
Crystallographic Combinations: Understanding Polymorphism and Approximate Symmetry in N-(1,3-Thiazol-2-yl)benzamide


Abstract

1. Introduction
2. Experimental Section
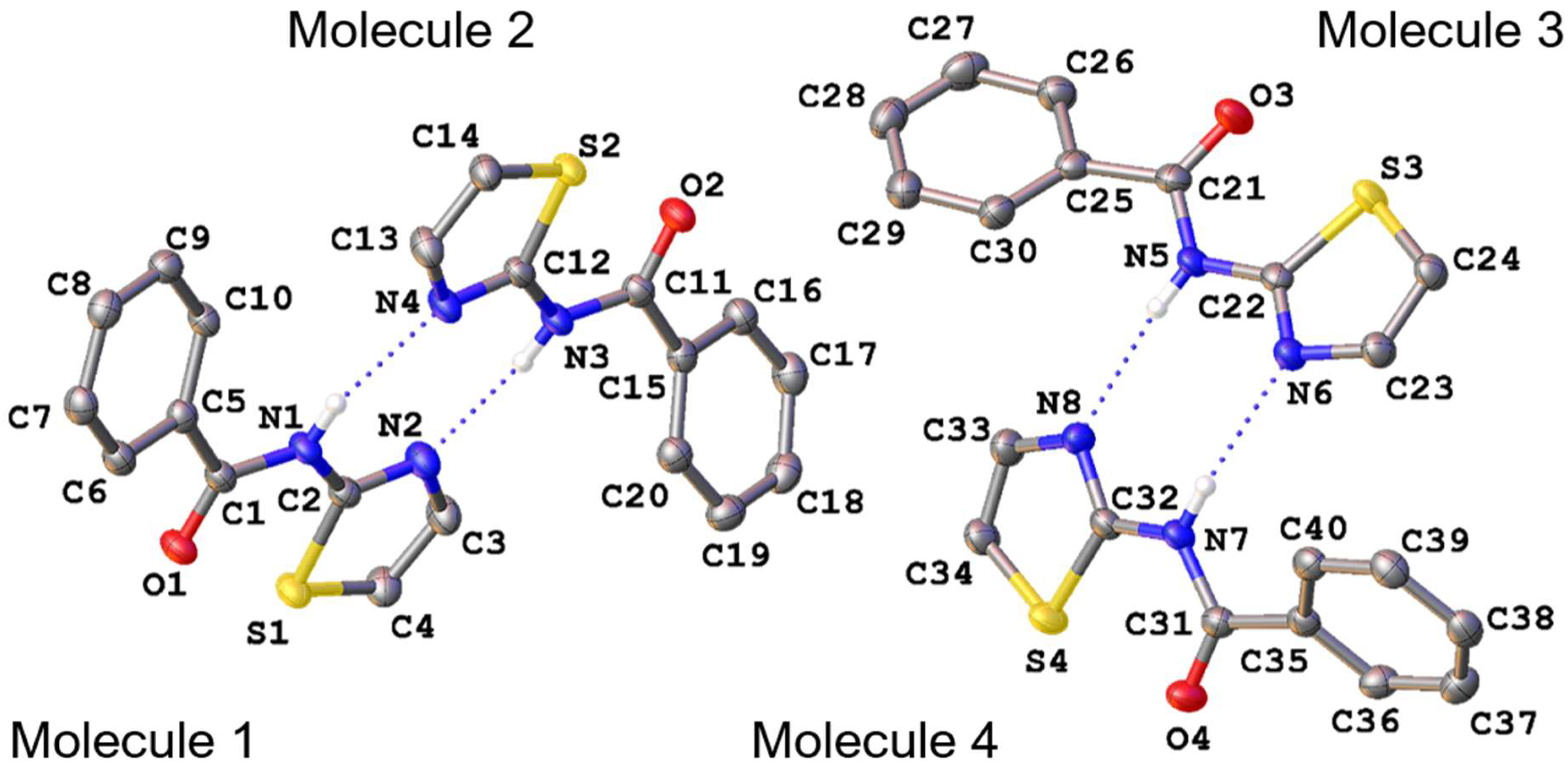
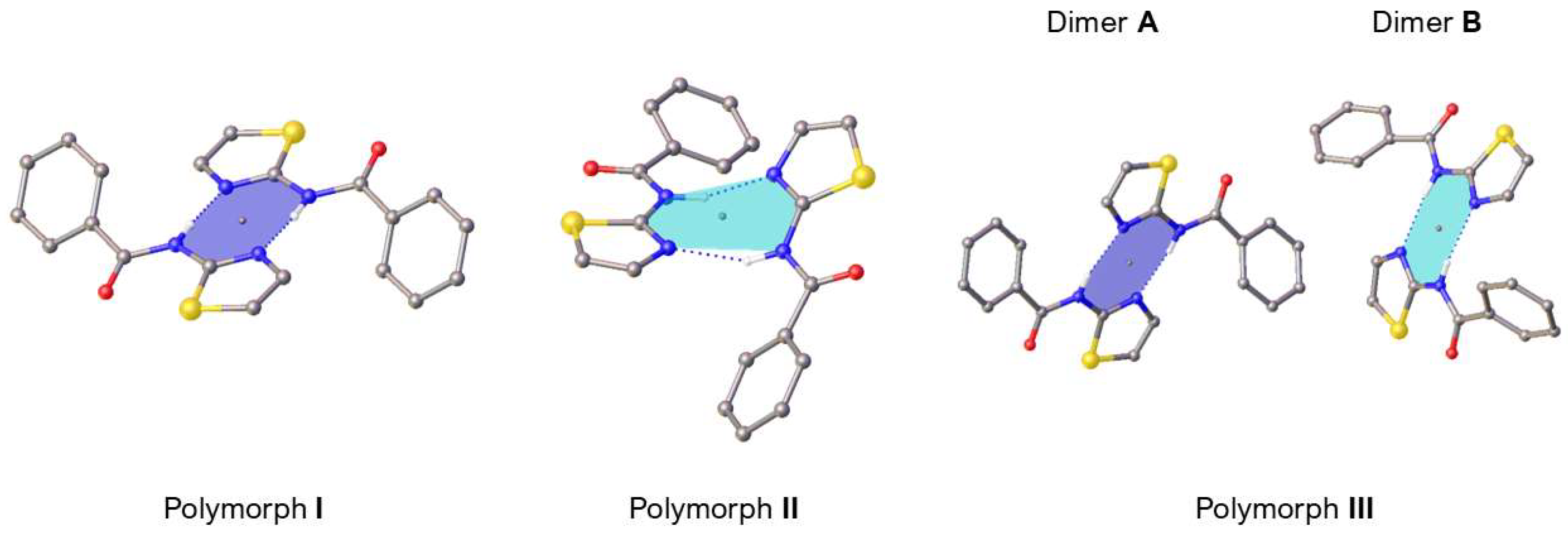
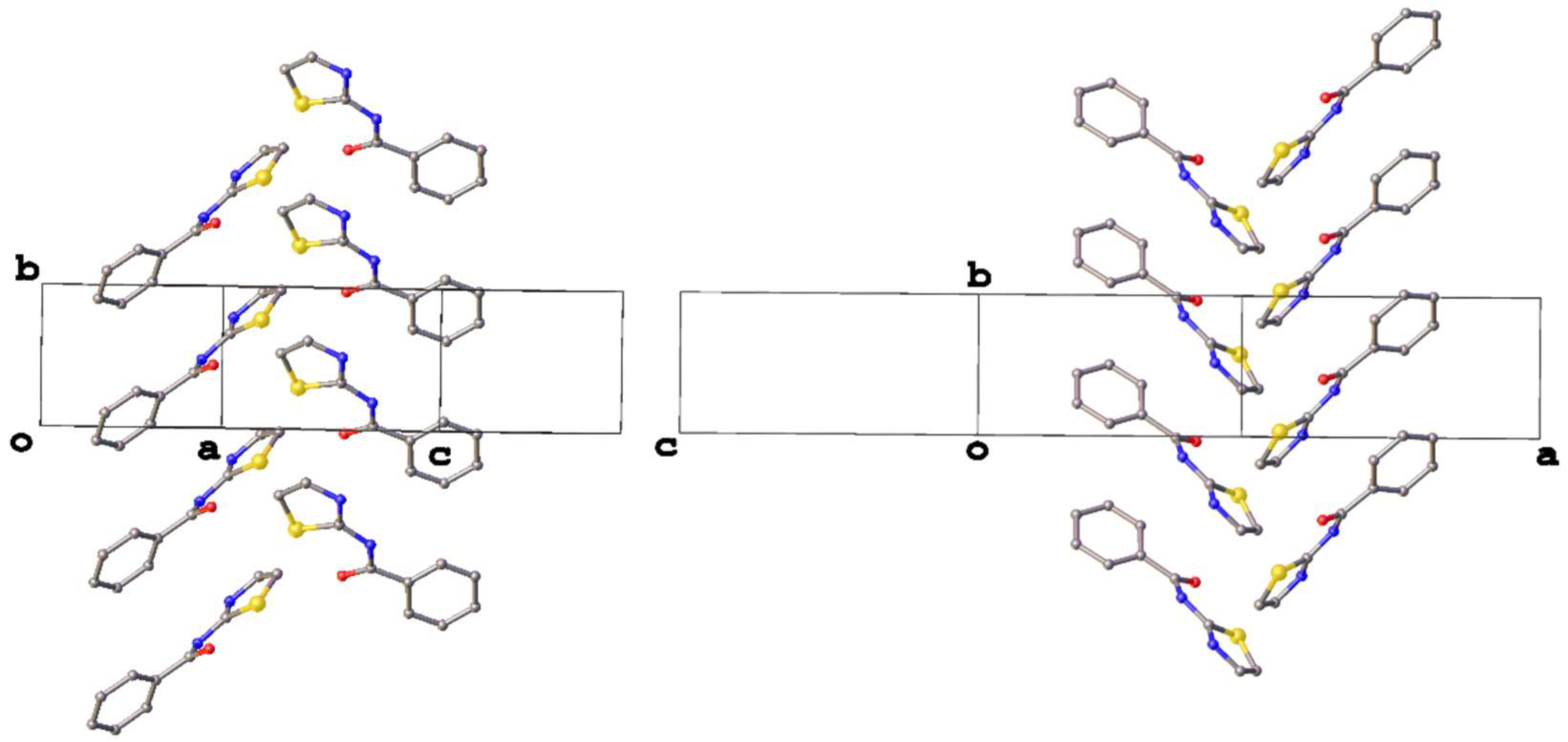
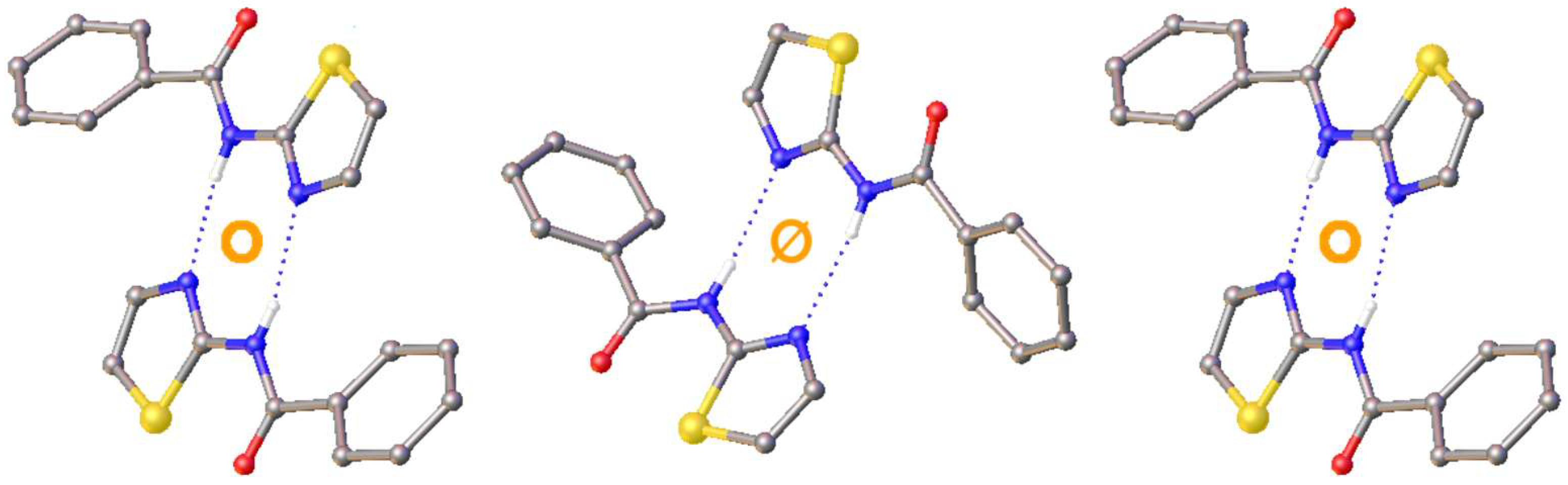
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brog, J.-P.; Chanez, C.-L.; Crochet, A.; Fromm, K.M. Polymorphism, what it is and how to identify it: A systematic review. RSC Adv. 2013, 3, 16905. [Google Scholar] [CrossRef]
- Cruz-Cabeza, A.J.; Bernstein, J. Conformational polymorphism. Chem. Rev. 2014, 114, 2170–2191. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Cabeza, A.J.; Reutzel-Edens, S.M.; Bernstein, J. Facts and fictions about polymorphism. Chem. Soc. Rev. 2015, 44, 8619–8635. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Cabeza, A.J.; Feeder, N.; Davey, R.J. Open questions in organic crystal polymorphism. Commun. Chem. 2020, 3, 142. [Google Scholar] [CrossRef] [PubMed]
- Nogueira, B.A.; Castiglioni, C.; Fausto, R. Color polymorphism in organic crystals. Commun. Chem. 2020, 3, 34. [Google Scholar] [CrossRef] [PubMed]
- Steed, K.M.; Steed, J.W. Packing problems: High Z′ crystal structures and their relationship to cocrystals, inclusion compounds, and polymorphism. Chem. Rev. 2015, 115, 2895–2933. [Google Scholar] [CrossRef] [PubMed]
- Brock, C.P. High-Z′ structures of organic molecules: Their diversity and organizing principles. Acta Crystallogr. B Struct. Sci. Cryst. Eng. Mater. 2016, 72, 807–821. [Google Scholar] [CrossRef] [PubMed]
- Waddell, P.G. A lot to unpack: A decade in high Z ′ crystal structures. CrystEngComm 2025, 27, 578–589. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Crystallogr. B Struct. Sci. Cryst. Eng. Mater. 2016, 72, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Clegg, W. Crystallographic curiosities: Polymorphism and structures with Z′ 1. Acta Crystallogr. C Struct. Chem. 2019, 75, 833–834. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.; Srivastava, N.; Kaushal, A.; Das, B.; Vashistha, A.; Kumar, L.; Kumar, R.; Kumar Yadav, A. Synthesis, in Silico Study and Biological Evaluation of N-(Benzothiazol/Thiazol-2-yl)benzamide Derivatives as Quorum Sensing Inhibitors against Pseudomonas aeruginosa. Chem. Biodivers. 2023, 20, e202300647. [Google Scholar] [CrossRef] [PubMed]
- Potopnyk, M.A.; Lytvyn, R.; Danyliv, Y.; Ceborska, M.; Bezvikonnyi, O.; Volyniuk, D.; Gražulevičius, J.V. N,O π-Conjugated 4-Substituted 1,3-Thiazole BF2 Complexes: Synthesis and Photophysical Properties. J. Org. Chem. 2018, 83, 1095–1105. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhang, Y.; Li, X.; Zhou, Y.; Sheng, W. One pot synthesis and crystal of N-benzoyl(benzo)thiazol-2-amine borodifluoride complexes (NBO) with aggregation induced luminescence properties. J. Mol. Struct. 2025, 1321, 139761. [Google Scholar] [CrossRef]
- Yadav, P.; Ballabh, A. N-(thiazol-2-yl)benzamide derivatives as a new series of supramolecular gelators: Role of methyl functionality and S⋯O interaction. J. Solid State Chem. 2020, 281, 121027. [Google Scholar] [CrossRef]
- Madjroh, N.; Mellou, E.; Davies, P.A.; Söderhielm, P.C.; Jensen, A.A. Discovery and functional characterization of N-(thiazol-2-yl)-benzamide analogs as the first class of selective antagonists of the Zinc-Activated Channel (ZAC). Biochem. Pharmacol. 2021, 193, 114782. [Google Scholar] [CrossRef] [PubMed]
- Abbasi, M.A.; Raza, H.; Rehman, A.U.; Siddiqui, S.Z.; Nazir, M.; Mumtaz, A.; Shah, S.A.A.; Seo, S.-Y.; Hassan, M. Synthesis, Antioxidant and In-Silico Studies of Potent Urease Inhibitors: N-(4-{(4-Methoxyphenethyl)-(substituted)aminosulfonyl}phenyl)acetamides. Drug Res. 2019, 69, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Iino, T.; Tsukahara, D.; Kamata, K.; Sasaki, K.; Ohyama, S.; Hosaka, H.; Hasegawa, T.; Chiba, M.; Nagata, Y.; Eiki, J.; et al. Discovery of potent and orally active 3-alkoxy-5-phenoxy-N-thiazolyl benzamides as novel allosteric glucokinase activators. Bioorg. Med. Chem. 2009, 17, 2733–2743. [Google Scholar] [CrossRef] [PubMed]
- In, E.-J.; Lee, Y.; Koppula, S.; Kim, T.-Y.; Han, J.-H.; Lee, K.-H.; Kang, T.-B. Identification and Characterization of NTB451 as a Potential Inhibitor of Necroptosis. Molecules 2018, 23, 2884. [Google Scholar] [CrossRef] [PubMed]
- Sams, A.G.; Mikkelsen, G.K.; Larsen, M.; Langgård, M.; Howells, M.E.; Schrøder, T.J.; Brennum, L.T.; Torup, L.; Jørgensen, E.B.; Bundgaard, C.; et al. Discovery of phosphoric acid mono-{2-(E/Z)-4-(3,3-dimethyl-butyrylamino)-3,5-difluoro-benzoylimino-thiazol-3-ylmethyl} ester (Lu AA47070): A phosphonooxymethylene prodrug of a potent and selective hA(2A) receptor antagonist. J. Med. Chem. 2011, 54, 751–764. [Google Scholar] [CrossRef] [PubMed]
- Satoh, A.; Nagatomi, Y.; Hirata, Y.; Ito, S.; Suzuki, G.; Kimura, T.; Maehara, S.; Hikichi, H.; Satow, A.; Hata, M.; et al. Discovery and in vitro and in vivo profiles of 4-fluoro-N-4-6-(isopropylamino)pyrimidin-4-yl-1,3-thiazol-2-yl-N-methylbenzamide as novel class of an orally active metabotropic glutamate receptor 1 (mGluR1) antagonist. Bioorg. Med. Chem. Lett. 2009, 19, 5464–5468. [Google Scholar] [CrossRef] [PubMed]
- Uto, Y.; Ogata, T.; Kiyotsuka, Y.; Miyazawa, Y.; Ueno, Y.; Kurata, H.; Deguchi, T.; Yamada, M.; Watanabe, N.; Takagi, T.; et al. Novel and potent inhibitors of stearoyl-CoA desaturase-1. Part II: Identification of 4-ethylamino-3-(2-hydroxyethoxy)-N-5-(3-trifluoromethylbenzyl)thiazol-2-ylbenzamide and its biological evaluation. Bioorg. Med. Chem. Lett. 2009, 19, 4159–4166. [Google Scholar] [CrossRef] [PubMed]
- van Muijlwijk-Koezen, J.E.; Timmerman, H.; Vollinga, R.C.; Frijtag von Drabbe Künzel, J.; de Groote, M.; Visser, S.; IJzerman, A.P. Thiazole and thiadiazole analogues as a novel class of adenosine receptor antagonists. J. Med. Chem. 2001, 44, 749–762. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Qiu, X.-L.; Zhou, L.; Zhu, J.; Chamberlin, R.; Lau, J.; Chen, P.-L.; Lee, W.-H. Small molecule targeting the Hec1/Nek2 mitotic pathway suppresses tumor cell growth in culture and in animal. Cancer Res. 2008, 68, 8393–8399. [Google Scholar] [CrossRef] [PubMed]
- Zonouzi, A.; Mirzazadeh, R.; Rahmani, H.; Ng, S.W. N-(1,3-Thia-zol-2-yl)benzamide. Acta Crystallogr. Sect. E Struct. Rep. Online 2009, 65, o817. [Google Scholar] [CrossRef] [PubMed]
- Galal, S.A.; Abdelsamie, A.S.; Tokuda, H.; Suzuki, N.; Lida, A.; Elhefnawi, M.M.; Ramadan, R.A.; Atta, M.H.E.; El Diwani, H.I. Part I: Synthesis, cancer chemopreventive activity and molecular docking study of novel quinoxaline derivatives. Eur. J. Med. Chem. 2011, 46, 327–340. [Google Scholar] [CrossRef] [PubMed]
- Barnikow, G.; Bödeker, J. Isothiocyanate. XXV. Umsetzungen von Benzoylisothiocyanat mit Amino-N-heterocyclen. J. Prakt. Chem. 1971, 313, 1148–1154. [Google Scholar] [CrossRef]
- Clark, R.C.; Reid, J.S. The analytical calculation of absorption in multifaceted crystals. Acta Crystallogr. A Found. Crystallogr. 1995, 51, 887–897. [Google Scholar] [CrossRef]
- Rigaku. CrysAlisPro R; Tokyo, Japan. 2024. Available online: https://rigaku.com/products/crystallography/x-ray-diffraction/crysalispro (accessed on 10 June 2025).
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Etter, M.C. Encoding and decoding hydrogen-bond patterns of organic compounds. Acc. Chem. Res. 1990, 23, 120–126. [Google Scholar] [CrossRef]
- Wood, P.A.; Allen, F.H.; Pidcock, E. Hydrogen-bond directionality at the donor H atom—Analysis of interaction energies and database statistics. CrystEngComm 2009, 11, 1563. [Google Scholar] [CrossRef]
- Brock, C.P. Prevalence and Significance of Approximate Symmetry in Organic Pc Structures. Helv. Chim. Acta 2023, 106, e202200170. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymorph I | Polymorph II | Polymorph III | |
|---|---|---|---|
| Empirical formula | C10H8N2OS | C10H8N2OS | C10H8N2OS |
| Formula weight | 204.24 | 203.243 | 204.24 |
| Temperature/K | 123 | 298 | 150 |
| Crystal system | Monoclinic | Monoclinic | Monoclinic |
| Space group | P21/c | C2/c | Pc |
| a/Å | 12.0142 (2) | 15.9169 (5) | 20.3396 (3) |
| b/Å | 5.0581 (1) | 10.0631 (4) | 5.07500 (10) |
| c/Å | 15.4090 (3) | 12.5115 (5) | 20.3274 (3) |
| α/° | 90 | 90 | 90 |
| β/° | 99.093 (1) | 97.7186 (14) | 116.755 (2) |
| γ/° | 90 | 90 | 90 |
| Volume/Å3 | 924.62 (3) | 1985.85 (13) | 1873.62 (6) |
| Z, Z′ | 4, 1 | 8, 1 | 8, 4 |
| ρcalcg/cm3 | 1.467 | 1.360 | 1.448 |
| μ/mm−1 | 0.313 | 0.29 | 2.786 |
| F(000) | 424.0 | 848.0 * | 848.0 |
| Radiation | MoKα (λ = 0.71073) | MoKα (λ = 0.71073) | CuKα (λ = 1.54184) |
| 2Θ range for data collection/° | 3.44 to 55 | 6.02 to 60.1 | 4.866 to 154.336 |
| Index ranges | −15 ≤ h ≤ 15, −6 ≤ k ≤ 6, −18 ≤ l ≤ 20 | −22 ≤ h ≤ 22, −13 ≤ k ≤ 14, −17 ≤ l ≤ 17 | −24 ≤ h ≤ 25, −5 ≤ k ≤ 6, −25 ≤ l ≤ 25 |
| Reflections collected | 6130 | 5493 | 32148 |
| Independent reflections | 2104 [Rint = 0.0164] | 3427 [Rint = 0.030] | 7102 [Rint = 0.0436] |
| Data/restraints/parameters | 2104/0/131 | 3427/0/127 | 7102/3/518 |
| Goodness-of-fit on F2 | 1.074 | 1.723 | 1.018 |
| Final R indices [I >= 2σ (I)] | R1 = 0.0290, wR2 = 0.0842 | R1 = 0.0400, wR2 = 0.0690 | R1 = 0.0269, wR2 = 0.0652 |
| Largest diff. peak/hole/e Å−3 | 0.37/−0.21 | 0.43/−0.51 | 0.23/−0.17 |
| Flack parameter | n/a | n/a | 0.374 (12) |
| Polymorph I | Polymorph II | Polymorph III | |
|---|---|---|---|
| C10-C5-C1-N1/° * | 37.5 (2) | 25.01 (4) | 25.8 (4) |
| C20-C15-C11-N3/° | −31.0 (4) | ||
| C30-C25-C21-N5/° | 22.9 (5) | ||
| C40-C35-C31-N7/° | 42.0 (4) | ||
| S1-C2-N1-C1/° ** | 3.8 (2) | −4.04 (2) | −2.3 (4) |
| S2-C12-N3-C11/° | 2.6 (4) | ||
| S3-C22-N5-C21/° | −5.4 (4) | ||
| S4-C32-N7-C31/° | 7.7 (4) |
| D—H⋯A | Polymorph I | Polymorph II |
|---|---|---|
| d(D—H)/Å | 0.88 (2) | 0.960 (2) |
| d(H⋯A)/Å | 2.04 (2) | 2.030 (2) |
| d(D⋯A)/Å | 2.922 (2) | 2.900 (2) |
| (D—H⋯A)/° | 173 (2) | 149.9 (2) |
| Dimer A | Dimer B | |||
|---|---|---|---|---|
| D—H⋯A | N1—H1⋯N4 | N3—H3A⋯N2 | N5—H5⋯N8 | N7—H7A⋯N6 |
| d(D—H)/Å | 0.81 (2) | 0.83 (4) | 0.82 (3) | 0.83 (3) |
| d(H⋯A)/Å | 2.22 (2) | 2.09 (4) | 2.16 (4) | 2.21 (3) |
| d(D⋯A)/Å | 3.014 (3) | 2.918 (3) | 2.977 (3) | 3.014 (3) |
| (D—H⋯A)/° | 168 (3) | 173 (3) | 172 (3) | 164 (3) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Voigt, J.C.; Hall, M.J.; Waddell, P.G. Crystallographic Combinations: Understanding Polymorphism and Approximate Symmetry in N-(1,3-Thiazol-2-yl)benzamide. Crystals 2025, 15, 657. https://doi.org/10.3390/cryst15070657
Voigt JC, Hall MJ, Waddell PG. Crystallographic Combinations: Understanding Polymorphism and Approximate Symmetry in N-(1,3-Thiazol-2-yl)benzamide. Crystals. 2025; 15(7):657. https://doi.org/10.3390/cryst15070657
Chicago/Turabian StyleVoigt, Johannes C., Michael J. Hall, and Paul G. Waddell. 2025. "Crystallographic Combinations: Understanding Polymorphism and Approximate Symmetry in N-(1,3-Thiazol-2-yl)benzamide" Crystals 15, no. 7: 657. https://doi.org/10.3390/cryst15070657
APA StyleVoigt, J. C., Hall, M. J., & Waddell, P. G. (2025). Crystallographic Combinations: Understanding Polymorphism and Approximate Symmetry in N-(1,3-Thiazol-2-yl)benzamide. Crystals, 15(7), 657. https://doi.org/10.3390/cryst15070657