
High-Pressure Crystal Structure and Unusual Magnetoresistance of a Single-Component Molecular Conductor [Pd(dddt)2] (dddt = 5,6-dihydro-1,4-dithiin-2,3-dithiolate)


Abstract
1. Introduction
2. Materials and Methods
2.1. High-Pressure Electrical Resistivity
2.2. High-Pressure Single Crystal Structure Determination
2.3. Tight-Binding Calculation
2.4. Magnetoresistance Measurements
3. Results
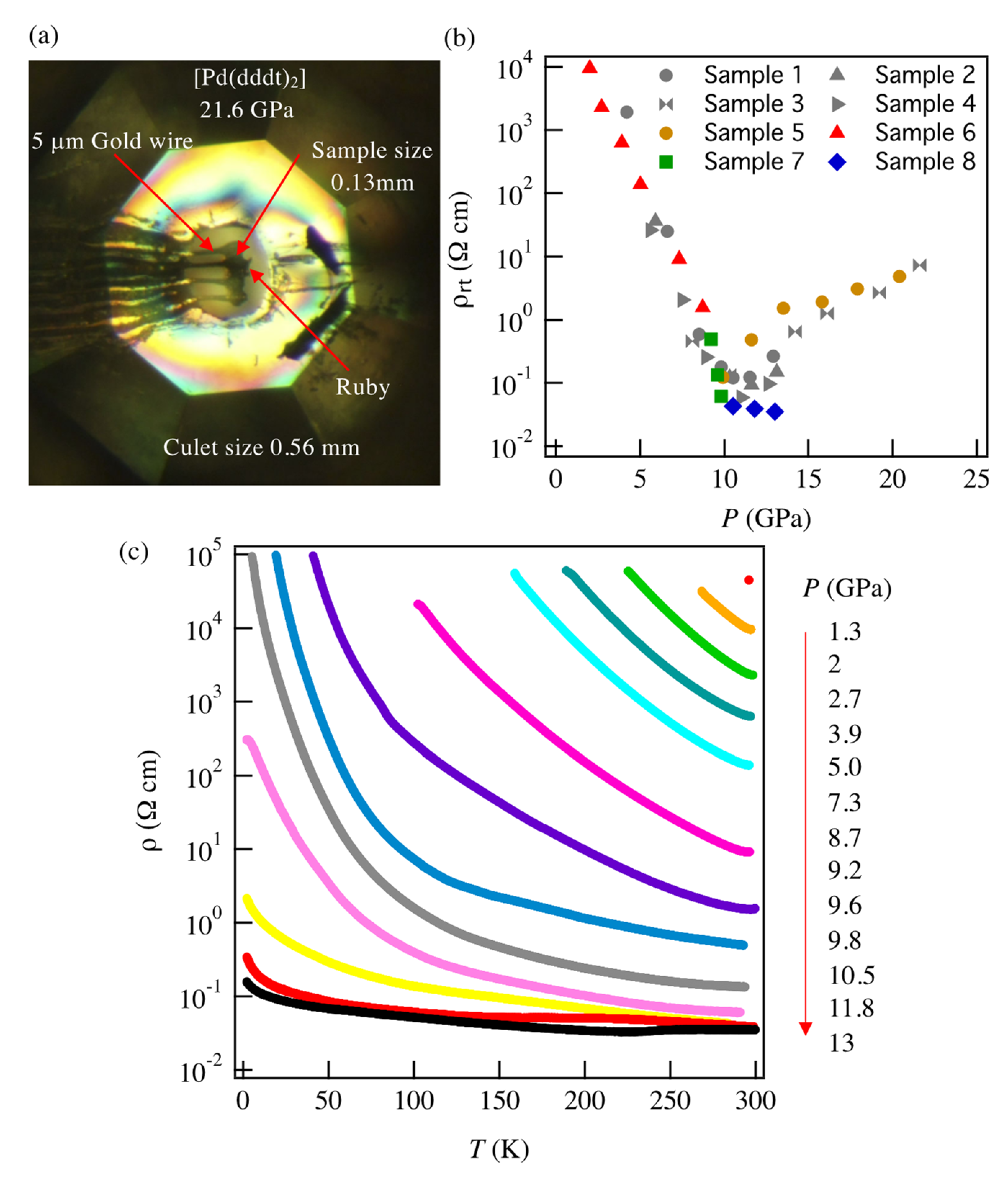
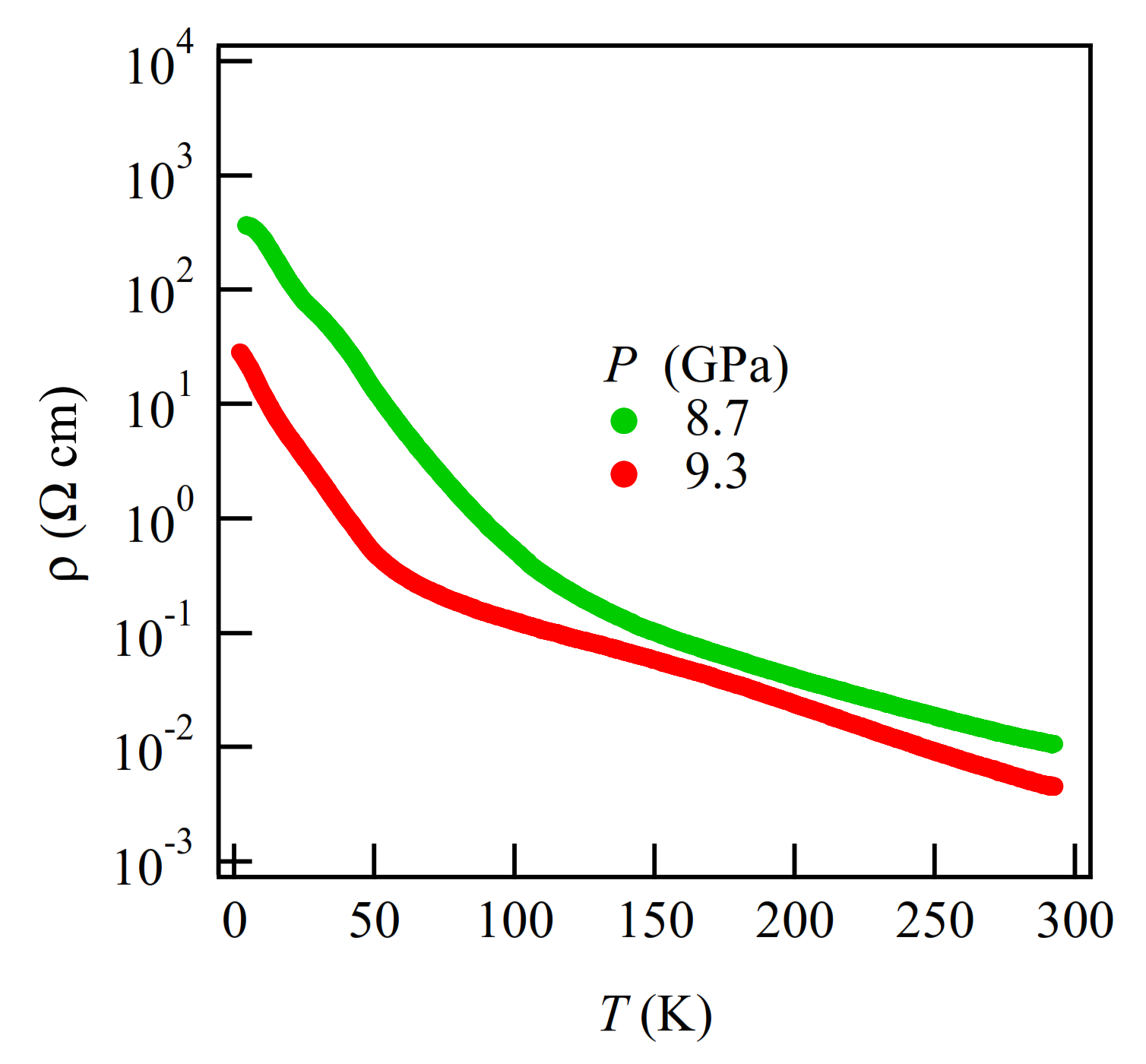
3.1. High-Pressure Electrical Properties
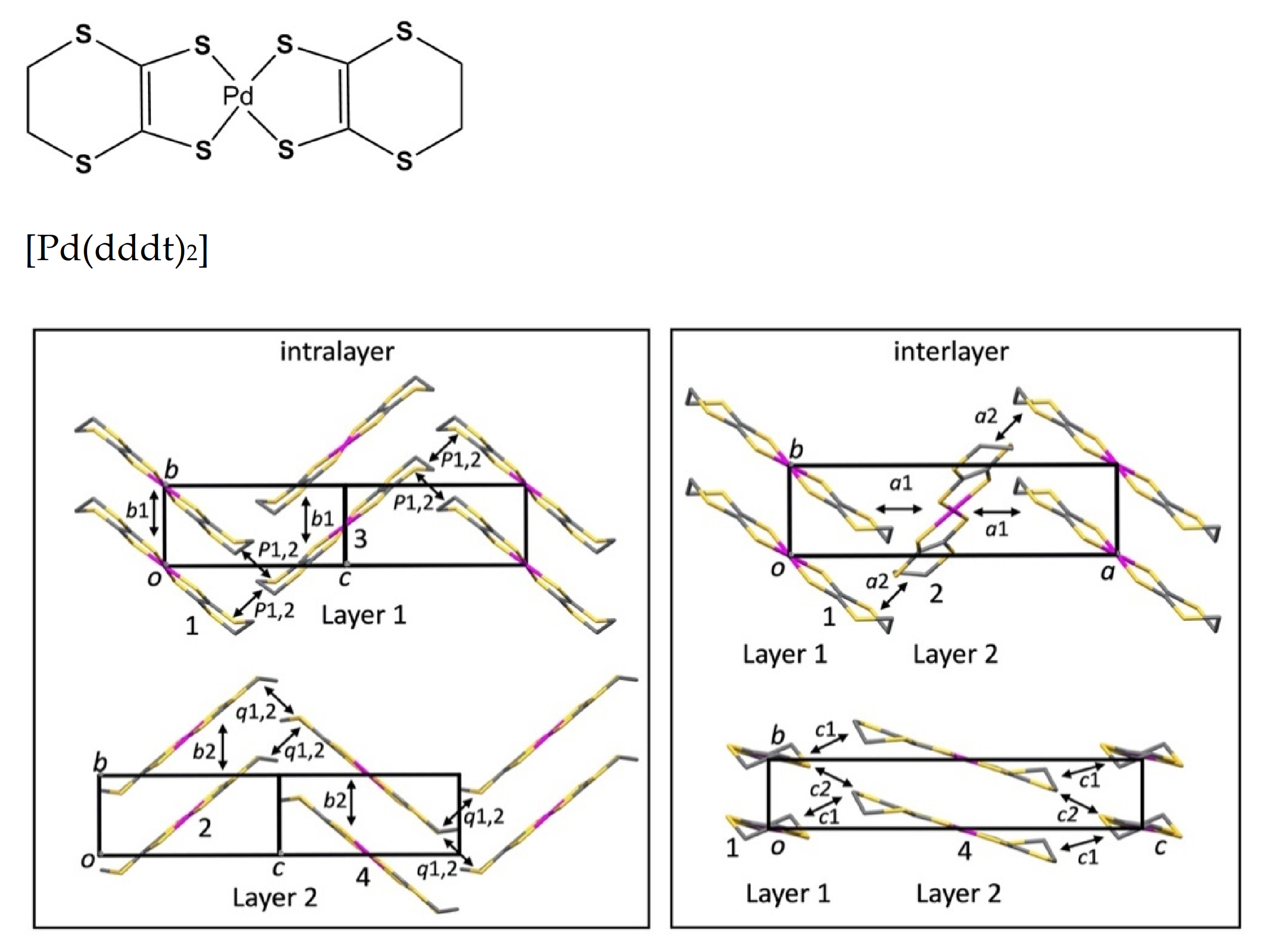
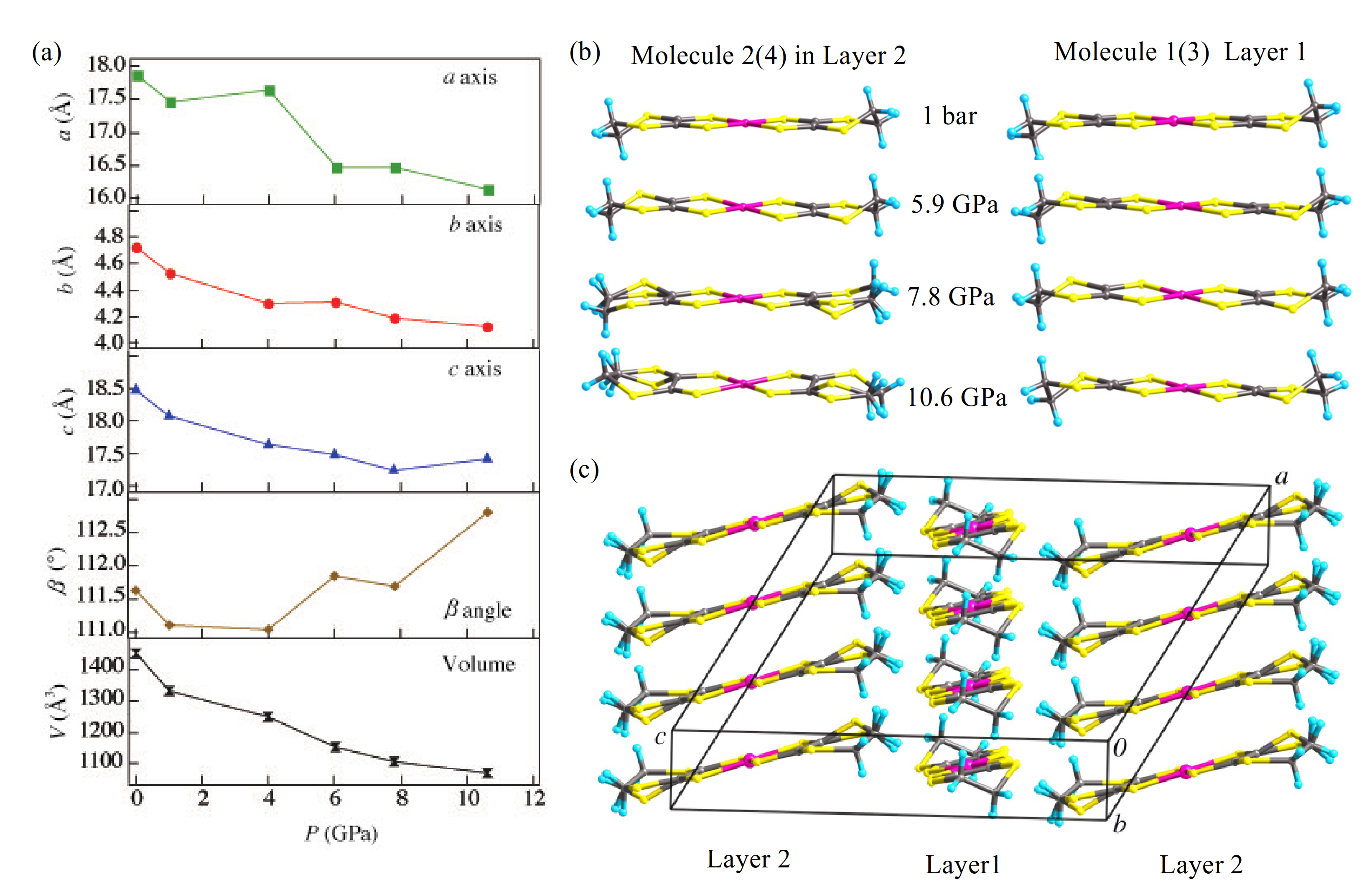
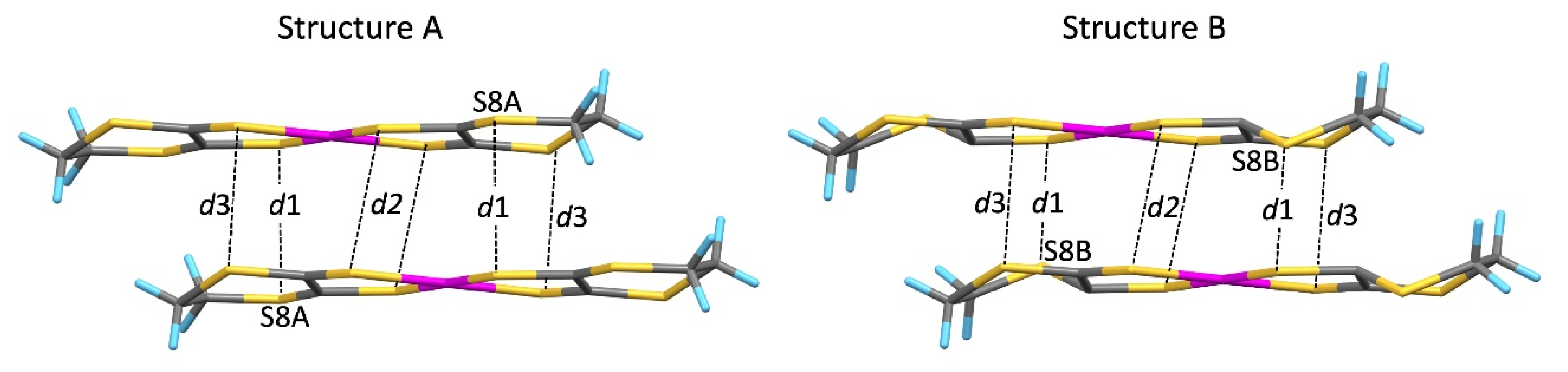
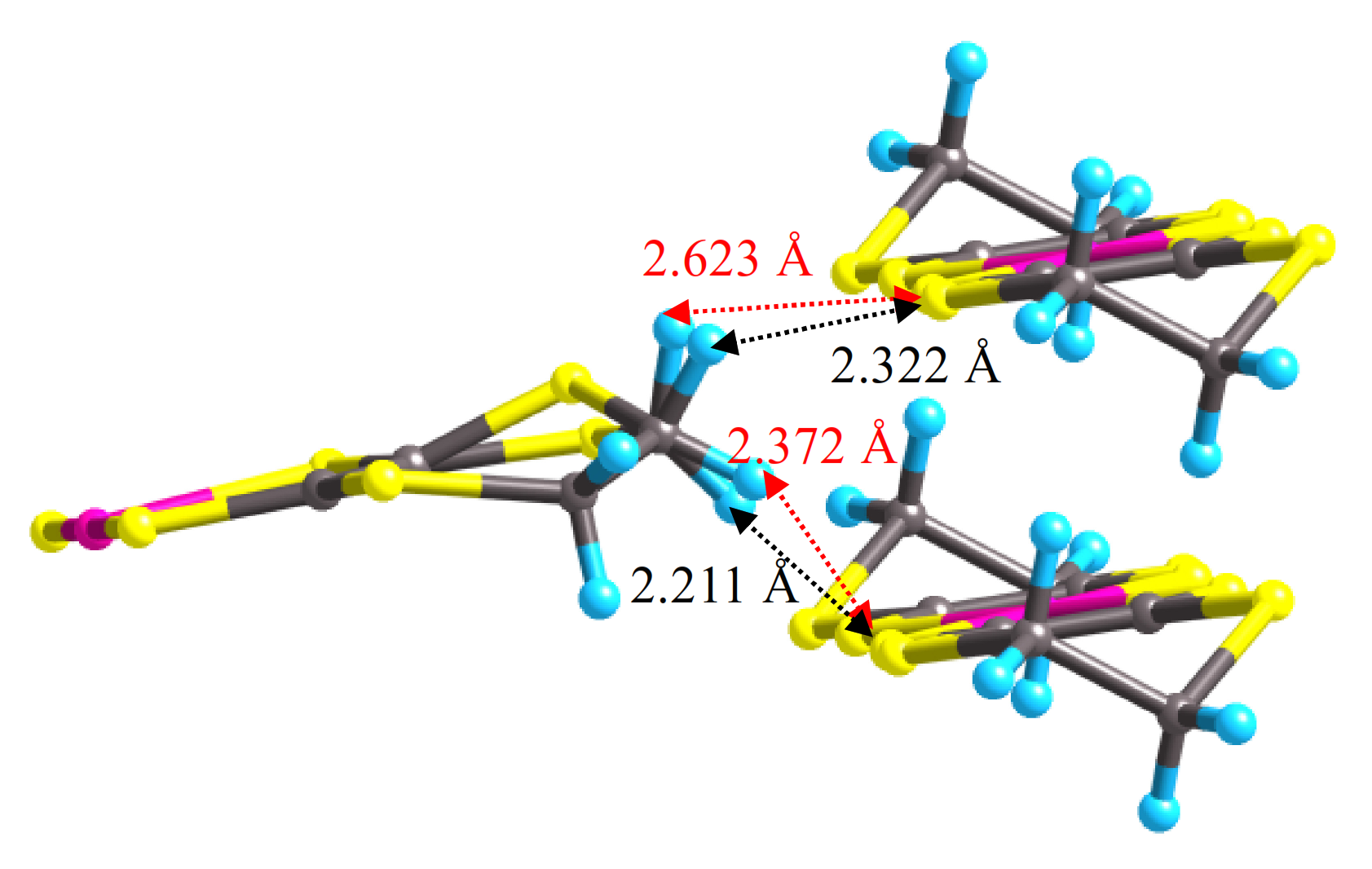
3.2. High-Pressure Single-Crystal Structure
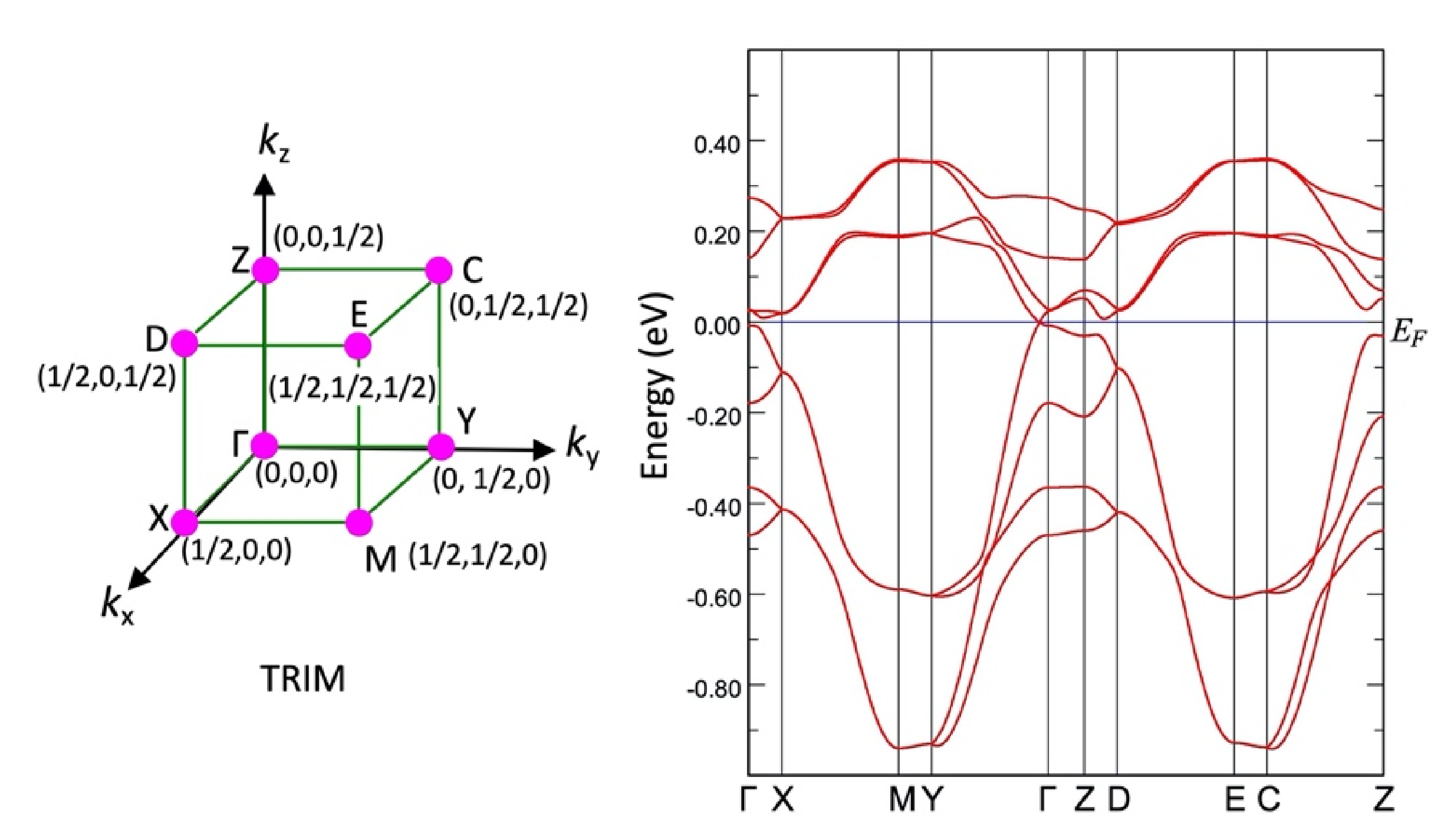
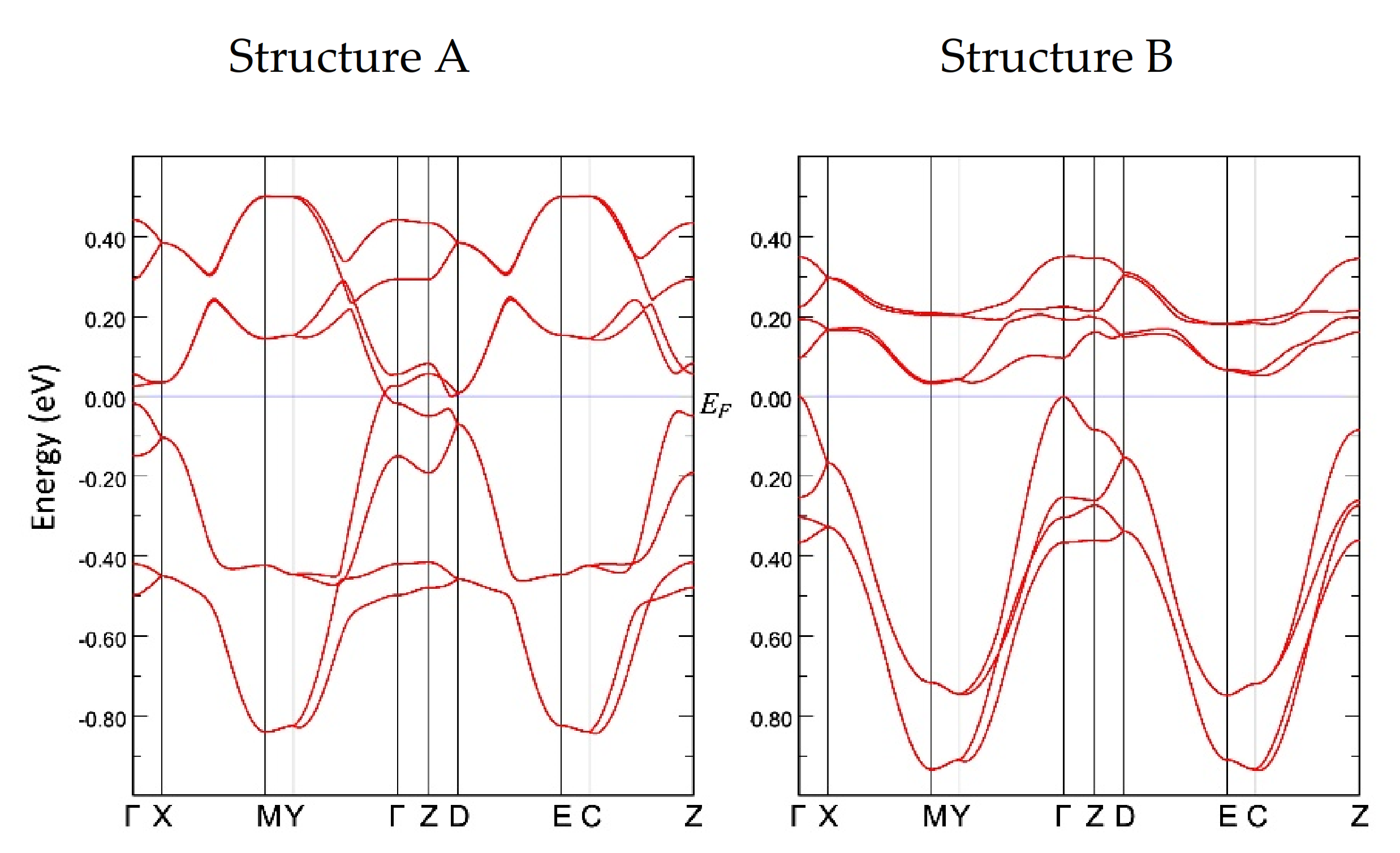
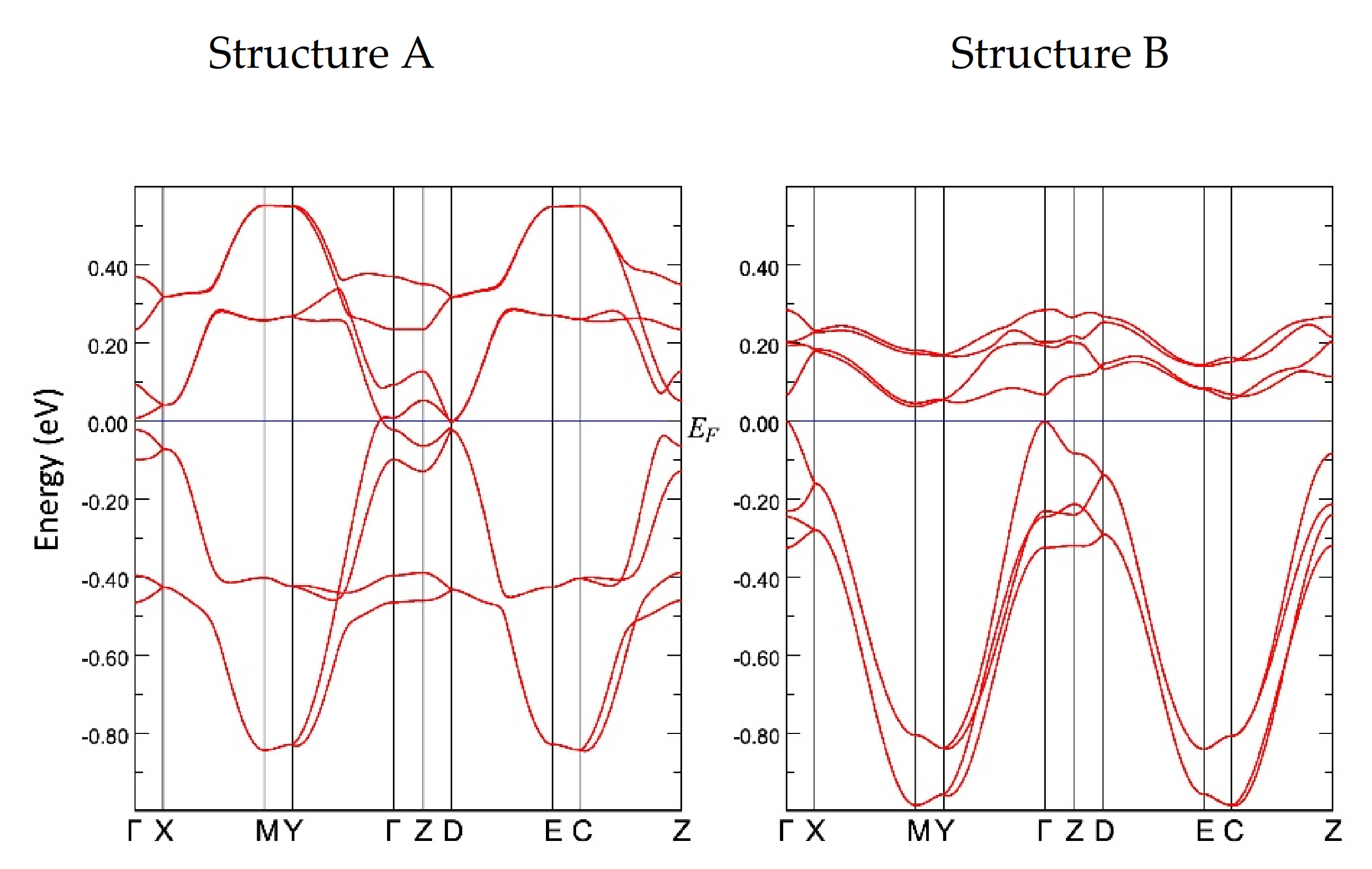
3.3. Band Structure
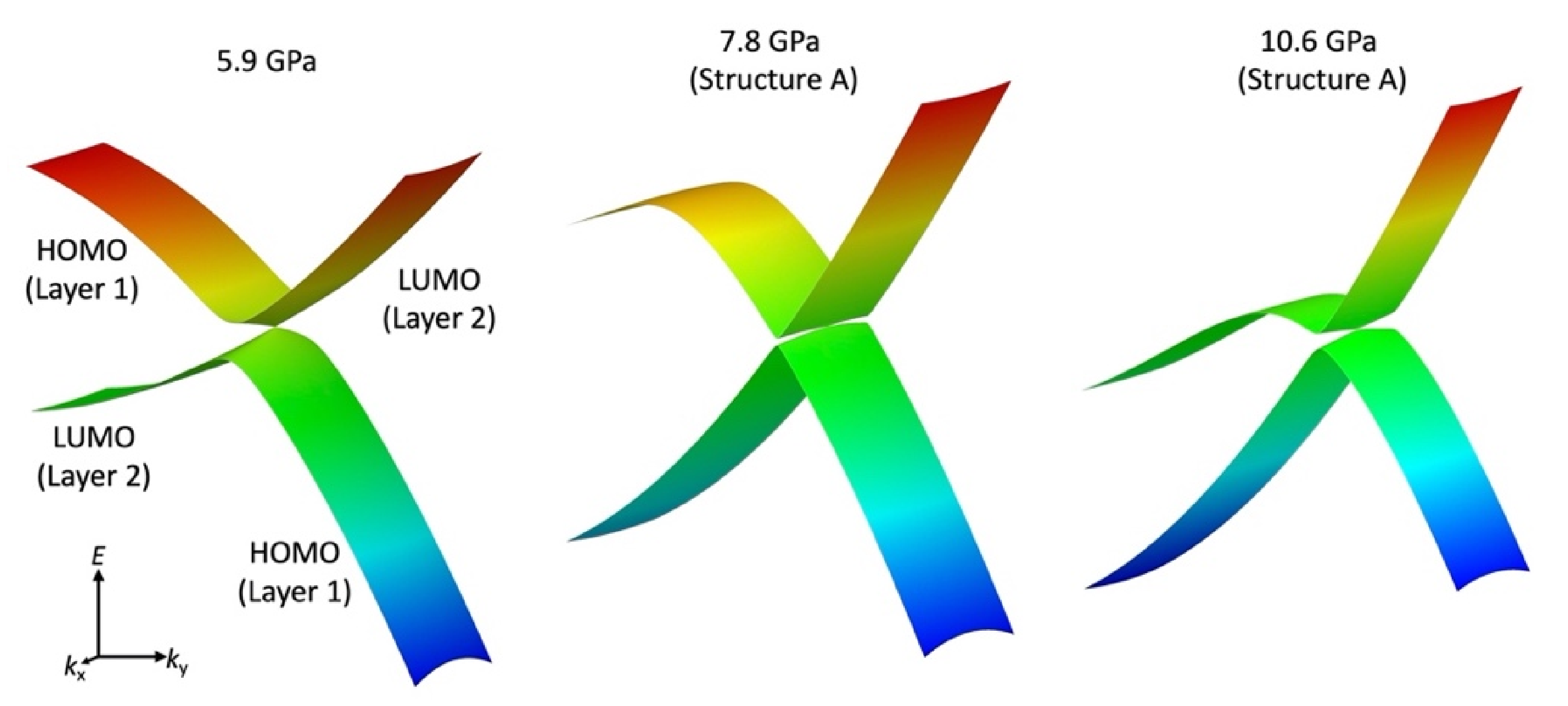
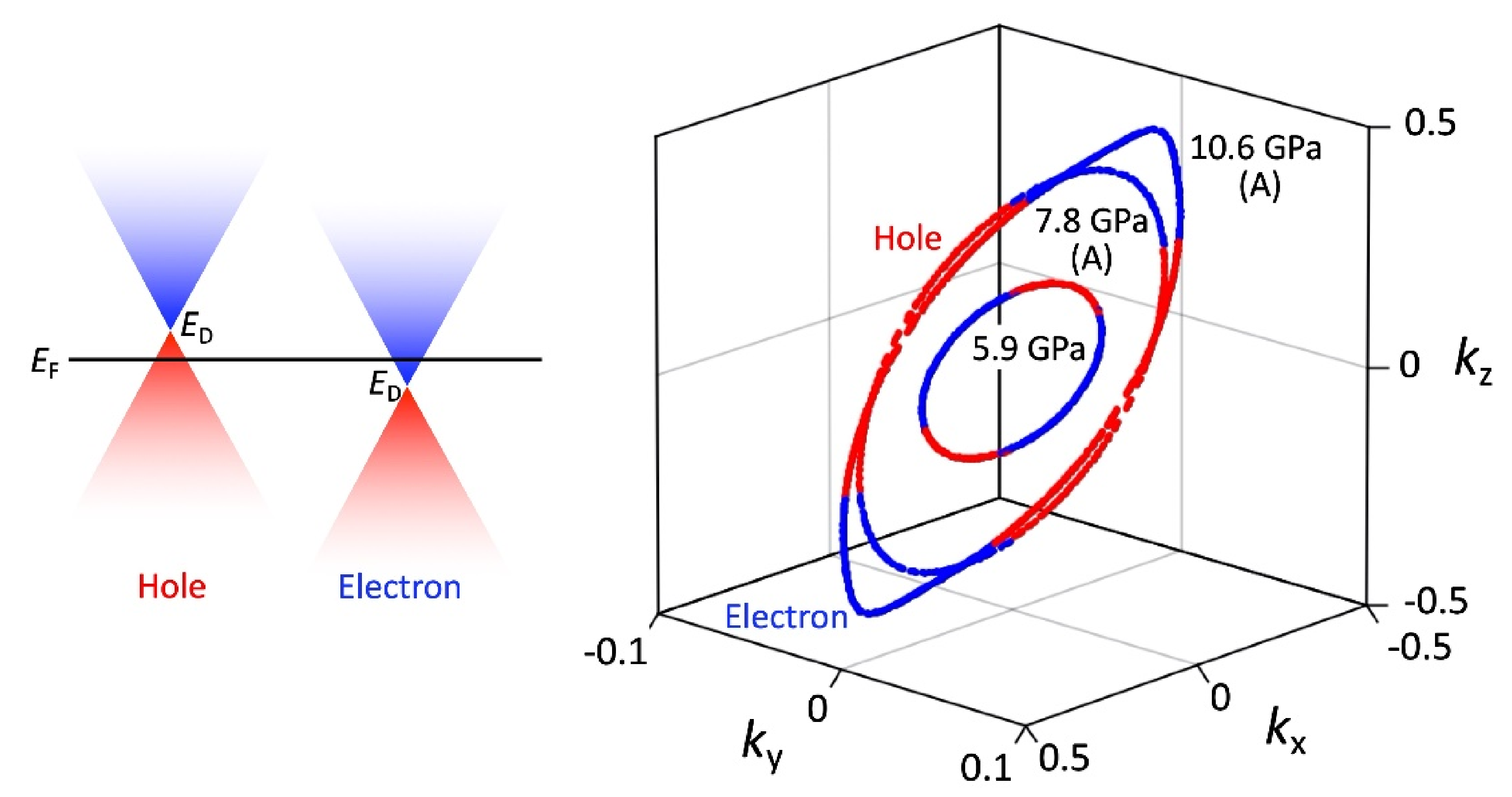
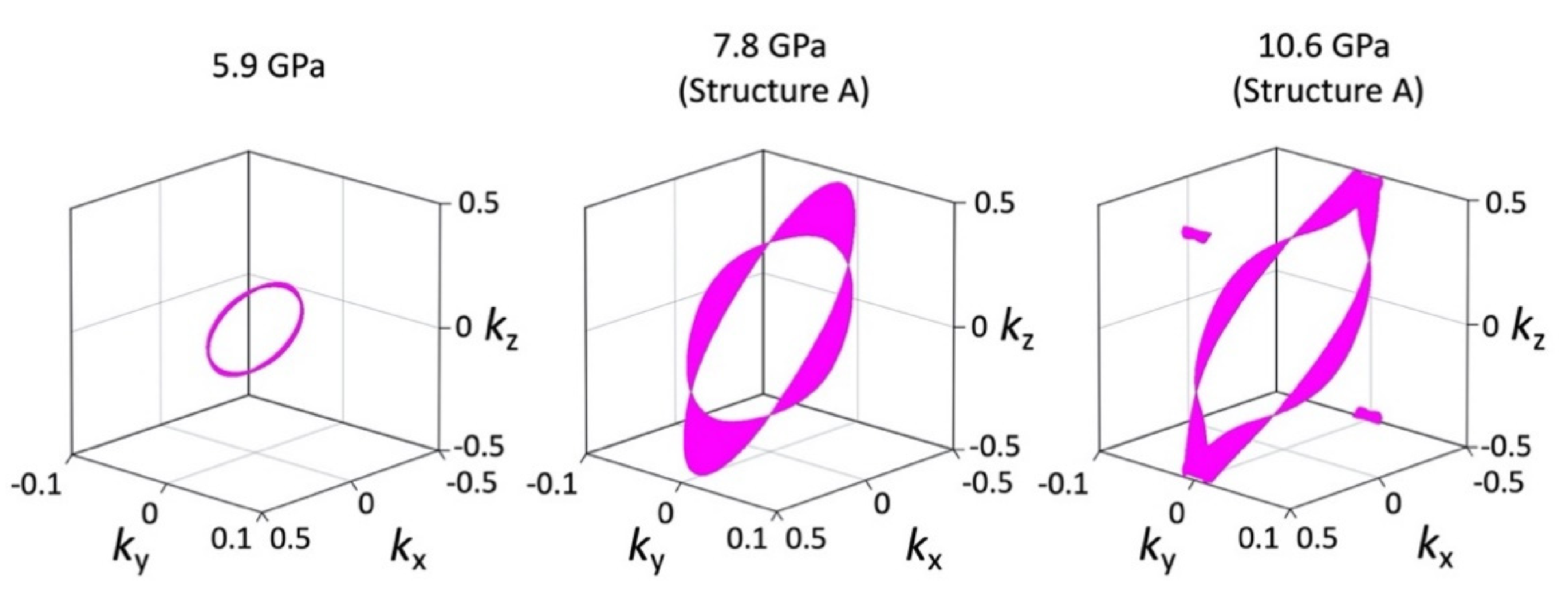
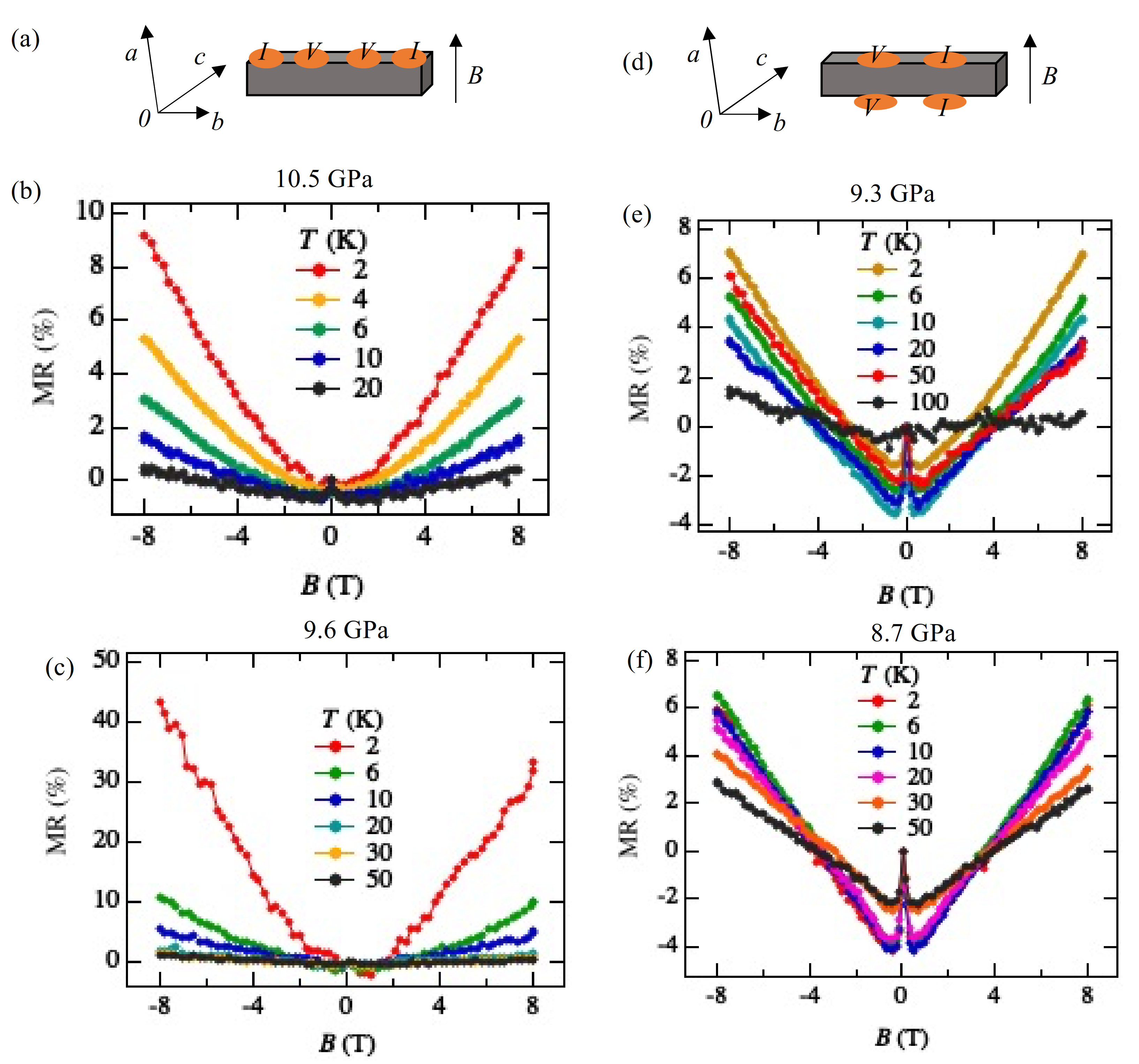
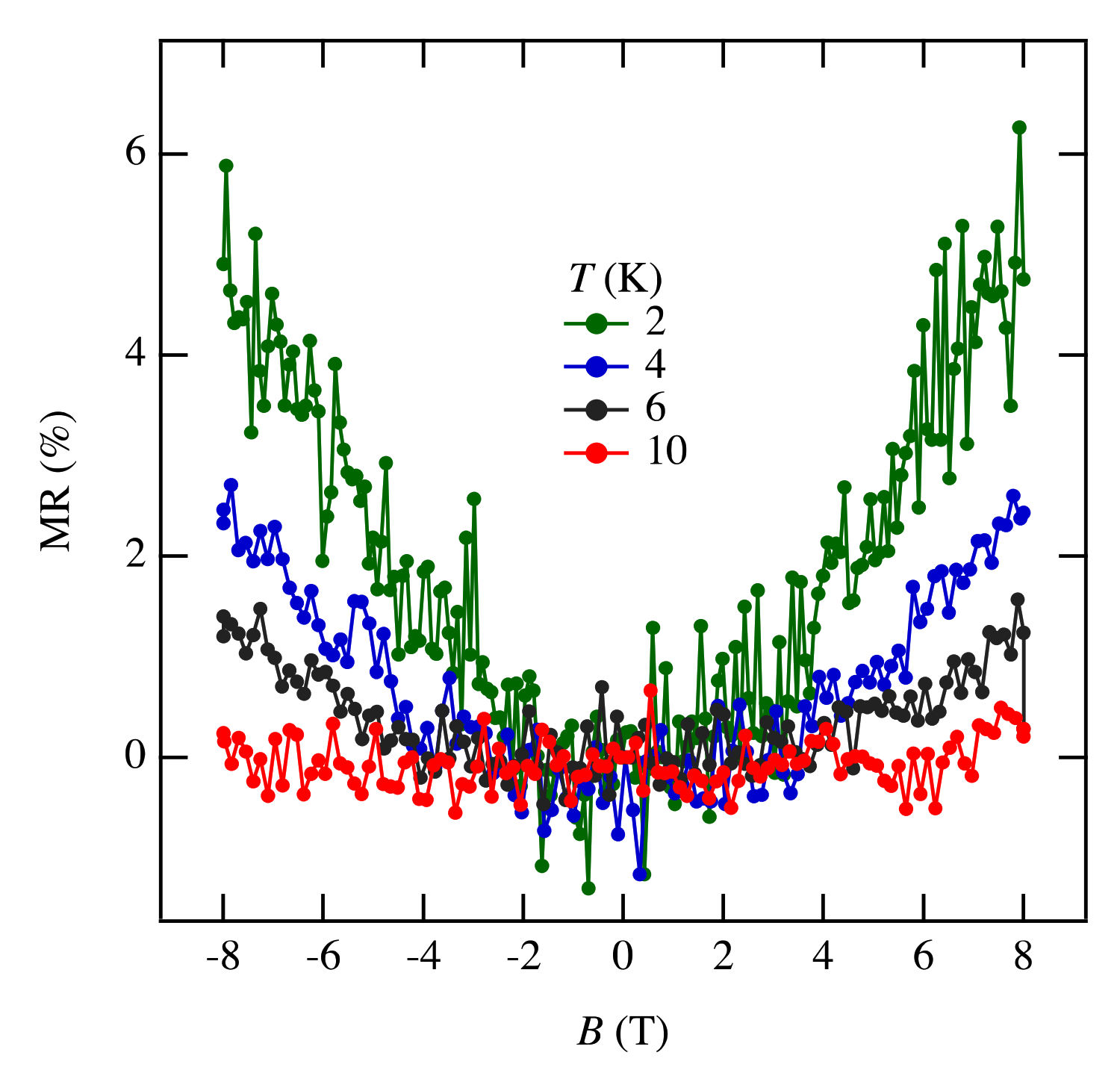
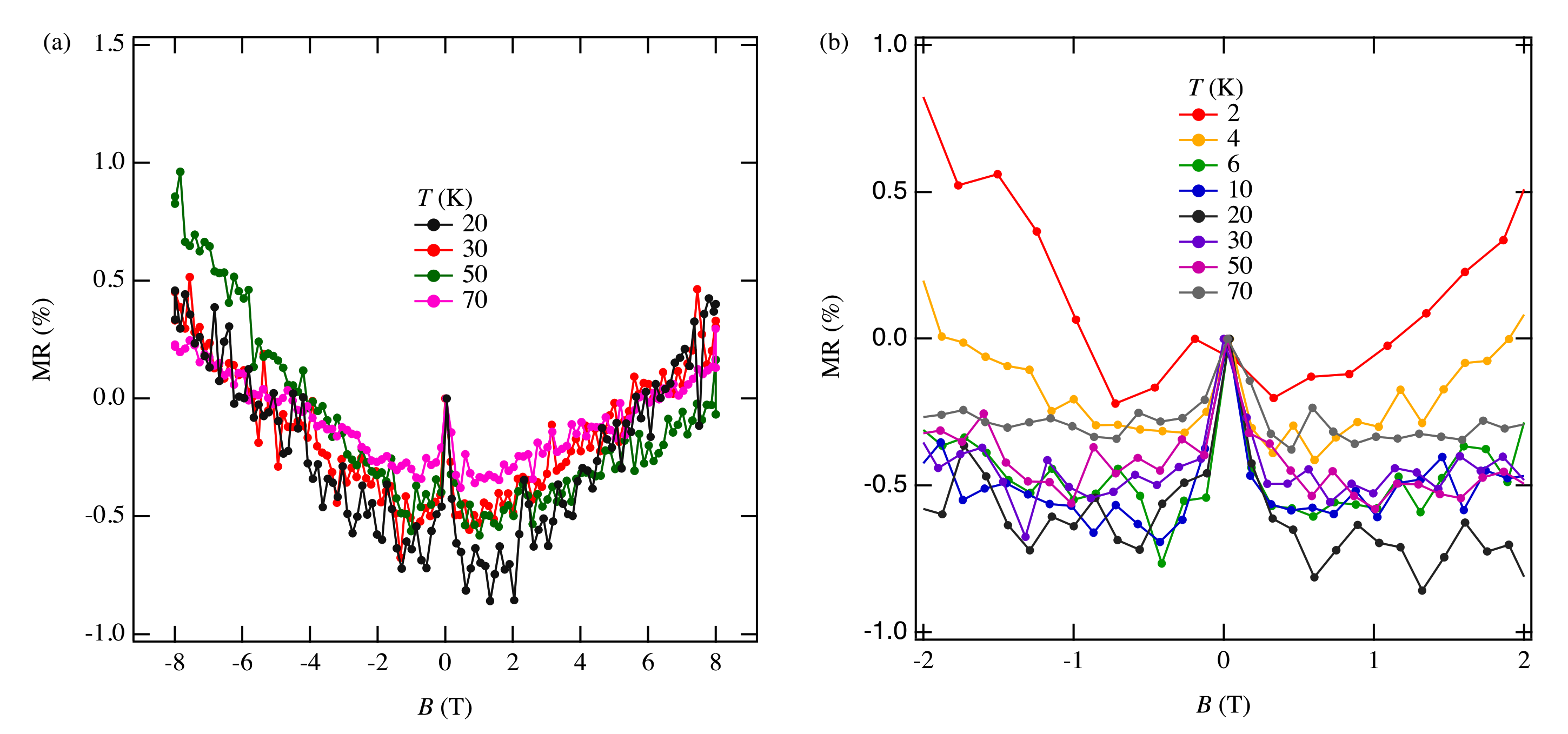
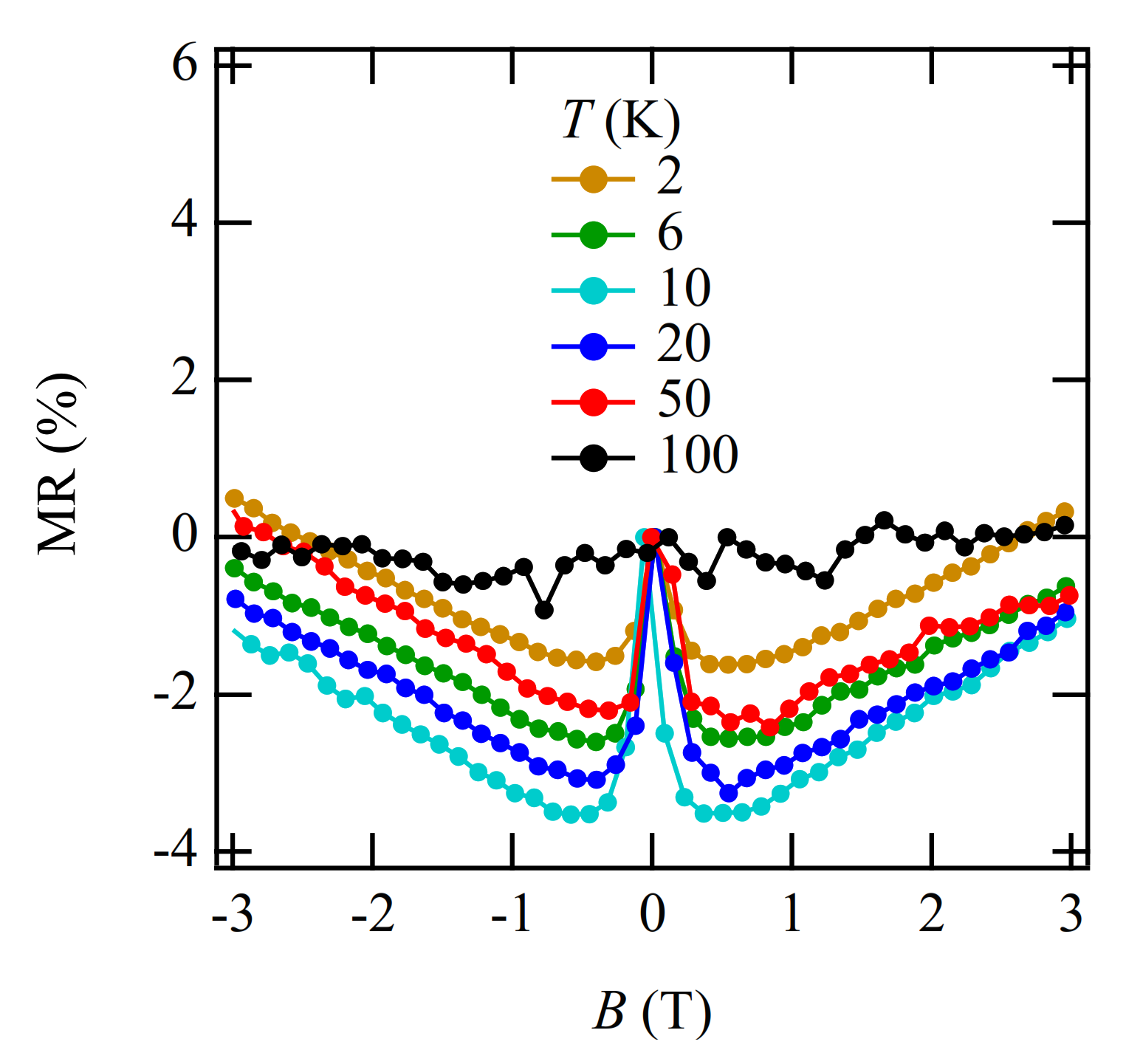
3.4. Magnetoresistance
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A





References
- Tanaka, H.; Okano, Y.; Kobayashi, H.; Suzuki, W.; Kobayashi, A. A three-dimensional synthetic metallic crystal composed of single-component molecules. Science 2001, 291, 285–287. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, W.; Fujiwara, E.; Kobayashi, A.; Fujishiro, Y.; Nishibori, E.; Takata, M.; Sakata, M.; Fujiwara, H.; Kobayashi, H. Highly conducting crystals based on single-component gold complexes with extended-TTF dithiolate ligands. J. Am. Chem. Soc. 2003, 125, 1486–1487. [Google Scholar] [CrossRef] [PubMed]
- Le Gal, Y.; Roisnel, T.; Auban-Senzier, P.; Bellec, N.; Íñiguez, J.; Canadell, E.; Lorcy, D. Stable metallic state of a neutral-radical single-component conductor at ambient pressure. J. Am. Chem. Soc. 2018, 140, 6998–7004. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Ishibashi, S.; Ishii, T.; Sekine, T.; Takehara, R.; Miyagawa, K.; Kanoda, K.; Nishibori, E.; Kobayashi, A. Single-component molecular conductor [Pt(dmdt)2]—A three-dimensional ambient-pressure molecular Dirac electron system. Chem. Commun. 2019, 55, 3327–3330. [Google Scholar] [CrossRef]
- Cui, H.; Brooks, J.S.; Kobayashi, A.; Kobayashi, H. Metallization of the single component molecular semiconductor [Ni(ptdt)2] under very high pressure. J. Am. Chem. Soc. 2009, 131, 6358–6359. [Google Scholar] [CrossRef]
- Zhou, B.; Idobata, Y.; Kobayashi, A.; Cui, H.; Kato, R.; Takagi, R.; Miyagawa, K.; Kanoda, K.; Kobayashi, H. Single-component molecular conductor [Cu(dmdt)2] with three-dimensionally arranged magnetic moments exhibiting a coupled electric and magnetic transition. J. Am. Chem. Soc. 2012, 134, 12724–12731. [Google Scholar] [CrossRef]
- Cui, H.; Tsumuraya, T.; Miyazaki, T.; Okano, Y.; Kato, R. Pressure-induced metallic conductivity in the single-component molecular crystal [Ni(dmit)2]. Eur. J. Inorg. Chem. 2014, 2014, 3837–3840. [Google Scholar] [CrossRef]
- Cui, H.; Kobayashi, H.; Ishibashi, S.; Sasa, M.; Iwase, F.; Kato, R.; Kobayashi, A. A single-component molecular superconductor. J. Am. Chem. Soc. 2014, 136, 7619–7622. [Google Scholar] [CrossRef]
- Tenn, N.; Bellec, N.; Jeannin, O.; Piekara-Sady, L.; Auban-Senzier, P.; Íñiguez, J.; Canadell, E.; Lorcy, D. A single-component molecular metal based on a thiazole dithiolate gold complex. J. Am. Chem. Soc. 2009, 131, 16961–16967. [Google Scholar] [CrossRef]
- Yzambart, G.; Bellec, N.; Nasser, G.; Jeannin, O.; Roisnel, T.; Fourmigué, M.; Auban-Senzier, P.; Íñiguez, J.; Canadell, E.; Lorcy, D. Anisotropic chemical pressure effects in single-component molecular metals based on radical dithiolene and diselenolene gold complexes. J. Am. Chem. Soc. 2012, 134, 17138–17148. [Google Scholar] [CrossRef]
- Kato, R.; Cui, H.; Tsumuraya, T.; Miyazaki, T.; Suzumura, Y. Emergence of the Dirac electron system in a single-component molecular conductor under high pressure. J. Am. Chem. Soc. 2017, 139, 1770–1773. [Google Scholar] [CrossRef]
- Cui, H.; Kato, R. Electrical properties of single-component molecular crystals under high pressure. Rev. H. Pres. Sci. Tech. 2018, 28, 217–224. [Google Scholar] [CrossRef]
- Novoselov, K.; Geim, A.; Morozov, S.; Jiang, D.; Grigorieva, M.; Dubonos, S.; Firsov, A. Two-dimensional gas of massless Dirac fermions in graphene. Nature 2005, 438, 197–200. [Google Scholar] [CrossRef]
- Hasan, M.; Kane, C. Colloquium: Topological insulators. Rev. Mod. Phys. 2010, 82, 3045–3067. [Google Scholar] [CrossRef]
- Huynh, K.K.; Tanabe, Y.; Tanigaki, K. Both electron and hole Dirac cone states in Ba(FeAs)2 confirmed by magnetoresistance. Phys. Rev. Lett. 2011, 106, 217004. [Google Scholar] [CrossRef]
- Tajima, N.; Sato, M.; Sugawara, S.; Kato, R.; Nishio, Y.; Kajita, K. Spin and valley splittings in multilayered massless Dirac fermion system. Phys. Rev. B 2010, 82, 121420(R). [Google Scholar] [CrossRef]
- Kato, R.; Suzumura, Y. Novel Dirac electron in single-component molecular conductor [Pd(dddt)2] (dddt =5,6-dihydro-1,4-dithiin-2,3-dithiolate). J. Phys. Soc. Jpn. 2017, 86, 064705. [Google Scholar] [CrossRef]
- Tsumuraya, T.; Kato, R.; Suzumura, Y. Effective hamiltonian of topological nodal line semimetal in single-component molecular conductor [Pd(dddt)2] from first-principles. J. Phys. Soc. Jpn. 2018, 87, 113701. [Google Scholar] [CrossRef]
- Evans, P.R.; Murshudov, G.N. How good are my data and what is the resolution? Acta Crystallogr. Sect. D Biol. Crystallogr. 2013, 69, 1204–1214. [Google Scholar] [CrossRef]
- Winn, M.D.; Ballard, C.C.; Cowtan, K.D.; Dodson, E.J.; Emsley, P.; Evans, P.R.; Keegan, R.M.; Krissinel, E.B.; Leslie, A.G.W.; McCoy, A.; et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. Sect. D Biol. Crystallogr. 2011, 67, 235–242. [Google Scholar] [CrossRef]
- Winter, G.; Waterman, D.G.; Parkhurst, J.M.; Brewster, A.S.; Gildea, R.J.; Gerstel, M.; Fuentes-Montero, L.; Vollmar, M.; Michels-Clark, T.; Young, I.D.; et al. DIALS: Implementation and evaluation of a new integration package. Acta Crystallogr. Sect. D Struct. Biol. 2018, 74, 85–97. [Google Scholar] [CrossRef]
- Evans, P. Scaling and assessment of data quality. Acta Crystallogr. Sect. D Biol. Crystallogr. 2006, 62, 72–82. [Google Scholar] [CrossRef]
- Winter, G. xia2: An expert system for macromolecular crystallography data reduction. J. Appl. Crystallogr. 2010, 43, 186–190. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Hoffmann, R. An Extended Hückel theory. I. hydrocarbons. J. Chem. Phys. 1963, 39, 1397. [Google Scholar] [CrossRef]
- Clementi, E.; Roetti, C. Roothaan-Hartree-Fock atomic wavefunctions: Basis functions and their coefficients for ground and certain excited states of neutral and ionized atoms, Z ≤ 54. At. Data Nucl. Data Tables 1974, 14, 177–478. [Google Scholar] [CrossRef]
- QuantumATK R-2020.09 Documentation, Manual of Atomic data. Available online: https://docs.quantumatk.com/manual/AtomicData.html#sect3-atomicdata-huckel-muller (accessed on 11 May 2021).
- Cui, H.; Tsumuraya, T.; Yeung, H.H.-M.; Coates, C.S.; Warren, M.R.; Kato, R. High pressure crystal structure and electrical properties of a single component molecular crystal [Ni(dddt)2] (dddt = 5,6-dihydro-1,4-dithiin-2,3-dithiolate). Molecules 2019, 24, 1843. [Google Scholar] [CrossRef]
- Cairns, A.B.; Goodwin, A.L. Negative linear compressibility. Phys. Chem. Chem. Phys. 2015, 17, 20449–20465. [Google Scholar] [CrossRef]
- Kato, R.; Cui, H.; Minamidate, T.; Yeung, H.H.-M.; Suzumura, Y. Electronic Structure of a Single-Component Molecular Conductor [Pd(dddt)2] (dddt = 5,6-dihydro-1,4-dithiin-2,3-dithiolate) under High Pressure. J. Phys. Soc. Jpn. 2020, 89, 124706. [Google Scholar] [CrossRef]
- Mori, T.; Kobayashi, A.; Sasaki, Y.; Kobayashi, H.; Saito, G.; Inokuchi, H. The intermolecular interaction of tetrathiafulvalene and bis(ethylenedithio) tetrathiafulvalene in organic metals. Calculation of orbital overlaps and models of energy-band Structures. Bull. Chem. Soc. Jpn. 1984, 57, 627–633. [Google Scholar] [CrossRef]
- Kato, R.; Suzumura, Y. A Tight-binding model of an ambient-pressure molecular Dirac electron system with open nodal lines. J. Phys. Soc. Jpn. 2020, 89, 044713. [Google Scholar] [CrossRef]
- Suzumura, Y.; Cui, H.; Kato, R. Conductivity and resistivity of Dirac electrons in single-component molecular conductor [Pd(dddt)2]. J. Phys. Soc. Jpn. 2018, 87, 084702. [Google Scholar] [CrossRef]
- Berry, M.V. Quantal phase factors accompanying adiabatic changes. Proc. R. Soc. Lond. Ser. A 1984, 392, 45–57. [Google Scholar]
- Stephen, G.M.; Lane, C.; Buda, G.; Graf, D.; Kaprzyk, S.; Barbiellini, B.; Bansil, A.; Heiman, D. Electrical and magnetic properties of thin films of the spin-filter material CrVTiAl. Phys. Rev. B 2019, 99, 224207. [Google Scholar] [CrossRef]
- Bodepudi, S.C.; Singh, A.P.; Pramanik, S. Current perpendicular to plane magnetoresistance in chemical vapor deposition grown multilayer graphene. Electronics 2013, 2, 315–331. [Google Scholar] [CrossRef]
- Liu, Y.; Lew, W.S.; Sun, L. Enhanced weak localization effect in few-layer graphene. Phys. Chem. Chem. Phys. 2011, 13, 20208–20214. [Google Scholar] [CrossRef]
- Heyd, J.; Peralta, J.E.; Scuseria, G.E.; Martin, R.L. Energy band gaps and lattice parameters evaluated with the Heyd-Scuseria-Ernzerhof screened hybrid functional. J. Chem. Phys. 2005, 123, 174101. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| H–H | L–L | H–L | ||
|---|---|---|---|---|
| b1 | 209.3 | −1.9 | −51.2 | Layer 1 |
| p1 (p) | 28.1 | −12.4 | 19.9 | |
| p2 | — | — | 17.1 | |
| b2 | 49.9 | −80.4 | −67.2 | Layer 2 |
| q1 (q) | 10.8 | 8.1 | 9.3 | |
| q2 | — | — | 9.2 | |
| a1 | −28.2 | 14.6 | −20.1 | Interlayer |
| a2 | 2.2 | 1.3 | −1.7 | |
| c1 | 15.4 | 12.7 | 14.1 | |
| c2 | −3.9 | 15.8 | −11.8 |
| H–H | L–L | H–L | ||
|---|---|---|---|---|
| b1 | 190.2 | 53.2 | −34.3 | Layer 1 |
| p1 (p) | 28.1 | −17.0 | 23.1 | |
| p2 | — | — | 20.5 | |
| b2 | −0.9 | −121.5 | −71.9 | Layer 2 |
| q1 (q) | 7.3 | 7.2 | 8.5 | |
| q2 | — | — | 6.1 | |
| a1 | −37.7 | 19.0 | −26.6 | Interlayer |
| a2 | 3.3 | 2.7 | −3.1 | |
| c1 | 9.1 | 6.0 | 7.3 | |
| c2 | 1.2 | 9.4 | −5.6 |
| H–H | L–L | H–L | ||
|---|---|---|---|---|
| b1 | 190.2 | 53.2 | −34.3 | Layer 1 |
| p1 (p) | 28.1 | −17.0 | 23.1 | |
| p2 | — | — | 20.5 | |
| b2 | 107.2 | −5.4 | −46.2 | Layer 2 |
| q1 (q) | 11.2 | 11.0 | 12.1 | |
| q2 | — | — | 10.2 | |
| a1 | −32.9 | 16.2 | −22.9 | Interlayer |
| a2 | 2.0 | 1.9 | −2.0 | |
| c1 | −7.7 | −15.2 | −12.6 | |
| c2 | −9.3 | 12.4 | −11.3 |
| H–H | L–L | H–L | ||
|---|---|---|---|---|
| b1 | 203.4 | 6.1 | −67.5 | Layer 1 |
| p1 (p) | 24.7 | −13.4 | 18.6 | |
| p2 | — | — | 17.7 | |
| b2 | −0.3 | −135.0 | −81.6 | Layer 2 |
| q1 (q) | 7.8 | 7.2 | 9.3 | |
| q2 | — | — | 5.9 | |
| a1 | −36.3 | 21.4 | −28.4 | Interlayer |
| a2 | 4.9 | 4.1 | −4.6 | |
| c1 | 7.6 | 4.8 | 6.0 | |
| c2 | −5.0 | 17.9 | −12.5 |
| H–H | L–L | H–L | ||
|---|---|---|---|---|
| b1 | 203.4 | 6.1 | −67.5 | Layer 1 |
| p1 (p) | 24.7 | −13.4 | 18.6 | |
| p2 | — | — | 17.7 | |
| b2 | 143.9 | 32.7 | −49.8 | Layer 2 |
| q1 (q) | 13.9 | 12.9 | 14.8 | |
| q2 | — | — | 12.2 | |
| a1 | −31.6 | 21.1 | −26.4 | Interlayer |
| a2 | 2.0 | 2.4 | −2.4 | |
| c1 | −6.4 | −15.3 | −11.7 | |
| c2 | −9.6 | 9.3 | −9.3 |
| Ambient Pressure | 5.9 GPa | 7.8 GPa | 10.6 GPa | |||
|---|---|---|---|---|---|---|
| Structure A | Structure B | Structure A | Structure B | |||
| d1 | 3.953 | 3.612 | 3.780 | 3.199 | 3.707 | 3.028 |
| d2 | 3.922 | 3.596 | 3.484 | 3.484 | 3.517 | 3.517 |
| d3 | 3.850 | 3.547 | 3.453 | 3.453 | 3.352 | 3.352 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cui, H.; Yeung, H.H.-M.; Kawasugi, Y.; Minamidate, T.; Saunders, L.K.; Kato, R. High-Pressure Crystal Structure and Unusual Magnetoresistance of a Single-Component Molecular Conductor [Pd(dddt)2] (dddt = 5,6-dihydro-1,4-dithiin-2,3-dithiolate). Crystals 2021, 11, 534. https://doi.org/10.3390/cryst11050534
Cui H, Yeung HH-M, Kawasugi Y, Minamidate T, Saunders LK, Kato R. High-Pressure Crystal Structure and Unusual Magnetoresistance of a Single-Component Molecular Conductor [Pd(dddt)2] (dddt = 5,6-dihydro-1,4-dithiin-2,3-dithiolate). Crystals. 2021; 11(5):534. https://doi.org/10.3390/cryst11050534
Chicago/Turabian StyleCui, Hengbo, Hamish H.-M. Yeung, Yoshitaka Kawasugi, Takaaki Minamidate, Lucy K. Saunders, and Reizo Kato. 2021. "High-Pressure Crystal Structure and Unusual Magnetoresistance of a Single-Component Molecular Conductor [Pd(dddt)2] (dddt = 5,6-dihydro-1,4-dithiin-2,3-dithiolate)" Crystals 11, no. 5: 534. https://doi.org/10.3390/cryst11050534
APA StyleCui, H., Yeung, H. H.-M., Kawasugi, Y., Minamidate, T., Saunders, L. K., & Kato, R. (2021). High-Pressure Crystal Structure and Unusual Magnetoresistance of a Single-Component Molecular Conductor [Pd(dddt)2] (dddt = 5,6-dihydro-1,4-dithiin-2,3-dithiolate). Crystals, 11(5), 534. https://doi.org/10.3390/cryst11050534