
First-Principles Investigation of the Effect of Vacancy Defects and Carbon Impurities on Thermal Conductivity of Uranium Mononitride (UN)

 ,
, 
Abstract
1. Introduction
2. Method
3. Results and Discussion
3.1. Structural Properties of Ideal UN
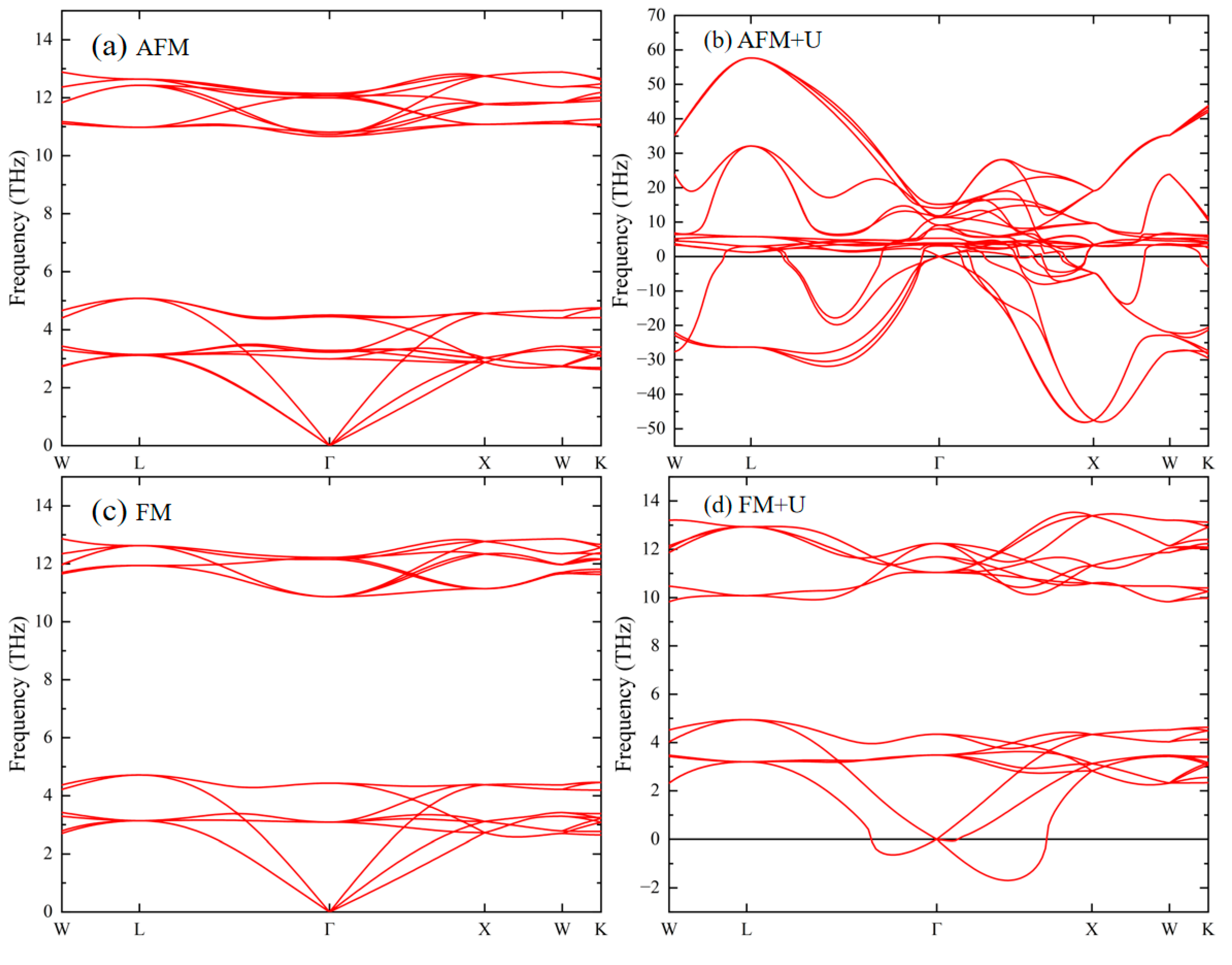
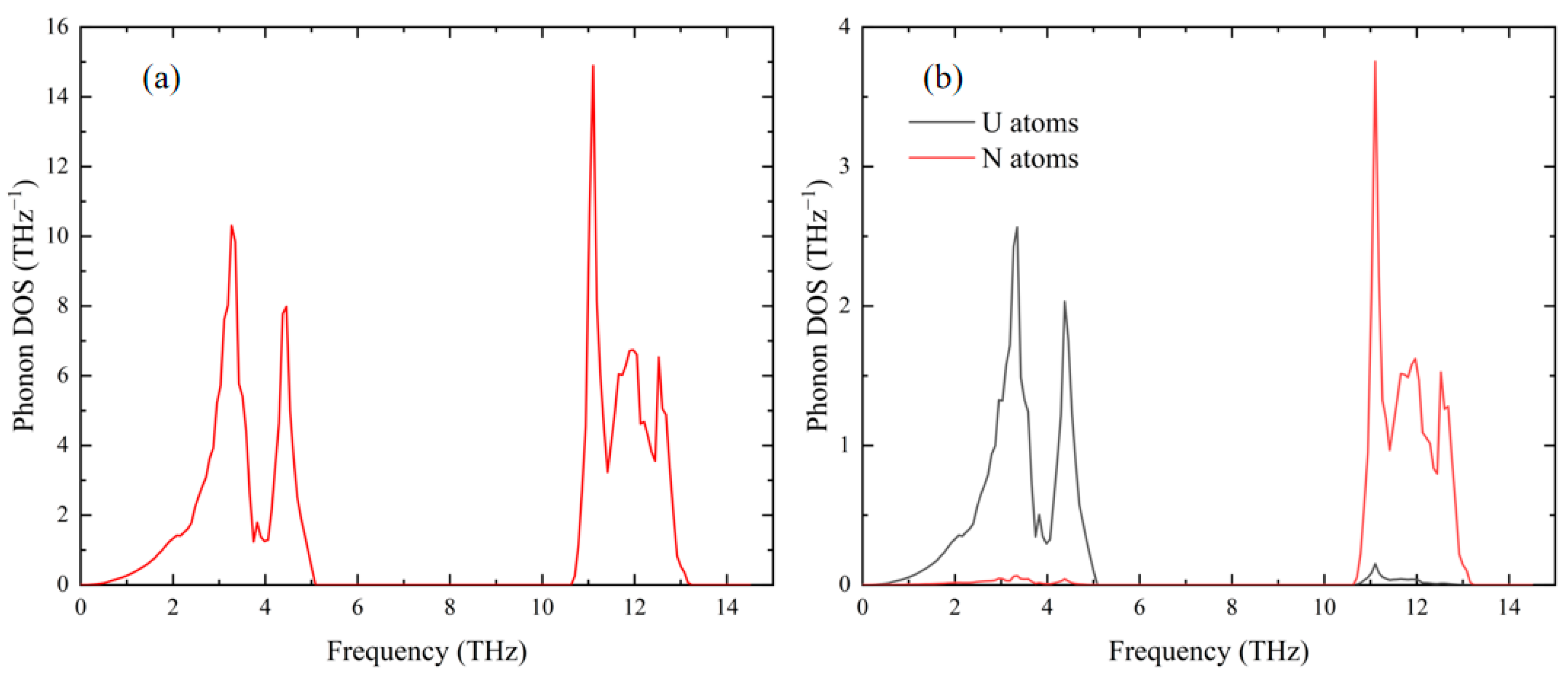
3.2. Phonon Properties of Ideal UN
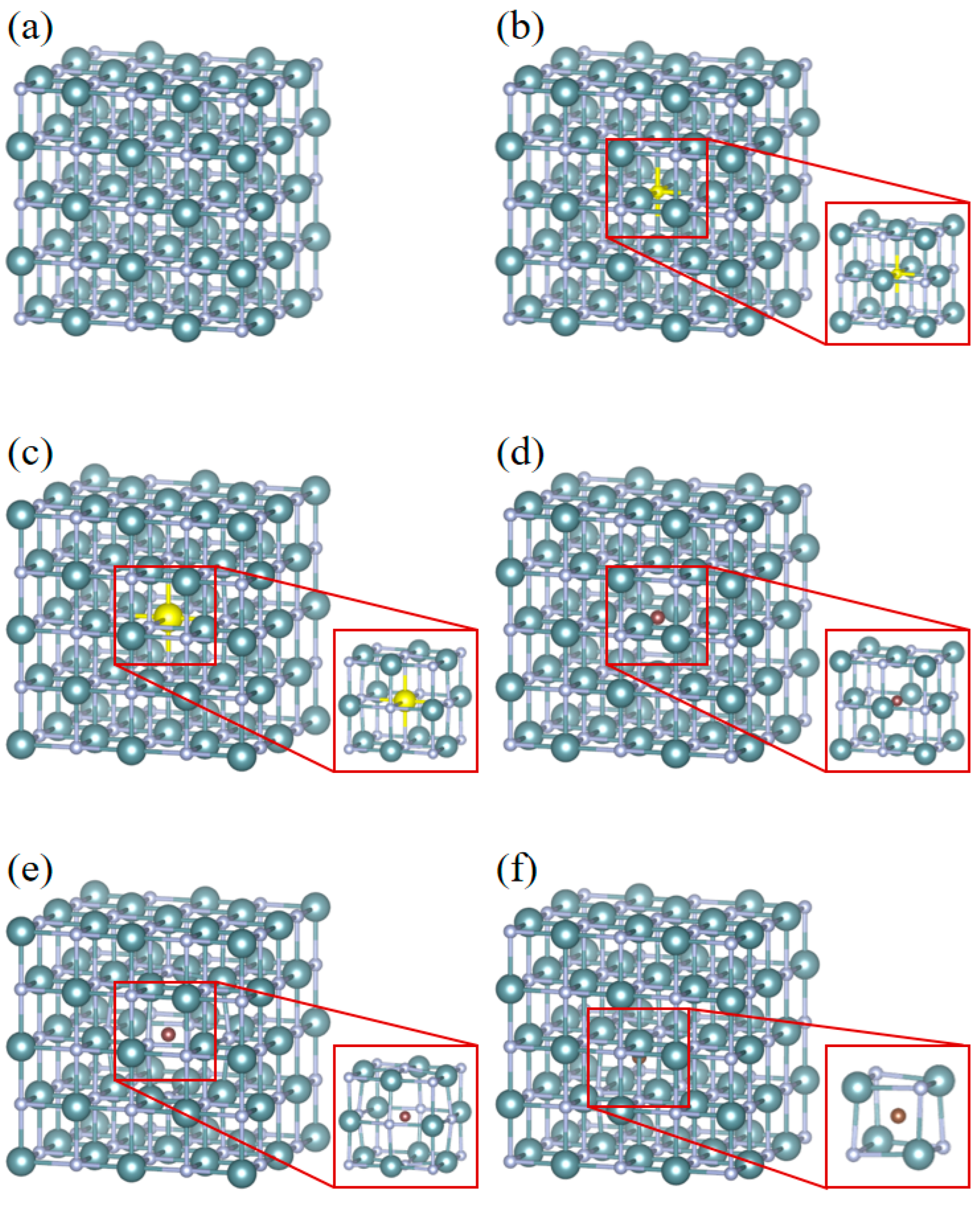
3.3. Energetics of Defect and Impurities
3.4. Mechanical Properties of Ideal and Defective UN
3.5. Thermal Transport Properties of Ideal and Defective UN
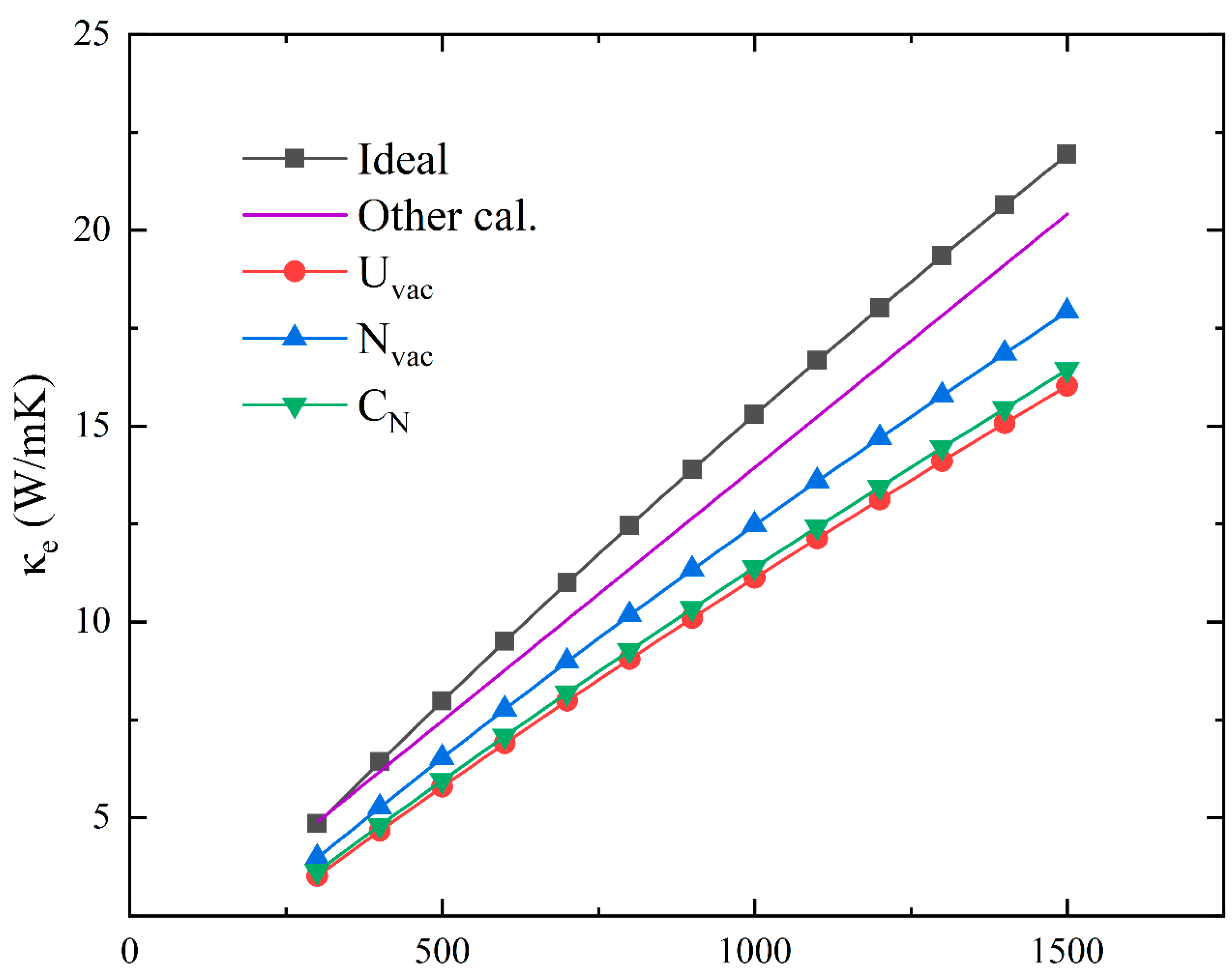
3.6. Electronic Thermal Conductivity of Ideal and Defective UN
3.7. Total Thermal Conductivity of Ideal and Defective UN
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhao, J.; Sun, D.; Xi, L.; Chen, P.; Zhao, J.; Wang, Y. Understanding xenon and vacancy behavior in UO2, UN and U3 Si2: A comparative DFT+ U study. Phys. Chem. Chem. Phys. 2023, 25, 14928–14941. [Google Scholar] [CrossRef]
- Fink, J.K.; Chasanov, M.G.; Leibowitz, L. Thermophysical properties of uranium dioxide. J. Nucl. Mater. 1981, 102, 17–25. [Google Scholar] [CrossRef]
- Qi, H.; Li, B.; Li, M.; Feng, S.; Hu, J.; Gong, H.; Ren, Q.; Liao, Y.; Xiao, H.; Zu, X. The effect of fission products Xe and Cs on the thermal conductivity of the U3 Si2 lattice: A first-principles study. J. Phys. Condens. Matter 2023, 35, 495701. [Google Scholar] [CrossRef] [PubMed]
- Li, D.S.; Garmestani, H.; Schwartz, J. Modeling thermal conductivity in UO2 with BeO additions as a function of microstructure. J. Nucl. Mater. 2009, 392, 22–27. [Google Scholar] [CrossRef]
- Szpunar, B.; Szpunar, J.A. Thermal conductivity of uranium nitride and carbide. Int. J. Nucl. Energy 2014, 2014, 178360. [Google Scholar] [CrossRef]
- Kocevski, V.; Rehn, D.A.; Cooper, M.W.D.; Andersson, D.A. First-principles investigation of uranium mononitride (UN): Effect of magnetic ordering, spin-orbit interactions and exchange correlation functional. J. Nucl. Mater. 2022, 559, 153401. [Google Scholar] [CrossRef]
- Lu, Y.; Wang, B.-T.; Li, R.-W.; Shi, H.; Zhang, P. Structural, electronic, and thermodynamic properties of UN: Systematic density functional calculations. J. Nucl. Mater. 2010, 406, 218–222. [Google Scholar] [CrossRef]
- Lan, J.-H.; Zhao, Z.-C.; Wu, Q.; Zhao, Y.-L.; Chai, Z.-F.; Shi, W.-Q. First-principles DFT+U modeling of defect behaviors in anti-ferromagnetic uranium mononitride. J. Appl. Phys. 2013, 114, 223516. [Google Scholar] [CrossRef]
- Lopes, D.A.; Claisse, A.; Olsson, P. Ab-initio study of C and O impurities in uranium nitride. J. Nucl. Mater. 2016, 478, 112–118. [Google Scholar] [CrossRef]
- Kocevski, V.; Rehn, D.A.; Terricabras, A.J.; Van Veelen, A.; Cooper, M.W.D.; Paisner, S.W.; Vogel, S.C.; White, J.T.; Andersson, D.A. Finite temperature properties of uranium mononitride. J. Nucl. Mater. 2023, 576, 154241. [Google Scholar] [CrossRef]
- Yang, L.; Kaltsoyannis, N. Incorporation of Kr and Xe in uranium mononitride: A density functional theory study. J. Phys. Chem. C 2021, 125, 26999–27008. [Google Scholar] [CrossRef]
- Samsel-Czekała, M.; Talik, E.; Du Plessis, P.D.V.; Troć, R.; Misiorek, H.; Sułkowski, C. Electronic structure and magnetic and transport properties of single-crystalline UN. Phys. Rev. B 2007, 76, 144426. [Google Scholar] [CrossRef]
- Yin, Q.; Kutepov, A.; Haule, K.; Kotliar, G.; Savrasov, S.Y.; Pickett, W.E. Electronic correlation and transport properties of nuclear fuel materials. Phys. Rev. B 2011, 84, 195111. [Google Scholar] [CrossRef]
- Szpunar, B.; Ranasinghe, J.I.; Malakkal, L.; Szpunar, J.A. First principles investigation of thermal transport of uranium mononitride. J. Phys. Chem. Solids 2020, 146, 109636. [Google Scholar] [CrossRef]
- Szpunar, B. First principles investigation of the electronic-thermal transport of ThN, UN, and ThC. Nucl. Mater. Energy 2022, 32, 101212. [Google Scholar] [CrossRef]
- Kurosaki, K.; Yano, K.; Yamada, K.; Uno, M.; Yamanaka, S. A molecular dynamics study of the thermal conductivity of uranium mononitride. J. Alloys Compd. 2000, 311, 305–310. [Google Scholar] [CrossRef]
- Webb, J.A.; Charit, I. Analytical determination of thermal conductivity of W–UO2 and W–UN CERMET nuclear fuels. J. Nucl. Mater. 2012, 427, 87–94. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 1993, 47, 558–561. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Hafner, J. Ab initio molecular-dynamics simulation of the liquid-metal–amorphous-semiconductor transition in germanium. Phys. Rev. B 1994, 49, 14251–14269. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Dudarev, S.L.; Botton, G.A.; Savrasov, S.Y.; Humphreys, C.J.; Sutton, A.P. Electron-energy-loss spectra and the structural stability of nickel oxide: An LSDA+U study. Phys. Rev. B 1998, 57, 1505–1509. [Google Scholar] [CrossRef]
- Togo, A. First-principles Phonon Calculations with Phonopy and Phono3py. J. Phys. Soc. Jpn. 2023, 92, 012001. [Google Scholar] [CrossRef]
- Togo, A.; Chaput, L.; Tadano, T.; Tanaka, I. Implementation strategies in phonopy and phono3py. J. Phys. Condens. Matter 2023, 35, 353001. [Google Scholar] [CrossRef] [PubMed]
- Madsen, G.K.H.; Carrete, J.; Verstraete, M.J. BoltzTraP2, a program for interpolating band structures and calculating semi-classical transport coefficients. Comput. Phys. Commun. 2018, 231, 140–145. [Google Scholar] [CrossRef]
- Morelli, D.T.; Slack, G.A. High Lattice Thermal Conductivity Solids. In High Thermal Conductivity Materials; Shindé, S.L., Goela, J.S., Eds.; Springer: New York, NY, USA, 2006; pp. 37–68. ISBN 978-0-387-22021-5. [Google Scholar] [CrossRef]
- Wang, V.; Xu, N.; Liu, J.-C.; Tang, G.; Geng, W.-T. VASPKIT: A user-friendly interface facilitating high-throughput computing and analysis using VASP code. Comput. Phys. Commun. 2021, 267, 108033. [Google Scholar] [CrossRef]
- Goncharov, V.G.; Liu, J.; Van Veelen, A.; Kriegsman, K.; Benmore, C.; Sun, C.; Kelly, S.; White, J.T.; Xu, H.; Guo, X. Energetics of oxidation and formation of uranium mononitride. J. Nucl. Mater. 2022, 569, 153904. [Google Scholar] [CrossRef]
- Adachi, J.; Kurosaki, K.; Uno, M.; Yamanaka, S.; Takano, M.; Akabori, M.; Minato, K. Mechanical properties at sub-microscale and macroscale of polycrystalline uranium mononitride. J. Nucl. Mater. 2009, 384, 6–11. [Google Scholar] [CrossRef]
- Mei, Z.G.; Stan, M.; Pichler, B. First-principles study of structural, elastic, electronic, vibrational and thermodynamic properties of UN. J. Nucl. Mater. 2013, 440, 63–69. [Google Scholar] [CrossRef]
- Bocharov, D.; Gryaznov, D.; Zhukovskii, Y.F.; Kotomin, E.A. DFT calculations of point defects on UN(001) surface. Surf. Sci. 2011, 605, 396–400. [Google Scholar] [CrossRef]
- Klipfel, M.; Van Uffelen, P. Ab initio modelling of volatile fission products in uranium mononitride. J. Nucl. Mater. 2012, 422, 137–142. [Google Scholar] [CrossRef]
- Li, Y.; Li, J.; Wu, W.; Gong, J.; Song, X.; Wang, Y.; Chen, Z. First-principles calculations to investigate mechanical, thermal, oxidation and hydrogen properties of Y, Zr and Nb alloyed U3Si2. Vacuum 2023, 215, 112269. [Google Scholar] [CrossRef]
- Waller, I. Dynamical Theory of Crystal Lattices by M. Born and K. Huang. Acta Crystallogr. 1956, 9, 837–838. [Google Scholar] [CrossRef]
- Kocevski, V.; Cooper, M.W.D.; Claisse, A.J.; Andersson, D.A. Development and application of a uranium mononitride (UN) potential: Thermomechanical properties and Xe diffusion. J. Nucl. Mater. 2022, 562, 153553. [Google Scholar] [CrossRef]
- Salleh, M.D.; MacDonald, J.E.; Saunders, G.A.; Du Plessis, P.D.V. Hydrostatic pressure dependences of elastic constants and vibrational anharmonicity of uranium nitride. J. Mater. Sci. 1986, 21, 2577–2580. [Google Scholar] [CrossRef]
- Hill, R. The elastic behaviour of a crystalline aggregate. Proc. Phys. Soc. A 1952, 65, 349–354. [Google Scholar] [CrossRef]
- Pugh, S.F. XCII. Relations between the elastic moduli and the plastic properties of polycrystalline pure metals. Lond. Edinb. Dublin Philos. Mag. J. Sci. 1954, 45, 823–843. [Google Scholar] [CrossRef]
- Ranganathan, S.I.; Ostoja-Starzewski, M. Universal elastic anisotropy index. Phys. Rev. Lett. 2008, 101, 055504. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Wang, Y.; Wang, Y.; Gao, M.; Chen, Y.; Chen, Z. First-principles study on the mechanical, thermal properties and hydrogen behavior of ternary V-Ni-M alloys. J. Mater. Sci. Technol. 2021, 70, 83–90. [Google Scholar] [CrossRef]
- Bannikov, V.V.; Shein, I.R.; Ivanovskii, A.L. Electronic structure, chemical bonding and elastic properties of the first thorium-containing nitride perovskite TaThN3. Phys. Status solidi (RRL)—Rapid Res. Lett. 2007, 1, 89–91. [Google Scholar] [CrossRef]
- Li, M.; Hu, J.; Gong, H.; Ren, Q.; Liao, Y.; Xiao, H.; Qiu, Q.; Feng, S.; Zu, X. First-principles study of point defects in U3 Si2: Effects on the mechanical and electronic properties. Phys. Chem. Chem. Phys. 2022, 24, 4287–4297. [Google Scholar] [CrossRef]
- Zhang, X.; Tang, J.; Deng, L.; Zhong, G.; Liu, X.; Li, Y.; Deng, H.; Hu, W. The effects of interstitial impurities on the mechanical properties of vanadium alloys: A first-principles study. J. Alloys Compd. 2017, 701, 975–980. [Google Scholar] [CrossRef]
- Bihan, T.L.; Idiri, M.; Heathman, S. New investigation of pressure-induced rhombohedral distortion of uranium nitride. J. Alloys Compd. 2003, 358, 120–125. [Google Scholar] [CrossRef]
- Moore, J.P.; Fulkerson, W.; Mcelroy, D.L. Thermal conductivity, electrical resistivity, and seebeck coefficient of uranium mononitride. J. Am. Ceram. Soc. 1970, 53, 76–82. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lattice Parameter (Å) | System Energy (eV/f.u.) | Magnetic Moment of U Atom () | ||
|---|---|---|---|---|
| This study | AFM | 4.858, 4.858, 4.882 | −22.176 | 1.050 |
| AFM + U | 4.962, 4.962, 4.848 | −20.608 | 1.538 | |
| FM | 4.871 | −22.229 | 1.347 | |
| FM + U | 4.920 | −20.585 | 1.792 | |
| Other cal. [6] | AFM | 4.853, 4.853, 4.872 | / | 0.99 |
| AFM + U | 4.898, 5.009, 4.956 | / | 1.83 | |
| FM | 4.861 | / | 1.21 | |
| FM + U | 4.939, 4.940, 4.940 | / | 1.79 | |
| Other cal. [11] | AFM | 4.865 | / | 1.05 |
| AFM + U | 4.927 | / | 1.59 | |
| FM | 4.868 | / | 1.25 | |
| FM + U | 4.916 | / | 1.71 | |
| Exp. [32] | 4.888 | / | / |
| This Work (eV) | Other Cal. [11] (eV) | Other Cal. [35] (eV) | |
|---|---|---|---|
| 3.36 | 3.17 | 3.74 | |
| 4.28 | 4.42 | 4.24 |
| (eV/Atom) | |
|---|---|
| −1.18 | |
| −1.29 | |
| −1.25 |
| (GPa) | (GPa) | (GPa) | ||
|---|---|---|---|---|
| Ideal | This work | 400.9 | 121.0 | 43.4 |
| Other cal. [33] | 404.6 | 124.2 | 45.0 | |
| Other cal. [38] | 416.5 | 115.1 | 71.0 | |
| Exp. [39] | 423.9 | 98.1 | 75.7 | |
| 373.1 | 111.0 | 36.3 | ||
| 371.6 | 116.5 | 39.4 | ||
| 409.3 | 123.9 | 45.7 |
| B (GPa) | G (GPa) | E (GPa) | |||
|---|---|---|---|---|---|
| Ideal | This work | 216.6 | 69.8 | 189.2 | 0.354 |
| Other cal. [33] | 217.7 | 72.4 | 195.1 | 0.350 | |
| Exp. [39] | 205.9 | 103.9 | 229 | 0.263 | |
| Exp. [47] | 194 | / | / | / | |
| 201.2 | 62.0 | 168.6 | 0.360 | ||
| 203.4 | 63.4 | 172.3 | 0.359 | ||
| 220.4 | 72.8 | 196.8 | 0.351 |
| (m/s) | (m/s) | (m/s) | (K) | |||
|---|---|---|---|---|---|---|
| Ideal | This work | 4616.47 | 2192.11 | 2466.10 | 2.17 | 301.79 |
| Exp. [32,39] | 4378 | 2507 | / | 1.98 | 282 | |
| 4473.05 | 2089.93 | 2353.04 | 2.22 | 287.01 | ||
| 4447.95 | 2087.29 | 2349.56 | 2.21 | 286.33 | ||
| 4670.15 | 2236.47 | 2514.90 | 2.14 | 307.98 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lan, Y.; Rui, T.; Ma, Z.; Lu, L.; Wang, Y.; Yu, Y.; Deng, M.; Lan, T.; Zhao, Z.; Wang, J.; et al. First-Principles Investigation of the Effect of Vacancy Defects and Carbon Impurities on Thermal Conductivity of Uranium Mononitride (UN). Crystals 2025, 15, 459. https://doi.org/10.3390/cryst15050459
Lan Y, Rui T, Ma Z, Lu L, Wang Y, Yu Y, Deng M, Lan T, Zhao Z, Wang J, et al. First-Principles Investigation of the Effect of Vacancy Defects and Carbon Impurities on Thermal Conductivity of Uranium Mononitride (UN). Crystals. 2025; 15(5):459. https://doi.org/10.3390/cryst15050459
Chicago/Turabian StyleLan, Yulin, Tianhao Rui, Zhuangzhuang Ma, Linyuan Lu, Yunhao Wang, Yang Yu, Mingxuan Deng, Tianxing Lan, Zhekang Zhao, Junjie Wang, and et al. 2025. "First-Principles Investigation of the Effect of Vacancy Defects and Carbon Impurities on Thermal Conductivity of Uranium Mononitride (UN)" Crystals 15, no. 5: 459. https://doi.org/10.3390/cryst15050459
APA StyleLan, Y., Rui, T., Ma, Z., Lu, L., Wang, Y., Yu, Y., Deng, M., Lan, T., Zhao, Z., Wang, J., Li, C., & Zhang, H. (2025). First-Principles Investigation of the Effect of Vacancy Defects and Carbon Impurities on Thermal Conductivity of Uranium Mononitride (UN). Crystals, 15(5), 459. https://doi.org/10.3390/cryst15050459