

Novel Quaternary Ammonium Aldimine Derivatives Featuring 3,4,5-Trimethoxy Phenyl Fragment: Synthesis, Crystal Structure and Evaluation of Antioxidant and Antibacterial Activity


Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. General Synthesis Procedure
2.2.1. Synthesis of 1-(Pyridin-4-yl)-N-(3,4,5-trimethoxyphenyl)methanimine (2)
2.2.2. Synthesis of 1-(2-(3-Nitrophenyl)-2-oxoethyl)-4-(((3,4,5-trimethoxyphenyl)imino)methyl)pyridin-1-ium bromide (3a)
2.2.3. Synthesis of 1-(2-(3,4-Dihydro-2H-benzo[b][1,4]dioxepin-7-yl)-2-oxoethyl)-4-(((3,4,5-trimethoxyphenyl)imino)methyl)pyridin-1-ium bromide (3b)
2.2.4. Synthesis of 1-(2-(Naphthalen-2-yl)-2-oxoethyl)-4-(((3,4,5-trimethoxyphenyl)imino)methyl)pyridin-1-ium bromide (3c)
2.2.5. Synthesis of 1-(2-([1,1′-Biphenyl]-4-yl)-2-oxoethyl)-4-(((3,4,5-trimethoxyphenyl)imino)methyl)pyridin-1-ium bromide (3d)
2.2.6. Synthesis of 1-(2-(2,5-Dimethoxyphenyl)-2-oxoethyl)-4-(((3,4,5-trimethoxyphenyl)imino)methyl)pyridin-1-ium bromide (3e)
2.3. Characterization Methods
2.3.1. Single-Crystal X-ray Diffraction (SCXRD) Analysis
2.3.2. Powder X-ray Diffraction (PXRD) Analysis
2.3.3. Nuclear Magnetic Resonance (NMR) Spectroscopy
2.3.4. Differential Scanning Calorimetry (DSC)
2.3.5. Fourier-Transform Infrared (FT-IR) Spectroscopy
2.3.6. High-Resolution Mass Spectrometry (HR-MS)
2.3.7. Antioxidative Activity Assays
2.3.8. Bacterial Susceptibility Testing—Disk-Diffusion and Broth Microdilution Methods
3. Results and Discussion
3.1. Synthesis
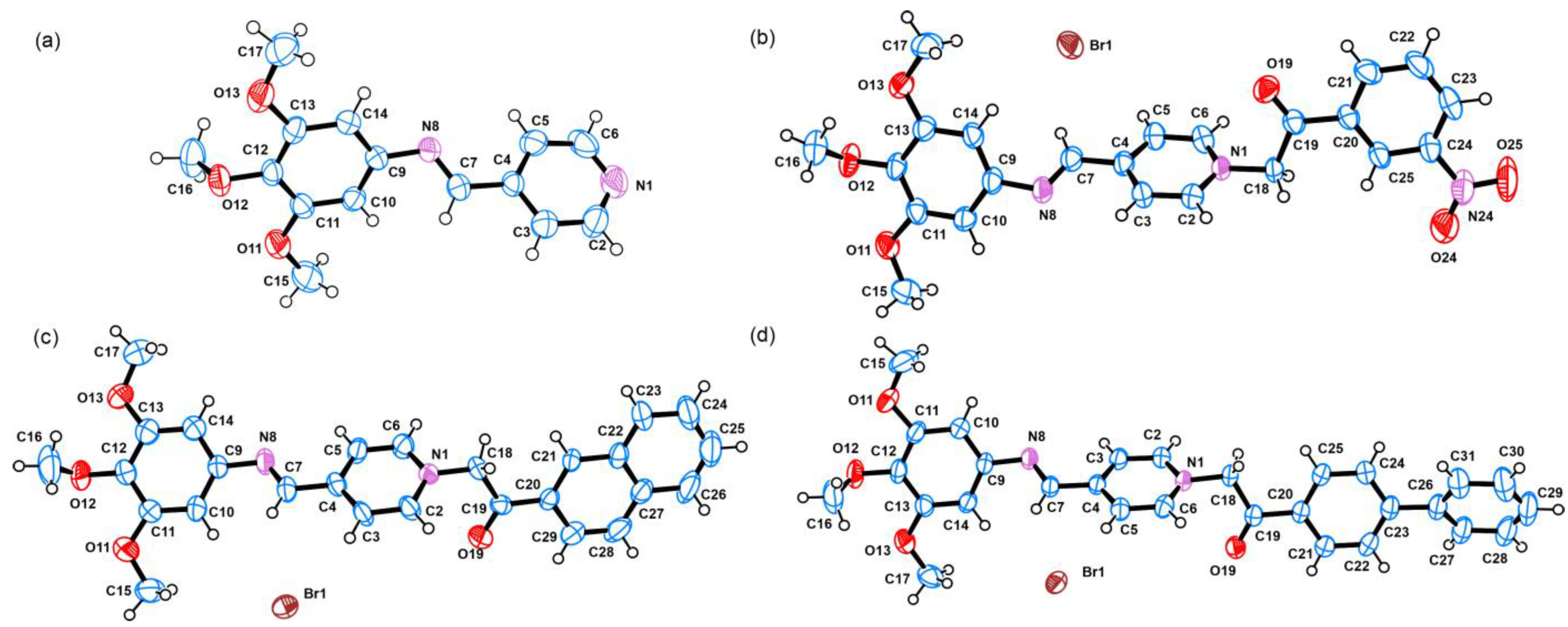
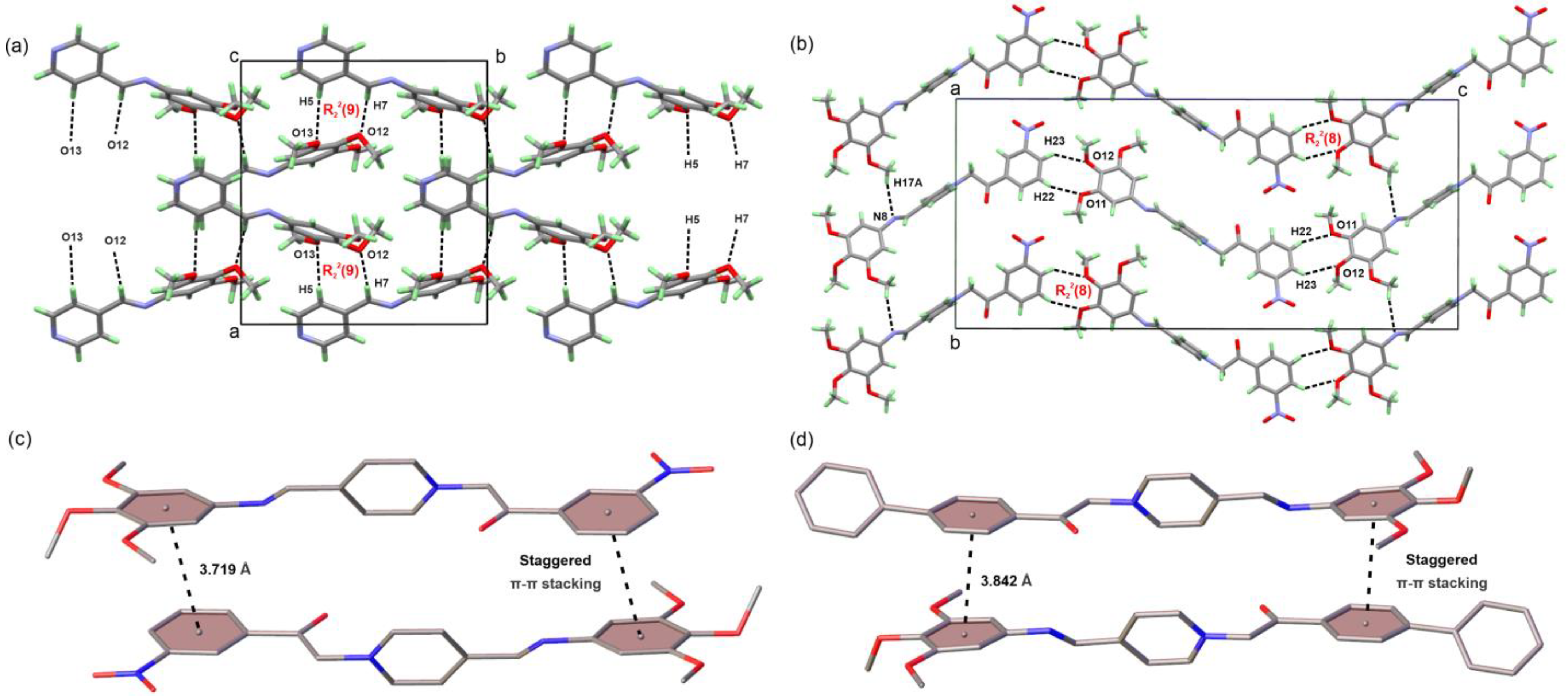
3.2. Crystal Structure Solution and Refinement
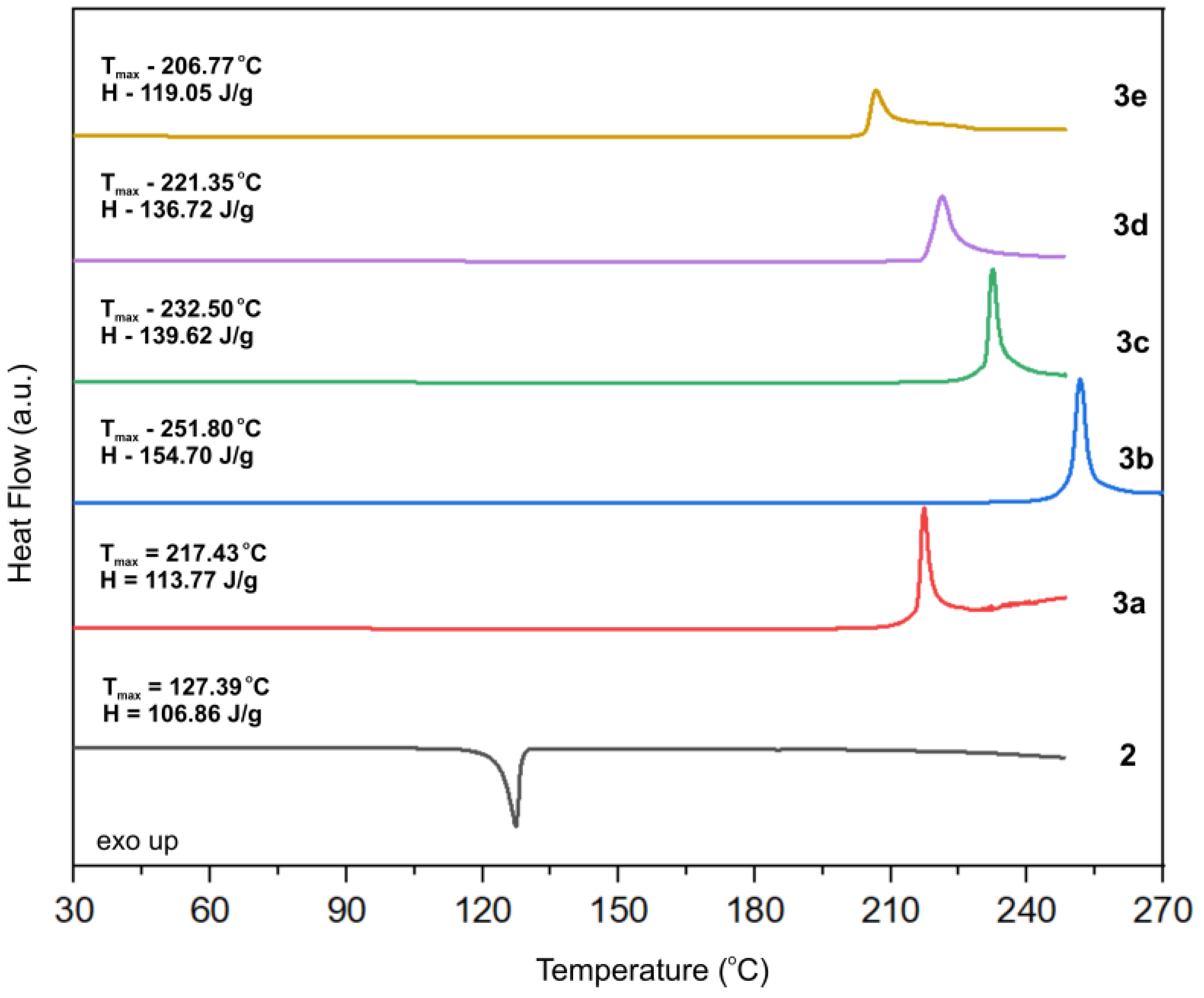
3.3. Differential Scanning Calorimetry Studies
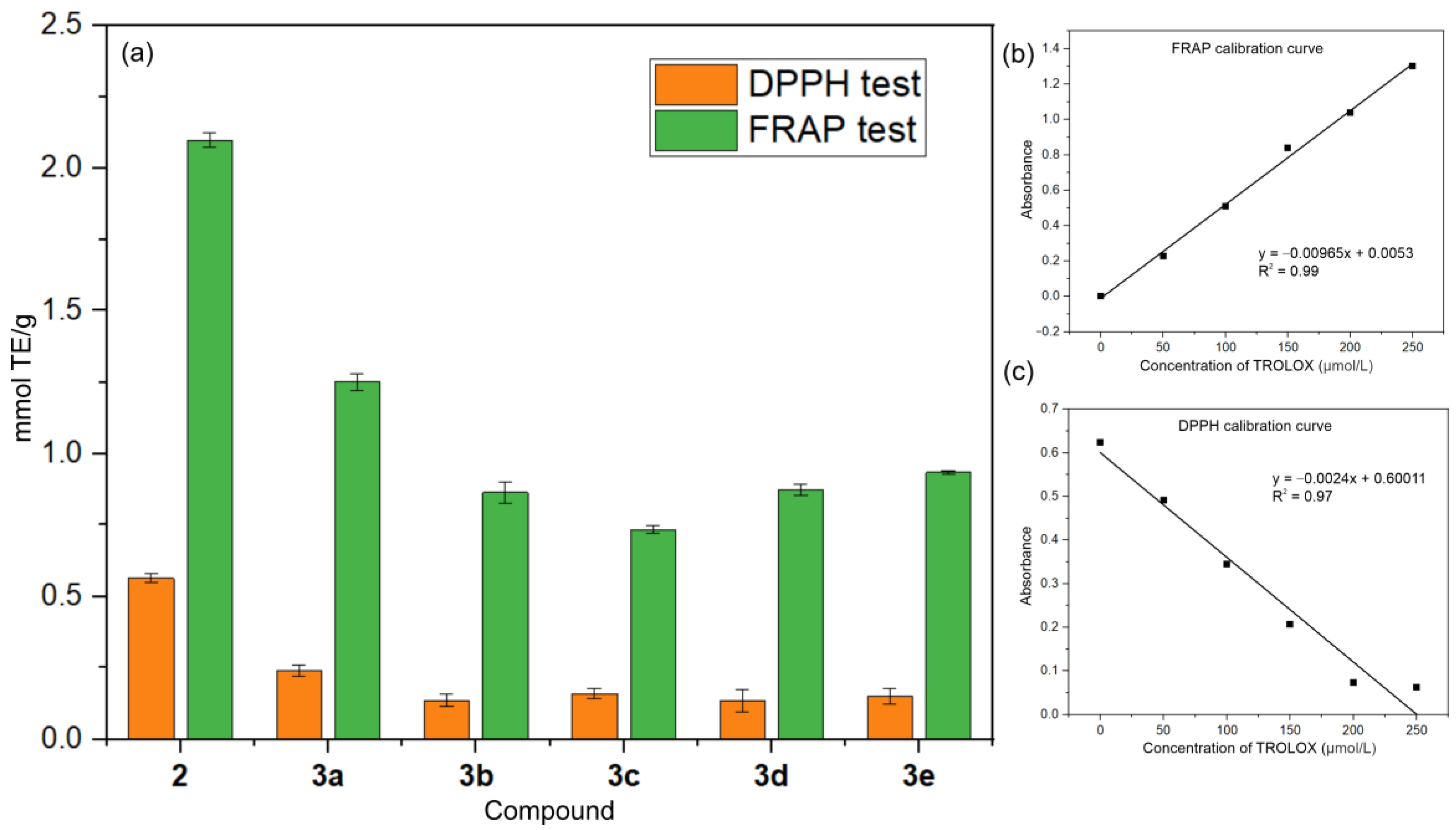
3.4. Antioxidant Activity Studies
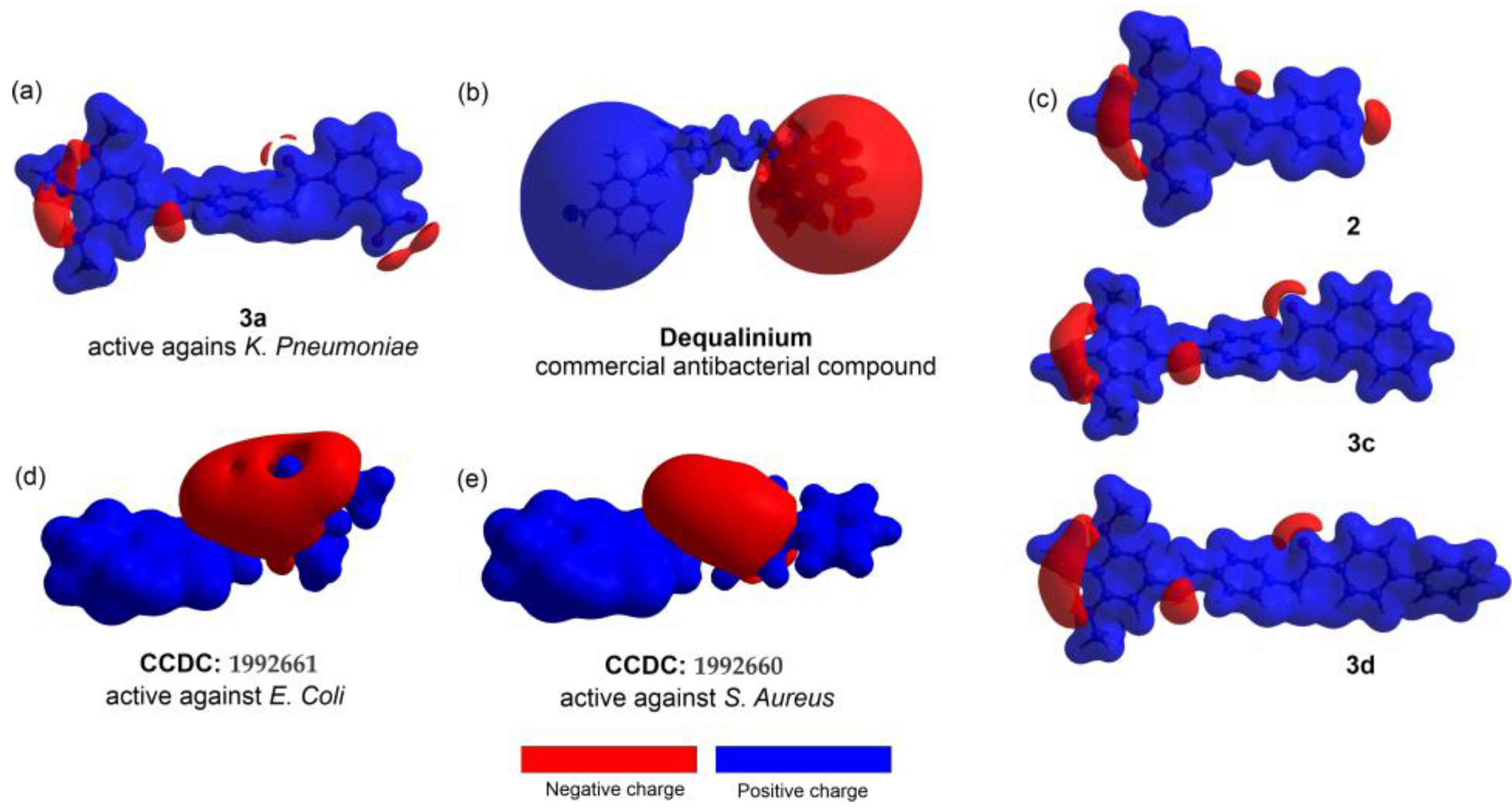
3.5. Antibacterial Susceptibility Testing and Activity
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Walsh, T.R.; Gales, A.C.; Laxminarayan, R.; Dodd, P.C. Antimicrobial Resistance: Addressing a Global Threat to Humanity. PLoS Med. 2023, 20, e1004264. [Google Scholar]
- Tangcharoensathien, V.; Sunicha, C.; Angkana, S. Complex Determinants of Inappropriate Use of Antibiotics. Bull. World Health Organ. 2018, 96, 141–144. [Google Scholar] [CrossRef]
- Brown, E.D.; Wright, G.D. Antibacterial Drug Discovery in the Resistance Era. Nature 2016, 529, 336–343. [Google Scholar] [CrossRef]
- Luepke, K.H.; Suda, K.J.; Boucher, H.; Russo, R.L.; Bonney, M.W.; Hunt, T.D.; Mohr, J.F., III. Past, Present, and Future of Antibacterial Economics: Increasing Bacterial Resistance, Limited Antibiotic Pipeline, and Societal Implications. Pharmacotherapy 2017, 37, 71–84. [Google Scholar] [CrossRef]
- Belete, T.M. Novel Targets to Develop New Antibacterial Agents and Novel Alternatives to Antibacterial Agents. Hum. Microbiome J. 2019, 11, 100052. [Google Scholar] [CrossRef]
- Simoes, N.G.; Bettencourt, A.F.; Monge, N.; Ribeiro, I.A.C. Novel Antibacterial Agents: An Emergent Need to Win the Battle against Infections. Mini Rev. Med. Chem. 2017, 17, 1364–1376. [Google Scholar] [CrossRef]
- Vila, J.; Moreno-Morales, J.; Ballesté-Delpierre, C. Current Landscape in the Discovery of Novel Antibacterial Agents. Clin. Microbiol. Infect. 2020, 26, 596–603. [Google Scholar] [CrossRef]
- Wright, G.D. Opportunities for Natural Products in 21 St Century Antibiotic Discovery. Nat. Prod. Rep. 2017, 34, 694–701. [Google Scholar] [CrossRef]
- Garcia-Gutierrez, E.; Mayer, M.J.; Cotter, P.D.; Narbad, A. Gut Microbiota as a Source of Novel Antimicrobials. Gut Microbes 2019, 10, 1–21. [Google Scholar] [CrossRef]
- Romano, G.; Costantini, M.; Sansone, C.; Lauritano, C.; Ruocco, N.; Ianora, A. Marine Microorganisms as a Promising and Sustainable Source of Bioactive Molecules. Mar. Environ. Res. 2017, 128, 58–69. [Google Scholar] [CrossRef]
- Walsh, C.T.; Wencewicz, T.A. Prospects for New Antibiotics: A Molecule-Centered Perspective. J. Antibiot. 2014, 67, 7–22. [Google Scholar] [CrossRef]
- Wright, P.M.; Seiple, I.B.; Myers, A.G. The Evolving Role of Chemical Synthesis in Antibacterial Drug Discovery. Angew. Chem. Int. Ed. 2014, 53, 8840–8869. [Google Scholar] [CrossRef]
- Mitcheltree, M.J.; Pisipati, A.; Syroegin, E.A.; Silvestre, K.J.; Klepacki, D.; Mason, J.D.; Terwilliger, D.W.; Testolin, G.; Pote, A.R.; Wu, K.J.Y.; et al. A Synthetic Antibiotic Class Overcoming Bacterial Multidrug Resistance. Nature 2021, 599, 507–512. [Google Scholar] [CrossRef]
- Sinn, E.; Harris, C.M. Schiff Base Metal Complexes as Ligands. Coord. Chem. Rev. 1969, 4, 391–422. [Google Scholar] [CrossRef]
- Yamada, S. Advancement in Stereochemical Aspects of Schiff Base Metal Complexes. Coord. Chem. Rev. 1999, 190, 537–555. [Google Scholar] [CrossRef]
- Bilyj, J.K.; Silajew, N.V.; Bernhardt, P.V. Nickel Coordination Chemistry of Bis(Dithiocarbazate) Schiff Base Ligands; Metal and Ligand Centered Redox Reactions. Dalton Trans. 2021, 50, 612–623. [Google Scholar] [CrossRef]
- Goel, A.; Malhotra, R. Efficient Detection of Picric Acid by Pyranone Based Schiff Base as a Chemosensor. J. Mol. Struct. 2022, 1249, 131619. [Google Scholar] [CrossRef]
- Jafari, H.; Ameri, E.; Rezaeivala, M.; Berisha, A.; Halili, J. Anti-Corrosion Behavior of Two N2O4 Schiff-Base Ligands: Experimental and Theoretical Studies. J. Phys. Chem. Solids 2022, 164, 110645. [Google Scholar] [CrossRef]
- Al Zoubi, W.; Ko, Y.G. Schiff Base Complexes and Their Versatile Applications as Catalysts in Oxidation of Organic Compounds: Part I. Appl. Organomet. Chem. 2017, 31, e3574. [Google Scholar] [CrossRef]
- Paşa, S.; Arslan, N.; Meriç, N.; Kayan, C.; Bingül, M.; Durap, F.; Aydemir, M. Boron Containing Chiral Schiff Bases: Synthesis and Catalytic Activity in Asymmetric Transfer Hydrogenation (Ath) of Ketones. J. Mol. Struct. 2020, 1200, 127064. [Google Scholar] [CrossRef]
- Gusev, A.N.; Kiskin, M.A.; Braga, E.V.; Kryukova, M.A.; Baryshnikov, G.V.; Karaush-Karmazin, N.N.; Minaeva, V.A.; Minaev, B.F.; Ivaniuk, K.; Stakhira, P. Schiff Base Zinc (II) Complexes as Promising Emitters for Blue Organic Light-Emitting Diodes. ACS Appl. Electron. Mater. 2021, 3, 3436–3444. [Google Scholar] [CrossRef]
- Nayak, A.; Naik, P.H.B.; Teja, H.B.; Kirthan, B.R.; Viswanath, R. Synthesis and Opto-Electronic Properties of Green Light Emitting Metal Schiff Base Complexes. Mol. Cryst. Liq. Cryst. 2021, 722, 67–75. [Google Scholar] [CrossRef]
- Imran, S.; Taha, M.; Ismail, N.H.; Khan, K.M.; Naz, F.; Hussain, M.; Tauseef, S. Synthesis of Novel Bisindolylmethane Schiff Bases and Their Antibacterial Activity. Molecules 2014, 19, 11722–11740. [Google Scholar] [CrossRef]
- Nair, R.; Shah, A.; Baluja, S.; Chanda, S. Synthesis and Antibacterial Activity of Some Schiff Base Complexes. J. Serb. Chem. Soc. 2006, 71, 733–744. [Google Scholar] [CrossRef]
- Yousif, E.; Majeed, A.; Al-Sammarrae, K.; Salih, N.; Salimon, J.; Abdullah, B. Metal Complexes of Schiff Base: Preparation, Characterization and Antibacterial Activity. Arab. J. Chem. 2017, 10, S1639–S1644. [Google Scholar] [CrossRef]
- Miloud, M.M.; El-Ajaily, M.M.; Al-Noor, T.H.; Al-Barki, N.S. Antifungal Activity of Some Mixed Ligand Complexes Incorporating Schiff Bases. J. Bacteriol. Mycol. 2020, 7, 1122. [Google Scholar]
- Joshi, R.; Kumari, A.; Singh, K.; Mishra, H.; Pokharia, S. Triorganotin (IV) Complexes of Schiff Base Derived from 1, 2, 4-Triazole Moiety: Synthesis, Spectroscopic Investigation, DFT Studies, Antifungal Activity and Molecular Docking Studies. J. Mol. Struct. 2020, 1206, 127639. [Google Scholar] [CrossRef]
- Hazra, S.; Paul, A.; Sharma, G.; Koch, B.; Guedes da Silva, M.F.C.; Pombeiro, A.J.L. Sulfonated Schiff Base Sn(IV) Complexes as Potential Anticancer Agents. J. Inorg. Biochem. 2016, 162, 83–95. [Google Scholar] [CrossRef]
- Gou, Y.; Li, J.; Fan, B.; Xu, B.; Zhou, M.; Yang, F. Structure and Biological Properties of Mixed-Ligand Cu(II) Schiff Base Complexes as Potential Anticancer Agents. Eur. J. Med. Chem. 2017, 134, 207–217. [Google Scholar] [CrossRef]
- Ramadhan, U.H.; Haddad, H.M.; Ezaria, Z.G. Synthesis of Schiff Bases Complexes as Anti-Inflammatory Agents. World J. Pharm. Pharm. Sci. 2016, 5, 98–108. [Google Scholar]
- Azam, M.; Al-Resayes, S.I.; Trzesowska-Kruszynska, A.; Kruszynski, R.; Shakeel, F.; Soliman, S.M.; Alam, M.; Khan, M.R.; Wabaidur, S.M. Zn(II) Complex Derived from Bidentate Schiff Base Ligand: Synthesis, Characterization, DFT Studies and Evaluation of Anti-Inflammatory Activity. J. Mol. Struct. 2020, 1201, 127177. [Google Scholar] [CrossRef]
- Jarrahpour, A.; Shirvani, P.; Sharghi, H.; Aberi, M.; Sinou, V.; Latour, C.; Brunel, J.M. Synthesis of Novel Mono- and Bis-Schiff Bases of Morpholine Derivatives and the Investigation of Their Antimalarial and Antiproliferative Activities. Med. Chem. Res. 2015, 24, 4105–4112. [Google Scholar] [CrossRef]
- Shaikh, I.; Travadi, M.; Jadeja, R.N.; Butcher, R.J.; Pandya, J.H. Crystal Feature and Spectral Characterization of Zn(II) Complexes Containing Schiff Base of Acylpyrazolone Ligand with Antimalarial Action. J. Indian Chem. Soc. 2022, 99, 100428. [Google Scholar] [CrossRef]
- Wang, Y.-Y.; Xu, F.-Z.; Zhu, Y.-Y.; Song, B.; Luo, D.; Yu, G.; Chen, S.; Xue, W.; Wu, J. Pyrazolo[3,4-d]pyrimidine Derivatives Containing a Schiff Base Moiety as Potential Antiviral Agents. Bioorg. Med. Chem. Lett. 2018, 28, 2979–2984. [Google Scholar] [CrossRef]
- Mansour, M.A.; AboulMagd, A.M.; Abdel-Rahman, H.M. Quinazoline-Schiff Base Conjugates: In Silico Study and ADMET Predictions as Multi-Target Inhibitors of Coronavirus (SARS-CoV-2) Proteins. RSC Adv. 2020, 10, 34033–34045. [Google Scholar] [CrossRef]
- Anouar, E.H.; Raweh, S.; Bayach, I.; Taha, M.; Baharudin, M.S.; Di Meo, F.; Hasan, M.H.; Adam, A.; Ismail, N.H.; Weber, J.-F.F. Antioxidant Properties of Phenolic Schiff Bases: Structure–Activity Relationship and Mechanism of Action. J. Comput. Aided Mol. Des. 2013, 27, 951–964. [Google Scholar] [CrossRef]
- Kumar, D.; Rawat, D.S. Synthesis and Antioxidant Activity of Thymol and Carvacrol Based Schiff Bases. Bioorg. Med. Chem. Lett. 2013, 23, 641–645. [Google Scholar]
- Garcia, M.T.; Kaczerewska, O.; Ribosa, I.; Brycki, B.; Materna, P.; Drgas, M. Biodegradability and Aquatic Toxicity of Quaternary Ammonium-Based Gemini Surfactants: Effect of the Spacer on Their Ecological Properties. Chemosphere 2016, 154, 155–160. [Google Scholar] [CrossRef]
- Kaneko, S.; Kumatabara, Y.; Shirakawa, S. A New Generation of Chiral Phase-Transfer Catalysts. Org. Biomol. Chem. 2016, 14, 5367–5376. [Google Scholar] [CrossRef]
- Zhuravlev, O.E.; Kaftanov, A.D.; Yulmasov, G.S.; Voronchikhina, L.I. Synthesis and Thermal Stability of Bis-Quaternary Ammonium Ionic Liquids with Inorganic Anions. Russ. J. Appl. Chem. 2023, 96, 395–401. [Google Scholar] [CrossRef]
- Yoganand, K.S.; Umapathy, M.J. Corrosion Inhibition Efficiency of Newly Synthesized Quaternary Ammonium Salt in 1M HCl. Indian J. Chem. Technol. 2022, 29, 68–74. [Google Scholar]
- Kasibhatla, S.; Amarante-Mendes, G.P.; Finucane, D.; Brunner, T.; Bossy-Wetzel, E.; Green, D.R. Acridine Orange/Ethidium Bromide (AO/EB) Staining to Detect Apoptosis. Cold Spring Harb. Protoc. 2006, 3, pdb.prot4493. [Google Scholar] [CrossRef]
- Kumar, R.; Saneja, A.; Panda, A.K. An Annexin V-FITC—Propidium Iodide-Based Method for Detecting Apoptosis in a Non-Small Cell Lung Cancer Cell Line. In Lung Cancer: Methods in Molecular Biology; Humana: New York, NY, USA, 2021; pp. 213–223. [Google Scholar]
- Feoktistova, M.; Geserick, P.; Leverkus, M. Crystal Violet Assay for Determining Viability of Cultured Cells. Cold Spring Harb. Protoc. 2016, 4, pdb.prot087379. [Google Scholar] [CrossRef]
- Basilico, N.; Migotto, M.; Ilboudo, D.P.; Taramelli, D.; Stradi, R.; Pini, E. Modified Quaternary Ammonium Salts as Potential Antimalarial Agents. Bioorg. Med. Chem. 2015, 23, 4681–4687. [Google Scholar] [CrossRef]
- Zhang, L.; Feng, X.-Z.; Xiao, Z.-Q.; Fan, G.-R.; Chen, S.-X.; Liao, S.-L.; Luo, H.; Wang, Z.-D. Design, Synthesis, Antibacterial, Antifungal and Anticancer Evaluations of Novel β-Pinene Quaternary Ammonium Salts. Int. J. Mol. Sci. 2021, 22, 11299. [Google Scholar] [CrossRef]
- Jin, G.; Xiao, F.; Li, Z.; Qi, X.; Zhao, L.; Sun, X. Design, Synthesis, and Dual Evaluation of Quinoline and Quinolinium Iodide Salt Derivatives as Potential Anticancer and Antibacterial Agents. ChemMedChem 2020, 15, 600–609. [Google Scholar] [CrossRef]
- Zhang, W.; Chang, Y.; Zhong, W.; Zhang, A.; Lin, Y. Antifungal Mechanisms of Polymeric Quaternary Ammonium Salts against Conidia of Fusarium oxysporum f. sp. cubense, Race 4. Eur. J. Plant Pathol. 2023, 165, 317–331. [Google Scholar] [CrossRef]
- Peng, Y.; Chang, J.; Xiao, Z.; Huang, J.; Xu, T.; Chen, S.; Fan, G.; Liao, S.; Wang, Z.; Luo, H. Synthesis and Antifungal Activity of Novel Tetrahydrogeranyl Quaternary Ammonium Salts. Nat. Prod. Commun. 2022, 17. [Google Scholar] [CrossRef]
- Masters, P.A.; O’Bryan, T.A.; Zurlo, J.; Miller, D.Q.; Joshi, N. Trimethoprim-Sulfamethoxazole Revisited. Arch. Intern. Med. 2003, 163, 402–410. [Google Scholar] [CrossRef]
- Allegra, C.J.; Chabner, B.A.; Tuazon, C.U.; Ogata-Arakaki, D.; Baird, B.; Drake, J.C.; Simmons, J.T.; Lack, E.E.; Shelhamer, J.H.; Balis, F. Trimetrexate for the Treatment of Pneumocystis Carinii Pneumonia in Patients with the Acquired Immunodeficiency Syndrome. New Engl. J. Med. 1987, 317, 978–985. [Google Scholar] [CrossRef]
- Vogelzang, N.J.; Weissman, L.B.; Herndon, J.E., 2nd; Antman, K.H.; Cooper, M.R.; Corson, J.M.; Green, M.R. Trimetrexate in Malignant Mesothelioma: A Cancer and Leukemia Group B Phase II Study. J. Clin. Oncol. 1994, 12, 1436–1442. [Google Scholar] [CrossRef]
- Sarris, A.H.; Phan, A.; Duvic, M.; Romaguera, J.; McLaughlin, P.; Mesina, O.; King, K.; Medeiros, L.J.; Rassidakis, G.Z.; Samuels, B. Trimetrexate in Relapsed T-Cell Lymphoma with Skin Involvement. J. Clin. Oncol. 2002, 20, 2876–2880. [Google Scholar] [CrossRef]
- Canel, C.; Moraes, R.M.; Dayan, F.E.; Ferreira, D. Podophyllotoxin. Phytochemistry 2000, 54, 115–120. [Google Scholar] [CrossRef]
- Zhang, W.; Gou, P.; Dupret, J.-M.; Chomienne, C.; Rodrigues-Lima, F. Etoposide, an Anticancer Drug Involved in Therapy-Related Secondary Leukemia: Enzymes at Play. Transl. Oncol. 2021, 14, 101169. [Google Scholar] [CrossRef]
- Yan, J.; Sun, J.; Zeng, Z. Teniposide Ameliorates Bone Cancer Nociception in Rats Via the P2X7 Receptor. Inflammopharmacology 2018, 26, 395–402. [Google Scholar] [CrossRef]
- Slobodnick, A.; Shah, B.; Pillinger, M.H.; Krasnokutsky, S. Colchicine: Old and New. Am. J. Med. 2015, 128, 461–470. [Google Scholar] [CrossRef]
- Nam, N.-H. Combretastatin A-4 Analogues as Antimitotic Antitumor Agents. Curr. Med. Chem. 2003, 10, 1697–1722. [Google Scholar] [CrossRef]
- Gerova, M.S.; Stateva, S.R.; Radonova, E.M.; Kalenderska, R.B.; Rusew, R.I.; Nikolova, R.P.; Chanev, C.D.; Shivachev, B.L.; Apostolova, M.D.; Petrov, O.I. Combretastatin A-4 Analogues with Benzoxazolone Scaffold: Synthesis, Structure and Biological Activity. Eur. J. Med. Chem. 2016, 120, 121–133. [Google Scholar] [CrossRef]
- Atanasov, G.; Rusew, R.I.; Gelev, V.M.; Chanev, C.D.; Nikolova, R.; Shivachev, B.L.; Petrov, O.I.; Apostolova, M.D. New Heterocyclic Combretastatin A-4 Analogs: Synthesis and Biological Activity of Styryl-2(3H)-Benzothiazolones. Pharmaceuticals 2021, 14, 1331. [Google Scholar] [CrossRef]
- Jian, X.-E.; Yang, F.; Jiang, C.-S.; You, W.-W.; Zhao, P.-L. Synthesis and Biological Evaluation of Novel Pyrazolo[3,4-b]Pyridines as Cis-Restricted Combretastatin A-4 Analogues. Bioorg. Med. Chem. Lett. 2020, 30, 127025. [Google Scholar] [CrossRef]
- Morales, S.; Guijarro, F.G.; Ruano, J.L.G.; Cid, M.B. A General Aminocatalytic Method for the Synthesis of Aldimines. J. Am. Chem. Soc. 2014, 136, 1082–1089. [Google Scholar] [CrossRef]
- Coates, J. Interpretation of Infrared Spectra, a Practical Approach. In Encyclopedia of Analytical Chemistry; Meyers, R.A., Ed.; Wiley: Chichester UK, 2000; Volume 12, pp. 10815–10837. [Google Scholar]
- Degen, I.A. Detection of the Methoxyl Group by Infrared Spectroscopy. Appl. Spectrosc. 1968, 22, 164–166. [Google Scholar] [CrossRef]
- Nandiyanto, A.B.D.; Oktiani, R.; Ragadhita, R. How to Read and Interpret FTIR Spectroscope of Organic Material. Indones. J. Sci. Technol. 2019, 4, 97–118. [Google Scholar] [CrossRef]
- Bruker, AXS. Inc. Apex 4. Bruker Advanced X-ray Solutions; Bruker AXS Inc.: Madison, WI, USA, 2022. [Google Scholar]
- Bruker, APEX. Saint and Sadabs; Bruker AXS Inc.: Madison, WI, USA, 2009. [Google Scholar]
- Sheldrick, G.M. Shelxt–Integrated Space-Group and Crystal-Structure Determination. Acta Crystallogr. Sect. A 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal Structure Refinement with Shelxl. Acta Crystallogr. Sect. C 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. Olex2: A Complete Structure Solution, Refinement and Analysis Program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Farrugia, L.J. Wingx and Ortep for Windows: An Update. J. Appl. Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Macrae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M. Mercury 4.0: From Visualization to Analysis, Design and Prediction. J. Appl. Crystallogr. 2020, 53, 226–235. [Google Scholar] [CrossRef]
- Brand-Williams, W.; Cuvelier, M.E.; Berset, C. Use of a Free Radical Method to Evaluate Antioxidant Activity. LWT-Food Sci. Technol. 1995, 28, 25–30. [Google Scholar] [CrossRef]
- Benzie, I.F.F.; Strain, J.J. The Ferric Reducing Ability of Plasma (FRAP) as a Measure of ‘Antioxidant Power’: The FRAP Assay. Anal. Biochem. 1996, 239, 70–76. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Arnett, E.M.; Reich, R. Electronic effects on the Menshutkin reaction. A complete kinetic and thermodynamic dissection of alkyl transfer to 3-and 4-substituted pyridines. J. Am. Chem. Soc. 1980, 102, 5892–5902. [Google Scholar] [CrossRef]
- Sola, M.; Lledos, A.; Duran, M.; Bertran, J.; Abboud, J.L.M. Analysis of solvent effects on the Menshutkin reaction. J. Am. Chem. Soc. 1991, 113, 2873–2879. [Google Scholar] [CrossRef]
- Castejon, H.; Wiberg, K.B. Solvent effects on methyl transfer reactions. 1. The Menshutkin reaction. J. Am. Chem. Soc. 1999, 121, 2139–2146. [Google Scholar] [CrossRef]
- Bernstein, J.; Davis, R.E.; Shimoni, L.; Chang, N.-L. Patterns in Hydrogen Bonding: Functionality and Graph Set Analysis in Crystals. Angew. Chem. Int. Ed. Engl. 1995, 34, 1555–1573. [Google Scholar] [CrossRef]
- Grell, J.; Bernstein, J.; Tinhofer, G. Graph-Set Analysis of Hydrogen-Bond Patterns: Some Mathematical Concepts. Acta Crystallogr. Sect. B Struct. Sci. 1999, 55, 1030–1043. [Google Scholar] [CrossRef] [PubMed]
- Denyer, S.P. Mechanisms of Action of Antibacterial Biocides. Int. Biodeterior. Biodegradation 1995, 36, 227–245. [Google Scholar] [CrossRef]
- Ioannou, C.J.; Hanlon, G.W.; Denyer, S.P. Action of Disinfectant Quaternary Ammonium Compounds against Staphylococcus aureus. Antimicrob. Agents Chemother. 2007, 51, 296–306. [Google Scholar] [CrossRef] [PubMed]
- Spackman, P.R.; Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer: A program for Hirshfeld surface analysis, visualization and quantitative analysis of molecular crystals. J. Appl. Cryst. 2021, 54, 1006–1011. [Google Scholar] [CrossRef] [PubMed]
- Rusew, R.; Kurteva, V.; Shivachev, B. Novel Quaternary Ammonium Derivatives of 4-Pyrrolidino Pyridine: Synthesis, Structural, Thermal, and Antibacterial Studies. Crystals 2020, 10, 339. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inhibitory Diameter (mm ± sd), 30 µg/Disk | |||||
|---|---|---|---|---|---|
| E. coli | S. aureus | K. pneumoniae | P. aeruginosa | ||
| 2 | – | – | 7.8 (0.3) | – | |
| 3a | – | – | 15.1 (0.3) | 9.4 (0.4) | |
| 3b | – | – | 11.9 (0.3) | 7.1 (0.4) | |
| 3c | – | – | 8.0 (0.2) | – | |
| 3d | – | – | 7.5 (0.2) | 7.2 (0.3) | |
| 3e | – | 9.5 (0.3) | 8.5 (0.4) | 9.5 (0.3) | |
| KAN | 18.1 (0.4) | 16.1 (0.4) | 18.3 (0.6) | 15.2 (0.3) | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rusew, R.; Georgieva, M.; Kurteva, V.; Shivachev, B. Novel Quaternary Ammonium Aldimine Derivatives Featuring 3,4,5-Trimethoxy Phenyl Fragment: Synthesis, Crystal Structure and Evaluation of Antioxidant and Antibacterial Activity. Crystals 2024, 14, 486. https://doi.org/10.3390/cryst14060486
Rusew R, Georgieva M, Kurteva V, Shivachev B. Novel Quaternary Ammonium Aldimine Derivatives Featuring 3,4,5-Trimethoxy Phenyl Fragment: Synthesis, Crystal Structure and Evaluation of Antioxidant and Antibacterial Activity. Crystals. 2024; 14(6):486. https://doi.org/10.3390/cryst14060486
Chicago/Turabian StyleRusew, Rusi, Mariya Georgieva, Vanya Kurteva, and Boris Shivachev. 2024. "Novel Quaternary Ammonium Aldimine Derivatives Featuring 3,4,5-Trimethoxy Phenyl Fragment: Synthesis, Crystal Structure and Evaluation of Antioxidant and Antibacterial Activity" Crystals 14, no. 6: 486. https://doi.org/10.3390/cryst14060486
APA StyleRusew, R., Georgieva, M., Kurteva, V., & Shivachev, B. (2024). Novel Quaternary Ammonium Aldimine Derivatives Featuring 3,4,5-Trimethoxy Phenyl Fragment: Synthesis, Crystal Structure and Evaluation of Antioxidant and Antibacterial Activity. Crystals, 14(6), 486. https://doi.org/10.3390/cryst14060486