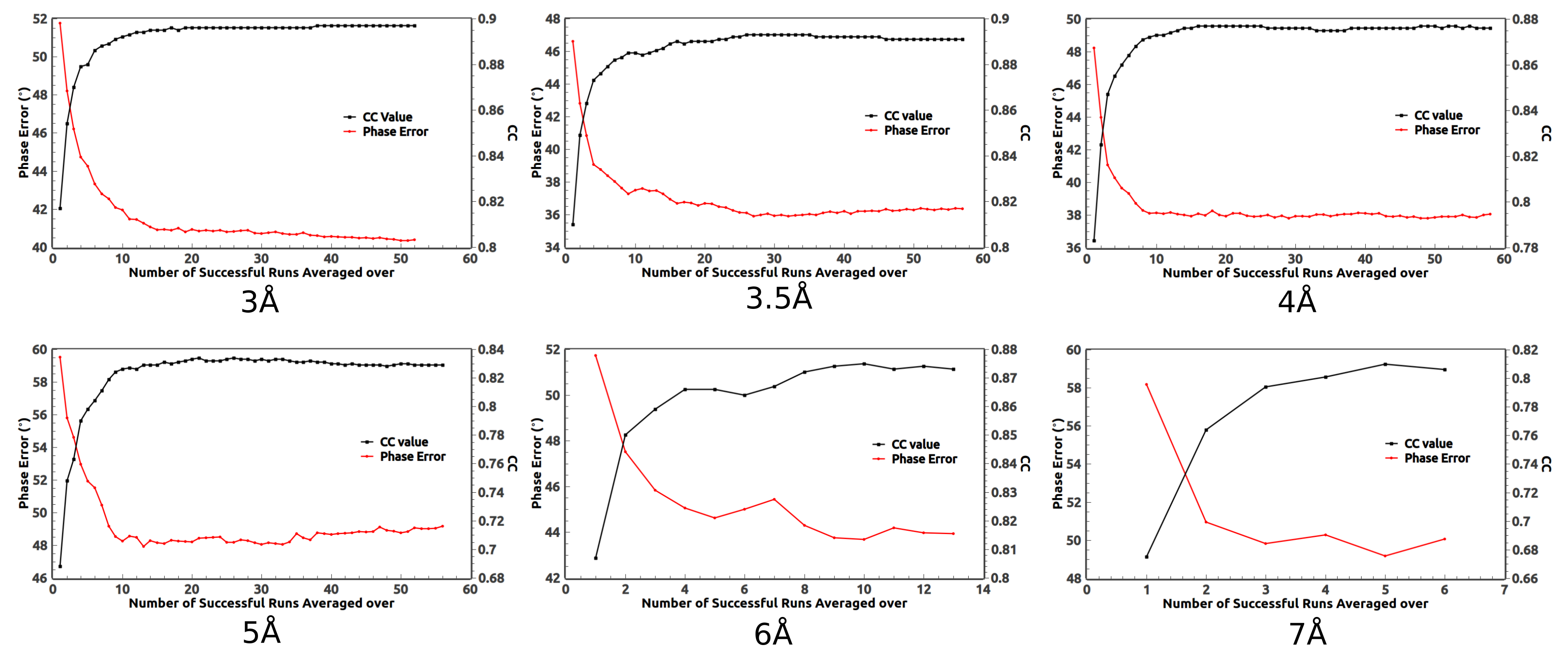
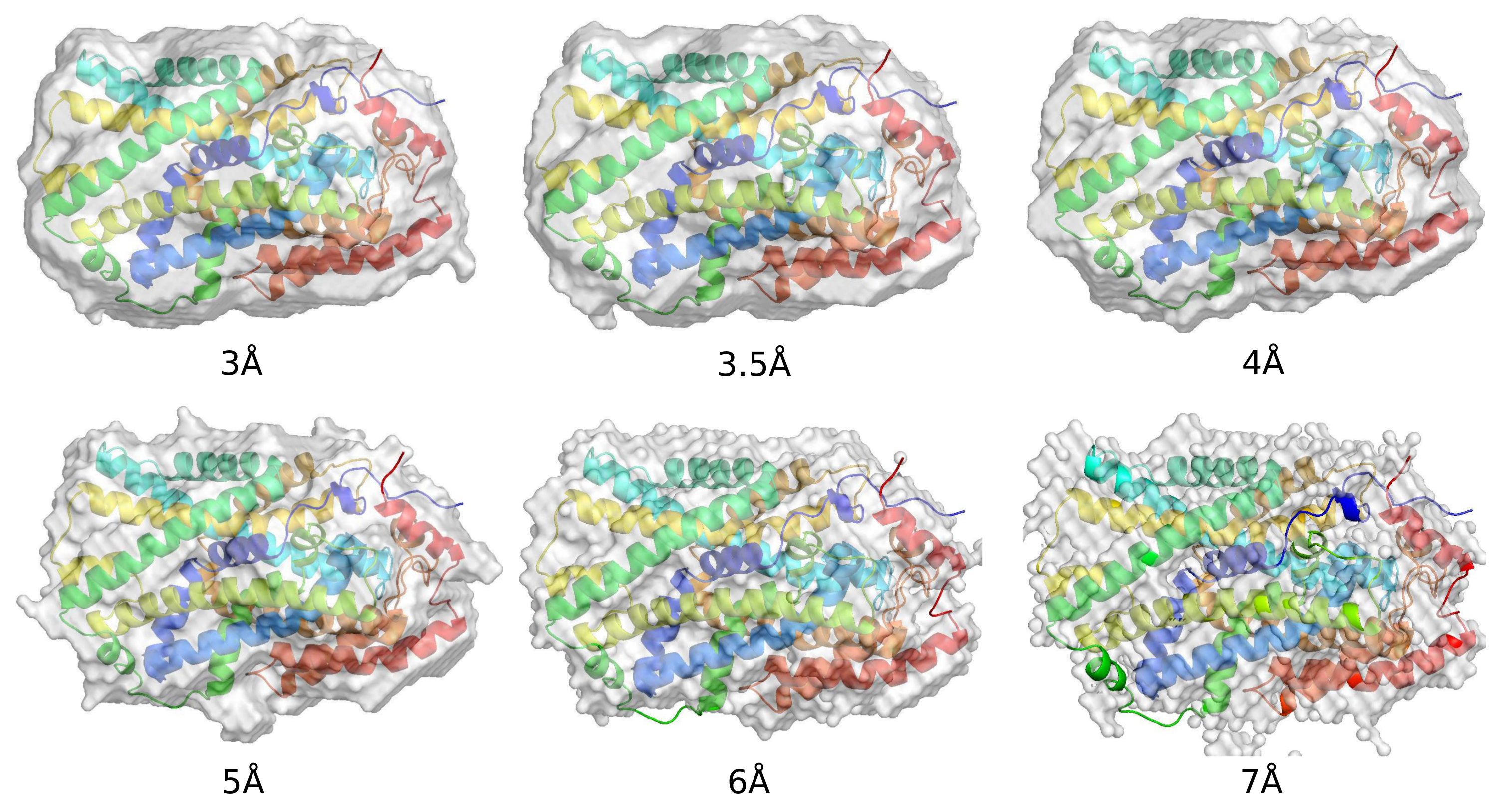
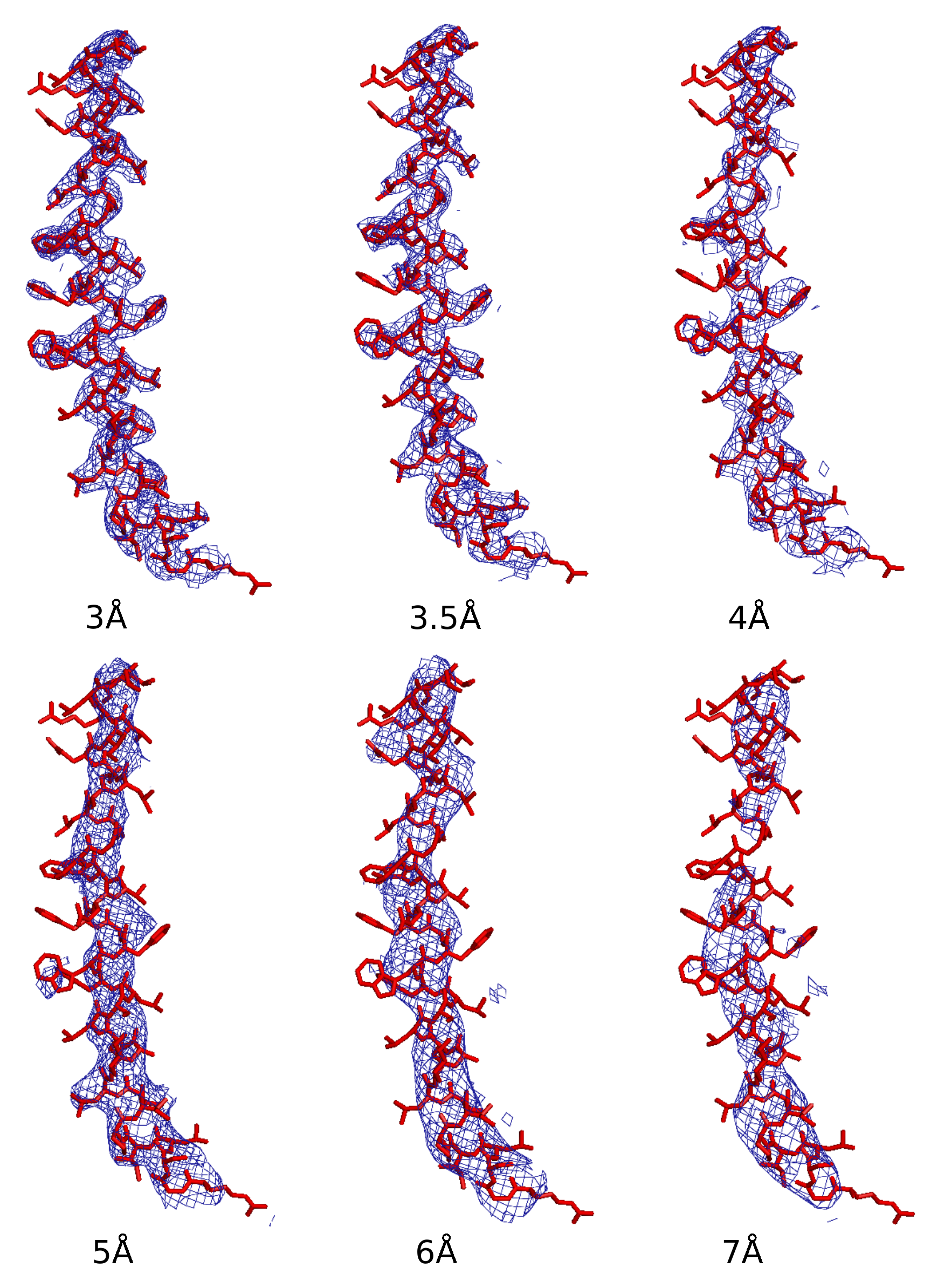
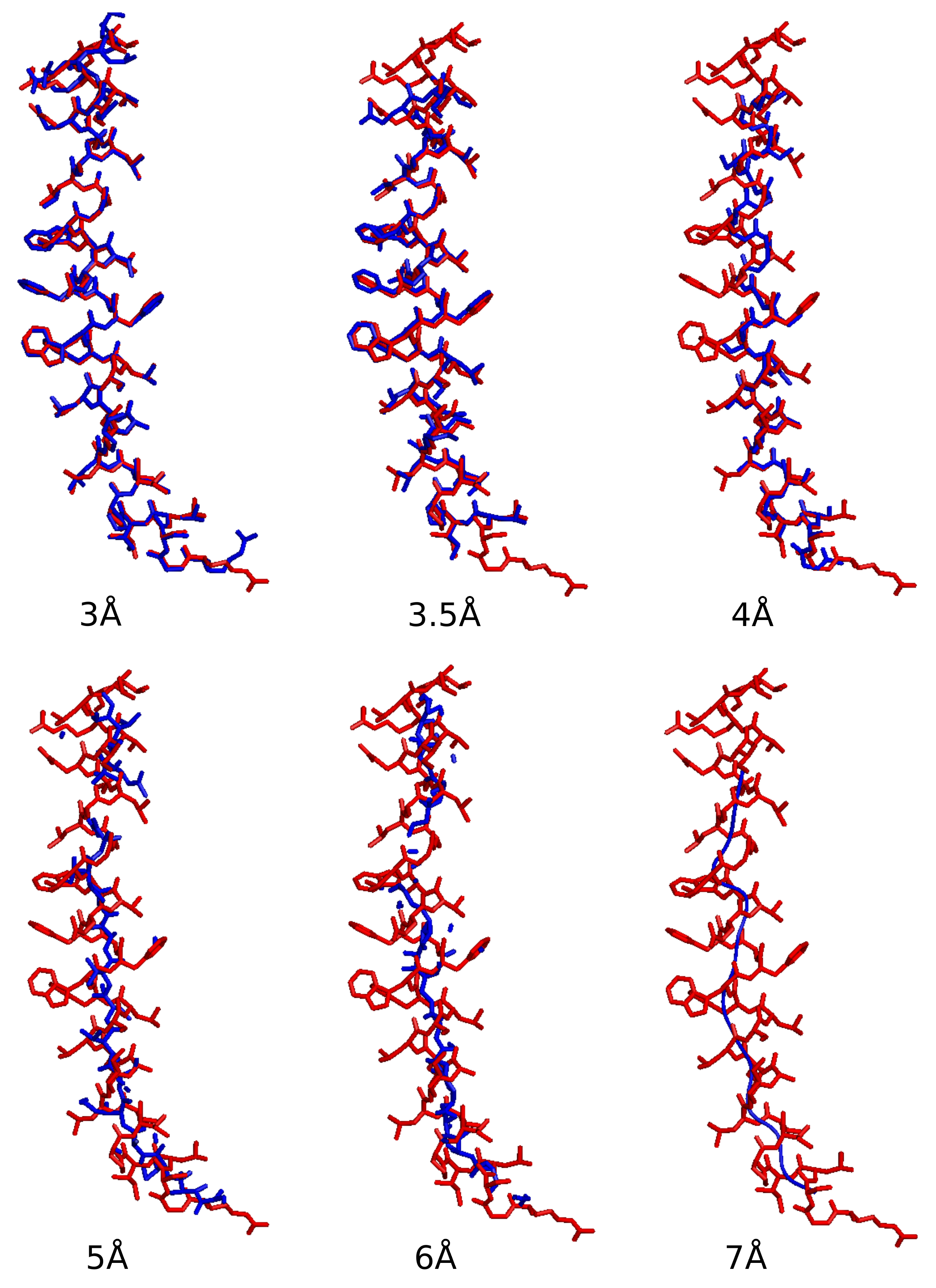
2. Ab Initio Phasing at Various Resolutions
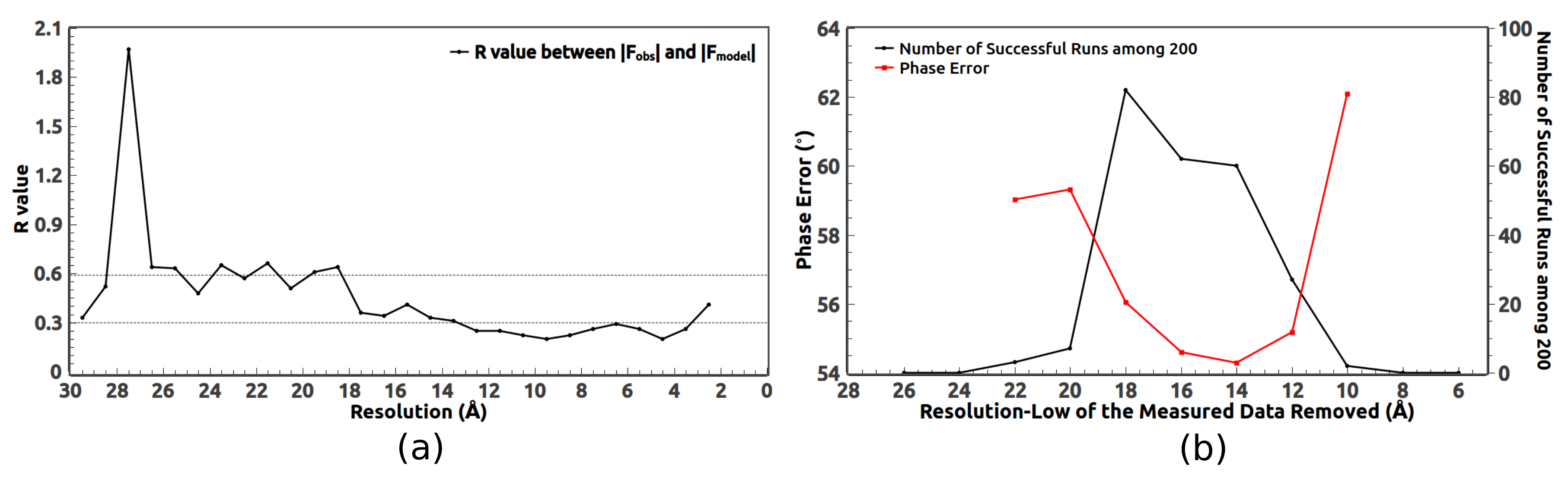
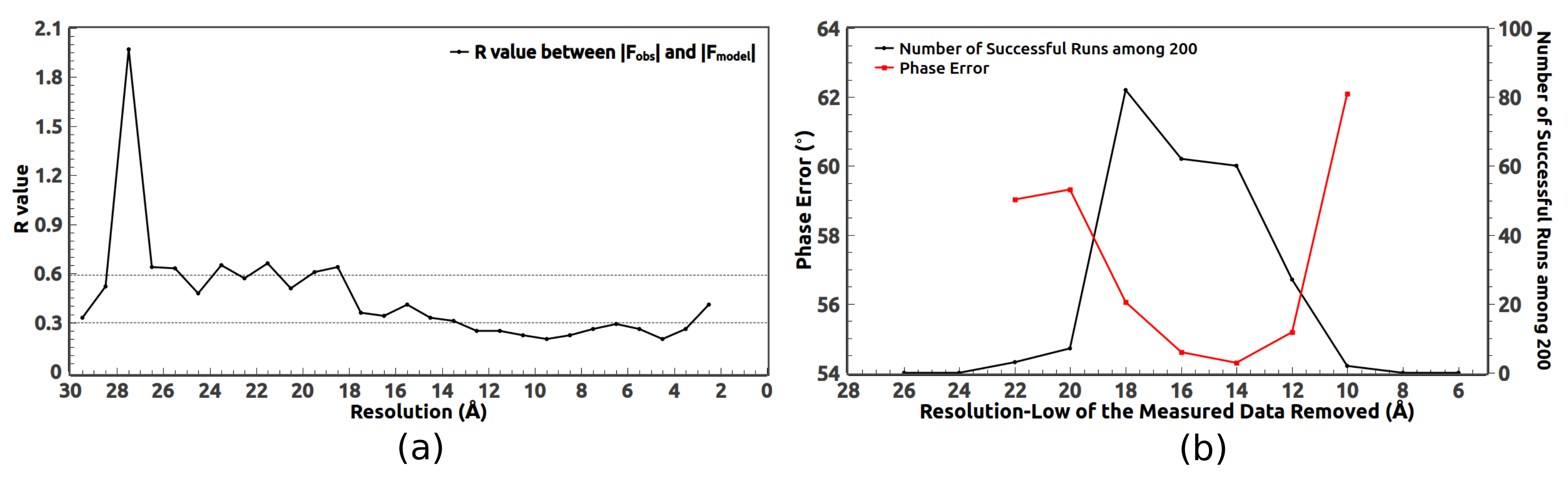
3. Removal of Very Low Resolution Data
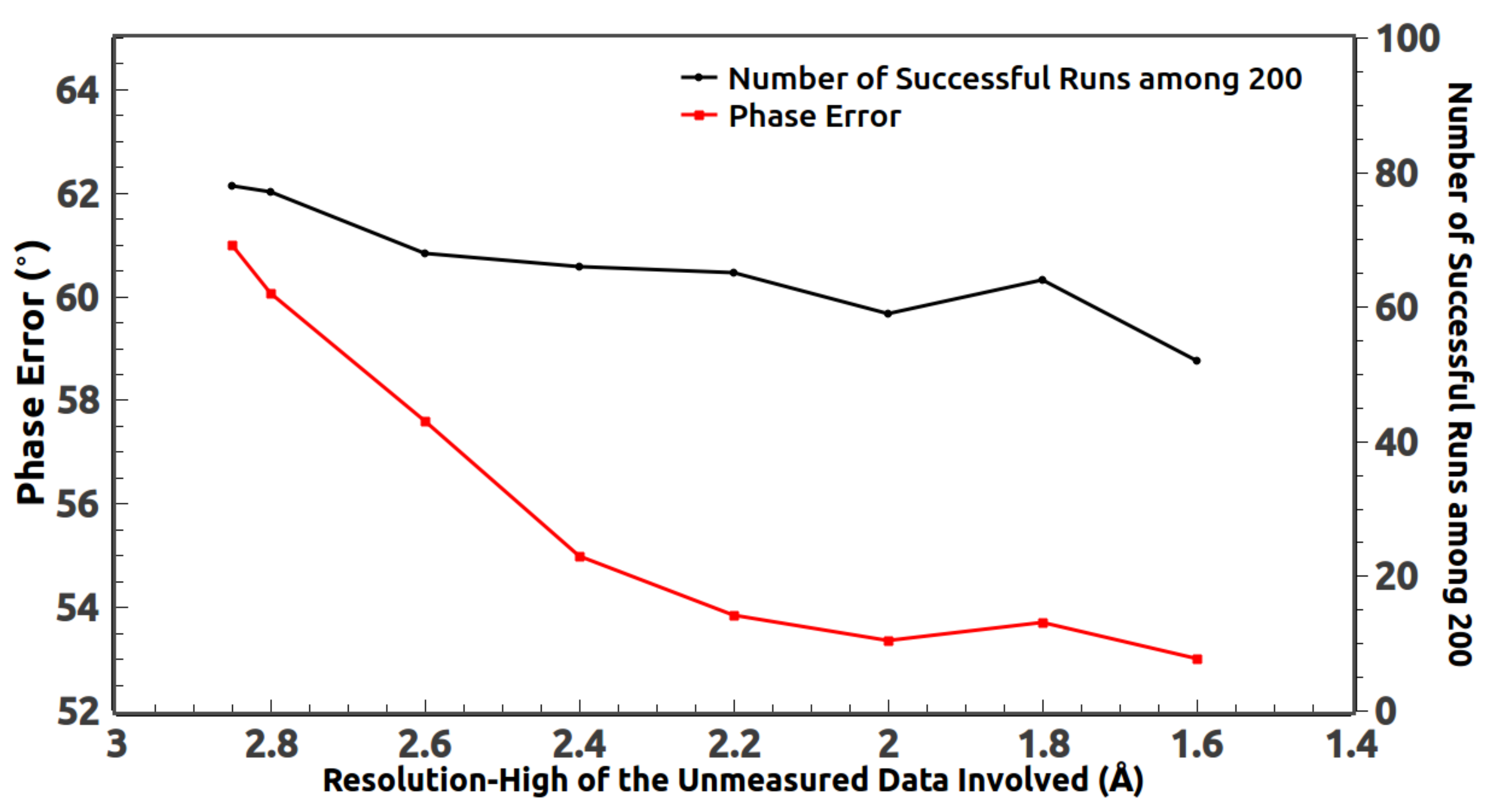
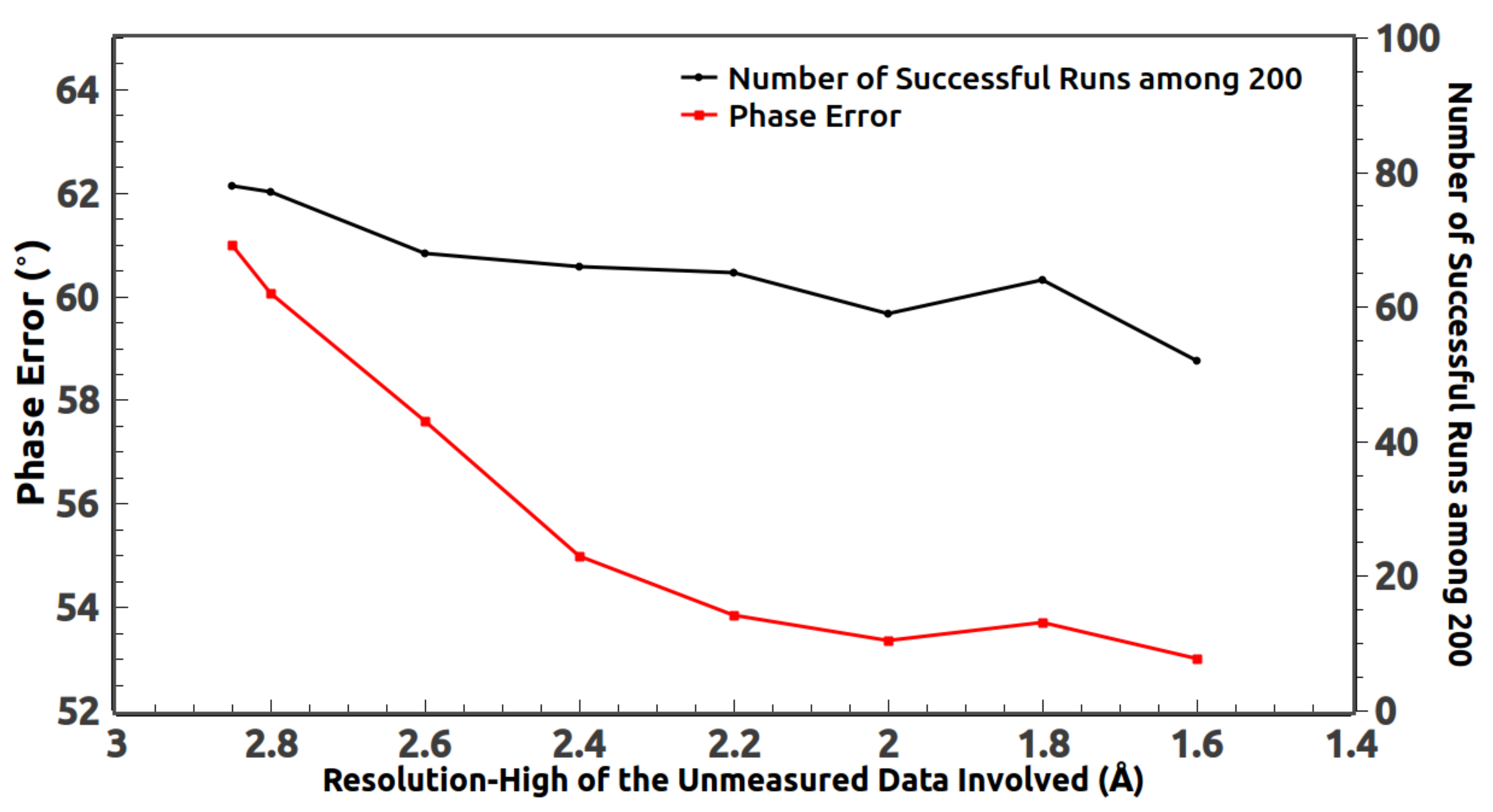
4. Inclusion of the Unmeasured High-Resolution Reflections
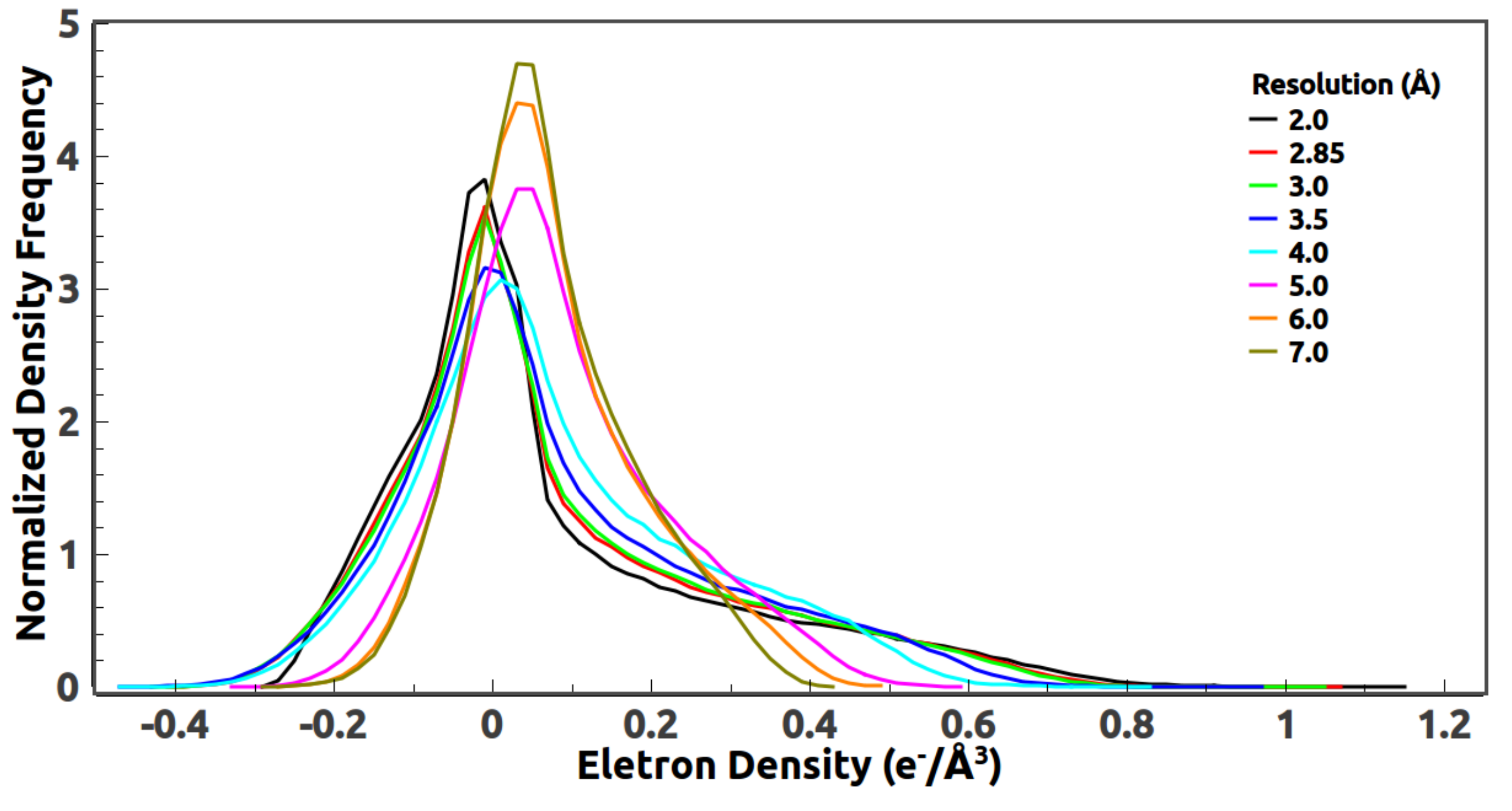
5. Optimal Resolution of the Reference Histogram
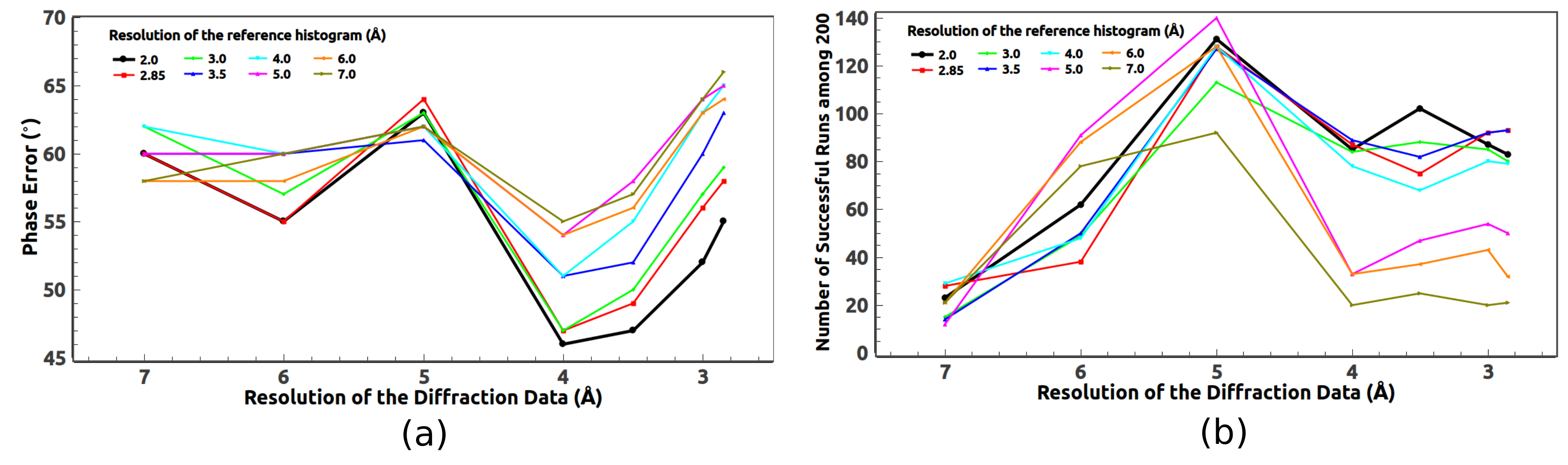
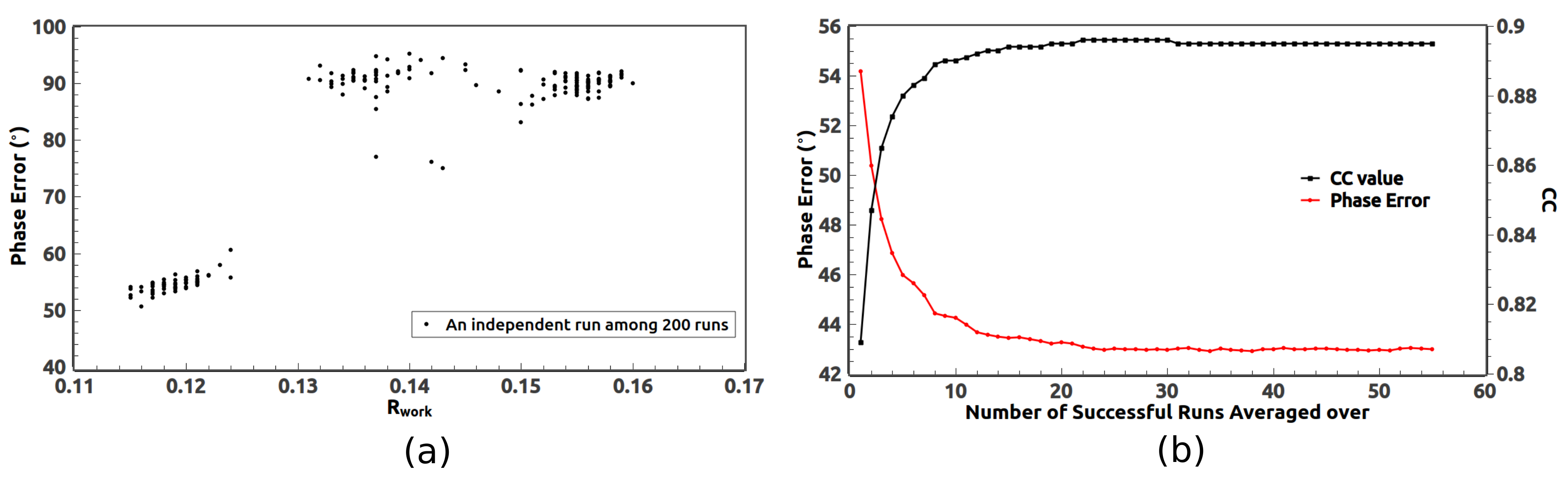
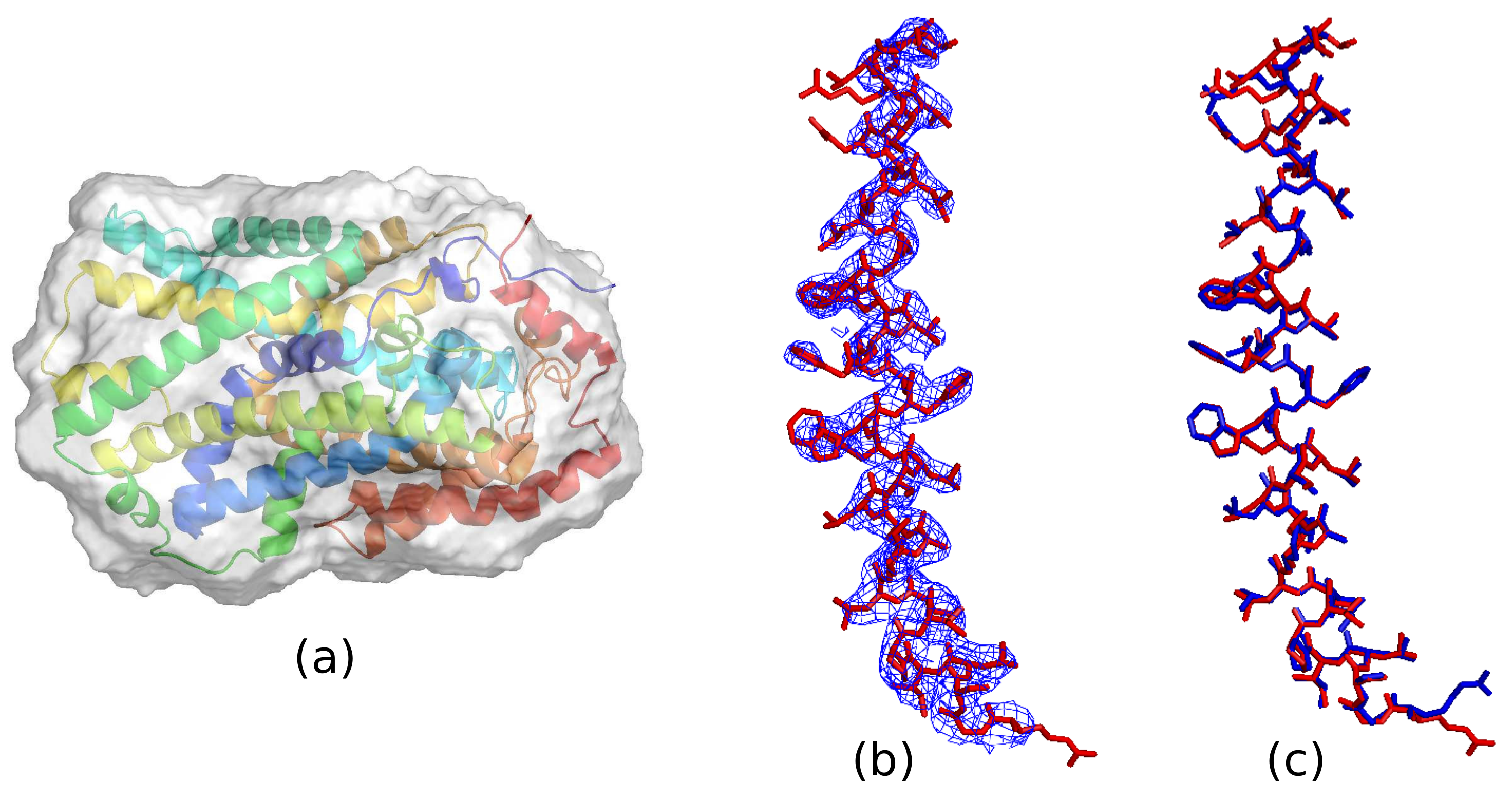
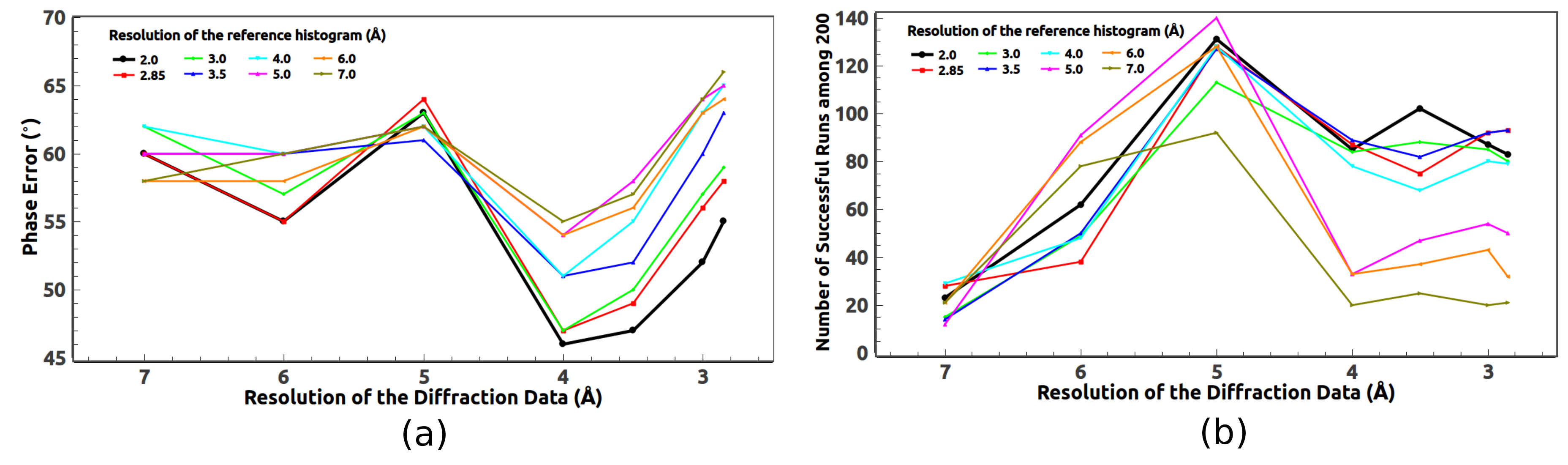
5.1. Optimal Resolution of the Reference Histogram Tested on the Observed Data
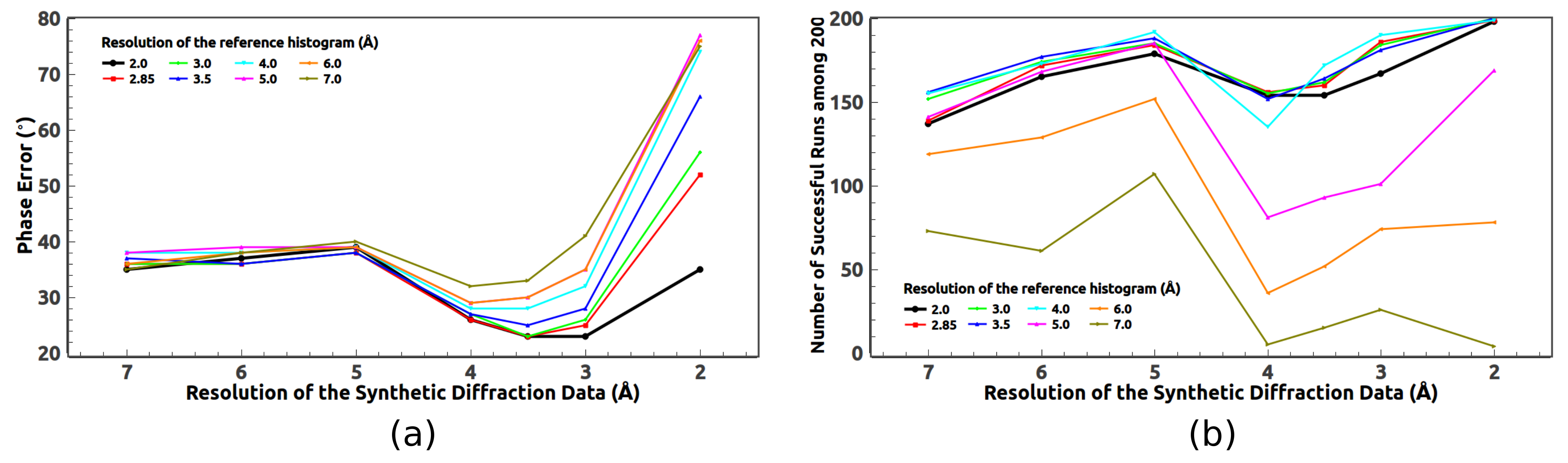
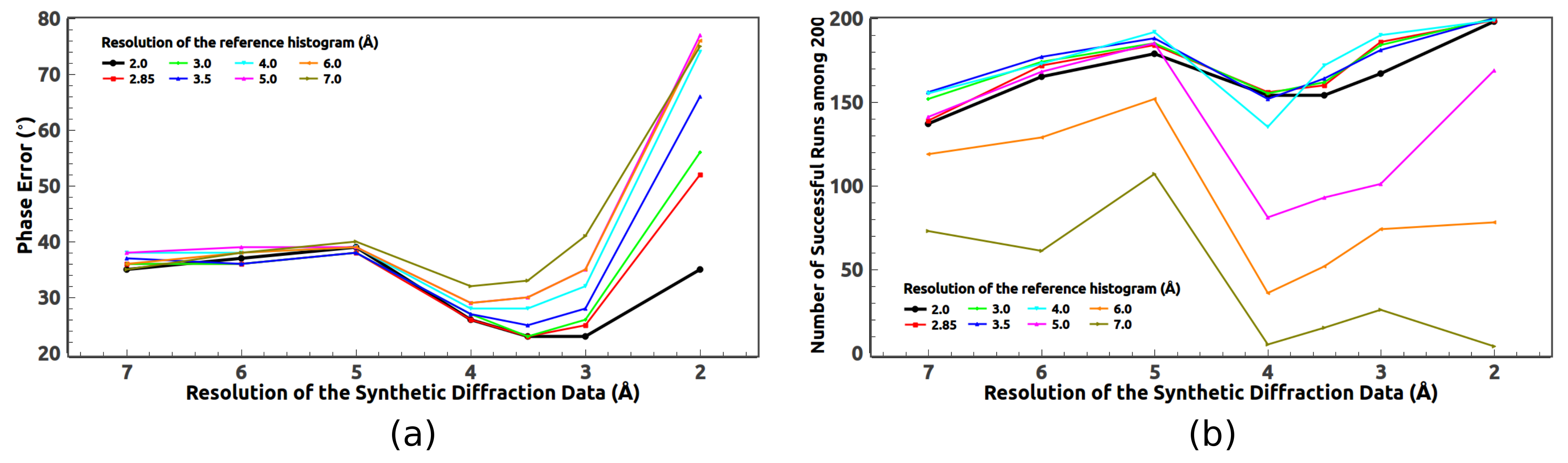
5.2. Optimal Resolution of the Reference Histogram Tested on the Synthetic Data
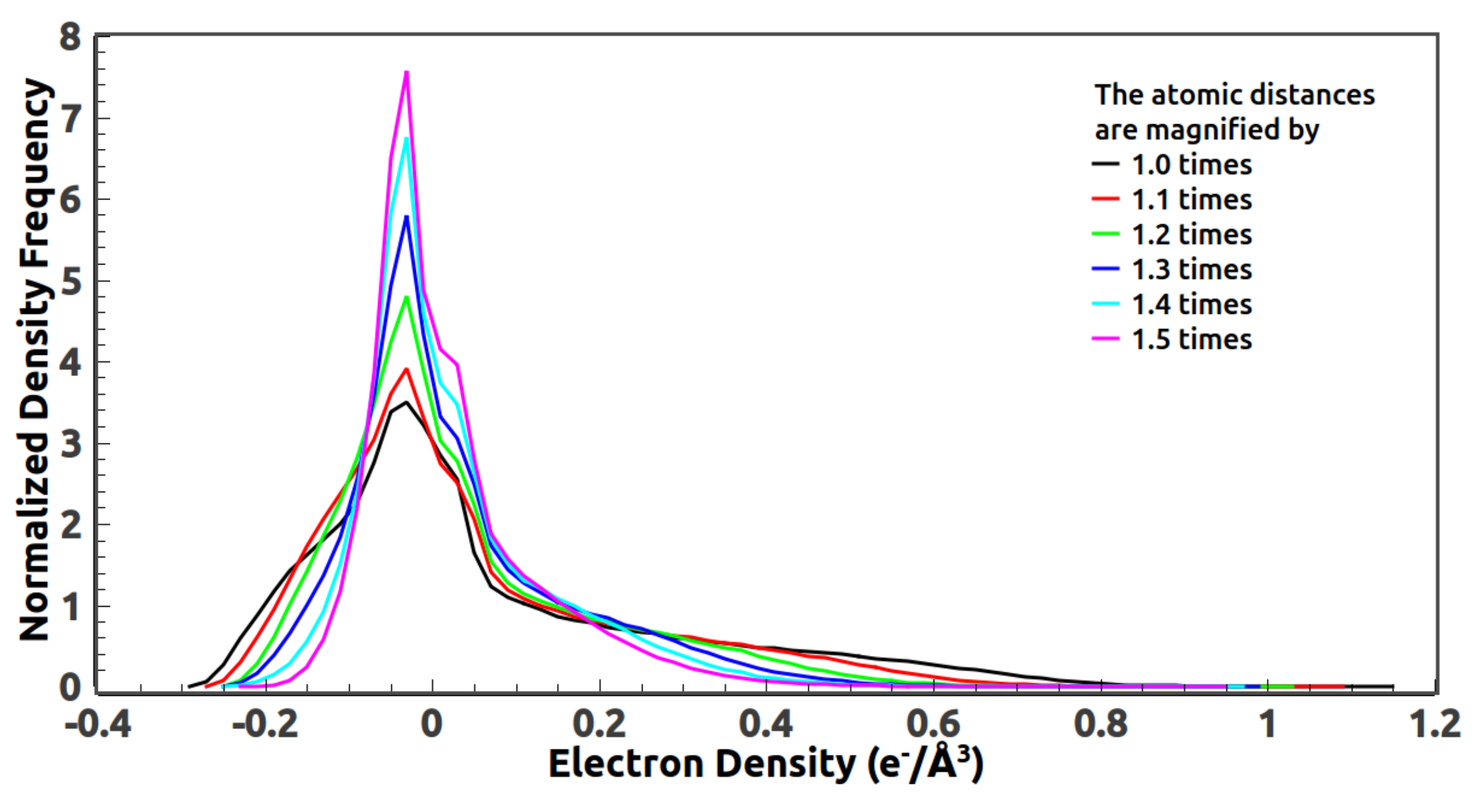
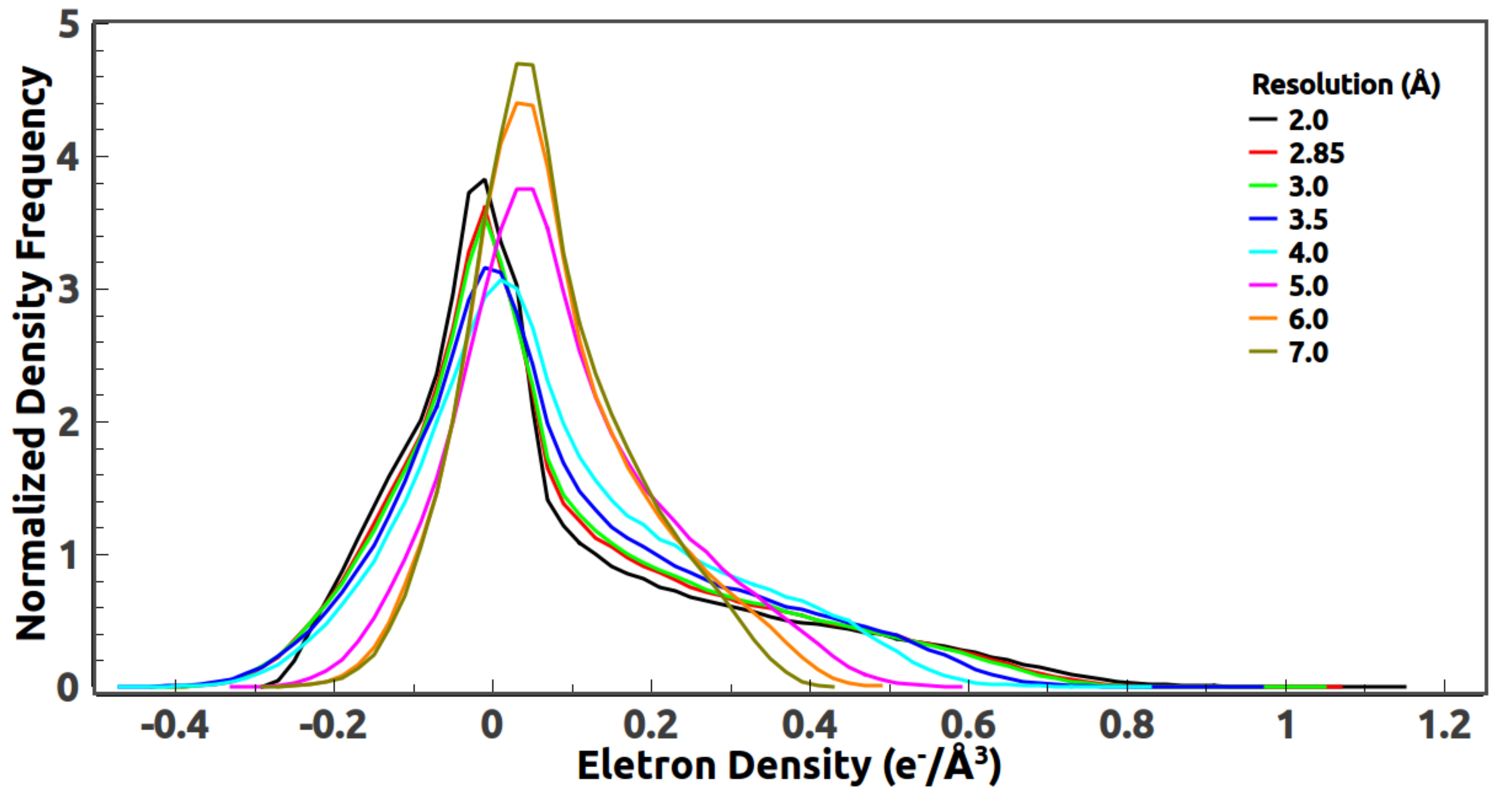
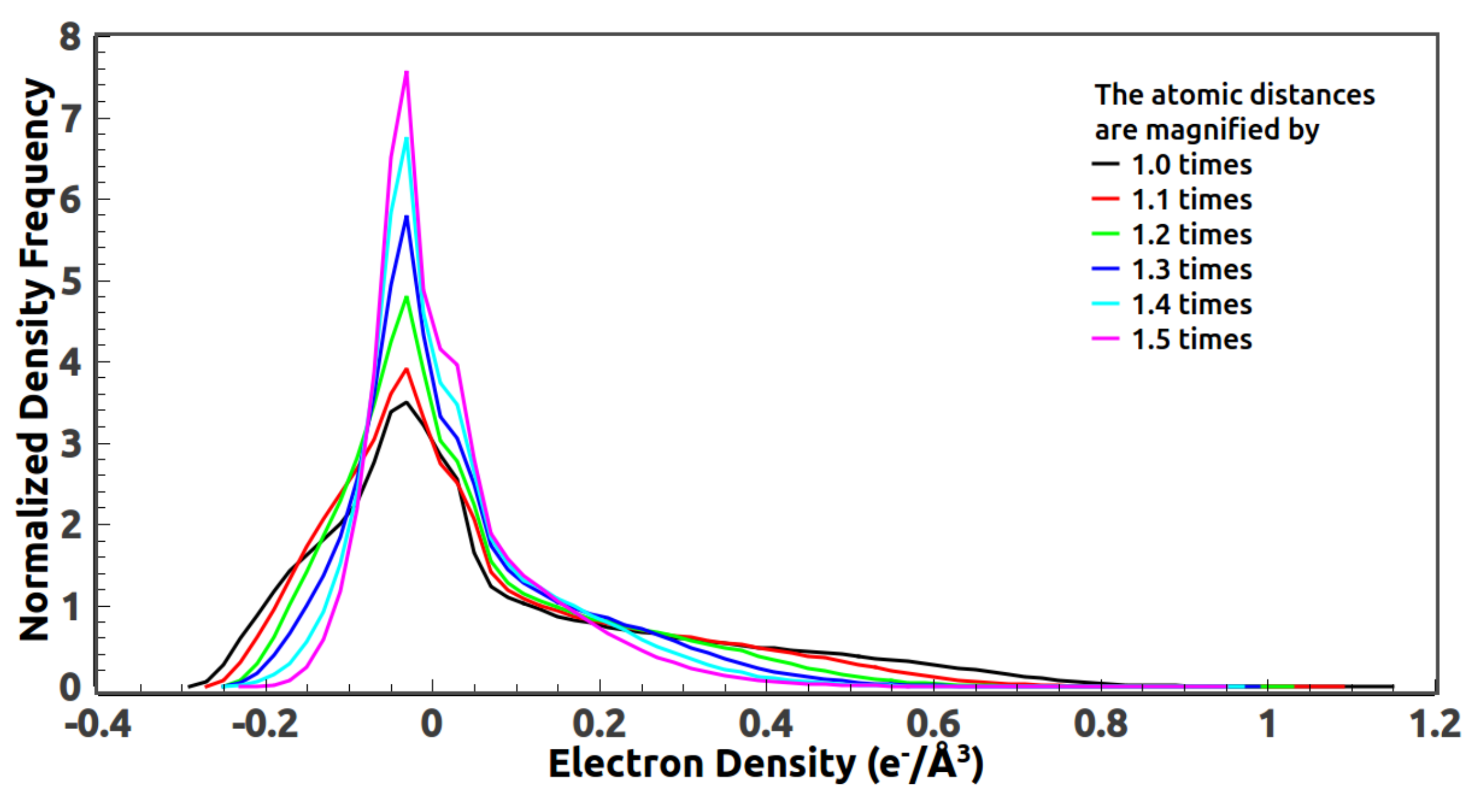
5.3. Reference Histogram Encodes the Information about Atomic Distance
6. Discussion
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Fienup, J.R. Reconstruction of an object from the modulus of its Fourier transform. Opt. Lett. 1978, 3, 27–29. [Google Scholar] [CrossRef] [PubMed]
- Fienup, J.R. Phase retrieval algorithms: A comparison. Appl. Opt. 1982, 21, 2758–2769. [Google Scholar] [CrossRef] [PubMed]
- Fienup, J.R. Reconstruction of a complex-valued object from the modulus of its Fourier transform using a support constraint. J. Opt. Soc. Am. A 1987, 4, 118–123. [Google Scholar] [CrossRef]
- Millane, R.P. Phase retrieval in crystallography and optics. J. Opt. Soc. Am. 1990, 7, 394–411. [Google Scholar] [CrossRef]
- Miao, J.; Sayer, D.; Chapman, H.N. Phase retrieval from the magnitude of the Fourier transforms of non-periodic objects. J. Opt. Soc. Am. 1998, 15, 1662–1669. [Google Scholar] [CrossRef]
- Elser, V. Phase retrieval by iterated projections. Acta Cryst. A 2003, 59, 201–209. [Google Scholar] [CrossRef]
- Marchesini, S.; He, H.; Chapman, H.N.; Hau-Riege, S.P.; Noy, A.; Howells, M.R.; Weierstall, U.; Spence, J.C.H. X-ray image reconstruction from a diffraction pattern alone. Phys. Rev. B 2003, 68, 140101. [Google Scholar] [CrossRef]
- Wu, J.S.; Weierstall, U.; Spence, J.C.H.; Koch, C.T. Iterative phase retrieval without support. Opt. Lett. 2004, 29, 2737. [Google Scholar] [CrossRef] [PubMed]
- Marchesini, S. Invited Article: A unified evaluation of iterative projection algorithms for phase retrieval. Rev. Sci. Instrum. 2007, 78, 011301. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.C.; Xu, R.; Dong, Y.H. Phase retrieval in protein crystallography. Acta Cryst. A 2012, 68, 256–265. [Google Scholar] [CrossRef] [PubMed]
- Millane, R.P.; Lo, V.L. Iterative projection algorithms in protein crystallography. I. Theory. Acta Cryst. A 2013, 69, 517–527. [Google Scholar] [CrossRef]
- Lo, V.L.; Kingston, R.L.; Millane, R.P. Iterative projection algorithms in protein crystallography. II. Application. Acta Cryst. A 2015, 71, 451–459. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Su, W.-P. Direct phasing of protein crystals with high solvent content. Acta Cryst. A 2015, 71, 92–98. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Fang, H.; Miller, M.D.; Phillips, G.N., Jr.; Su, W.-P. Improving the efficiency of molecular replacement by utilizing a new iterative transform phasing algorithm. Acta Cryst. A 2016, 72, 539–547. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Su, W.-P. Improving the convergence rate of a hybrid input-output phasing algorithm by varying the reflection data weight. Acta Cryst. A 2018, 74, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Rossmann, M.G. Ab initio phase determination and phase extension using non-crystallographic symmetry. Curr. Opin. Struct. Biol. 1995, 5, 650–655. [Google Scholar] [CrossRef]
- Giacovazzo, C.; Siliqi, D.; Zanotti, G. The ab initio crystal structure solution of proteins by direct methods. III. The phase extension process. Acta Cryst. A 1995, 51, 177–188. [Google Scholar] [CrossRef]
- Lunin, V.Y.; Lunina, N.L.; Petrova, T.E.; Urzhumtsev, A.G.; Podjarny, A.D. On the ab initio solution of the phase problem for macromolecules at very low resolution. II. Generalized likelihood based approach to cluster discrimination. Acta Cryst. D 1998, 54, 726–734. [Google Scholar] [CrossRef]
- Lunin, V.Y.; Lunina, N.L.; Petrova, T.E.; Skovoroda, T.P.; Urzhumtsev, A.G.; Podjarny, A.D. Low-resolution ab initio phasing: Problems and advances. Acta Cryst. D 2000, 56, 1223–1232. [Google Scholar] [CrossRef]
- Carpenter, E.P.; Beis, K.; Cameron, A.D.; Iwata, S. Overcoming the challenges of membrane protein crystallography. Curr. Opin. Struct. Biol. 2008, 18, 581–586. [Google Scholar] [CrossRef] [PubMed]
- Caliandro, R.; Carrozzini, B.; Cascarano, G.L.; De Caro, L.; Giacovazzo, C.; Siliqi, D. Phasing at resolution higher than the experimental resolution. Acta Cryst. D 2005, 61, 556–565. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Caliandro, R.; Carrozzini, B.; Cascarano, G.L.; De Caro, L.; Giacovazzo, C.; Siliqi, D. Ab initio phasing at resolution higher than experimental resolution. Acta Cryst. D 2005, 61, 1080–1087. [Google Scholar] [CrossRef] [PubMed]
- Caliandro, R.; Carrozzini, B.; Cascarano, G.L.; De Caro, L.; Giacovazzo, C.; Siliqi, D. Advances in the free lunch method. J. Appl. Cryst. 2007, 40, 931–937. [Google Scholar] [CrossRef]
- Zhang, K.Y.J.; Main, P. Histogram matching as a new density modification technique for phase refinement and extension of protein molecules. Acta Cryst. A 1990, 46, 41–46. [Google Scholar] [CrossRef]
- Zhang, K.Y.J.; Main, P. The use of Sayre’s equation with solvent flattening and histogram matching for phase extension and refinement of protein structures. Acta Cryst. A 1990, 46, 377–381. [Google Scholar] [CrossRef]
- Millane, R.P.; Stroud, W.J. Reconstructing symmetric images from their undersampled Fourier intensities. J. Opt. Soc. Am. A 1997, 14, 568–579. [Google Scholar] [CrossRef]
- Van der Plas, J.L.; Millane, R.P. Ab-initio phasing in protein crystallography. Proc. SPIE 2000, 4123, 249–260. [Google Scholar] [CrossRef]
- Brünger, A.T. Free R value: A novel statistical quantity for assessing the accuracy of crystal structures. Nature 1992, 355, 472–475. [Google Scholar] [CrossRef]
- Fourier Transform Functions. Intel® Math Kernel Library 11.3 Reference Manual; Intel Corporation: Santa Clara, CA, USA, 2015; pp. 1911–1962. [Google Scholar]
- Wang, B.C. Resolution of phase ambiguity in macromolecular crystallography. Methods Enzymol. 1985, 115, 90–112. [Google Scholar] [CrossRef] [PubMed]
- Leslie, A.G.W. A reciprocal-space method for calculating a molecular envelope using the algorithm of B.C. Wang. Acta Cryst. A 1987, 43, 134–136. [Google Scholar] [CrossRef]
- Terwilliger, T.C. Reciprocal-space solvent flattening. Acta Cryst. D 1999, 55, 1863–1871. [Google Scholar] [CrossRef]
- Abrahams, J.P.; Leslie, A.G.W. Methods used in the structure determination of bovine mitochondrial F1 ATPase. Acta Cryst. D 1996, 52, 30–42. [Google Scholar] [CrossRef] [PubMed]
- Weyand, S.; Shimamura, T.; Yajima, S.; Suzuki, S.; Mirza, O.; Krusong, K.; Carpenter, E.P.; Rutherford, N.G.; Hadden, J.M.; O’Reilly, J.; et al. Structure and molecular mechanism of a nucleobase-cation-symport-1 family transporter. Science 2008, 322, 709–713. [Google Scholar] [CrossRef] [PubMed]
- Vaguine, A.A.; Richelle, J.; Wodak, S.J. SFCHECK: A unified set of procedure for evaluating the quality of macromolecular structure-factor data and their agreement with atomic model. Acta Cryst. D 1999, 55, 191–205. [Google Scholar] [CrossRef] [PubMed]
- Winn, M.D.; Ballard, C.C.; Cowtan, K.D.; Dodson, E.J.; Emsley, P.; Evans, P.R.; Keegan, R.M.; Krissinel, E.B.; Leslie, A.G.W.; McCoy, A.; et al. Overview of the CCP4 suite and current developments. Acta. Cryst. D 2011, 67, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Boggavarapu, R.; Jeckelmann, J.M.; Harder, D.; Ucurum, Z.; Fotiadis, D. Role of electrostatic interactions for ligand recognition and specificity of peptide transporters. BMC Biol. 2015, 13, 58. [Google Scholar] [CrossRef] [PubMed]
- Langer, G.G.; Cohen, S.X.; Lamzin, V.S.; Perrakis, A. Automated macromolecular model building for X-ray crystallography using ARP/wARP version 7. Nat. Protoc. 2008, 3, 1171–1179. [Google Scholar] [CrossRef] [PubMed]
- Terwilliger, T.C.; Grosse-Kunstleve, R.W.; Afonine, P.V.; Moriarty, N.W.; Zwart, P.H.; Hung, L.-W.; Read, R.J.; Adams, P.D. Iterative model building, structure refinement and density modification with the PHENIX AutoBuild wizard. Acta Cryst. D 2008, 64, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Adams, P.D.; Afonine, P.V.; Bunkóczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.-W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Cryst. D 2010, 66, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Su, W.-P. Retrieving low- and medium-resolution structural features of macromolecules directly from the diffraction intensities—A real-space approach to the X-ray phase problem. Acta Cryst. A 2008, 64, 625–630. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Data ( Å) | Successful Runs in 200 | Final Phase Error | Final CC Value | Model Completeness % |
|---|---|---|---|---|
| 2.85 | 55 | 43 | 0.89 | 81 |
| 3.0 | 52 | 40 | 0.90 | 84 |
| 3.5 | 57 | 36 | 0.89 | 74 |
| 4.0 | 58 | 38 | 0.88 | 56 |
| 5.0 | 56 | 48 | 0.83 | 34 |
| 6.0 | 13 | 44 | 0.87 | 34 |
| 7.0 | 6 | 49 | 0.81 | 24 |
| 8.0 | 0 | na | na | na |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiang, M.; He, H.; Cheng, Y.; Su, W.-P. Resolution Dependence of an Ab Initio Phasing Method in Protein X-ray Crystallography. Crystals 2018, 8, 156. https://doi.org/10.3390/cryst8040156
Jiang M, He H, Cheng Y, Su W-P. Resolution Dependence of an Ab Initio Phasing Method in Protein X-ray Crystallography. Crystals. 2018; 8(4):156. https://doi.org/10.3390/cryst8040156
Chicago/Turabian StyleJiang, Mengchao, Hongxing He, Yunpeng Cheng, and Wu-Pei Su. 2018. "Resolution Dependence of an Ab Initio Phasing Method in Protein X-ray Crystallography" Crystals 8, no. 4: 156. https://doi.org/10.3390/cryst8040156
APA StyleJiang, M., He, H., Cheng, Y., & Su, W.-P. (2018). Resolution Dependence of an Ab Initio Phasing Method in Protein X-ray Crystallography. Crystals, 8(4), 156. https://doi.org/10.3390/cryst8040156