
Neutral and Ionic Form of (Benzylthio)Acetic Acid in Novel Aminopyrimidine Based Multi-Component Crystalline Phases


Abstract
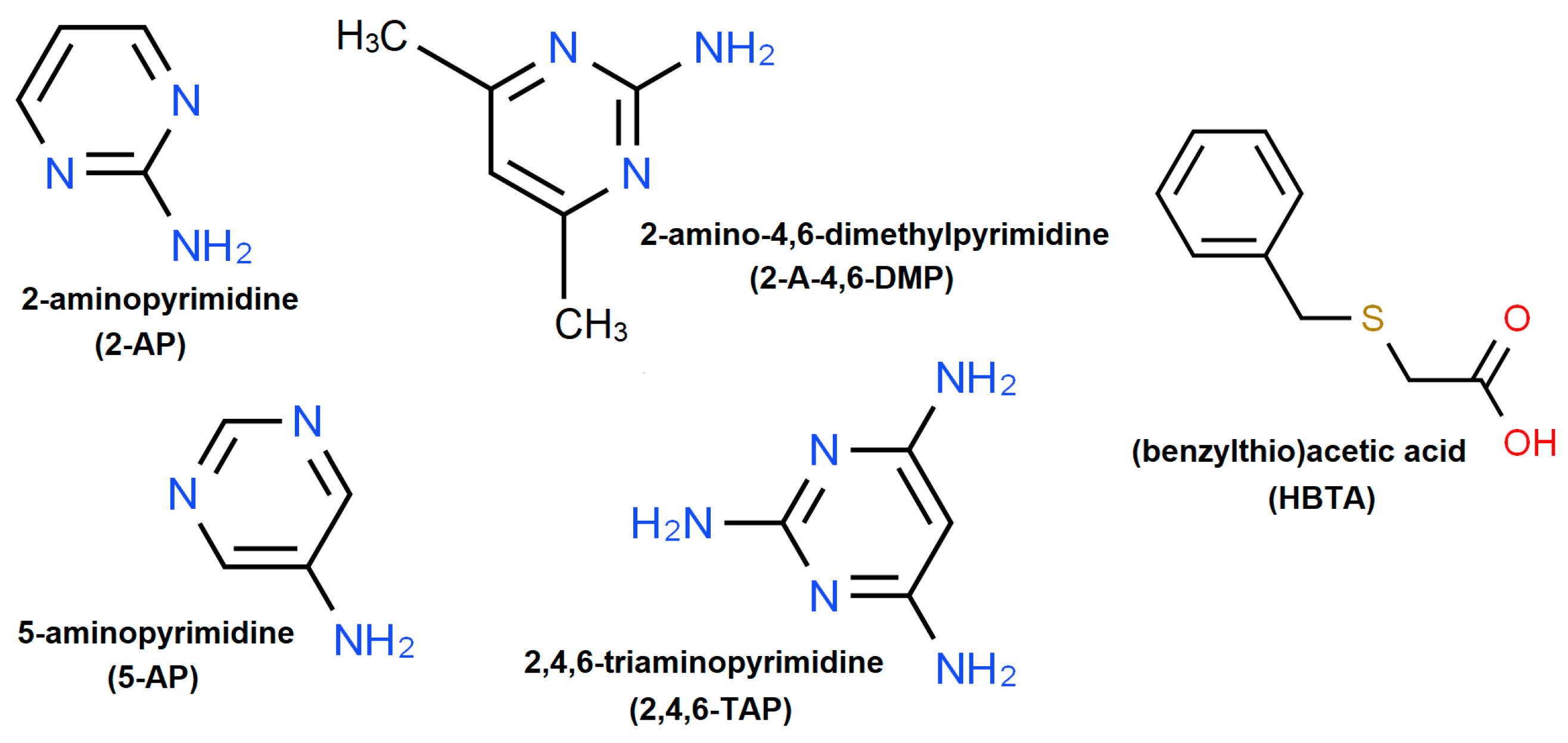
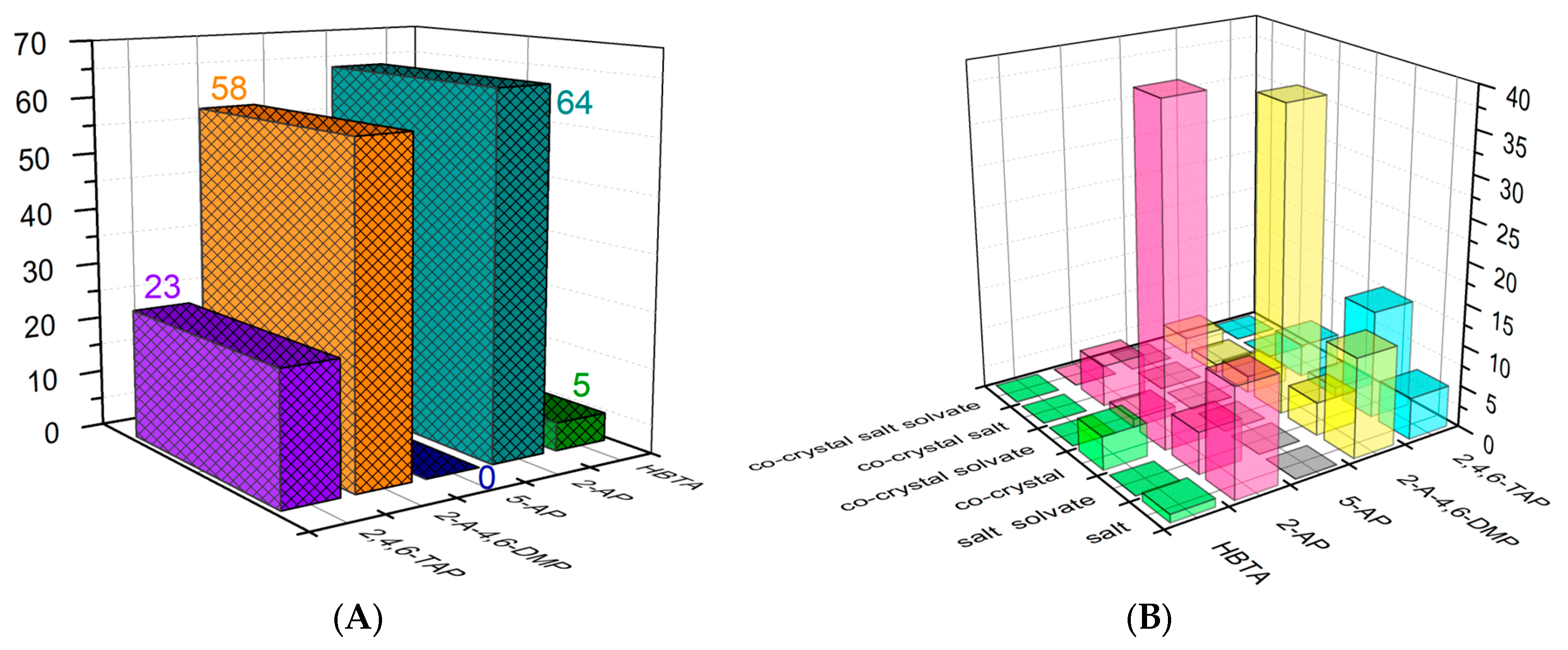
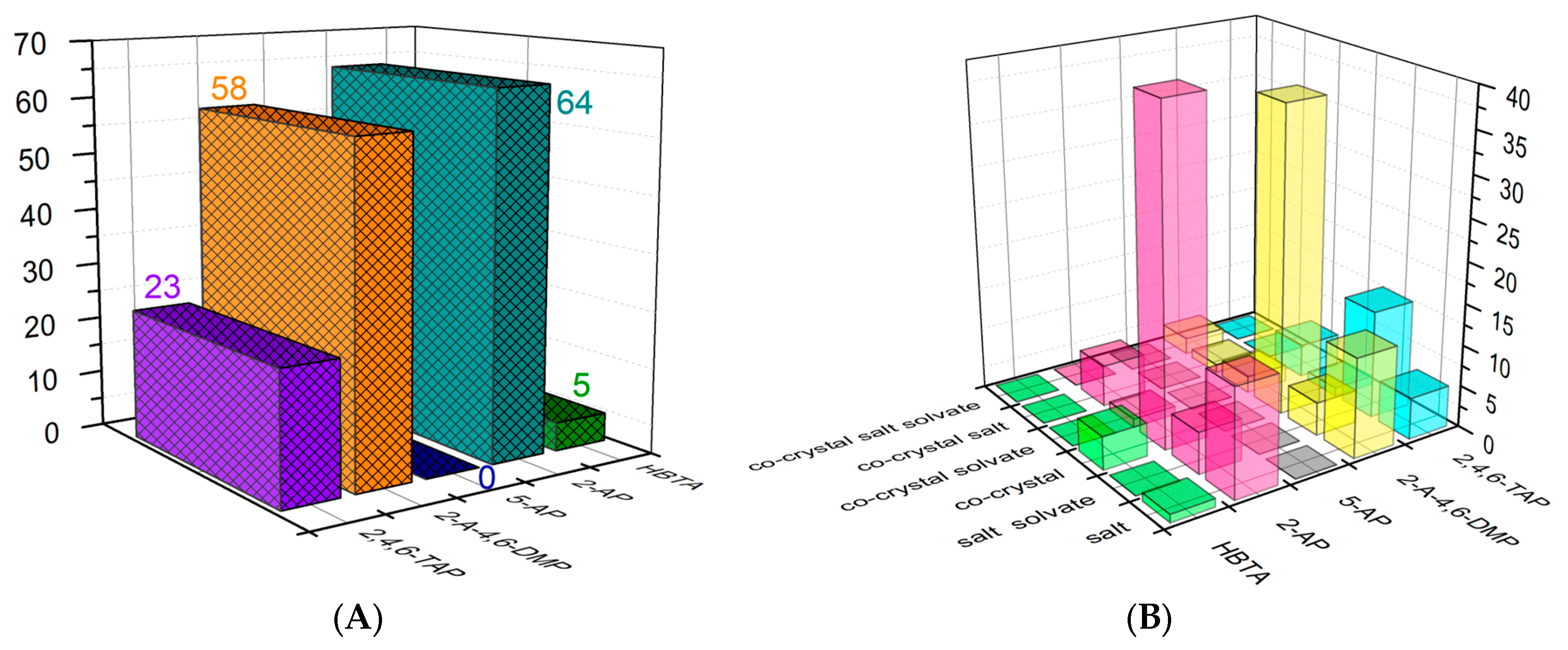
:1. Introduction
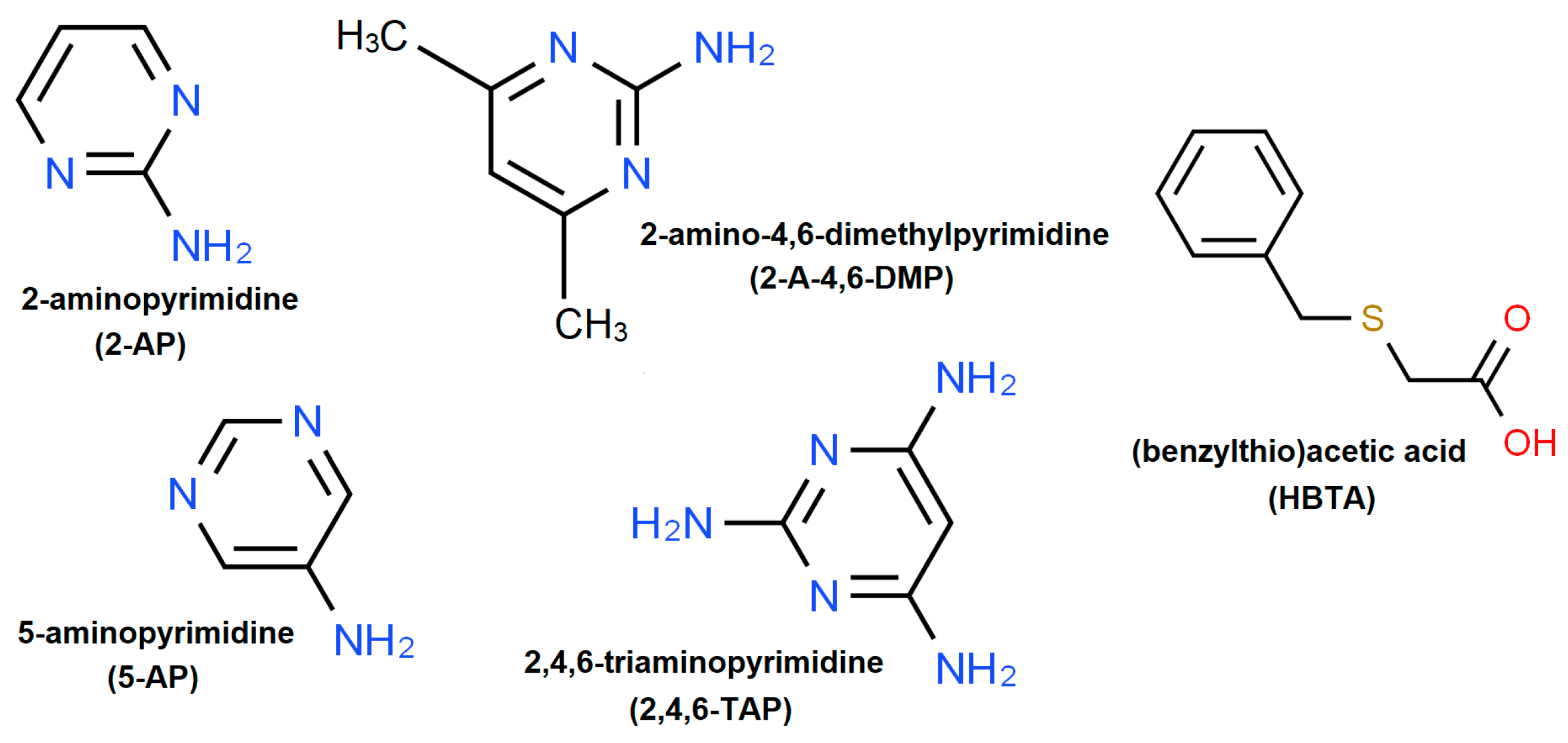
2. Materials and Methods
2.1. Single Crystal Preparation by Solution Co-Crystallization
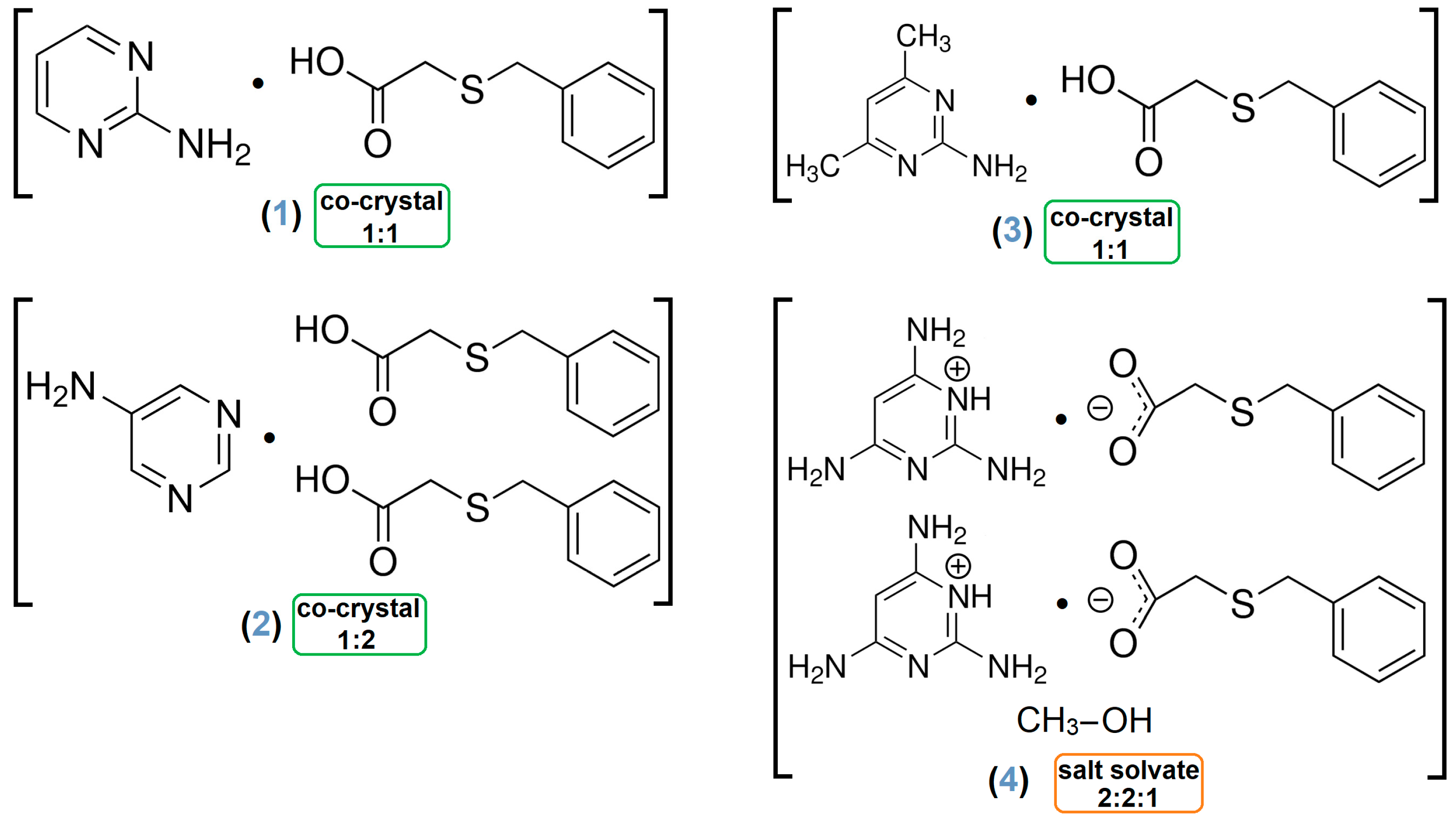
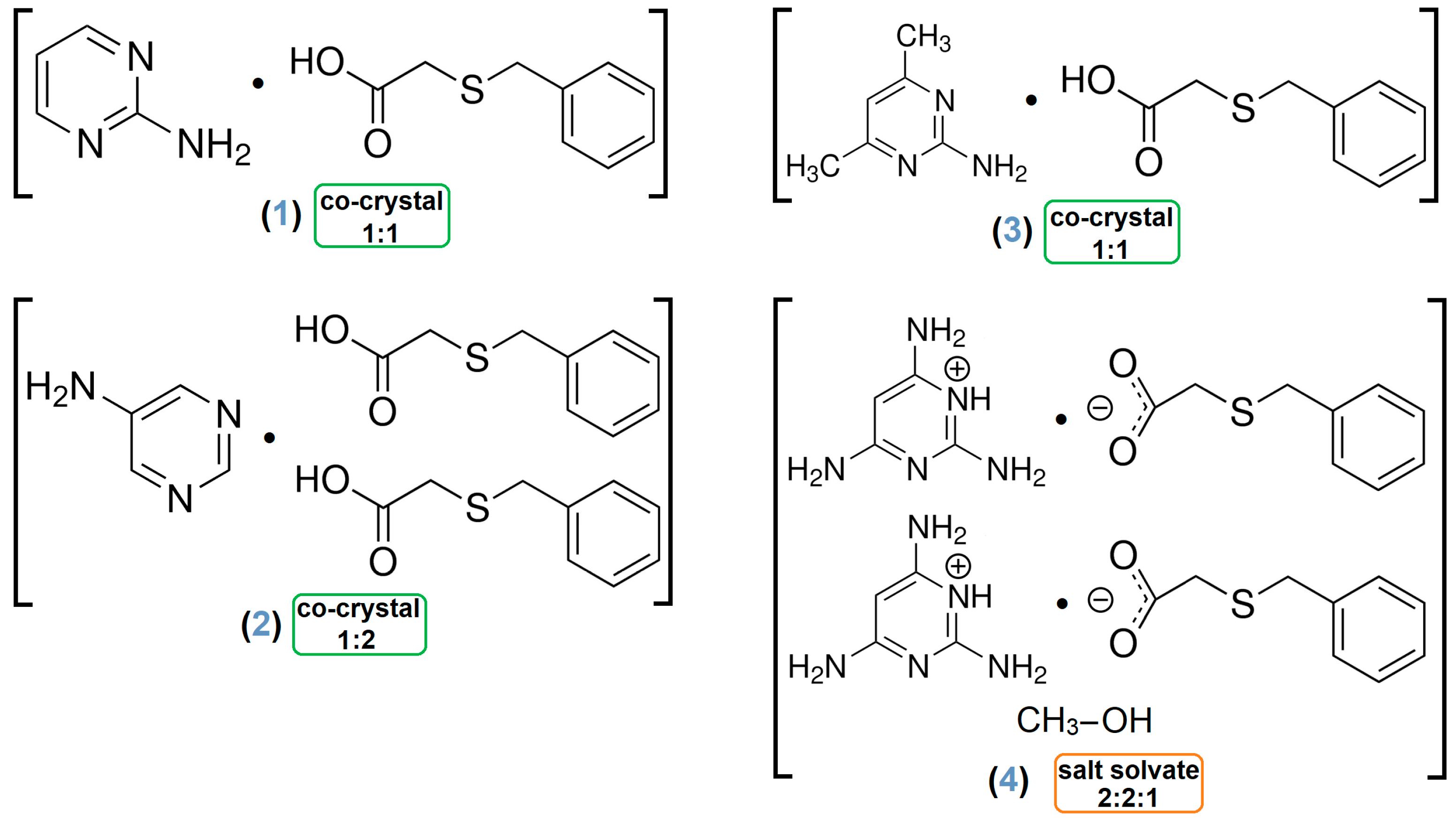
- Adduct [2-AP·HBTA] (1)
- Yield for 1: 0.251 g (90.61%); Elemental analysis results for [C4H5N3·C9H10O2S] (MW: 277.34 g mol−1). Calcd (%): C, 56.30; H, 5.45; N, 15.15; S, 11.56. Found (%): C, 56.61; H, 5.34; N, 15.28; S, 11.39.
- Architecture [5-AP·2(HBTA)] (2)
- Yield for 2: 0.228 g (82.31%); Elemental analysis results for [C4H5N3·2(C9H10O2S)] (MW: 459.56 g mol−1). Calcd (%): C, 57.50; H, 5.48; N, 9.14; S, 13.95. Found (%): C, 57.32; H, 5.57; N, 9.31; S, 14.06.
- Association [2-A-4,6-DMP·HBTA] (3)
- Yield for 3: 0.285 g (93.44%); Elemental analysis results for [C6H9N3·C9H10O2S] (MW: 305.39 g mol−1). Calcd (%): C, 58.99; H, 6.27; N, 13.76; S, 10.50. Found (%): C, 59.17; H, 6.15; N, 13.62; S, 10.74.
- Assembly [2(2,4,6-TAP+)·2(BTA−)·MeOH] (4)
- Yield for 4: 0.269 g (87.62%); Elemental analysis results for [2(C4H8N5)·2(C9H9O2S)·CH4O] (MW: 646.79 g mol−1). Calcd (%): C, 50.14; H, 5.92; N, 21.66; S, 9.91. Found (%): C, 50.02; H, 5.81; N, 21.84; S, 10.08.
2.2. Single-Crystal X-ray Diffraction

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compund | 1 [2-AP·HBTA] | 2 [5-AP·2(HBTA)] | 3 [2-A-4,6-DMP·HBTA] | 4 [2(2,4,6-TAP⁺)·2(BTA−)·MeOH] |
| Chemical formula | [C4H5N3·C9H10O2S] | [C4H5N3·2(C9H10O2S)] | [C6H9N3·C9H10O2S] | [2(C4H8N5⁺)·2(C9H9O2S−)·CH3OH] |
| Formula weight | 277.34 | 459.58 | 305.39 | 646.79 |
| T [K] | 120(2) | 100(2) | 295(2) | 120(2) |
| Crystal system | triclinic | monoclinic | triclinic | triclinic |
| Space group | P-1 | I2/a | P-1 | P-1 |
| a [Ĺ] | 5.473(2) | 11.2873(8) | 7.3644(9) | 11.9766(12) |
| b [Ĺ] | 8.720(3) | 9.1219(6) | 8.2883(10) | 12.2477(11) |
| c [Ĺ] | 14.262(4) | 22.7196(17) | 13.5264(12) | 12.7081(12) |
| α [°] | 83.08(3) | 90.00 | 82.748(9) | 99.661(8) |
| β [°] | 82.62(3) | 103.110(7) | 87.375(9) | 111.581(9) |
| γ [°] | 80.40(3) | 90.00 | 75.153(10) | 106.054(8) |
| V [Ĺ3] | 662.1(4) | 2278.3(3) | 791.60(16) | 1588.2(3) |
| Z | 2 | 8 | 2 | 2 |
| Dcalc. [g cm−3] | 1.391 | 1.340 | 1.281 | 1.352 |
| µ [mm−1] | 0.246 | 0.267 | 0.212 | 0.221 |
| Crystal color and shape | colorless plate | colorless plate | colorless plate | colorless block |
| Crystal size [mm] | 0.21 × 0.56 × 0.02 | 0.23 × 0.55 × 0.05 | 0.58 × 0.52 × 0.18 | 0.11 × 0.20 × 0.31 |
| θ range [°] | 2.895–27.477 | 2.902–27.485 | 2.560–27.485 | 2.582–27.484 |
| F(000) | 292 | 968 | 324 | 684 |
| Reflections measured unique | 5060 3040 | 8584 2607 | 6170 3637 | 13,063 7280 |
| Observed data [I > 2σ(I)] | 2382 | 2252 | 2317 | 4470 |
| Rint | 0.0274 | 0.0263 | 0.0324 | 0.0592 |
| Completeness to θmax | 0.999 | 0.999 | 0.999 | 0.999 |
| Goodness-of-fit on F2 | 1.017 | 1.048 | 1.034 | 1.017 |
| R1, wR2 [I > 2σ(I)] | 0.0421, 0.0906 | 0.0313, 0.0752 | 0.0534, 0.1299 | 0.0672, 0.1158 |
| R1, wR2 (all data) | 0.0621, 0.1023 | 0.0391, 0.0799 | 0.0893, 0.1528 | 0.1250, 0.1417 |
| Residual density [e Å−3] | 0.289, −0.252 | 0.264, −0.271 | 0.305, −0.204 | 0.345, −0.326 |
| Deposition no | 2304064 | 2304065 | 2304066 | 2304067 |
2.3. Instrumentation and Measurement Methodology
3. Results and Discussion
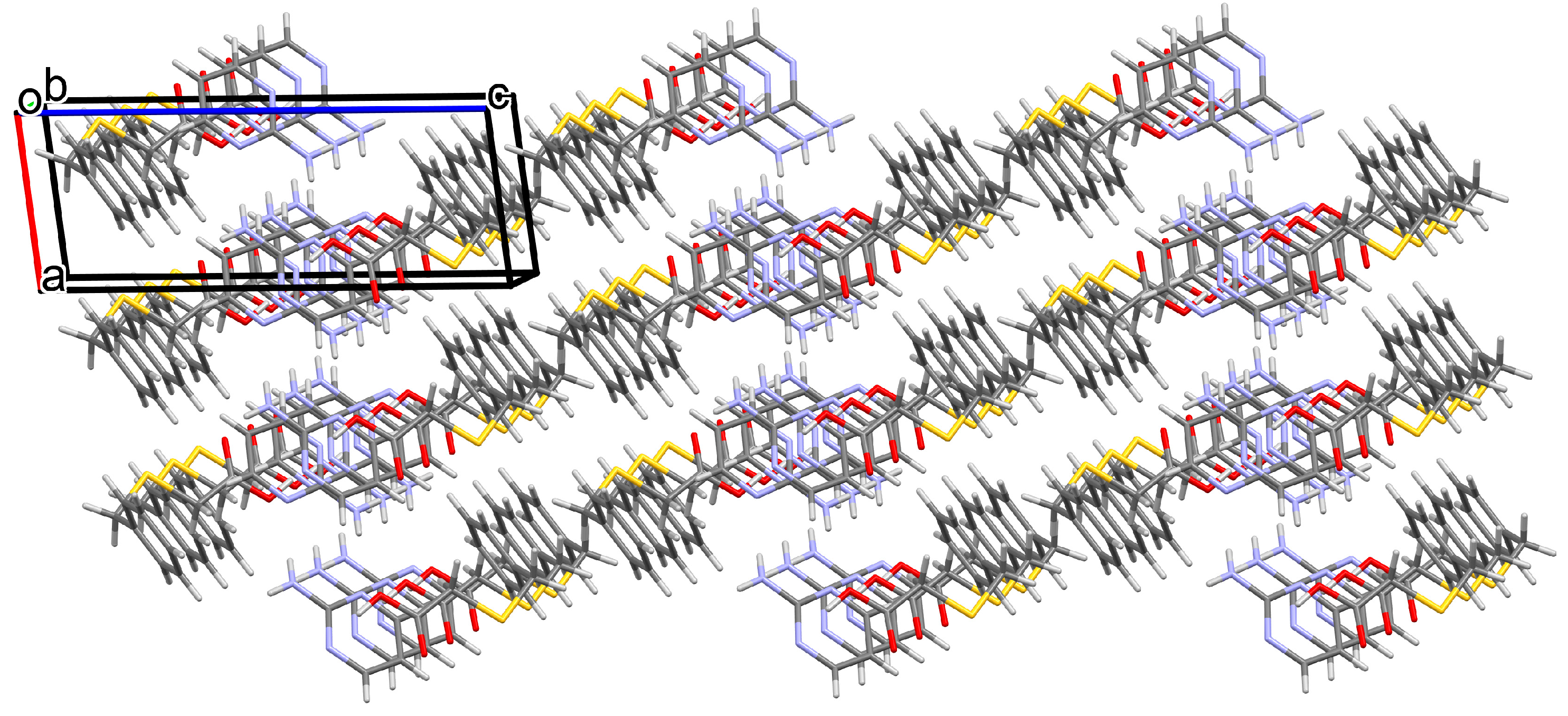
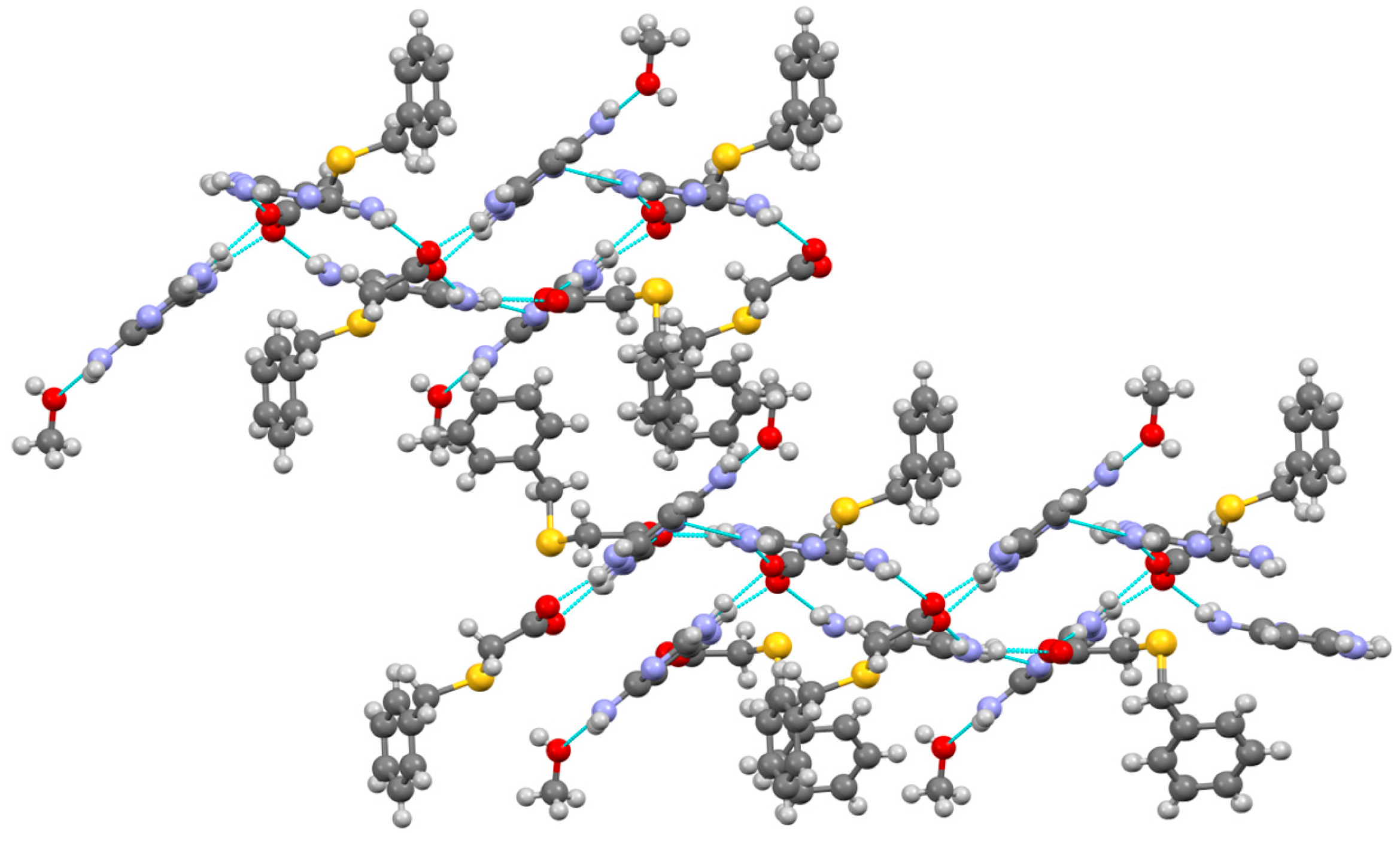
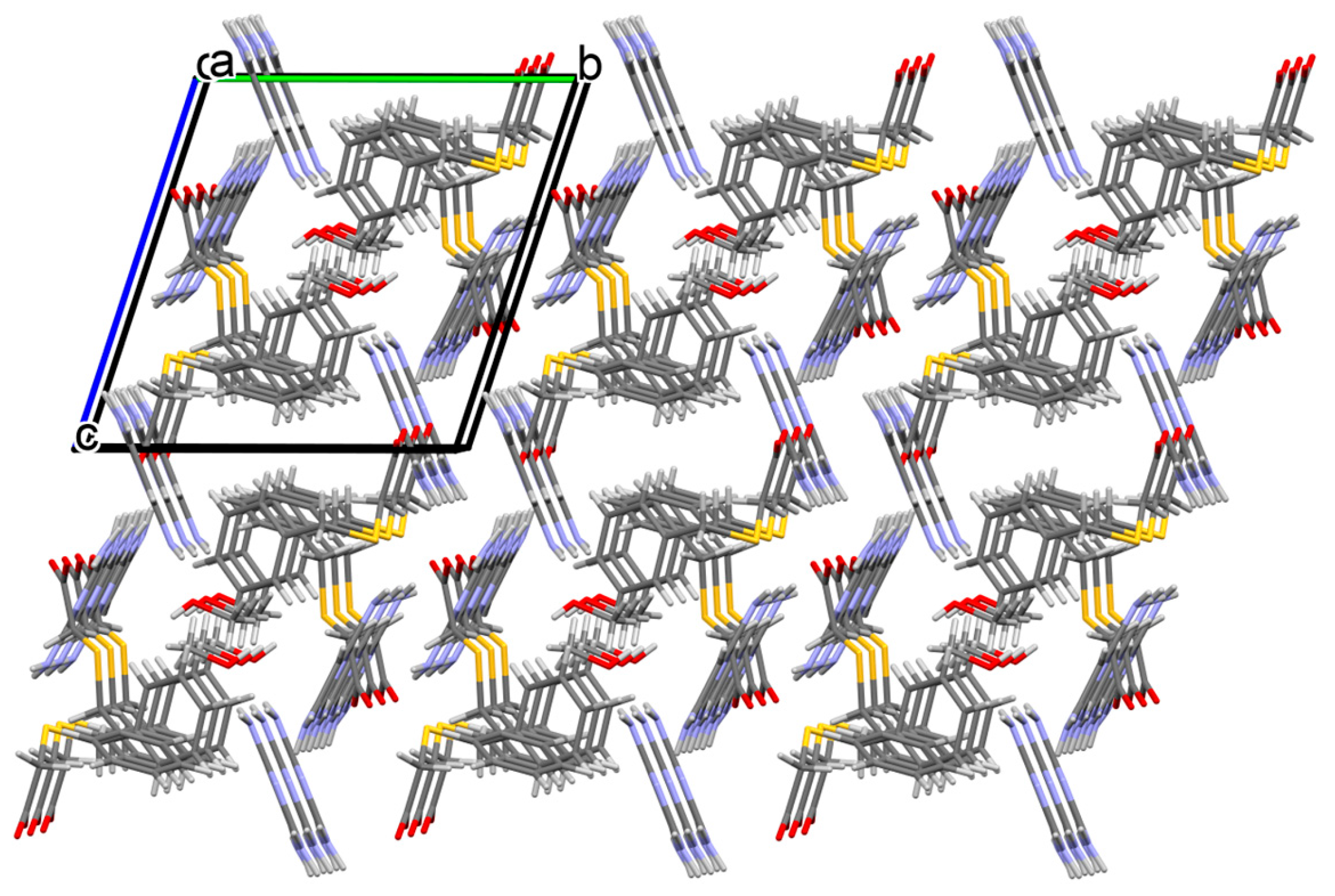
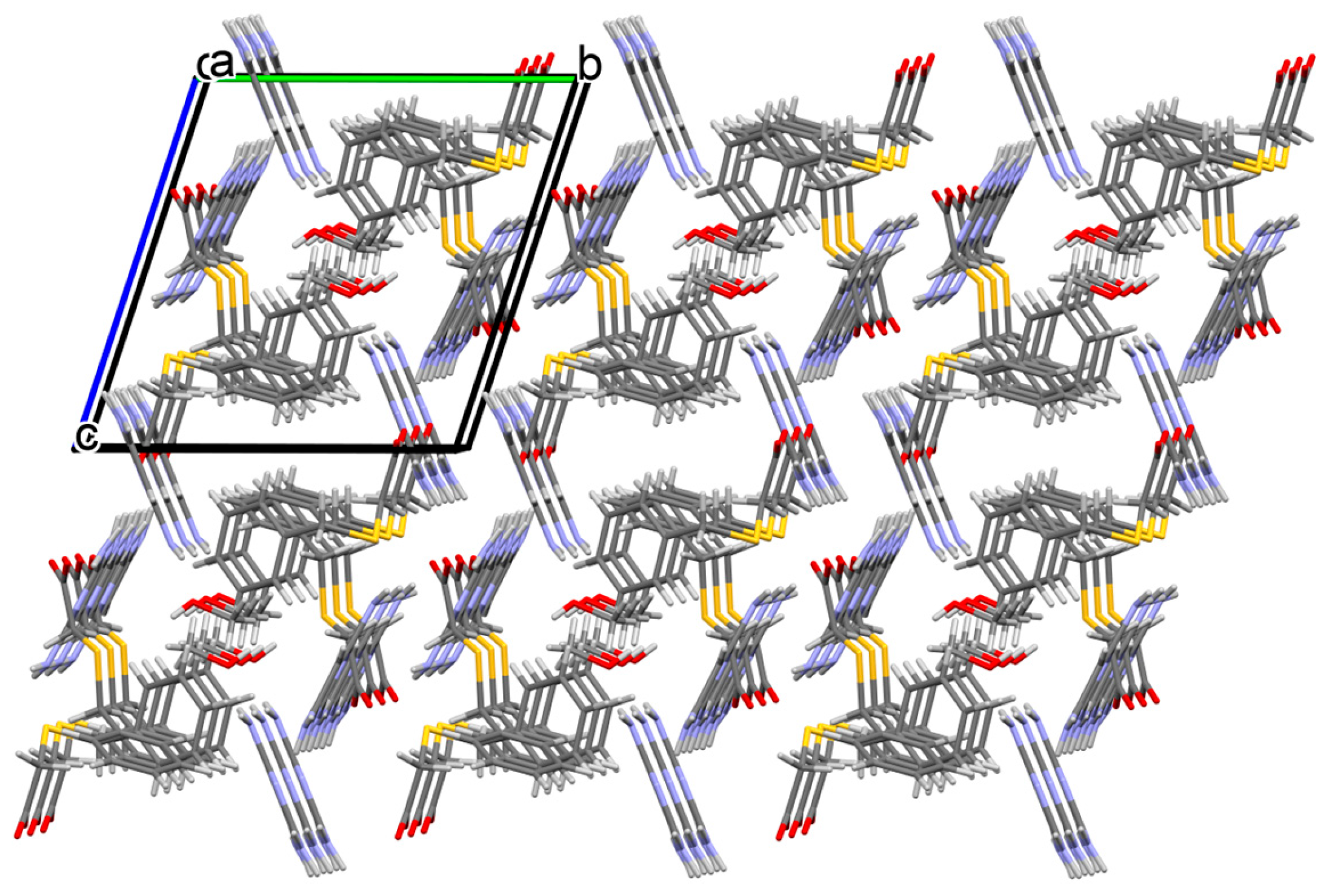
3.1. Structural and Supramolecular Characteristics
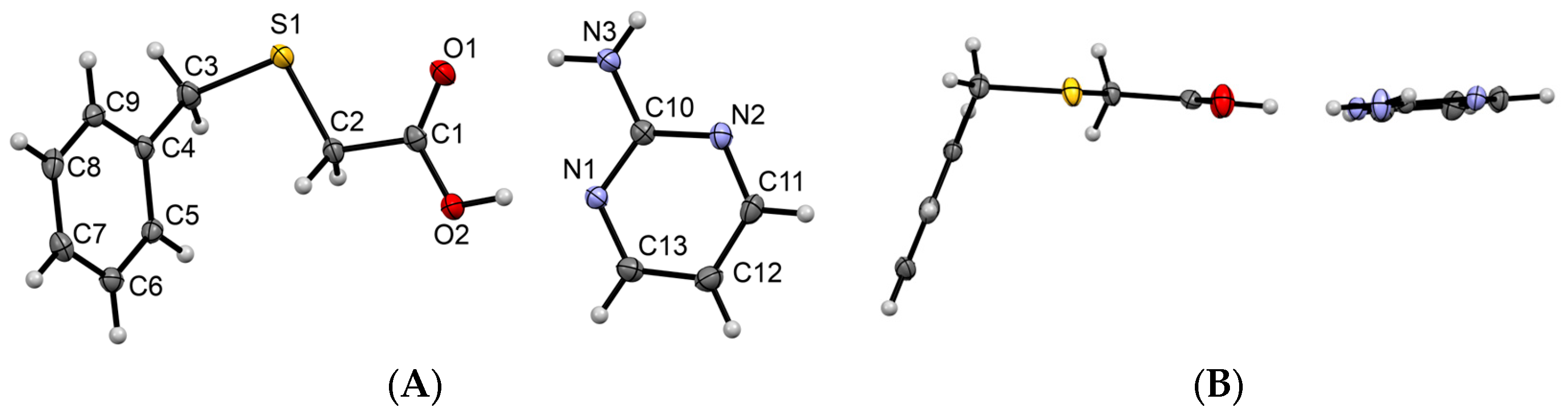
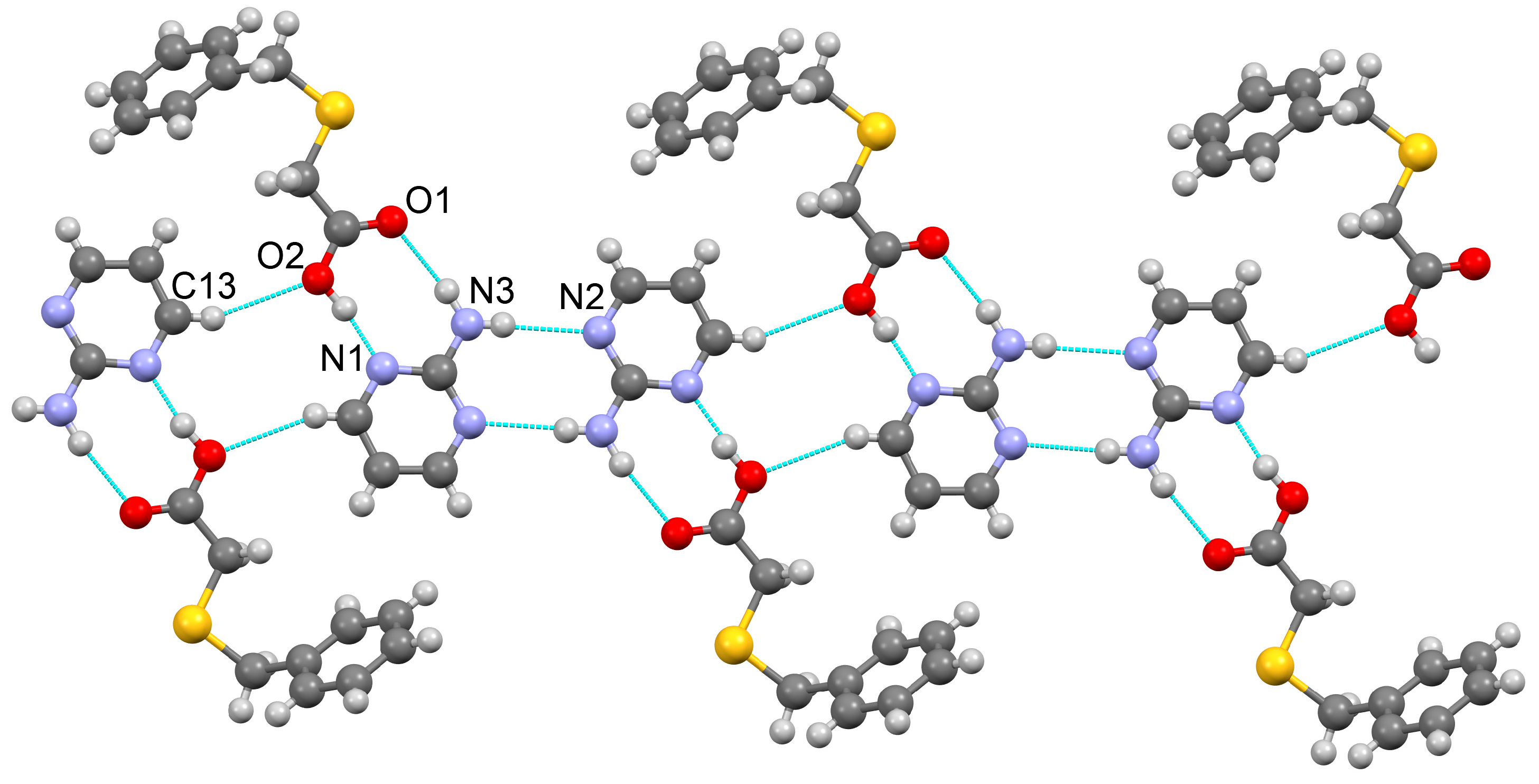
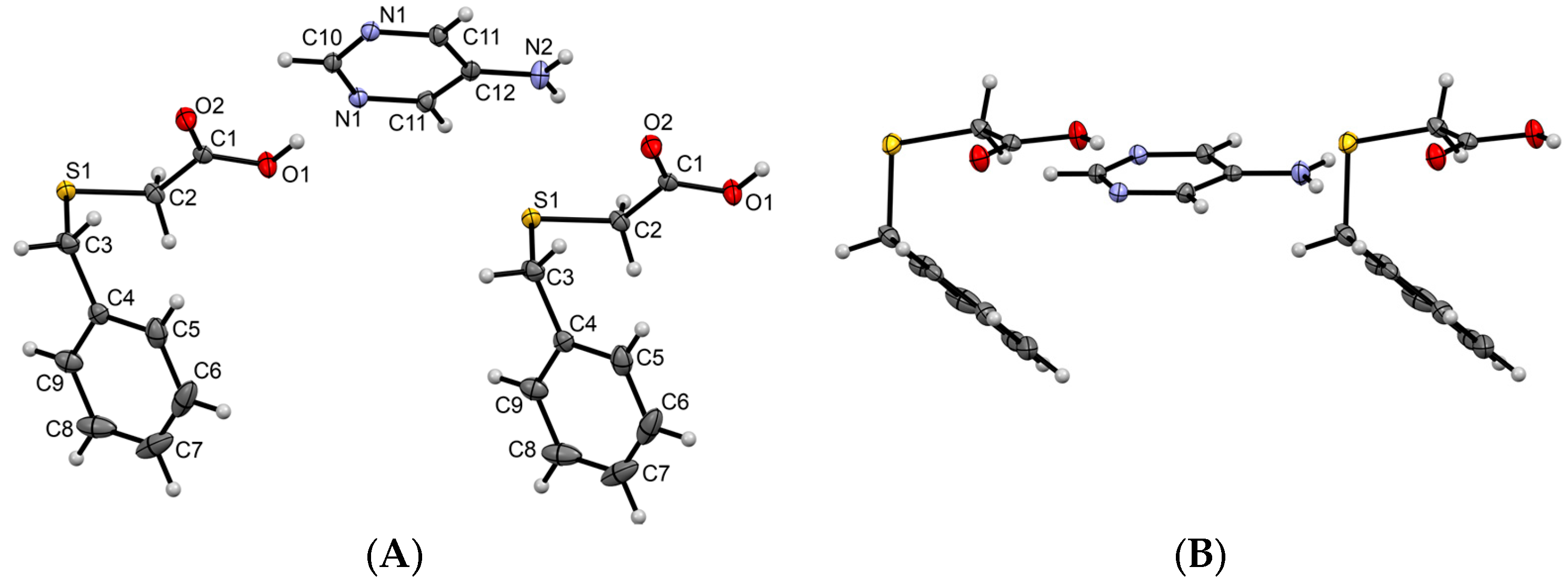
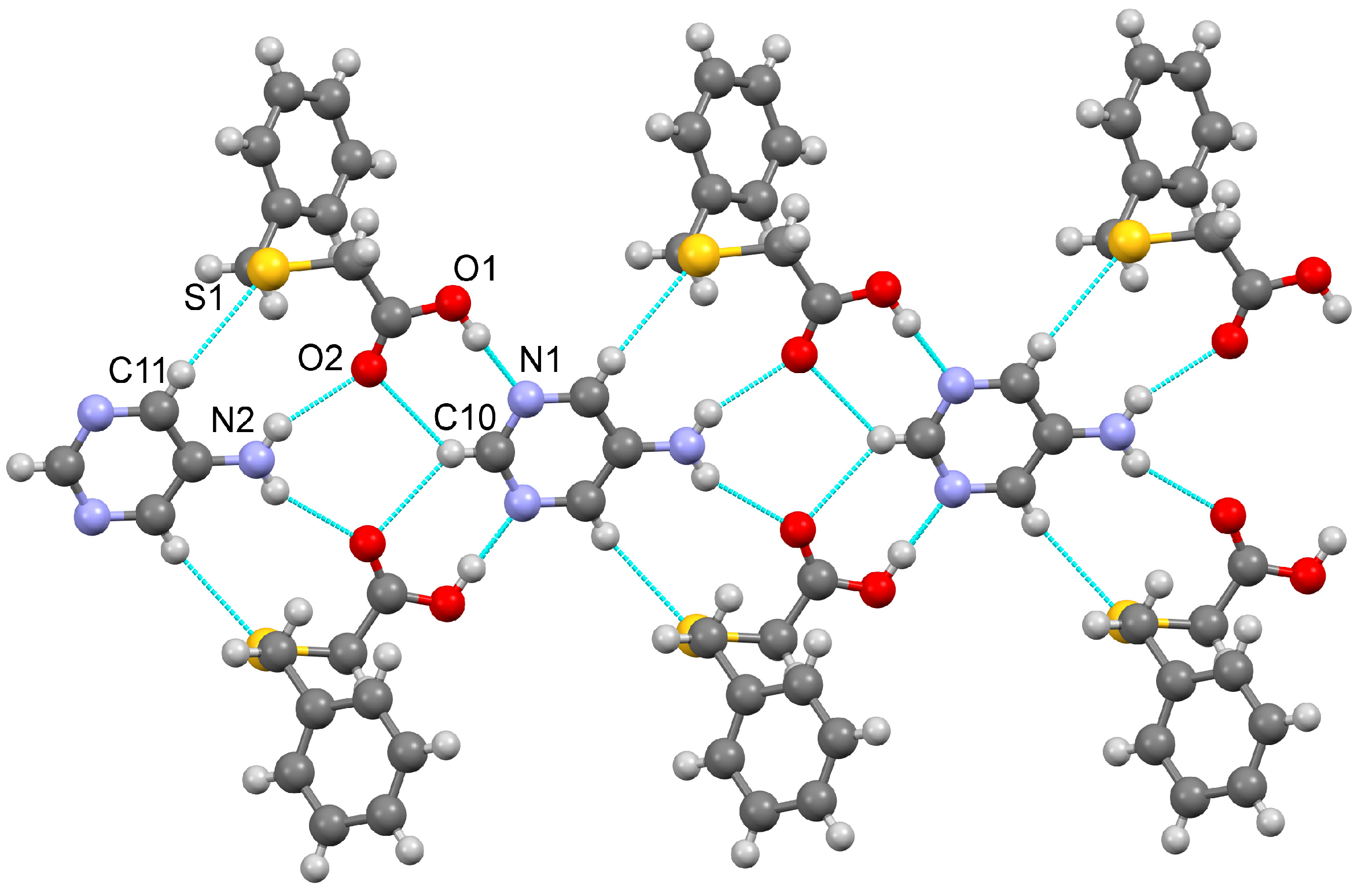
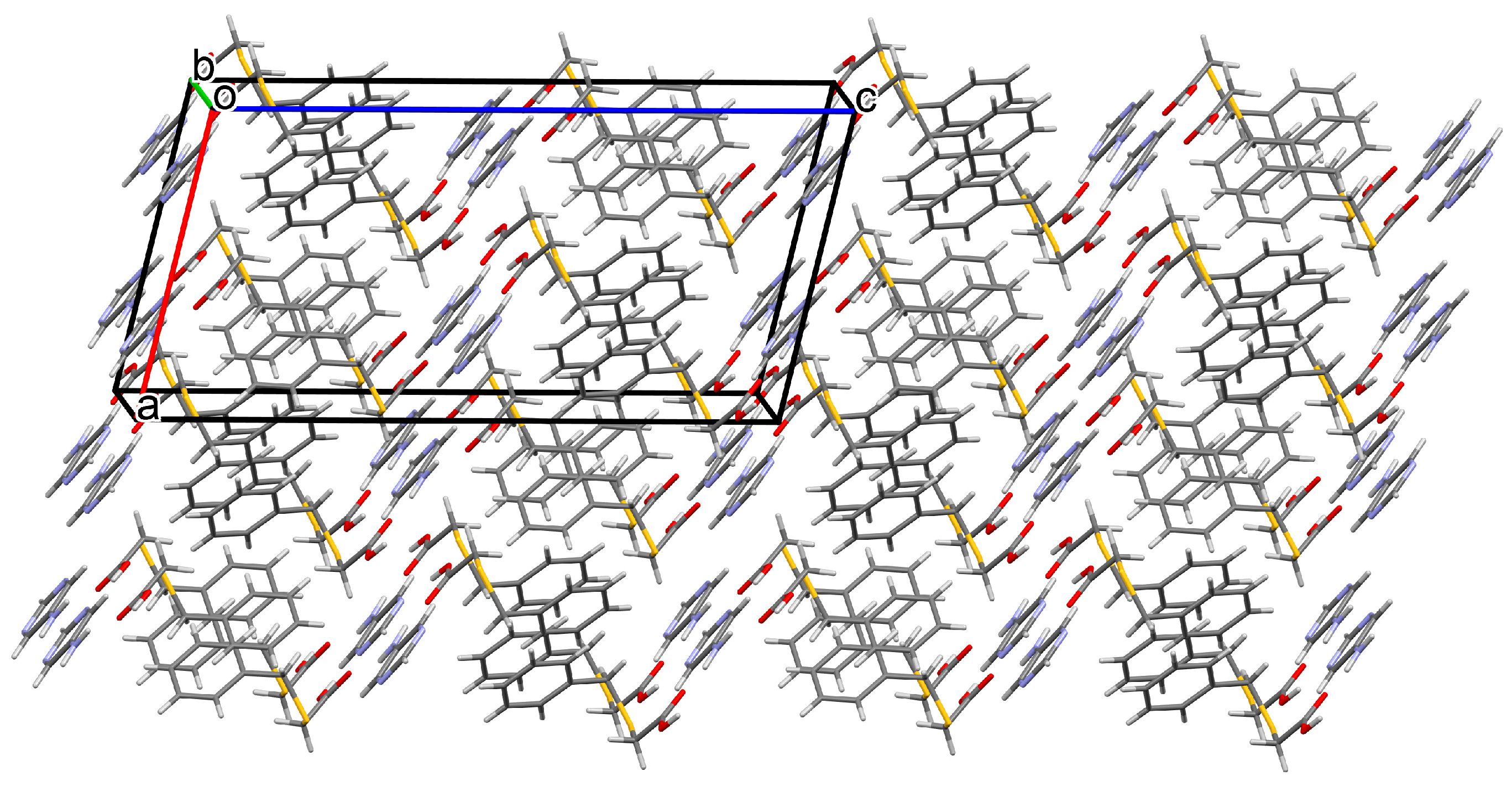
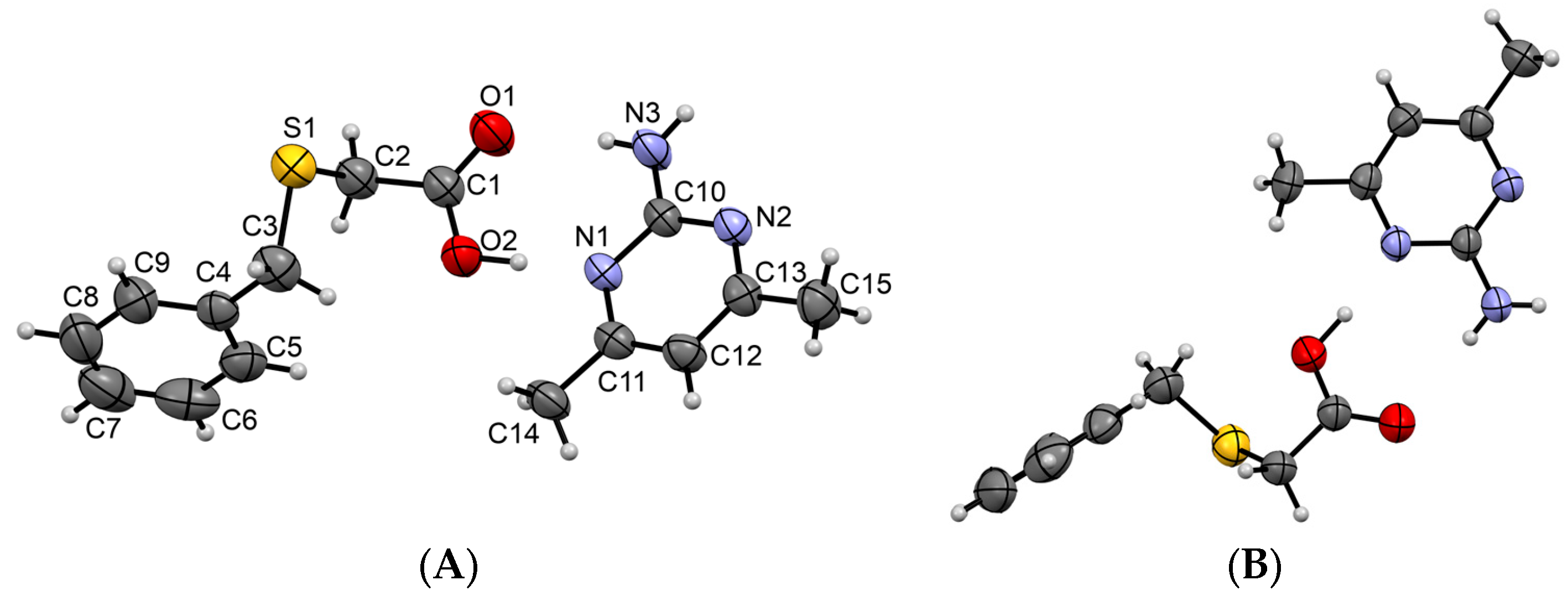
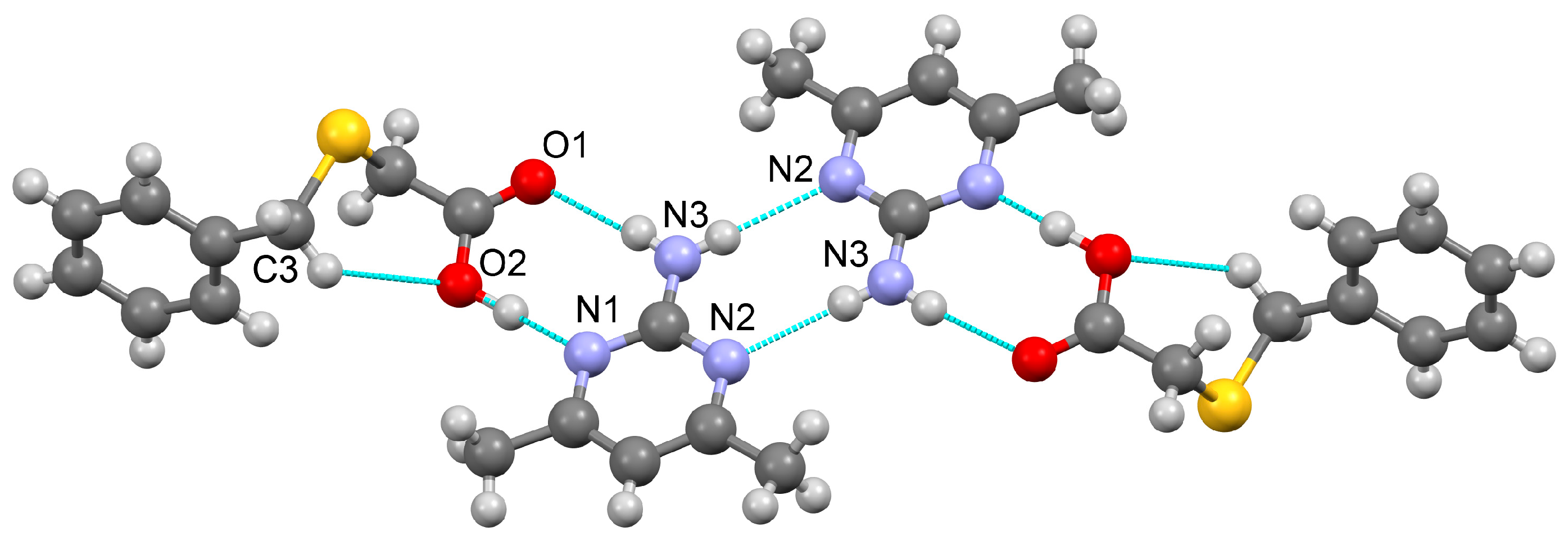
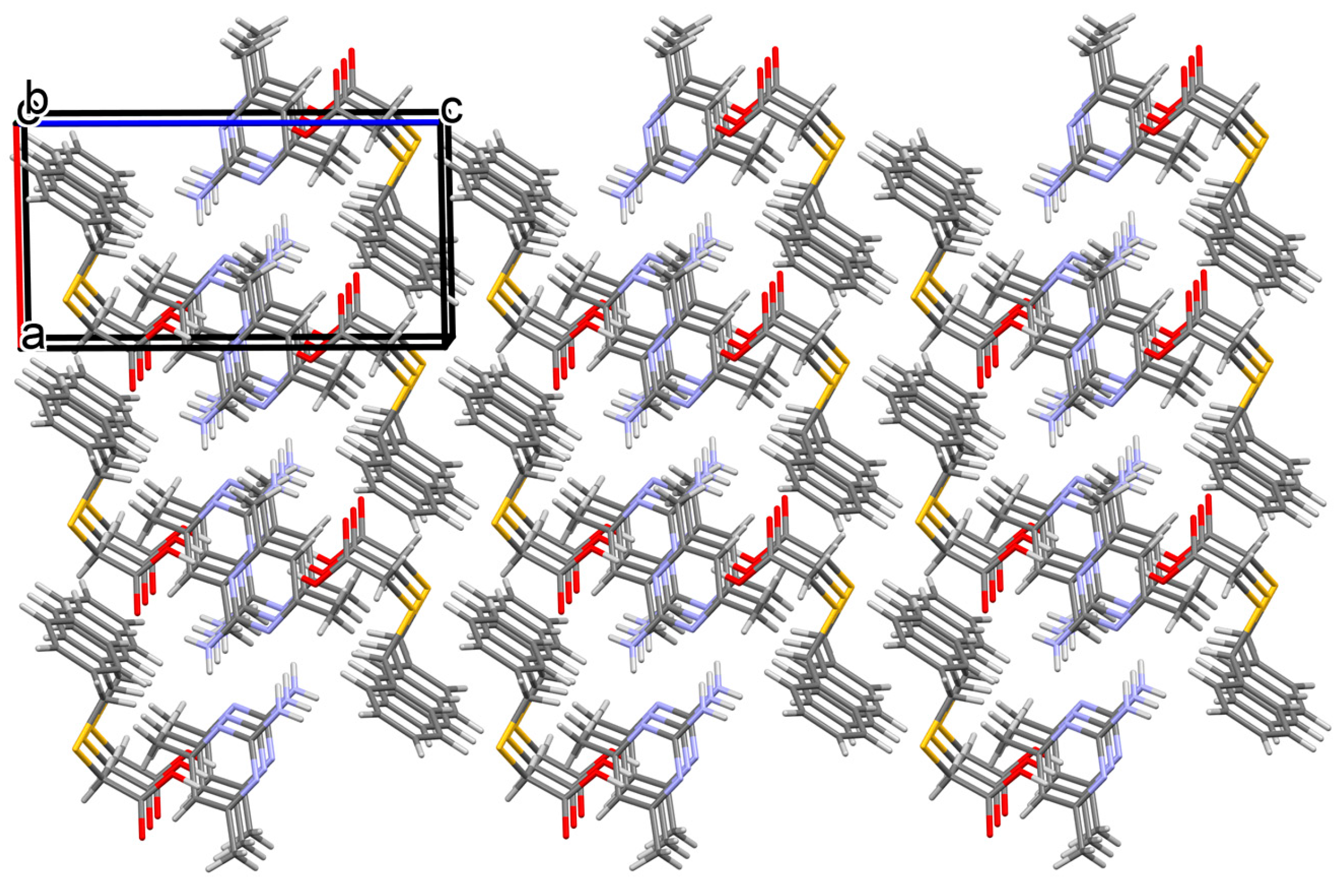
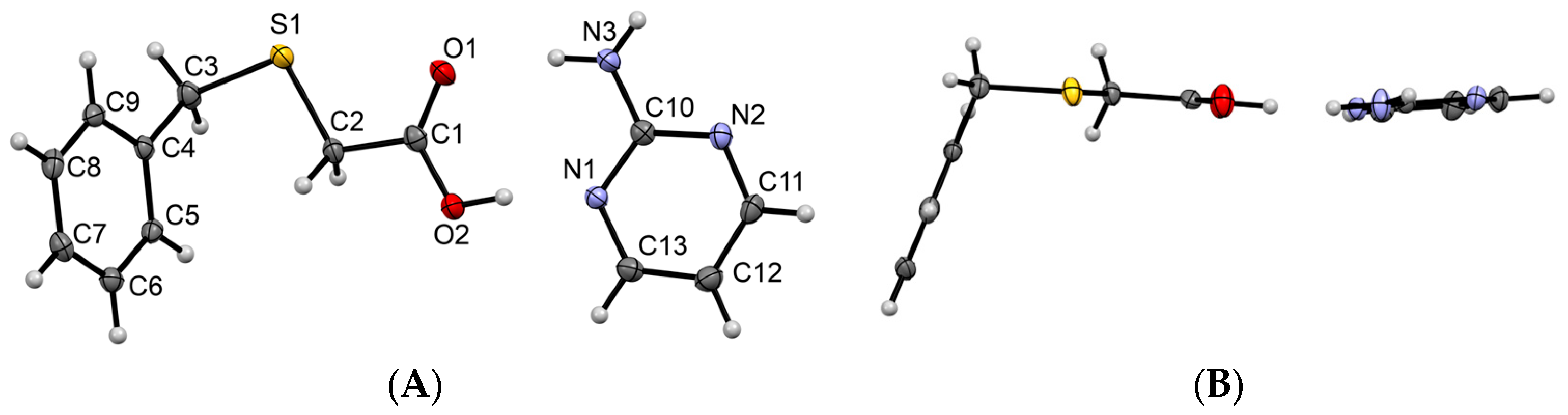
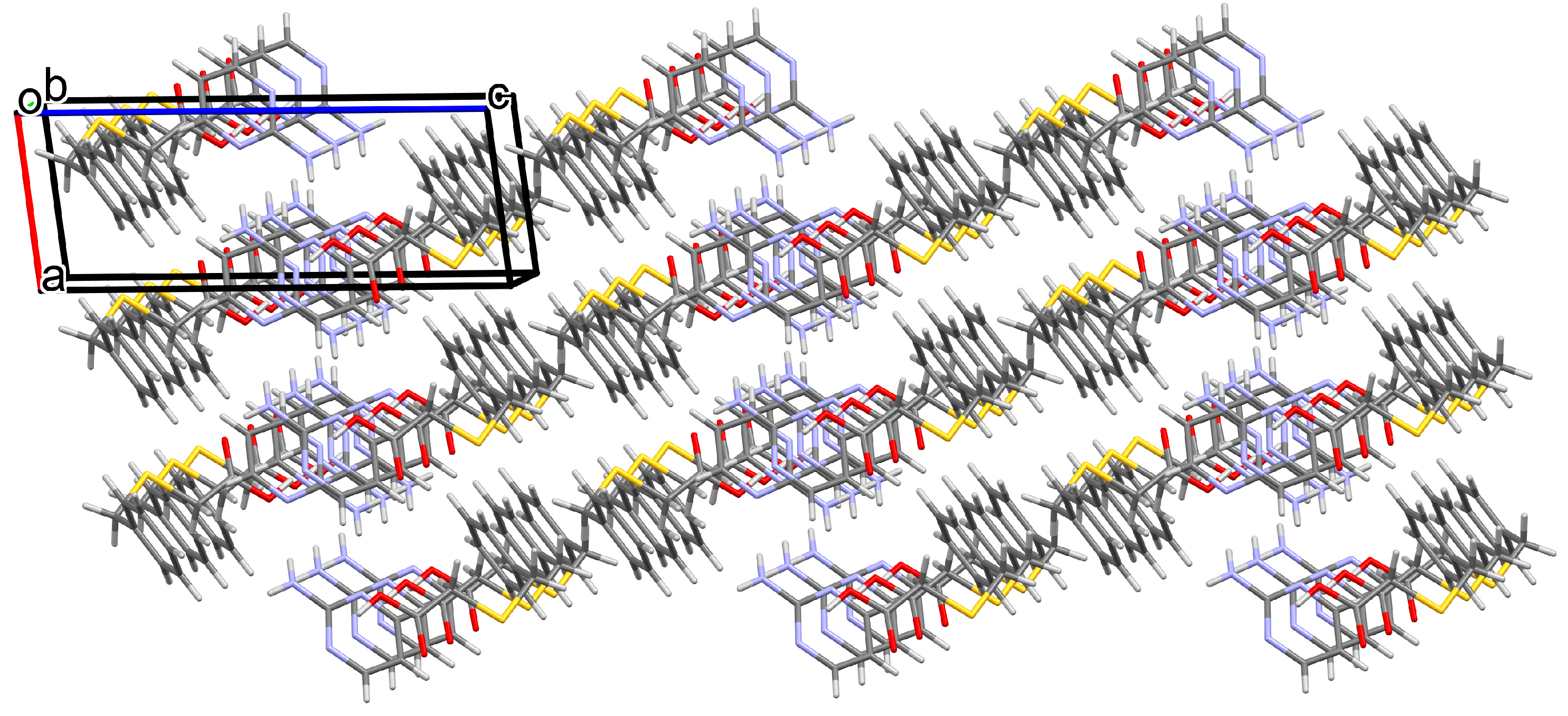
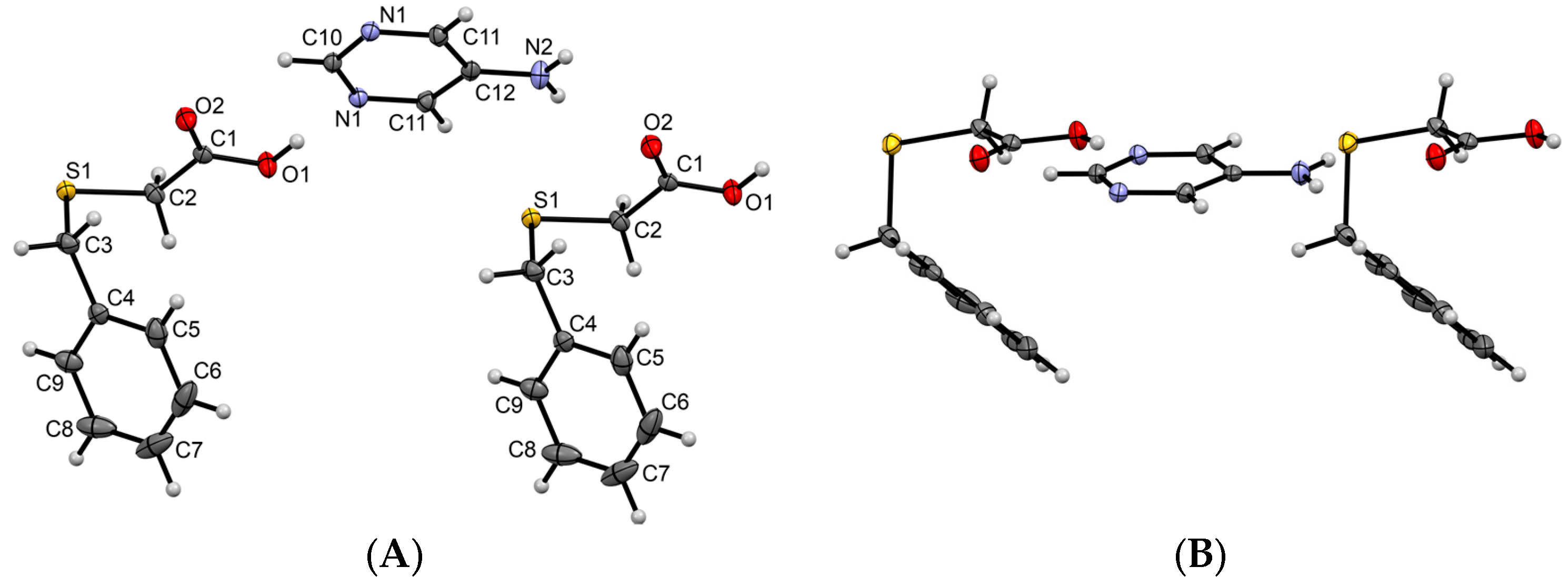
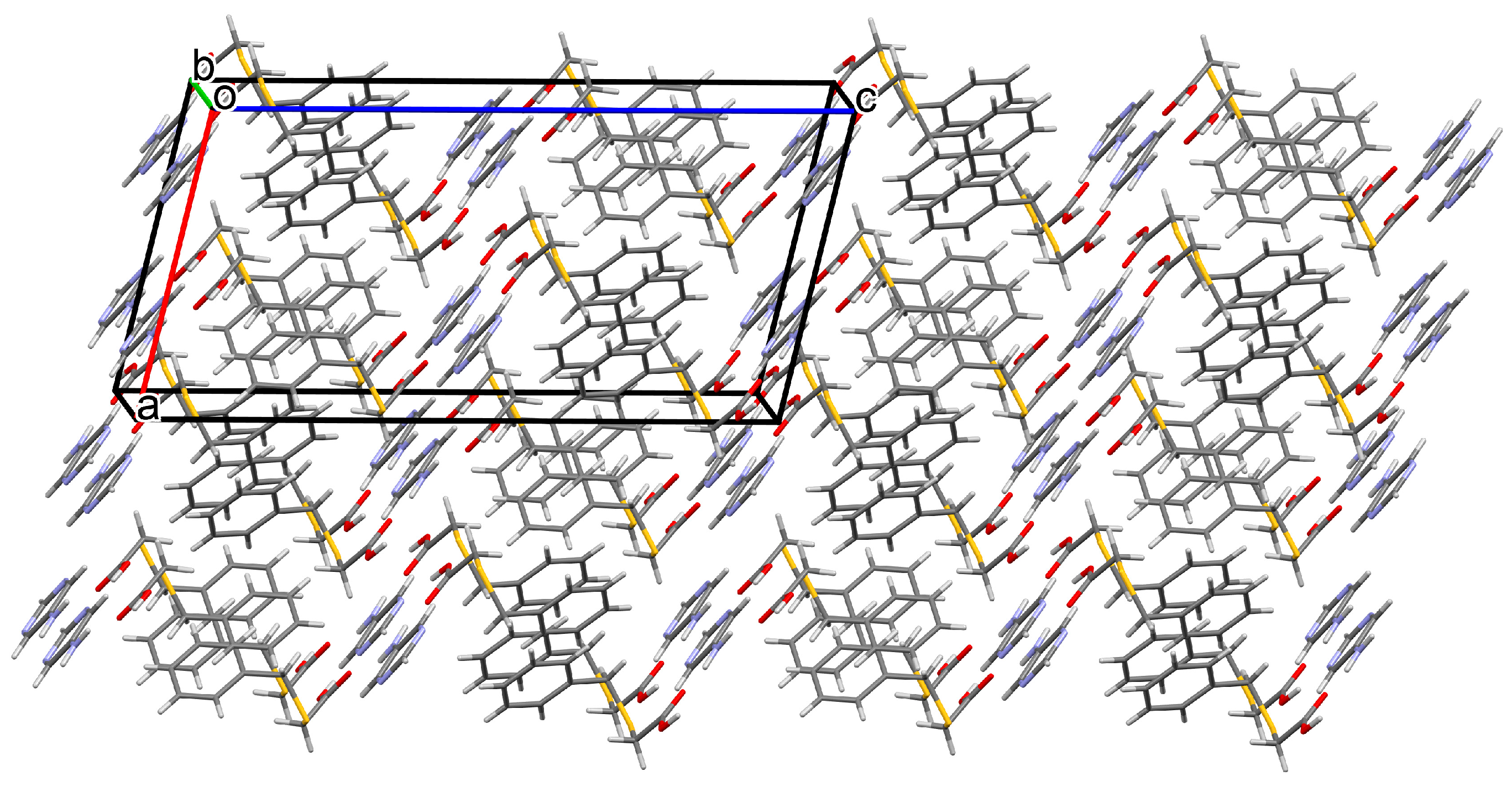
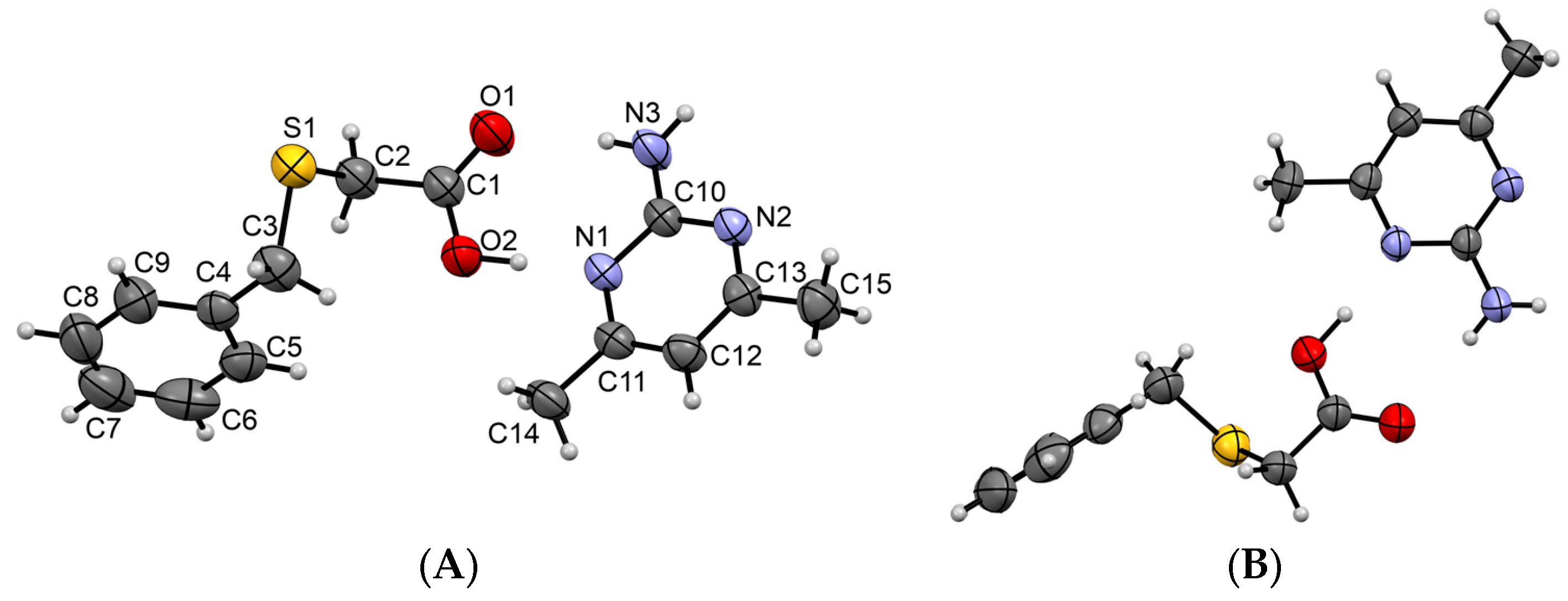
3.1.1. Co-Crystal Obtained from 2-AP and HBTA (1)
3.1.2. Co-Crystal Formed between 5-AP and HBTA (2)
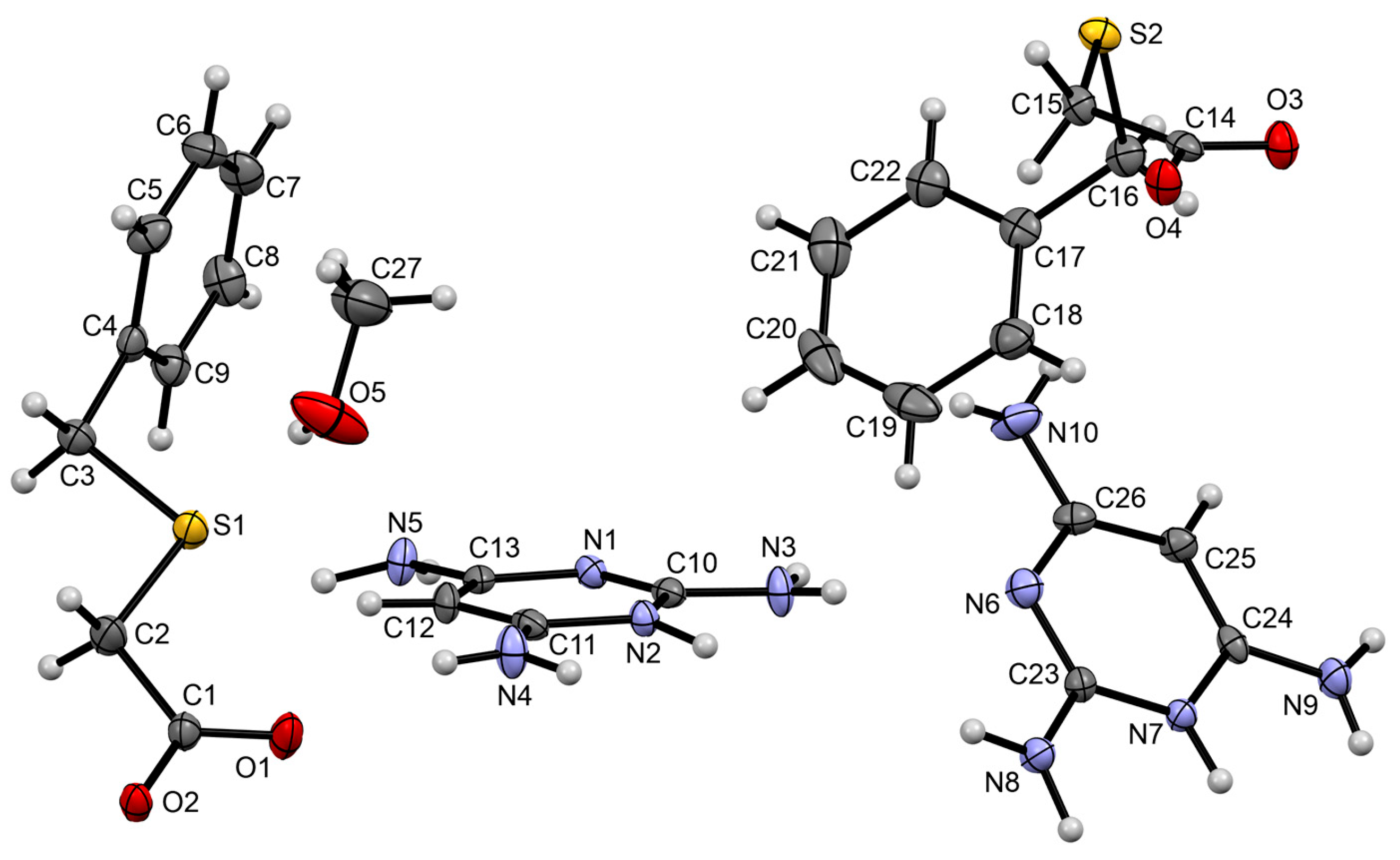
3.1.3. Co-Crystal Based on 2-A-4,6-DMP and HBTA (3)
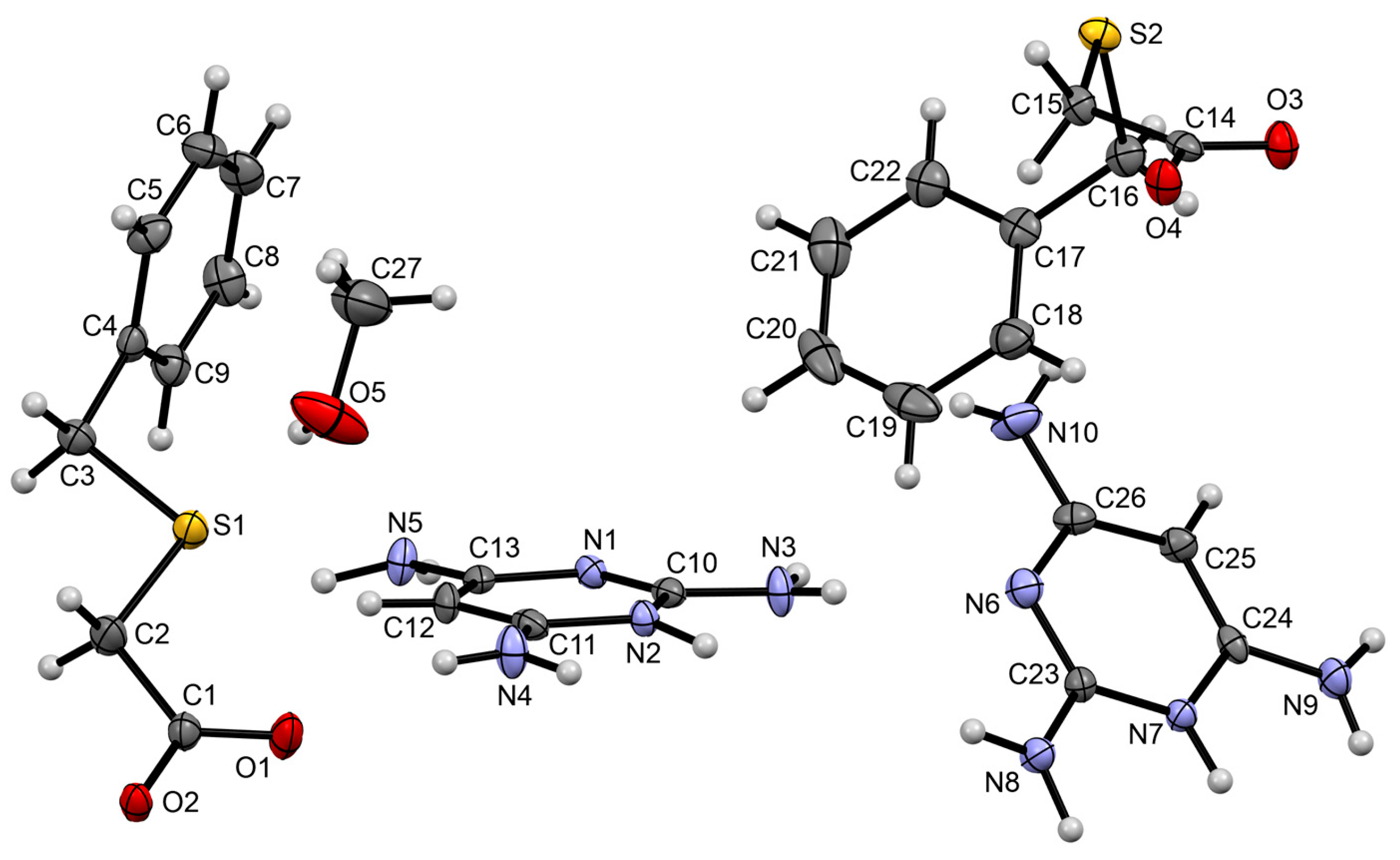
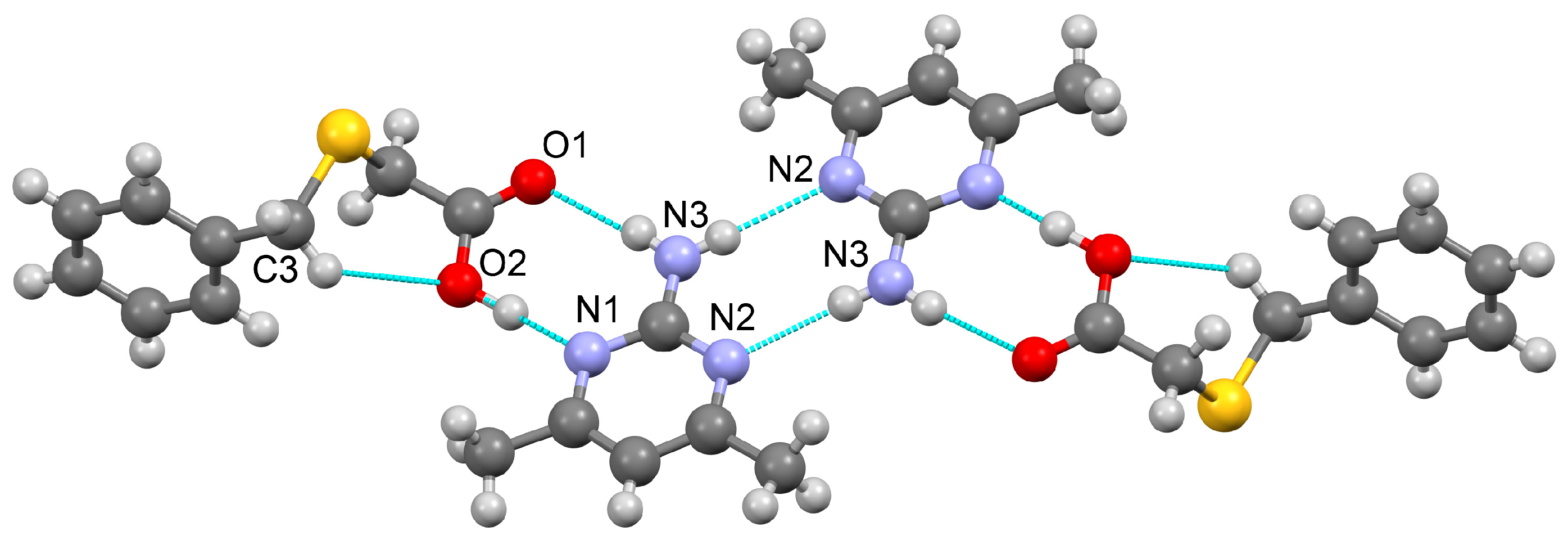
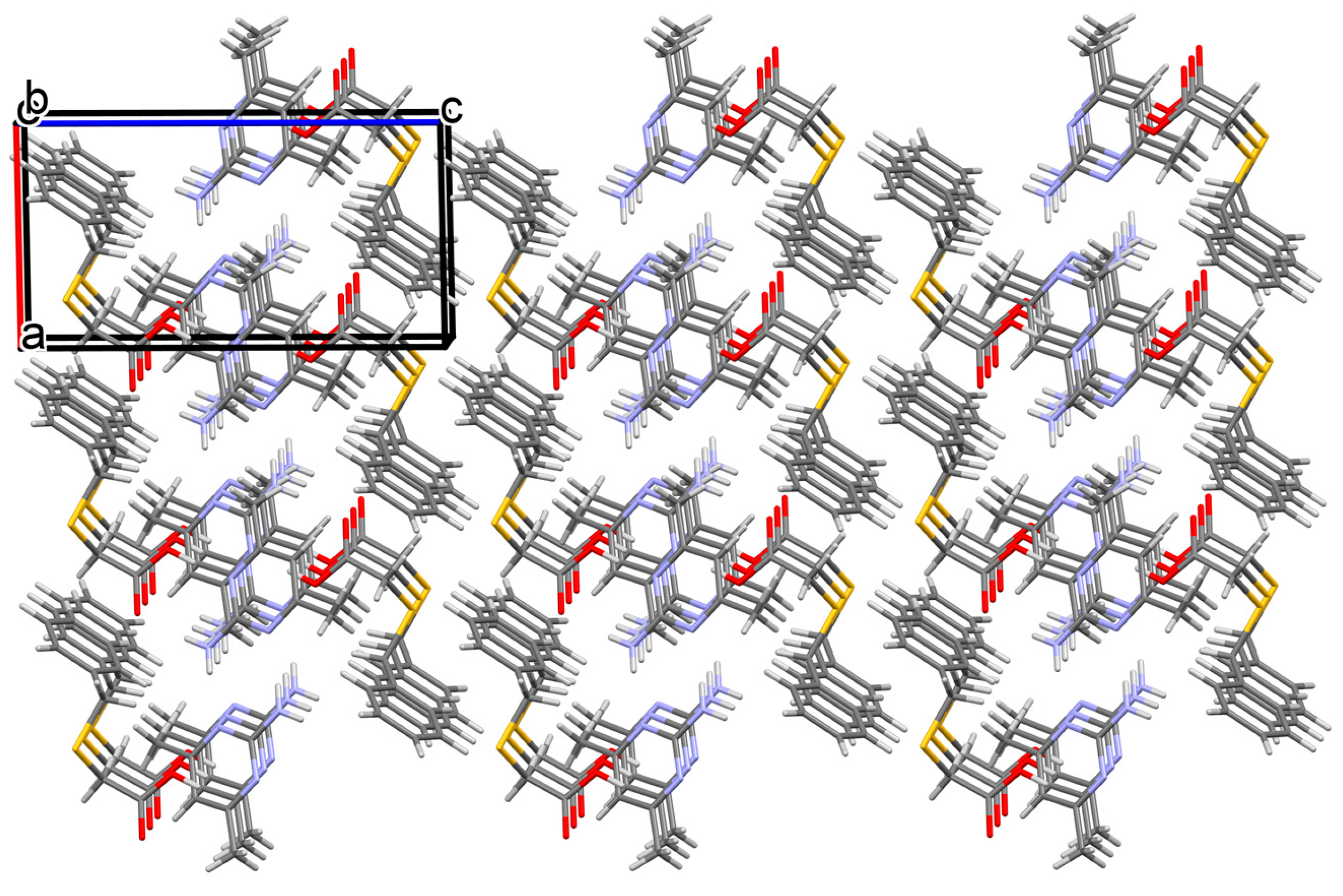
3.1.4. Salt Methanol Solvate Derived from 2,4,6-TAP and HBTA (4)
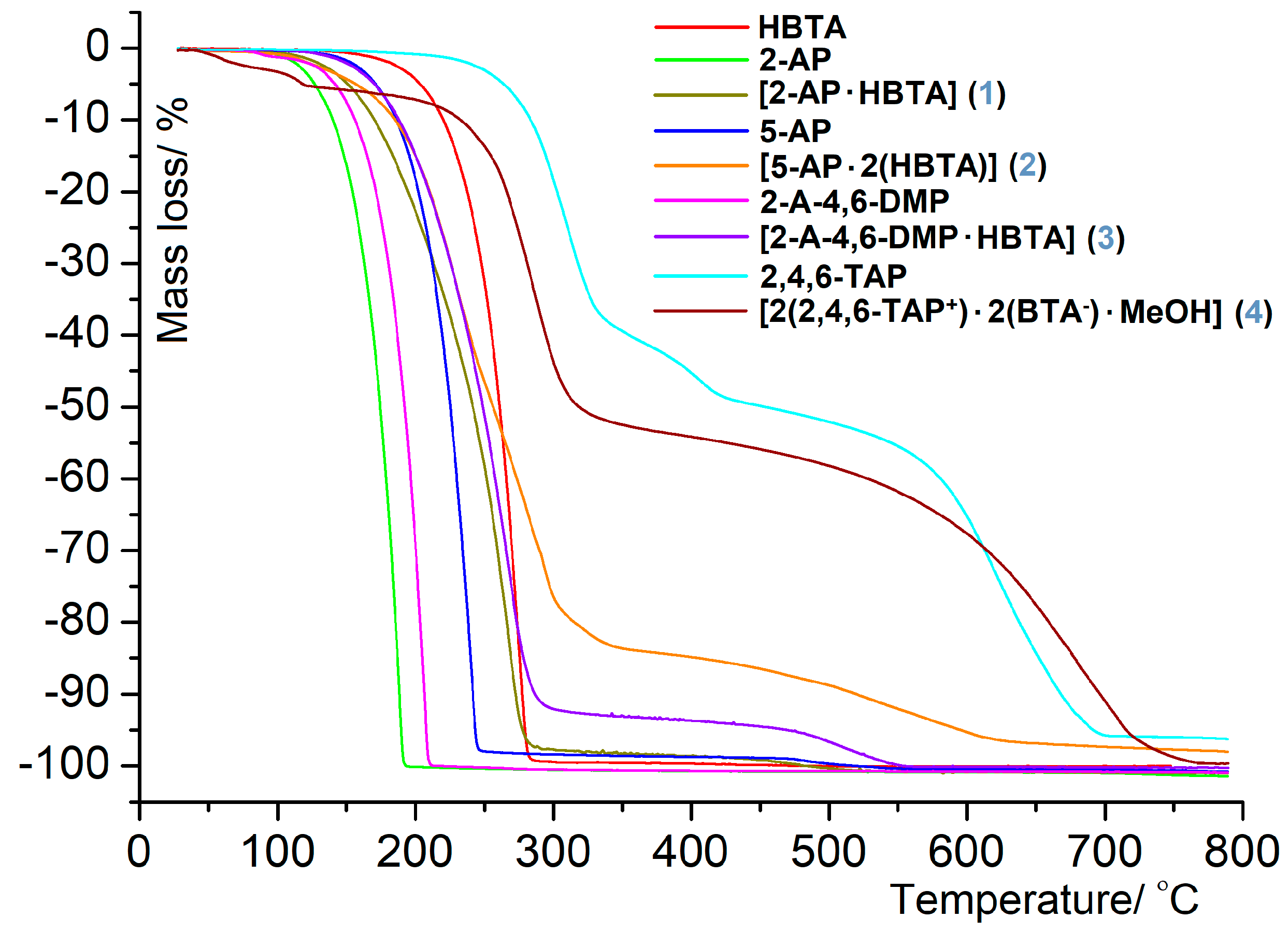
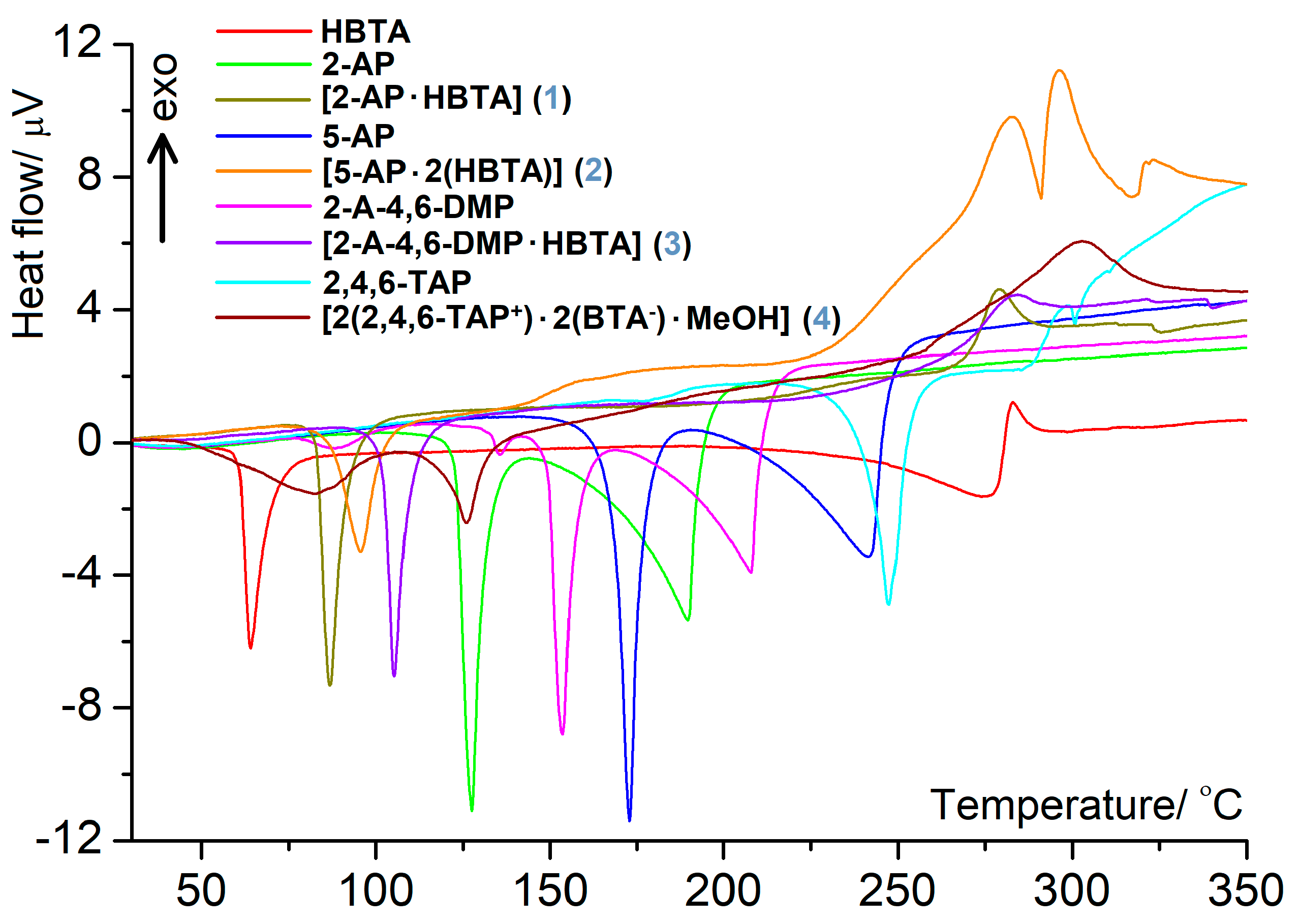
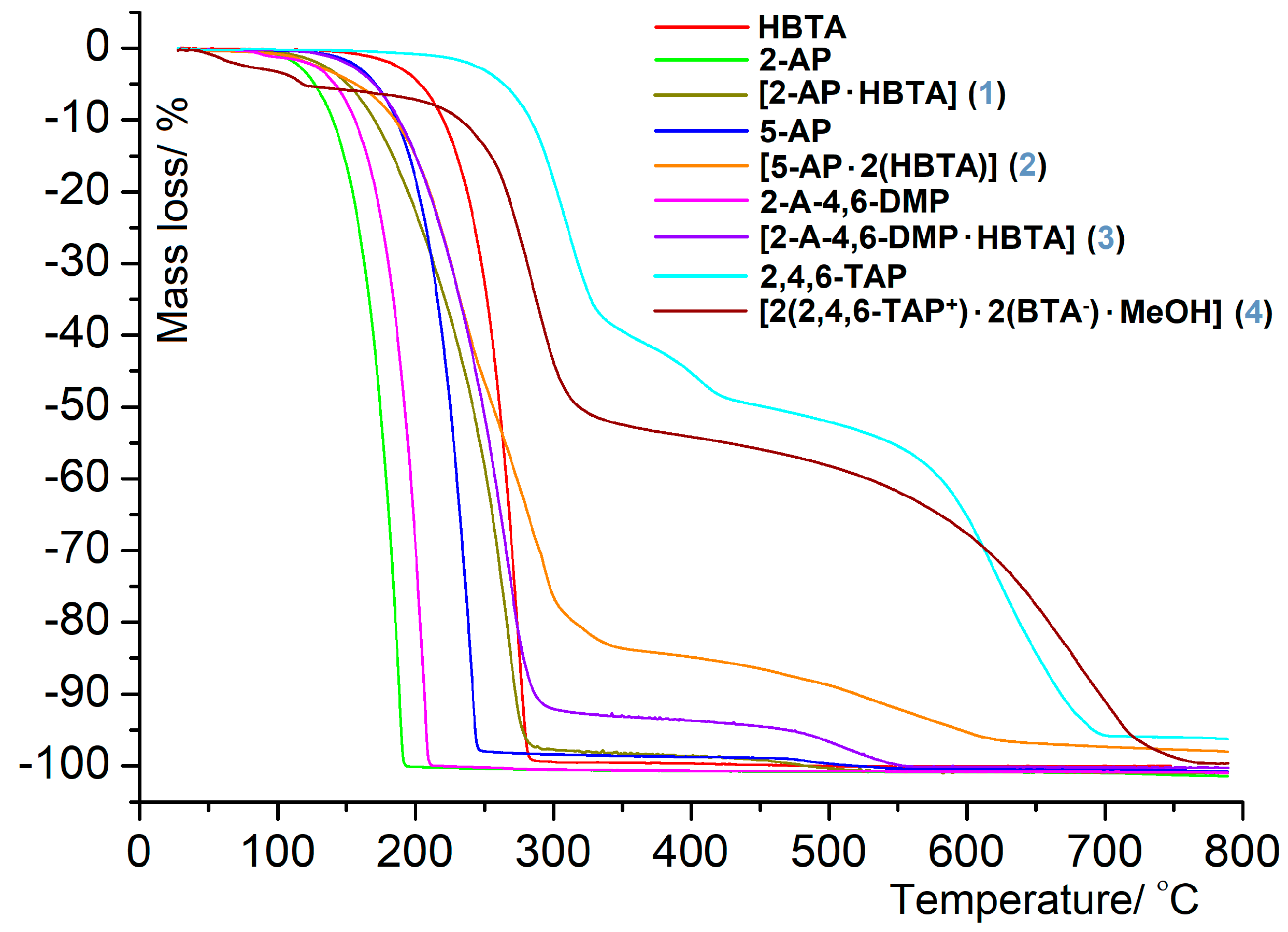
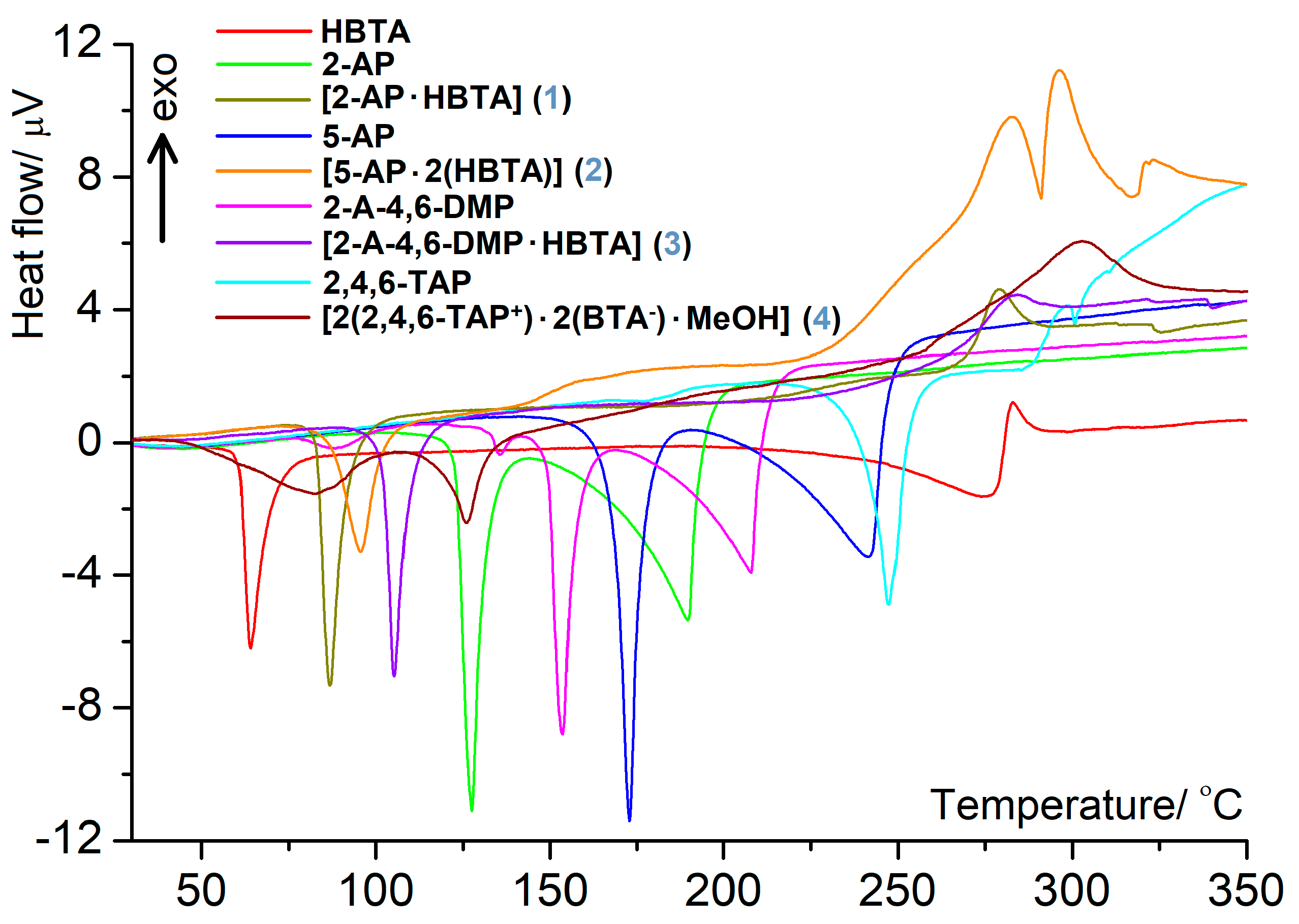
3.2. Thermal Behavior in the Air Atmosphere
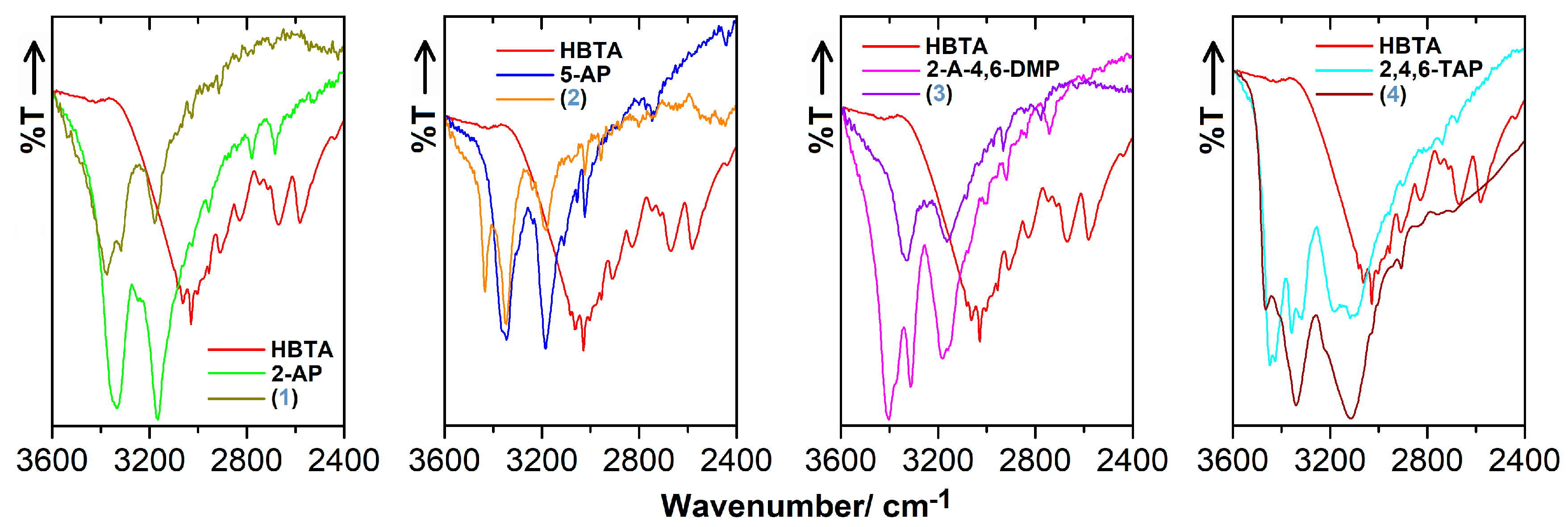
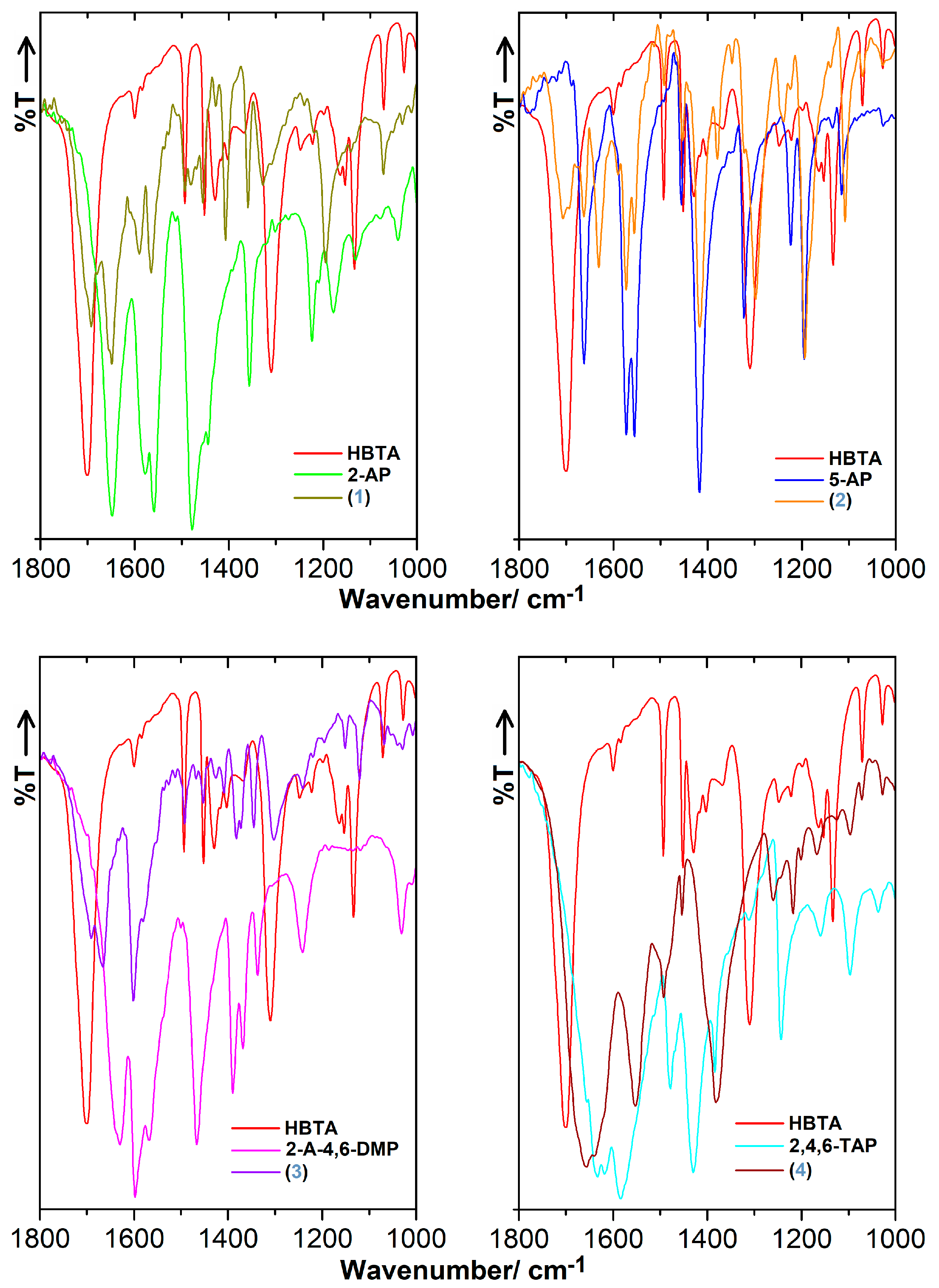
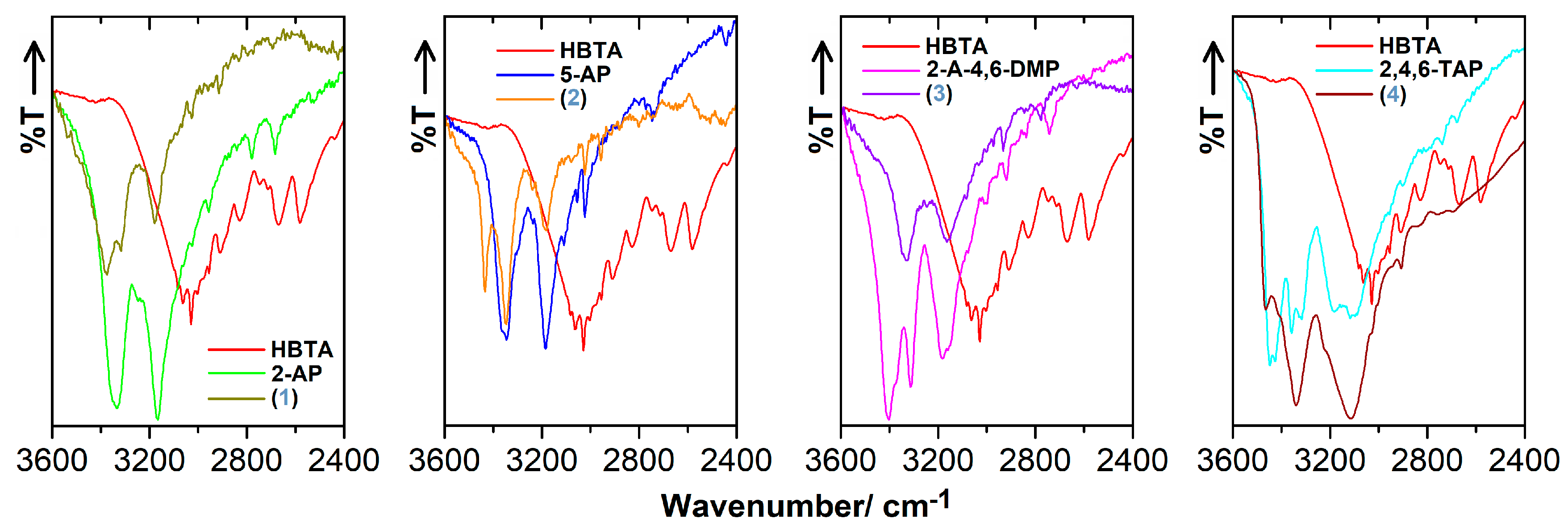
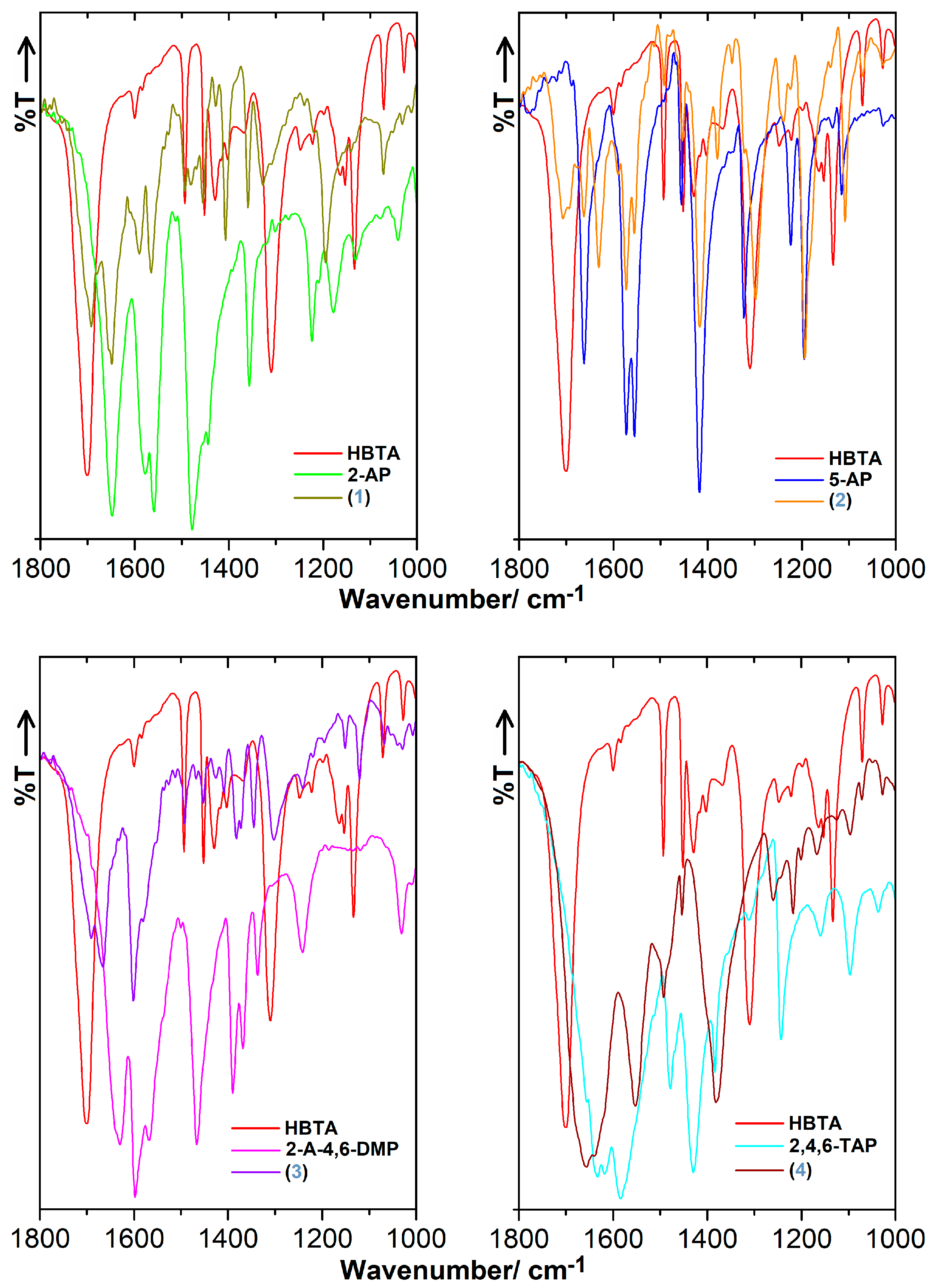
3.3. FT-IR Characteristics
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Ebenezer, S.; Muthiah, P.T. Design of co-crystals/salts of aminopyrimidines and carboxylic acids through recurrently occurring synthons. Cryst. Growth Des. 2012, 12, 3766–3785. [Google Scholar] [CrossRef]
- Oruganti, M.; Nechipadappu, S.K.; Khade, P.A.; Trivedi, D.R. Solid-state versatility of the molecular salts/cocrystals of 2-chloro-4-nitrobenzoic acid: A case study on halogen bonds. ACS Omega 2017, 2, 7146–7162. [Google Scholar] [CrossRef] [PubMed]
- Mirzaei, M.; Sadeghi, F.; Molčanov, K.; Zaręba, J.K.; Gomila, R.M. Recurrent supramolecular motifs in a series of acid–base adducts based on pyridine-2,5-dicarboxylic acid N-oxide and organic bases: Inter- and intramolecular hydrogen bonding. Cryst. Growth Des. 2020, 20, 1738–1751. [Google Scholar] [CrossRef]
- Odiase, I.; Nicholson, C.E.; Ahmad, R.; Cooper, J.; Yufit, D.S.; Cooper, S.J. Three cocrystals and a cocrystal salt of pyrimidin-2-amine and glutaric acid. Acta Crystallogr. 2015, C71, 276–283. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhou, J.; Wu, Y.; Wang, P.; Jin, S.; Lu, Y. Noncovalent-bonded 2D-3D supramolecular adducts from 6-methylpyridine-3-carboxamide and carboxylic acids. J. Mol. Struct. 2022, 1264, 133256. [Google Scholar] [CrossRef]
- Wu, R.; Yu, Y.; Guo, M.; Jin, S.; Wang, D. Eight salts of 4-dimethylaminopyridine and organic acids by H-bonds and some noncovalent associations. J. Mol. Struct. 2021, 1230, 129850. [Google Scholar] [CrossRef]
- Xu, W.; Hu, K.; Lu, Y.; Ye, H.; Jin, S.; Li, M.; Guo, M.; Wang, D. The crystal structures of ten supramolecular salts of benzylamine and organic acids. J. Mol. Struct. 2020, 1219, 128554. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Crystallogr. 2016, B72, 171–179. [Google Scholar] [CrossRef]
- Bruno, I.J.; Cole, J.C.; Edgington, P.R.; Kessler, M.; Macrae, C.F.; McCabe, P.; Pearson, J.; Taylor, R. New software for searching the Cambridge Structural Database and visualizing crystal structures. Acta Crystallogr. 2002, B58, 389–397. [Google Scholar] [CrossRef]
- Grothe, E.; Meekes, H.; Vlieg, E.; ter Horst, J.H.; de Gelder, R. Solvates, salts, and cocrystals: A proposal for a feasible classification system. Cryst. Growth Des. 2016, 16, 3237–3243. [Google Scholar] [CrossRef]
- Jin, S.; Wang, D.; Liang, S.; Chen, S. Crystal and molecular structure of two organic acid–base adducts from 2-aminopyrimidine and carboxylic acids. J. Chem. Crystallogr. 2012, 42, 759–766. [Google Scholar] [CrossRef]
- Eshtiagh-Hosseini, H.; Yousefi, Z.; Mirzaei, M. 2-Aminopyrimidinium hydrogen oxalate monohydrate. Acta Crystallogr. 2009, E65, o2816. [Google Scholar] [CrossRef] [PubMed]
- Miyan, L.; Adam, A.M.A.; Refat, M.S.; Alsuhaibani, A.M. 2-Aminopyrimidine-oxalic acid liquid-liquid charge-transfer interactions: Synthesis, spectroscopic characterizations, and the effect of temperature. J. Mol. Liq. 2022, 365, 120106. [Google Scholar] [CrossRef]
- Eshtiagh-Hosseini, H.; Mahjoobizadeh, M.; Mirzaei, M. 2-Aminopyrimidinium 4-hydroxypyridinium-2,6-dicacoxylate monohydrate. Acta Crystallogr. 2010, E66, o2210. [Google Scholar]
- Smith, G.; Wermuth, U.D.; Healy, P.C. Hydrogen bonding in proton-transfer compounds of 5-sulfosalicylic acid with ortho-substituted monocyclic hetroaromatic Lewis bases. J. Chem. Crystallogr. 2006, 36, 841–849. [Google Scholar] [CrossRef]
- Smith, G.; Wermuth, U.D.; Healy, P.C.; White, J.M. Structure-making with 3,5-dinitrosalicylic acid. II.* The proton-transfer compounds of 3,5-dinitrosalicylic acid with the monocyclic heteroaromatic amines. Aust. J. Chem. 2003, 56, 707–713. [Google Scholar] [CrossRef]
- Kobayashi, K.; Shirasaka, T.; Yamaguchi, K.; Sakamoto, S.; Horn, E.; Furukawa, N. Molecular capsule constructed by multiple hydrogen bonds: Self-assembly of cavitand tetracarboxylic acid with 2-aminopyrimidine. Chem. Commun. 2000, 41–42. [Google Scholar] [CrossRef]
- Skovsgaard, S.; Bond, A.D. Co-crystallization of benzoic acid derivatives with N-containing bases in solution and by mechanical grinding: Stoichiometric variants, polymorphism and twinning. CrystEngComm 2009, 11, 444–453. [Google Scholar] [CrossRef]
- Lynch, D.E.; Smith, G.; Freney, D.; Byriel, K.A.; Kennard, H.L. Molecular cocrystals of carboxylic acids. XV* Preparation and characterization of heterocyclic base adducts with a series of carboxylic acids, and the crystal structures of the adducts of 2-aminopyrimidine with 2,6-dihydroxybenzoic acid, 4-aminobenzoic acid, phenoxyacetic acid, (2,4-dichlorophenoxy)acetic acid, (3,4-dichloro)phenoxyacetic acid and salicylic acid, and 2-aminopyridine with 2,6-dihydroxybenzoic acid. Aust. J. Chem. 1994, 47, 1097–1115. [Google Scholar]
- Lynch, D.E.; Barfield, J.; Frost, J.; Antrobus, R.; Simmons, J. Conformational comparisons between phenoxyacetic acid derivatives in adducts and those in the free form. Part 2. Cryst. Eng. 2003, 6, 109–122. [Google Scholar] [CrossRef]
- Ostasz, A.; Łyszczek, R.; Mazur, L.; Tarasiuk, B. Co-crystal formation between 2-amino-4,6-dimethylpyrimidine and a new p-xylylene-bis(thioacetic) acid. CrystEngComm 2014, 16, 10262–10272. [Google Scholar] [CrossRef]
- Adsmond, D.A.; Sinha, A.S.; Khandavilli, U.B.R.; Maguire, A.R.; Lawrence, S.E. Design and synthesis of ternary cocrystals using carboxyphenols and two complementary acceptor compounds. Cryst. Growth Des. 2016, 16, 59–69. [Google Scholar] [CrossRef]
- Zong, Y.; Shao, H.; Pang, Y.; Wang, D.; Liu, K.; Wang, L. Multicomponent hydrogen-bonding organic solids constructed from 6-hydroxy-2-napthoic acid and N-heterocycles: Synthesis, structural characterization and synthon discussion. J. Mol. Struct. 2016, 1115, 187–198. [Google Scholar] [CrossRef]
- Singh, M.P.; Tarai, A.; Baruah, J.B. Neuytal, zwitterions, ionic forms of 5-aminoisophthalic acid in cocrystals, salts and their optical properties. ChemistrySelect 2019, 4, 5427–5436. [Google Scholar] [CrossRef]
- Thirumurugan, R.; Anitha, K. Experimental and theoretical investigations on a hydrous organic salt of 2,4,6-triaminopyrimidine-1,3-diium L-tartarate monohydrate (TTM). J. Mol. Struct. 2021, 1237, 130368. [Google Scholar] [CrossRef]
- Sangavi, M.; Kumaraguru, N.; McMillen, C.D.; Butcher, R.J. Supramolecular interactions in some organic hydrated 2,4,6-triaminopyrimidinium carboxylate and sulfate salts. Acta Crystallogr. 2023, C79, 435–442. [Google Scholar] [CrossRef]
- Abidi, S.S.A.; Azim, Y.; Gupta, A.K.; Pradeep, C.P. Cocrystals of indole-3-acetic acid and indole-3-butyric acid: Synthesis, structural characterization and Hirshfeld surface analysis. J. Mol. Struct. 2018, 1166, 202–213. [Google Scholar] [CrossRef]
- Mohana, M.; Muthiah, P.T.; McMillen, C.D.; Butcher, R.J. Supramolecular interactions in salts/cocrystals involving pyrimidine derivatives of sulfonate/carboxylic acid. Acta Crystallogr. 2023, C79, 61–67. [Google Scholar] [CrossRef]
- Xing, P.; Li, Q.; Li, Y.; Wang, K.; Zhang, Q.; Wang, L. Organic salts formed by 2,4,6-triaminopyrimidine and selected carboxylic acids via variety of hydrogen bonds: Synthons cooperation, and crystal structures. J. Mol. Struct. 2017, 1136, 59–68. [Google Scholar] [CrossRef]
- Mapp, L.K.; Coles, S.J.; Aitipamula, S. Novel solid forms of lonidamine: Crystal structures and physicochemical properties. CrystEngComm 2017, 19, 2925–2935. [Google Scholar] [CrossRef]
- Li, Z.; Li, Y.; Du, F. Crystal structure of 2,4,6-pyrimidinetriamine-trifluoroacetate(1:1), C4H8N5+C2F3O2−, C6H8F3N5O2. Z. Kristallogr. NCS 2012, 227, 519–520. [Google Scholar]
- Garg, U.; Azim, Y.; Kar, A.; Pradeep, C.P. Cocrystals/salt of 1-naphthaleneacetic acid and utilizing Hirshfeld surface calculations for acid−aminopyrimidine synthons. CrystEngComm 2020, 22, 2978–2989. [Google Scholar] [CrossRef]
- Pedireddi, V.R.; Chatterjee, S.; Ranganathan, A.; Rao, C.N.R. A study of supramolecular hydrogen bonded complexes formed by aliphatic dicarboxylic acids with azaaromatic donors. Tetrahedron 1998, 54, 9457–9474. [Google Scholar] [CrossRef]
- Sienkiewicz-Gromiuk, J.; Tarasiuk, B.; Mazur, L. New organic single crystal of (benzylthio)acetic acid: Synthesis, crystal structure, spectroscopic (ATR-FTIR, 1H and 13C NMR) and thermal characterization. J. Mol. Struct. 2016, 1110, 65–71. [Google Scholar] [CrossRef]
- Sienkiewicz-Gromiuk, J.; Drzewiecka-Antonik, A. The first noncovalent-bonded supramolecular frameworks of (benzylthio)acetic acid with proline compounds, isonicotinamide and tryptamine. Molecules 2022, 27, 8203. [Google Scholar] [CrossRef] [PubMed]
- CrysAlis PRO; v.1.171.37.35g; Agilent Technologies Ltd.: Oxford, UK, 2014.
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. 2008, A64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. 2015, C71, 3–8. [Google Scholar]
- Farrugia, L.J. WinGX and ORTEP for Windows: An update. J. Appl. Cryst. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Macrae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M.; et al. Mercury 4.0: From visualization to analysis, design and prediction. J. Appl. Crystallogr. 2020, 53, 226–235. [Google Scholar] [CrossRef]
- Singaravelan, K.; Chandramohan, A.; Madhankumar, S.; Enoch, M.V.; Vinitha, G. Structural characterization, computational and biological studies of a new third order NLO (1:1) organic adduct: 2-Aminopyrimidine:3-nitrophthalic acid. J. Mol. Struct. 2019, 1194, 57–65. [Google Scholar] [CrossRef]
- Sienkiewicz-Gromiuk, J. DFT approach to (benzylthio)acetic acid: Conformational search. Molecular (monomer and dimer) structure, vibrational spectroscopy and some electronic properties. Spectrochim. Acta A 2018, 189, 116–128. [Google Scholar] [CrossRef] [PubMed]
- Prabavathi, N.; Nilufer, A.; Krishnakumar, V.; Akilandeswari, L. Spectroscopic, electronic structure and natural bond analysis of 2-aminopyrimidine and 4-aminopyrazolo[3,4-d]pyrimidine: A comparative study. Spectrochim. Acta A 2012, 96, 226–241. [Google Scholar] [CrossRef] [PubMed]
- Rospenk, M.; Koll, A. Self-assembly of 2-aminopyrimidines in nonpolar solvents. J. Mol. Struct. 2007, 844–845, 232–241. [Google Scholar] [CrossRef]
- Fu, W.-W.; Liu, Y.; Huang, G.; Zhu, X.-M. 4,6-dimethylpyrimidin-2-amine. Acta Crystallogr. 2013, E69, o32. [Google Scholar] [CrossRef]
- Lin, C.-H.; Guo, H.-M.; Jian, F.-F. Crystal structure of 2-amino-4,6-dimethylpyrimidine hydrate, (C6H9N3)10‧H2O. Z. Kristallogr. NCS 2008, 223, 511–512. [Google Scholar] [CrossRef]
- Li, Z.; Huang, J.; Meng, A.; Zheng, B. Crystal Structure, energy band and optical properties of benzoic acid—2,amino-4,6-dimethylpyrimidine (1:1) co-crystals. J. Struct. Chem. 2010, 51, 53–59. [Google Scholar] [CrossRef]
- Sundaraganesan, N.; Joshua, B.D.; Meganathan, C.; Sebastian, S. Vibrational spectroscopic studies supported by HF/DFT calculations of 2,4,6-triaminopyrimidine. Indian J. Chem. 2008, A47, 821–829. [Google Scholar]
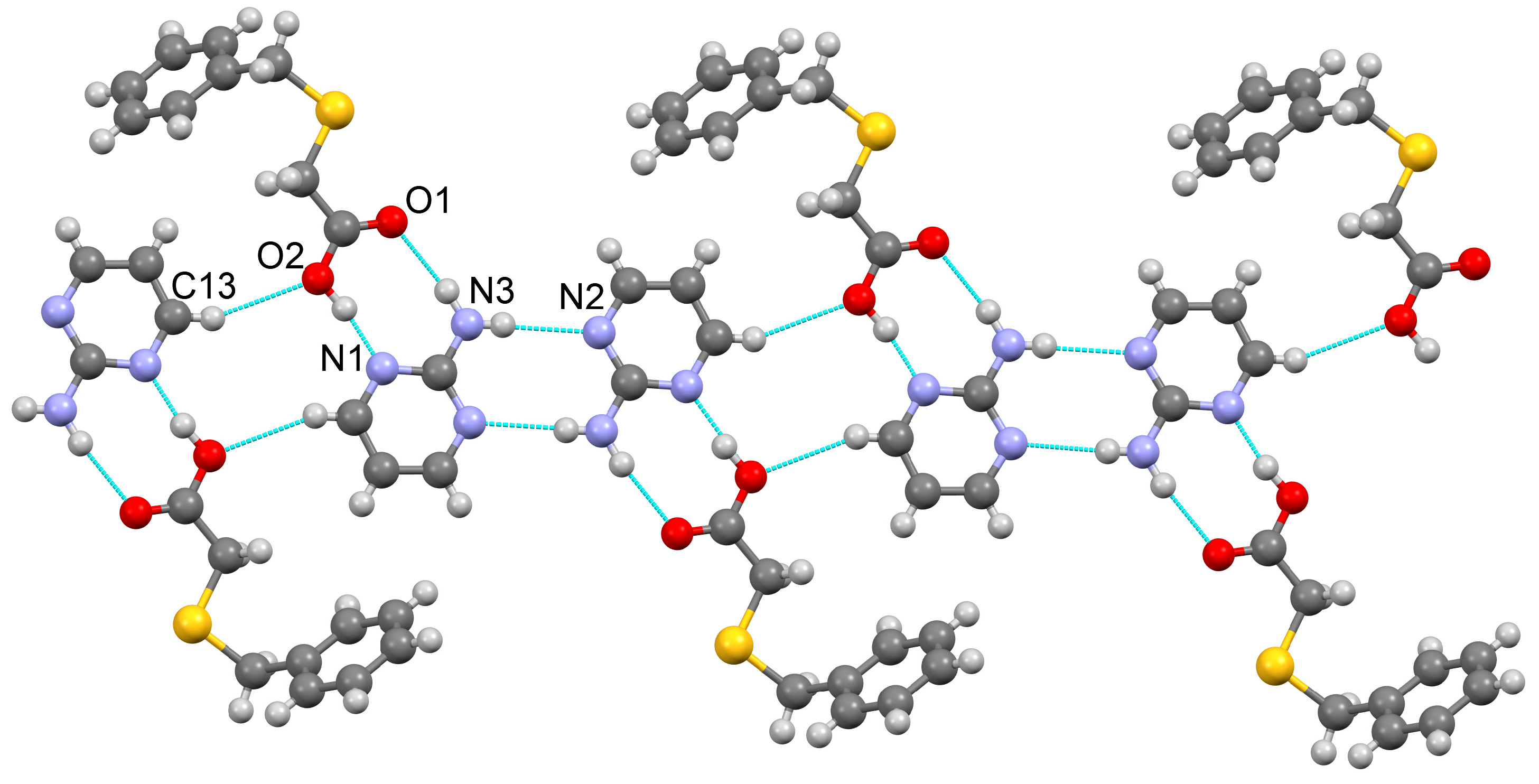
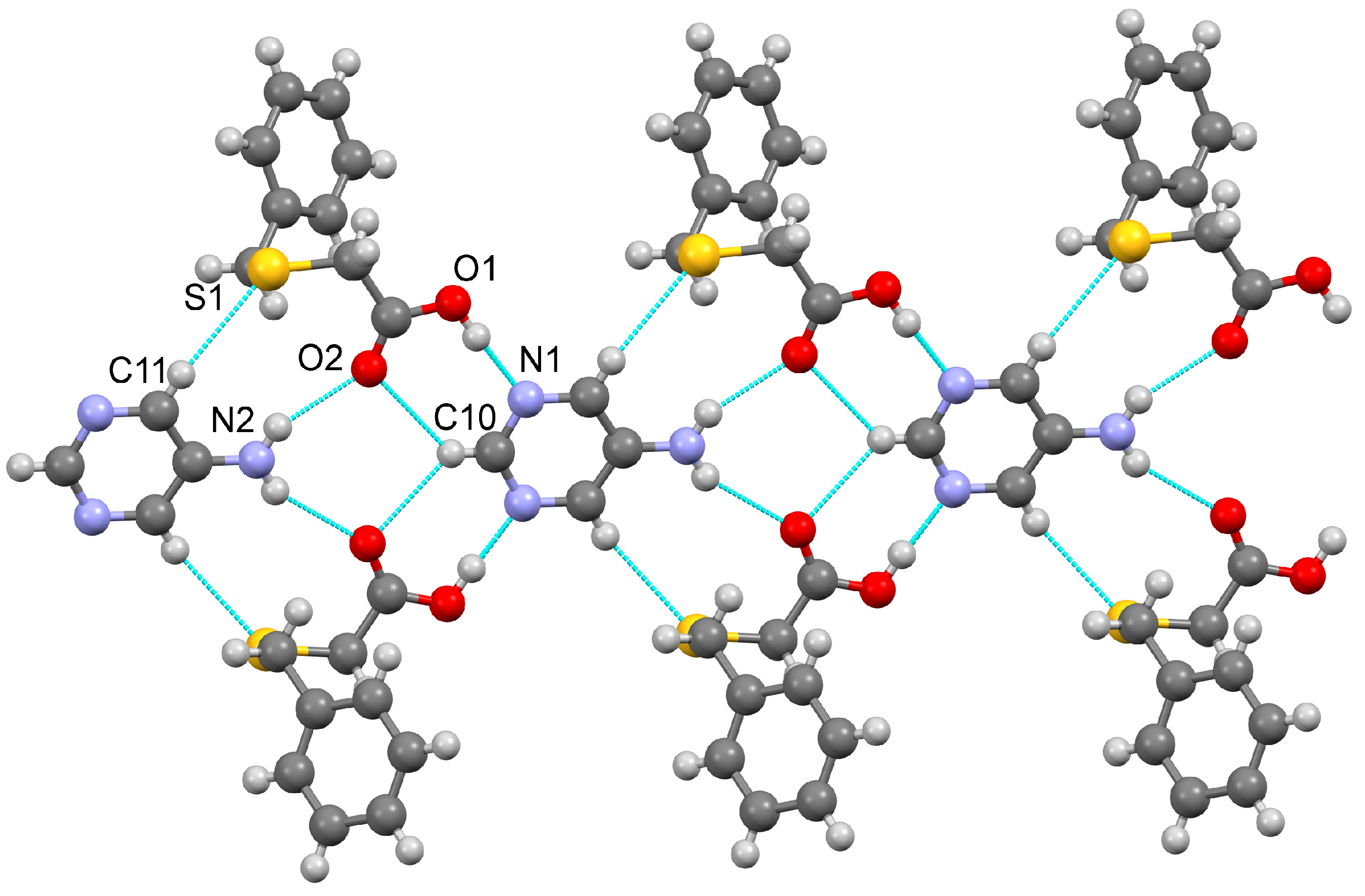

| D–H···A * | D–H [Å] | H···A [Å] | D···A [Å] | D–H···A [°] | * Symmetry Code for A |
|---|---|---|---|---|---|
| [2-AP·HBTA] (1) | |||||
| O2–H1O2···N1 | 0.96(3) | 1.65(3) | 2.595(2) | 169(3) | x, y + 1, z |
| N3–H1N3···O1 | 0.91(2) | 2.09(3) | 2.978(2) | 167(2) | x, y − 1, z |
| N3–H2N3···N2 | 0.82(2) | 2.27(2) | 3.090(3) | 176(2) | −x−1, −y + 1, −z + 1 |
| C13–H13···O2 | 0.93 | 2.66 | 3.531(3) | 157.1 | −x + 1, −y + 1, −z + 1 |
| [5-AP·2(HBTA)] (2) | |||||
| O1–H1O1···N1 | 0.90(2) | 1.74(2) | 2.6361(14) | 176(2) | x, y − 1, z |
| N2–H1N2···O2 | 0.847(16) | 2.268(17) | 3.0225(16) | 148.6(15) | |
| C10–H10···O2 | 0.93 | 2.56 | 3.2693(17) | 133.1 | x, y + 1, z |
| C10–H10···O2 | 0.93 | 2.56 | 3.2693(17) | 133.1 | −x + 3/2, y + 1, −z |
| C11–H11···S1 | 0.93 | 2.84 | 3.7208(13) | 158.5 | |
| [2-A-4,6-DMP·HBTA] (3) | |||||
| O2–H1O2···N1 | 1.01(3) | 1.64(3) | 2.645(2) | 170(3) | |
| N3–H1N3···O1 | 0.81(2) | 2.11(3) | 2.916(3) | 172(2) | |
| N3–H2N3···N2 | 0.83(2) | 2.20(2) | 3.036(3) | 177(2) | −x − 1, −y + 2, −z + 1 |
| C3–H3A···O2 | 0.97 | 2.54 | 3.249(3) | 129.5 | |
| [2(2,4,6-TAP+)·2(BTA−)·MeOH] (4) | |||||
| N2–H1N2···O4 | 0.95(3) | 1.74(4) | 2.684(3) | 176(3) | −x − 1, −y + 1, −z |
| N7–H1N7···O2 | 0.93(4) | 1.80(4) | 2.724(3) | 173(3) | −x − 1, −y, −z |
| N3–H2N3···O2 | 0.90(3) | 2.24(3) | 3.028(3) | 146(2) | x−1, y, z |
| N4–H1N4···O3 | 0.85(4) | 1.99(4) | 2.842(4) | 179(3) | −x − 1, −y + 1, −z |
| N4–H2N4···O1 | 0.83(3) | 2.10(3) | 2.906(4) | 164(3) | |
| N5–H2N5···O2 | 0.94(3) | 2.03(4) | 2.921(3) | 158(3) | −x, −y, −z + 1 |
| N8–H1N8···O4 | 0.89(4) | 2.07(4) | 2.957(3) | 176(3) | −x − 1, −y + 1, −z |
| N8–H2N8···O1 | 0.95(3) | 1.91(3) | 2.853(4) | 169(3) | −x − 1, −y, −z |
| N9–H2N9···O3 | 0.85(4) | 2.03(4) | 2.877(3) | 173(4) | −x − 2, −y + 1, −z |
| N3–H1N3···N6 | 0.82(4) | 2.23(4) | 2.947(4) | 146(3) | |
| N5–H1N5···N1 | 0.84(3) | 2.41(3) | 3.147(4) | 146(3) | −x − 1, −y, −z + 1 |
| N9–H1N9···N3 | 0.83(3) | 2.48(4) | 3.249(4) | 155(3) | −x − 2, −y, −z |
| N10–H2N10···O5 | 0.89(3) | 1.98(3) | 2.835(4) | 161(3) | x − 1, y, z |
| N5–H1N5···S2 | 0.84(3) | 2.91(3) | 3.534(3) | 133(3) | −x − 1, −y + 1, −z + 1 |
| O5–H1O5···S1 | 0.75(5) | 2.56(5) | 3.297(3) | 165(4) | |
| C12–H12···S1 | 0.93 | 2.81 | 3.646(3) | 150.5 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sienkiewicz-Gromiuk, J.; Drzewiecka-Antonik, A. Neutral and Ionic Form of (Benzylthio)Acetic Acid in Novel Aminopyrimidine Based Multi-Component Crystalline Phases. Crystals 2023, 13, 1628. https://doi.org/10.3390/cryst13121628
Sienkiewicz-Gromiuk J, Drzewiecka-Antonik A. Neutral and Ionic Form of (Benzylthio)Acetic Acid in Novel Aminopyrimidine Based Multi-Component Crystalline Phases. Crystals. 2023; 13(12):1628. https://doi.org/10.3390/cryst13121628
Chicago/Turabian StyleSienkiewicz-Gromiuk, Justyna, and Aleksandra Drzewiecka-Antonik. 2023. "Neutral and Ionic Form of (Benzylthio)Acetic Acid in Novel Aminopyrimidine Based Multi-Component Crystalline Phases" Crystals 13, no. 12: 1628. https://doi.org/10.3390/cryst13121628
APA StyleSienkiewicz-Gromiuk, J., & Drzewiecka-Antonik, A. (2023). Neutral and Ionic Form of (Benzylthio)Acetic Acid in Novel Aminopyrimidine Based Multi-Component Crystalline Phases. Crystals, 13(12), 1628. https://doi.org/10.3390/cryst13121628