Calcium Phosphate Nanoparticle Precipitation by a Continuous Flow Process: A Design of Experiment Approach

 ,
, 
 , ,
, , 
Abstract
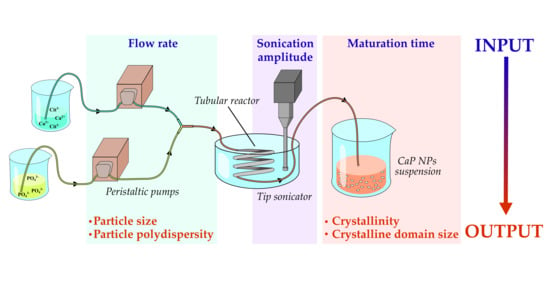
:
1. Introduction
2. Materials and Methods
2.1. Materials
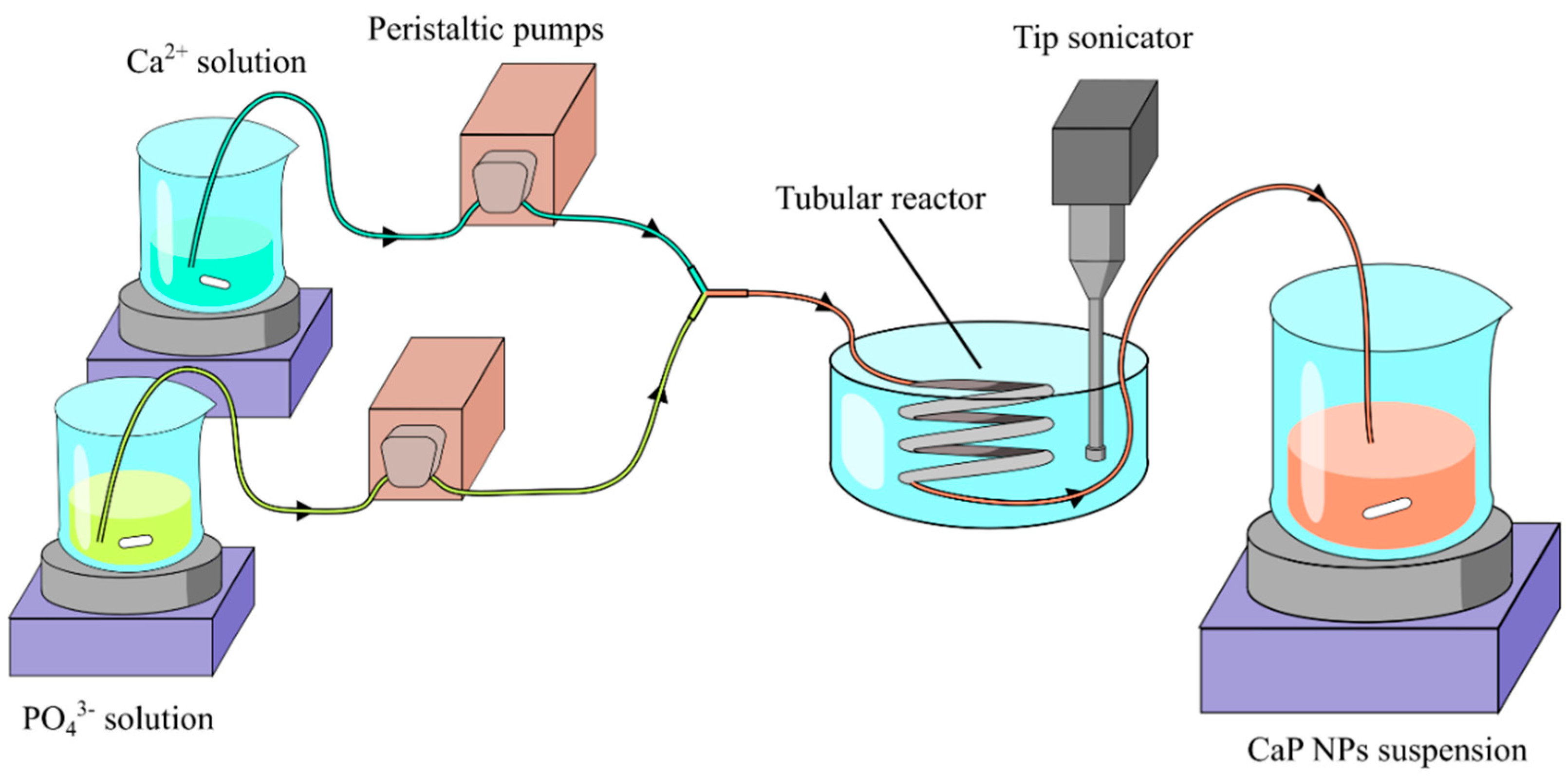
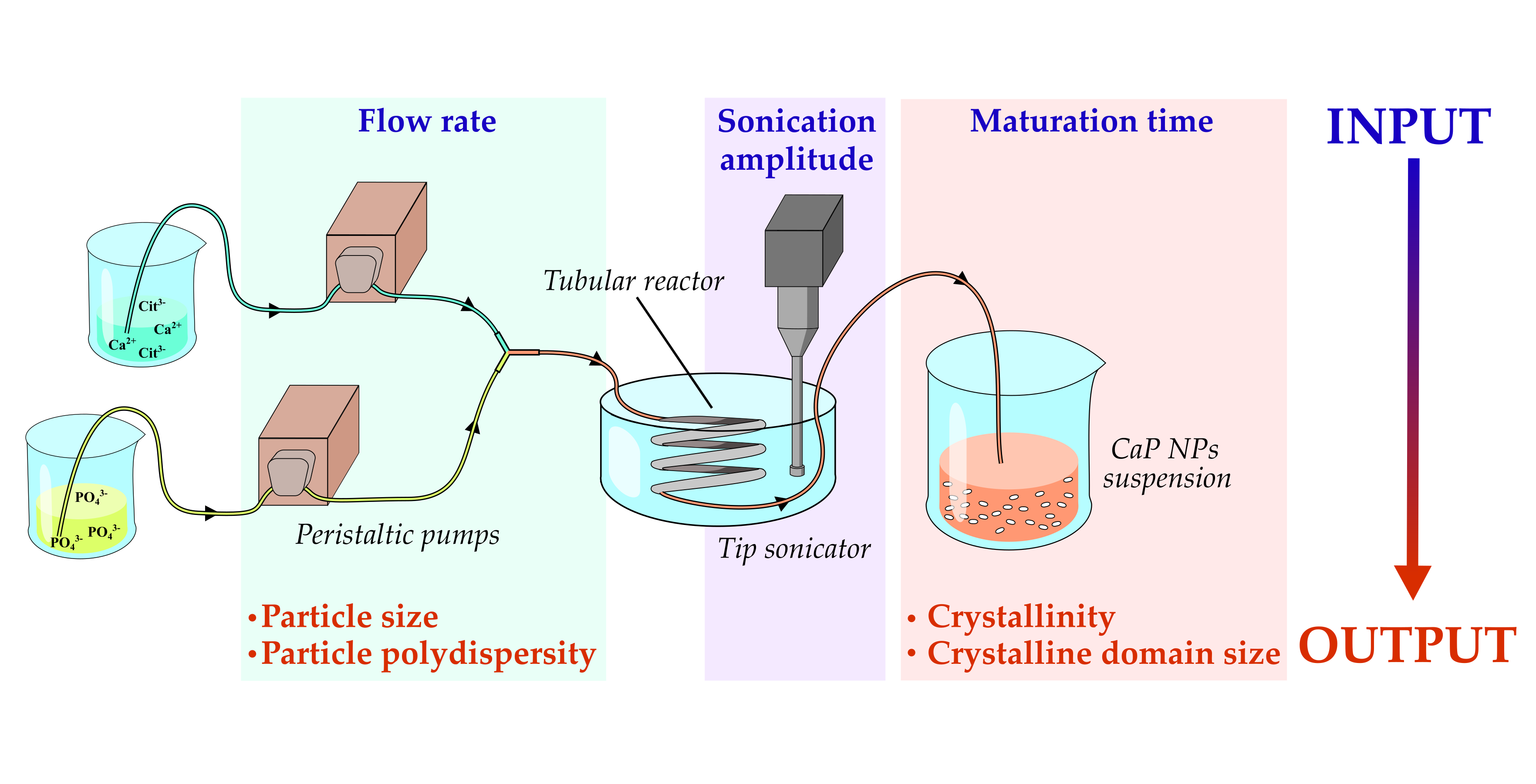
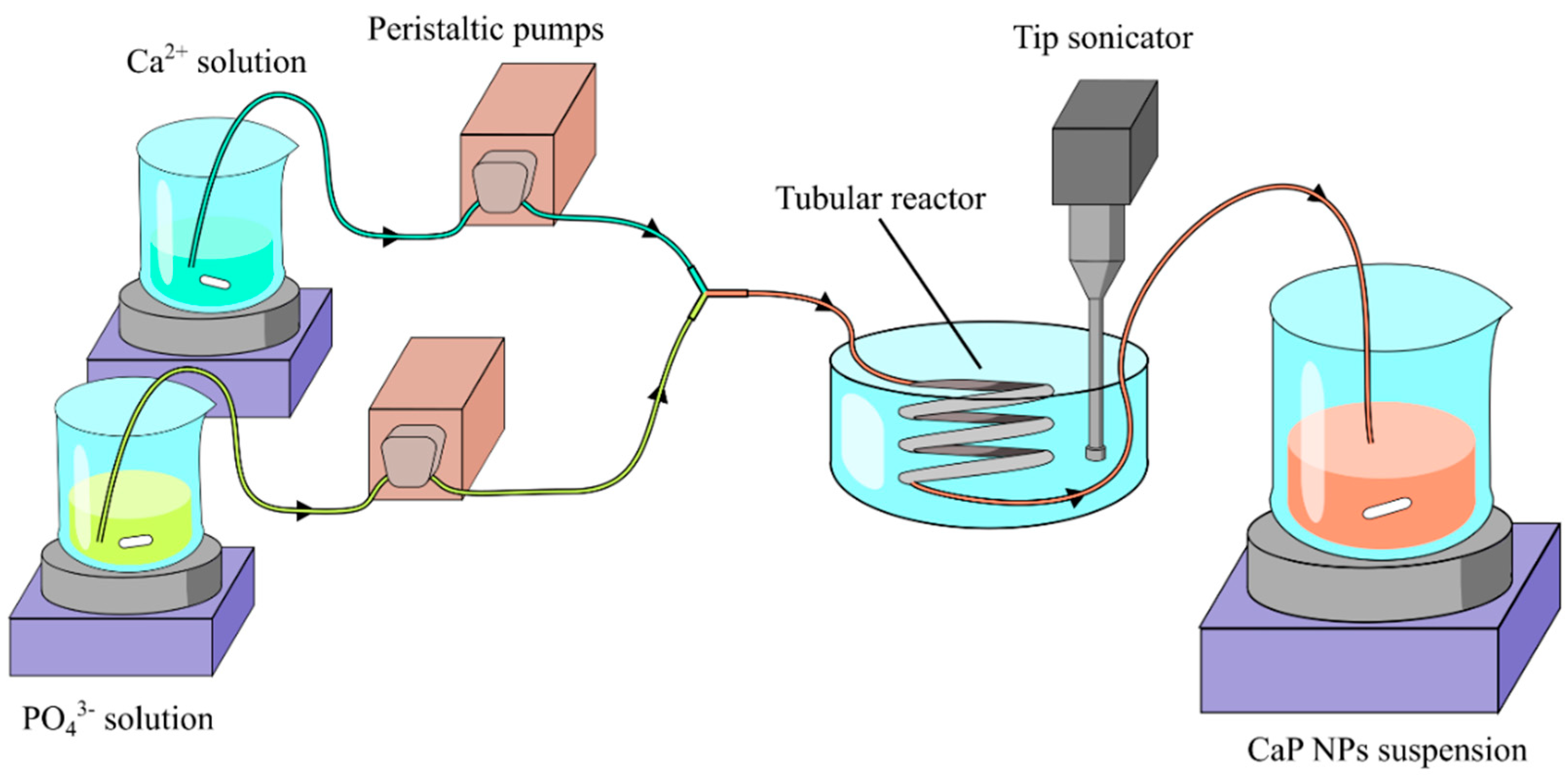
2.2. Description of the Continuous Flow Tube Type Reactor
2.3. Calcium Phosphate Nanoparticles Preparation
2.4. Design of Experiment
2.5. Chemical, Morphological and Structural Characterization of the Samples
3. Results and Discussions
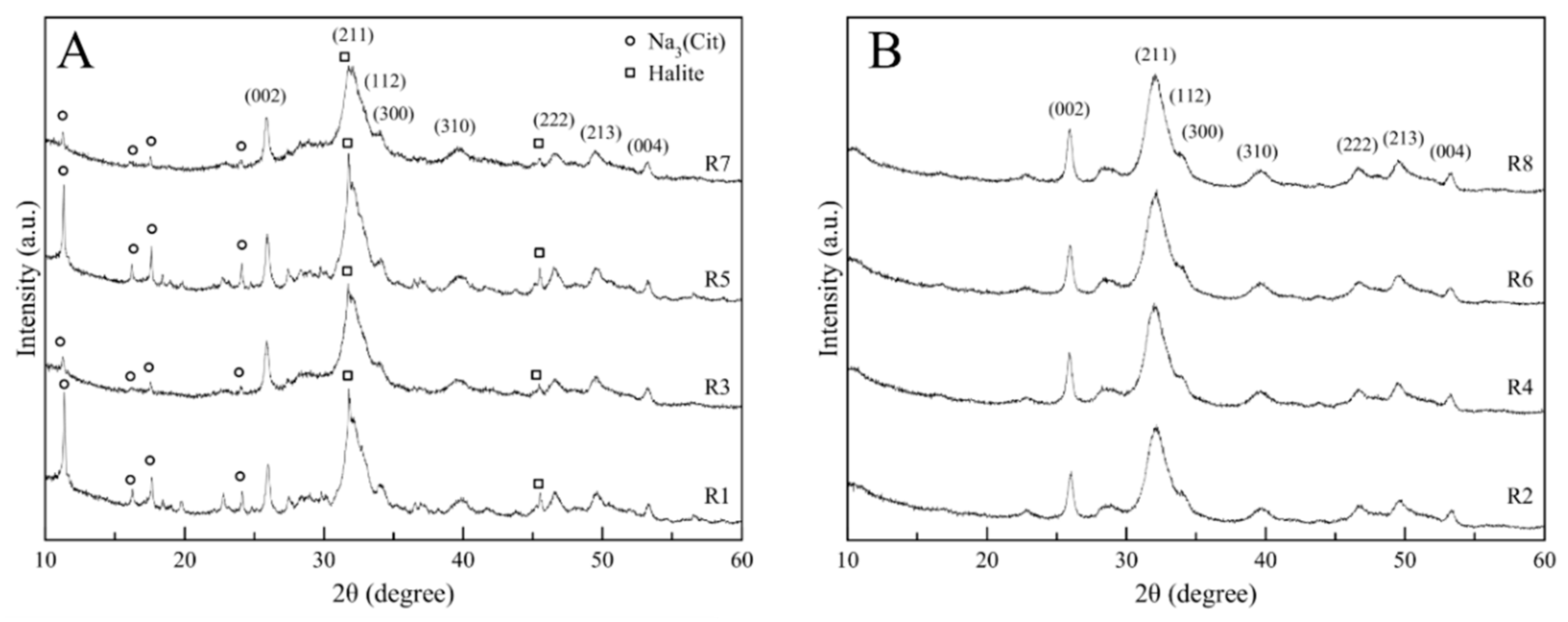
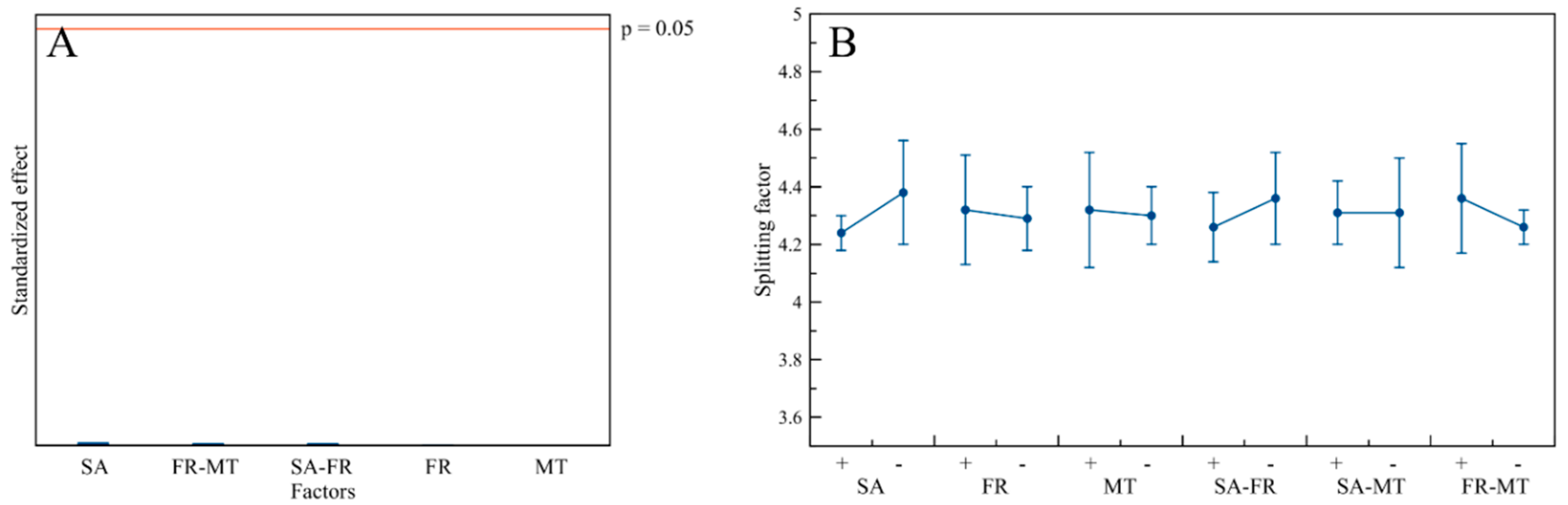
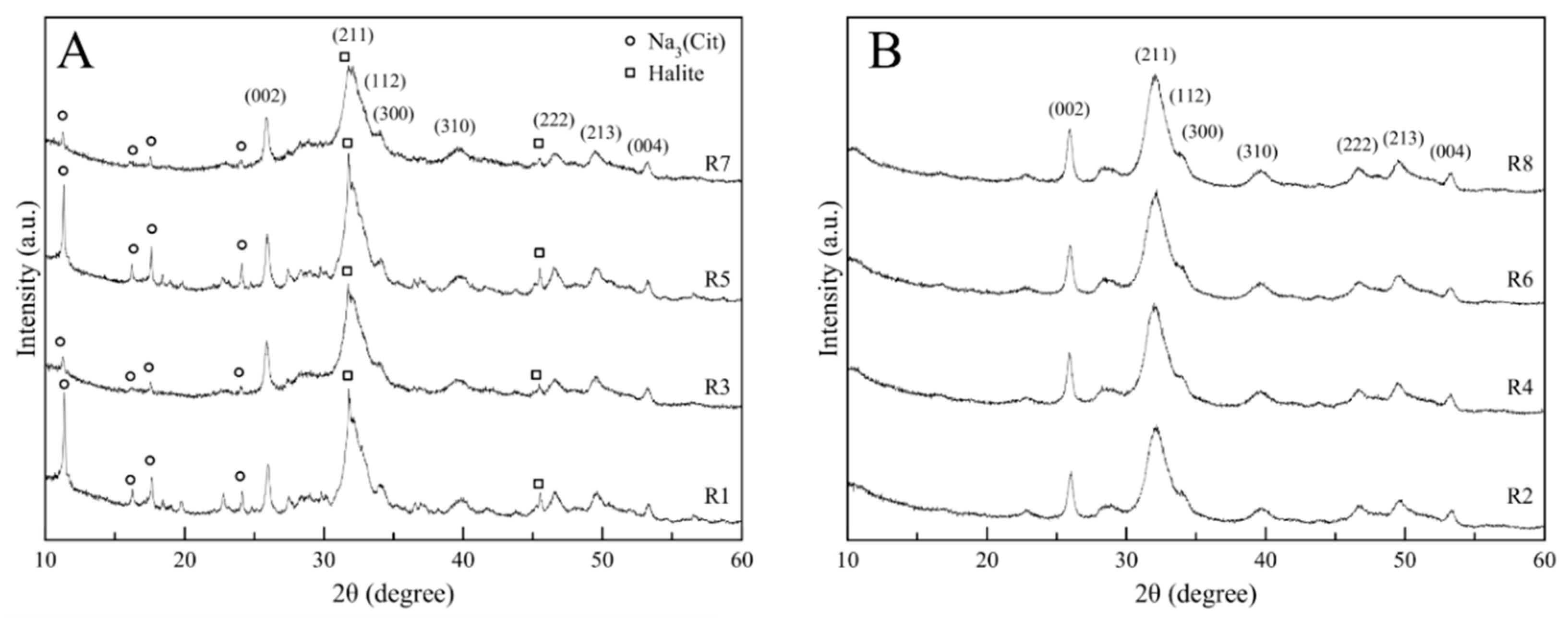
3.1. Crystallographic Features and Product Purity
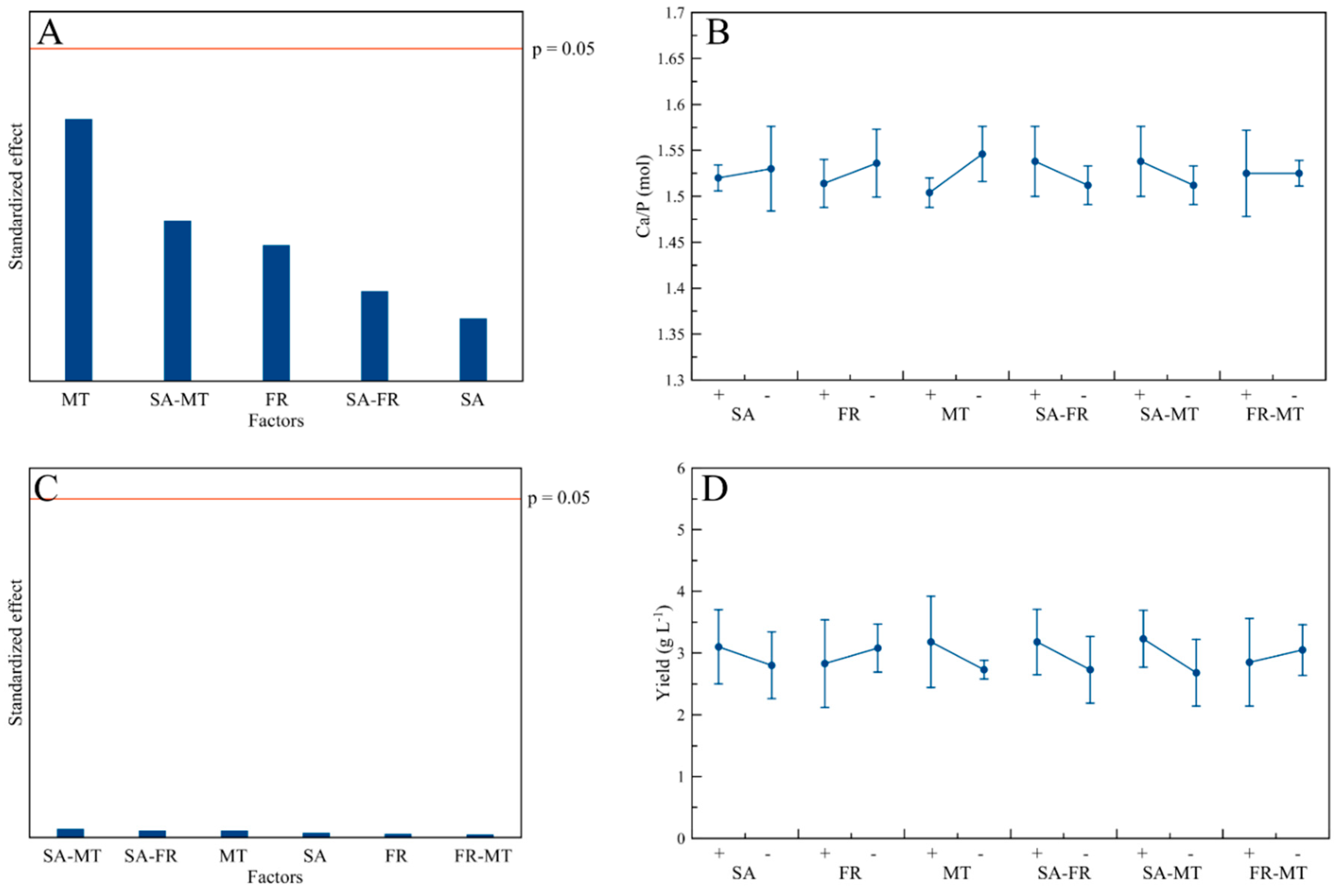
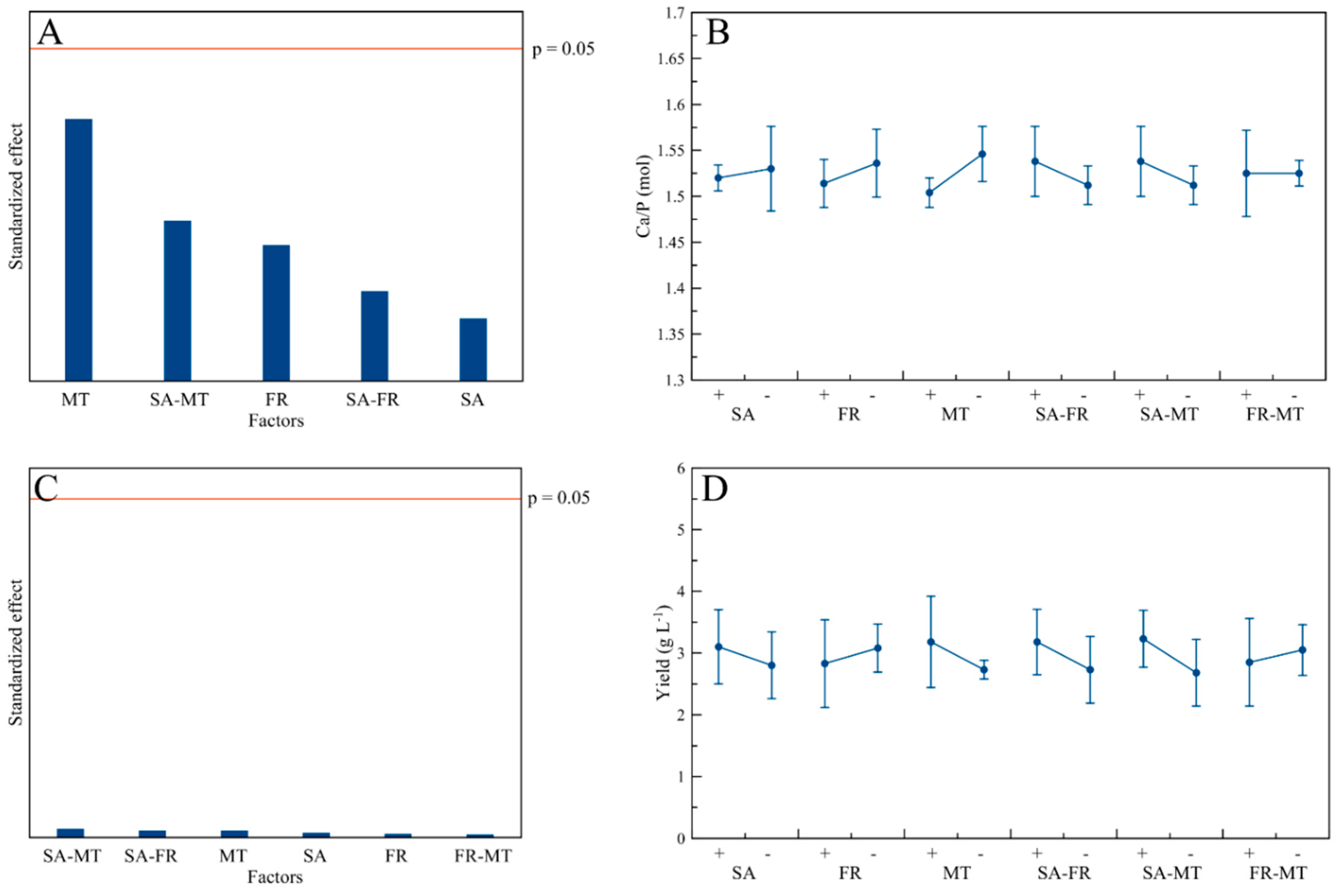
3.2. Ca/P Ratio and Yield
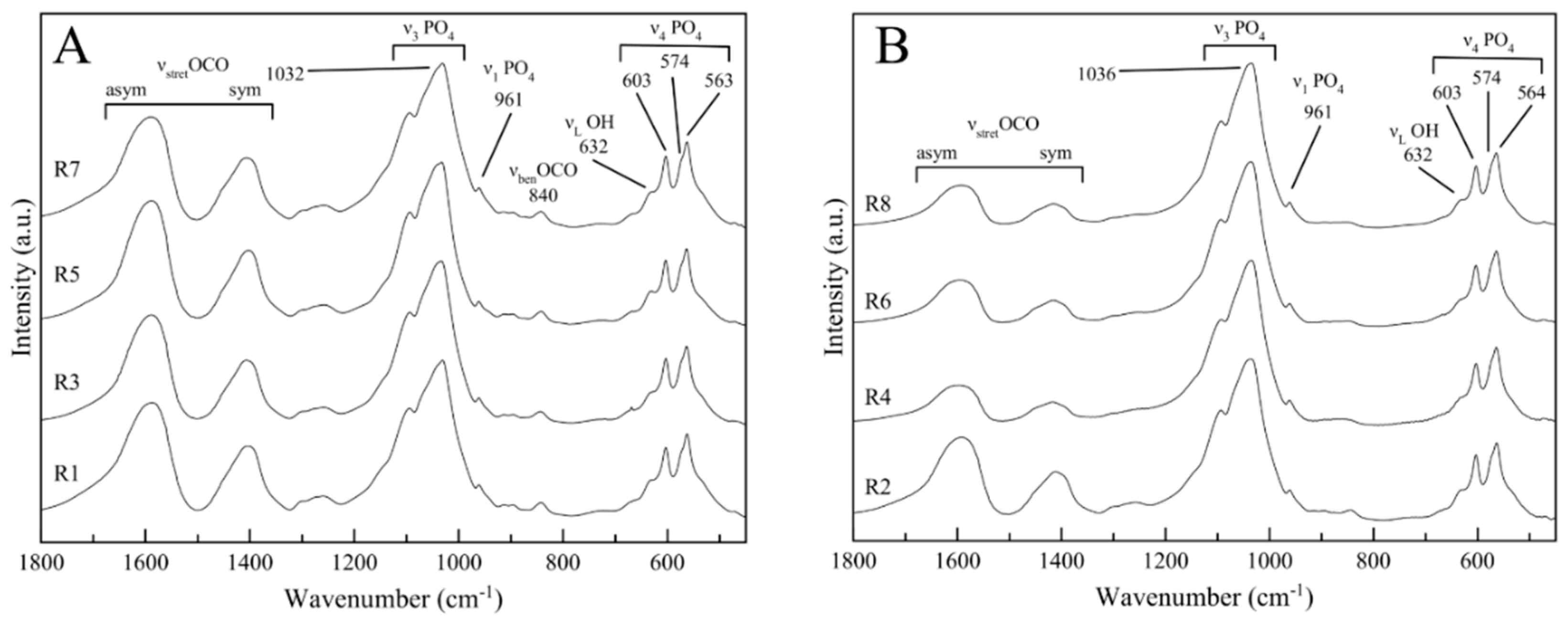
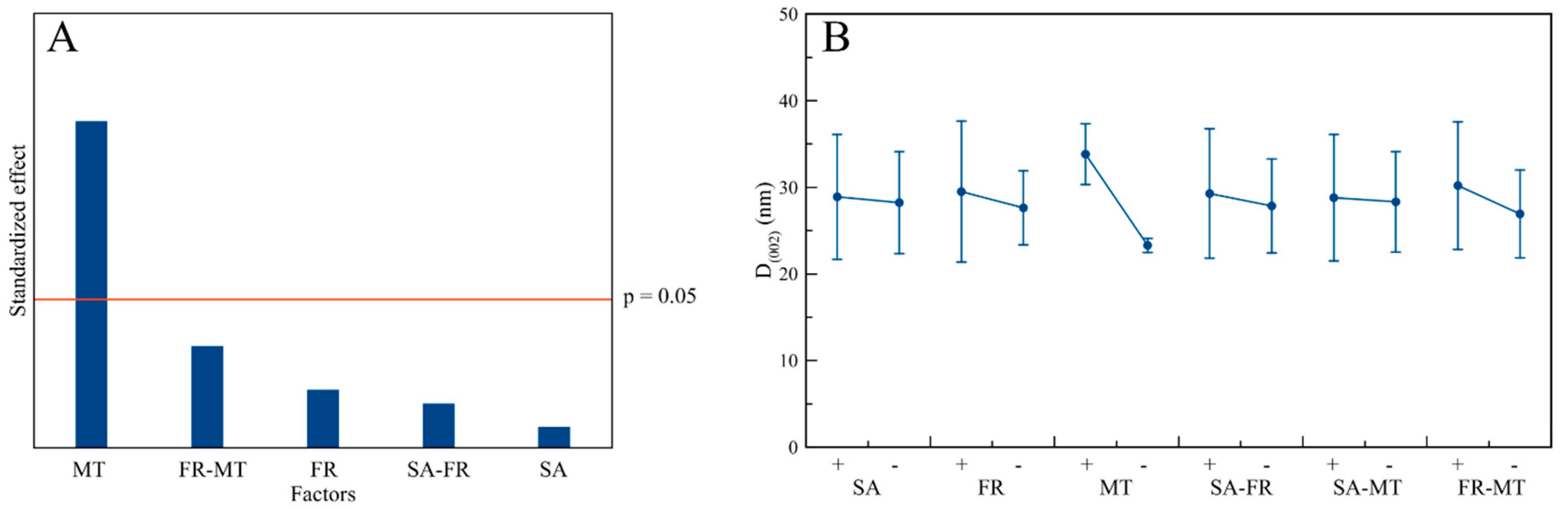
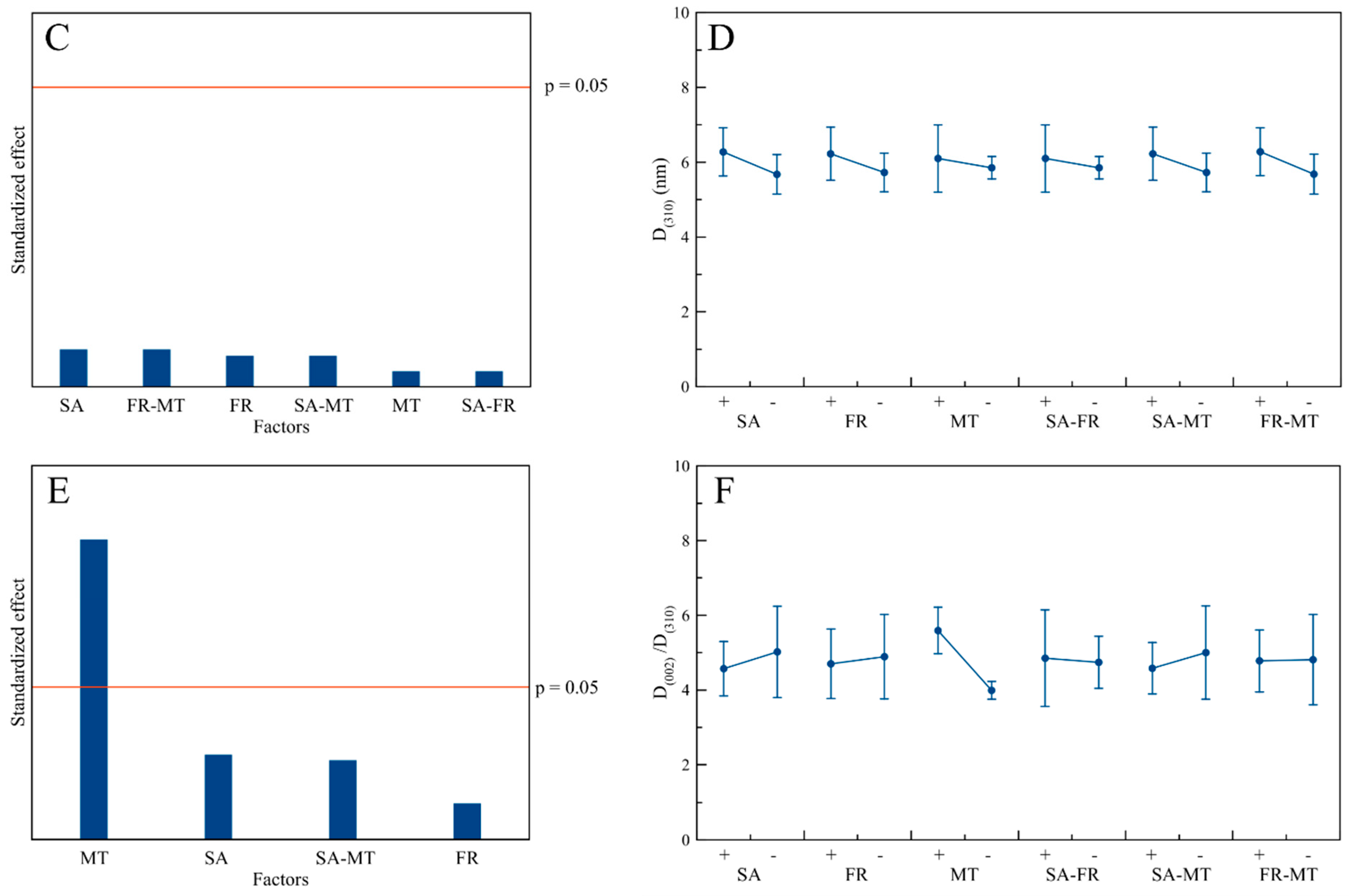
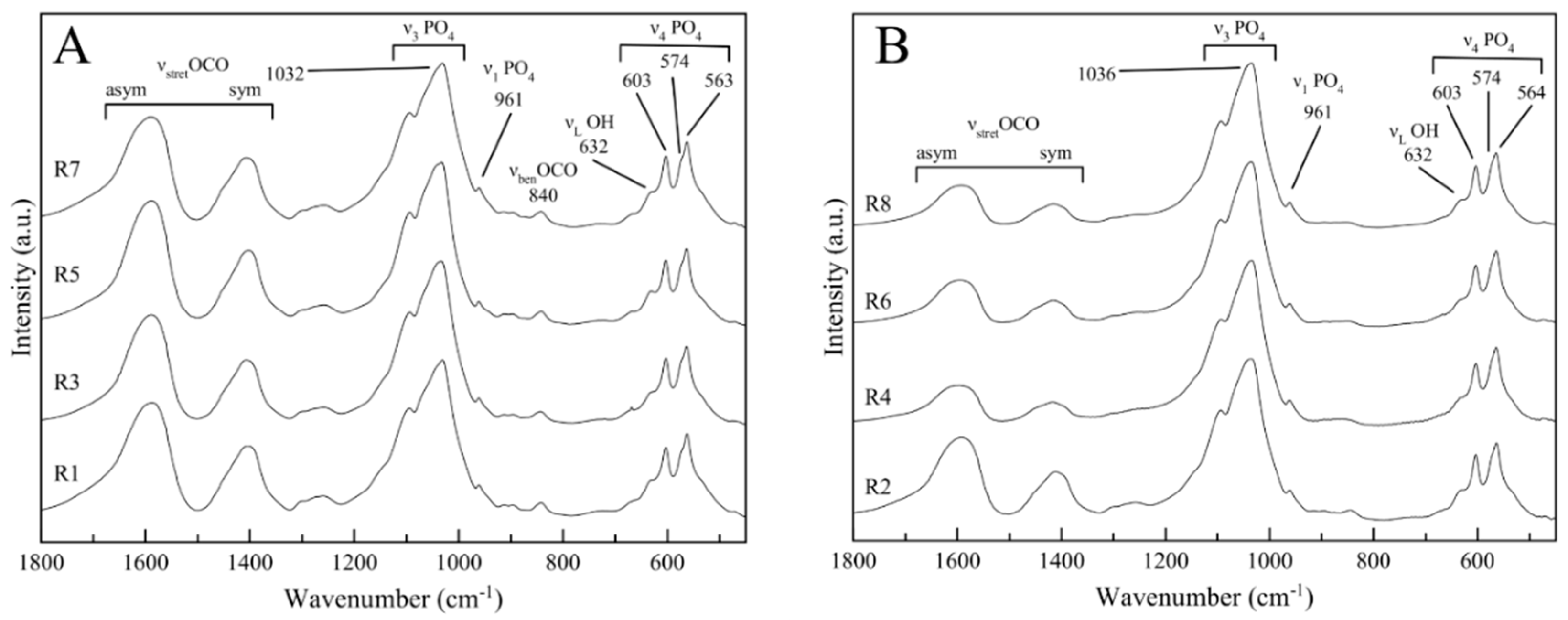
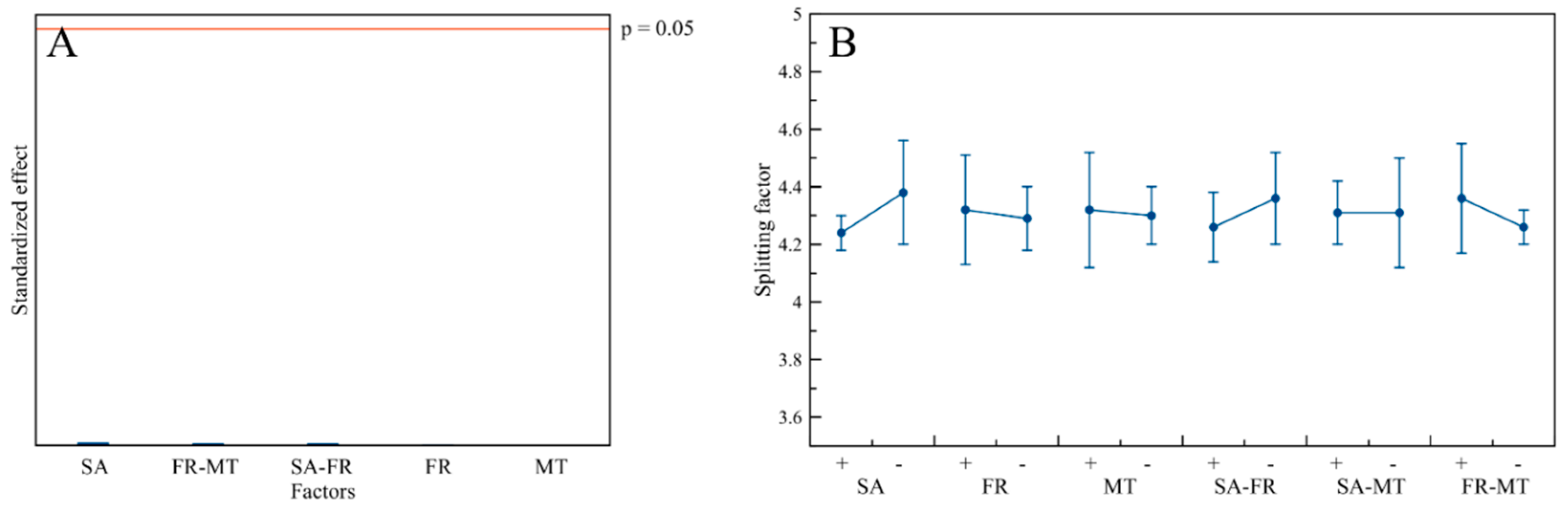
3.3. Structural Characterization
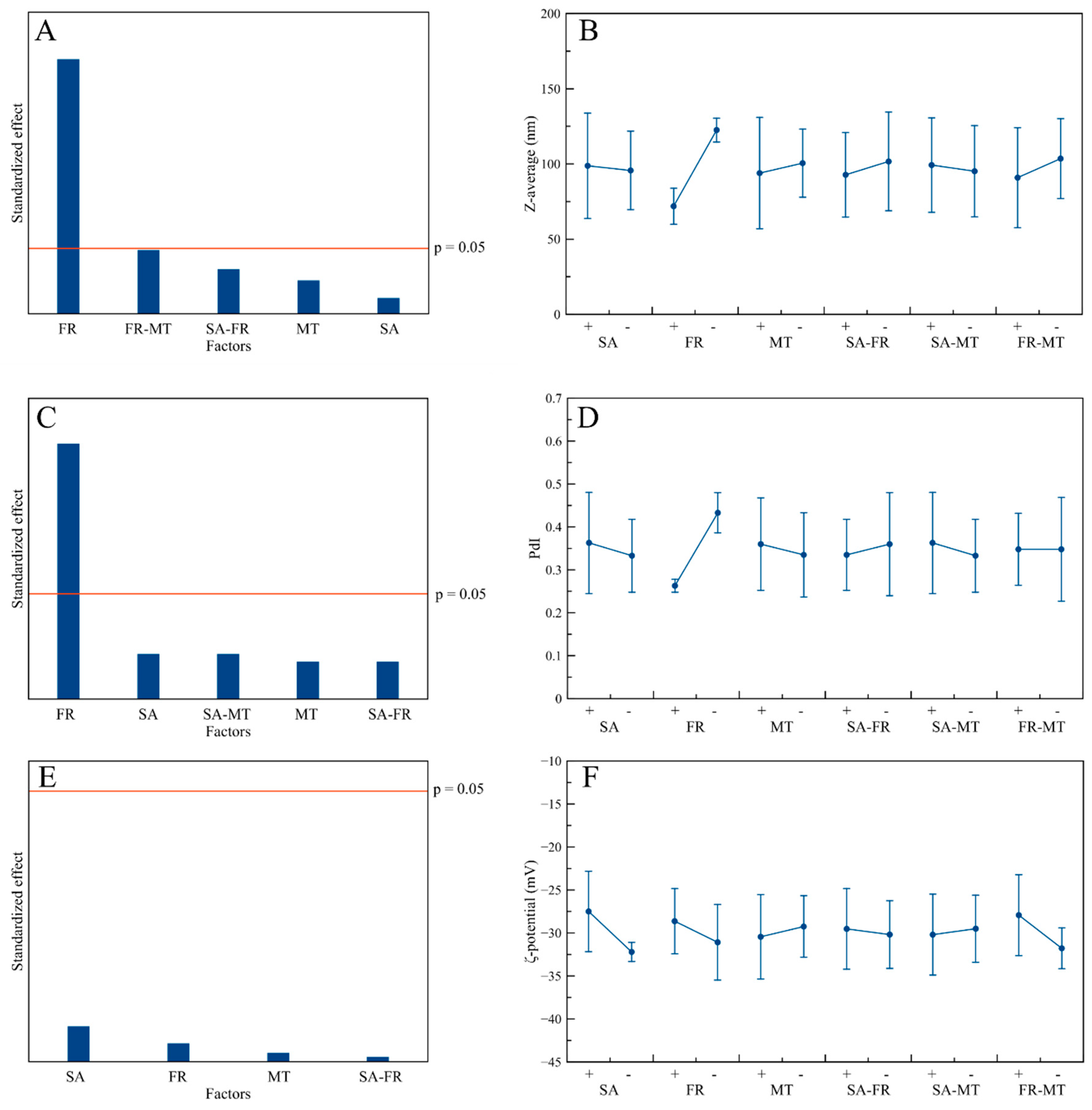
3.4. Hydrodynamic Diameter, PdI, and ζ-Potential
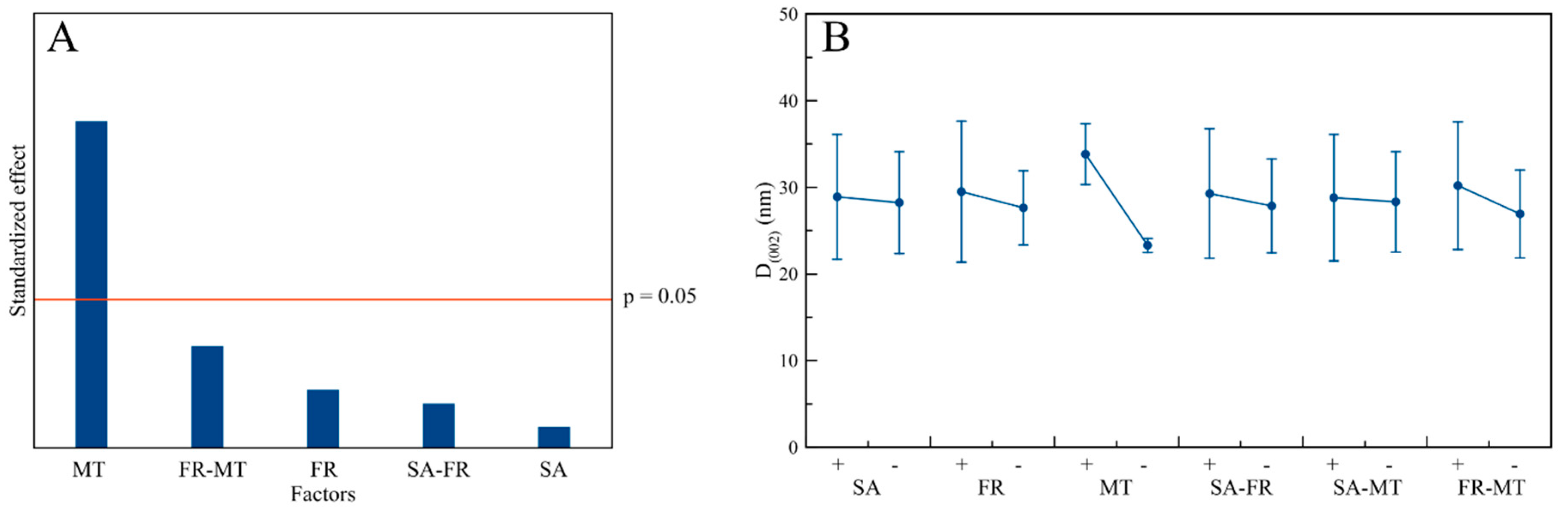
3.5. Influence of the Process Factors on CaP NPs’ Physico-Chemical Properties
3.6. Morphological Investigation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Wagner, V.; Dullaart, A.; Bock, A.-K.; Zweck, A. The emerging nanomedicine landscape. Nat. Biotechnol. 2006, 24, 1211–1217. [Google Scholar] [CrossRef] [PubMed]
- Arico, A.S.; Bruce, P.; Scrosati, B.; Tarascon, J.-M.; Van Schalkwijk, W. Nanostructured materials for advanced energy conversion and storage devices. In Materials for Sustainable Energy: A Collection of Peer-Reviewed Research and Review Articles from Nature Publishing Group; World Scientific: Singapore, 2011; pp. 148–159. [Google Scholar]
- Sattler, K.D. Handbook of Nanophysics: Nanoelectronics and Nanophotonics; CRC Press: Boca Raton, FL, USA, 2010. [Google Scholar]
- Wang, P.; Lombi, E.; Zhao, F.-J.; Kopittke, P.M. Nanotechnology: A new opportunity in plant sciences. Trends Plant Sci. 2016, 21, 699–712. [Google Scholar] [CrossRef]
- Agarwal, A.; Joshi, H. Application of nanotechnology in the remediation of contaminated groundwater: A short review. Recent Res. Sci. Technol. 2010, 2, 51–57. [Google Scholar]
- Dorozhkin, S.V. Bioceramics of calcium orthophosphates. Biomaterials 2010, 31, 1465–1485. [Google Scholar] [CrossRef] [PubMed]
- Dorozhkin, S.V.; Epple, M. Biological and Medical Significance of Calcium Phosphates. Angew. Chem. Int. Ed. 2002, 41, 3130–3146. [Google Scholar] [CrossRef]
- Uskoković, V.; Wu, V.M. Calcium phosphate as a key material for socially responsible tissue engineering. Materials 2016, 9, 434. [Google Scholar] [CrossRef]
- Uskoković, V. Ion-doped hydroxyapatite: An impasse or the road to follow? Ceram. Int. 2020, 46, 11443–11465. [Google Scholar] [CrossRef]
- Miragoli, M.; Ceriotti, P.; Iafisco, M.; Vacchiano, M.; Salvarani, N.; Alogna, A.; Carullo, P.; Ramirez-Rodríguez, G.B.; Patrício, T.; Degli Esposti, L.; et al. Inhalation of peptide-loaded nanoparticles improves heart failure. Sci. Transl. Med. 2018, 10, eaan6205. [Google Scholar] [CrossRef] [Green Version]
- Di Mauro, V.; Iafisco, M.; Salvarani, N.; Vacchiano, M.; Carullo, P.; Ramírez-Rodríguez, G.B.; Patrício, T.; Tampieri, A.; Miragoli, M.; Catalucci, D. Bioinspired negatively charged calcium phosphate nanocarriers for cardiac delivery of MicroRNAs. Nanomedicine 2016, 11, 891–906. [Google Scholar] [CrossRef] [Green Version]
- Victor, S.P.; Kumar, T.S.S. Tailoring calcium-deficient hydroxyapatite nanocarriers for enhanced release of antibiotics. J. Biomed. Nanotechnol. 2008, 4, 203–209. [Google Scholar] [CrossRef]
- Victor, S.P.; Paul, W.; Jayabalan, M.; Sharma, C.P. Supramolecular hydroxyapatite complexes as theranostic near-infrared luminescent drug carriers. CrystEngComm 2014, 16, 9033–9042. [Google Scholar] [CrossRef]
- Iafisco, M.; Drouet, C.; Adamiano, A.; Pascaud, P.; Montesi, M.; Panseri, S.; Sarda, S.; Tampieri, A. Superparamagnetic iron-doped nanocrystalline apatite as a delivery system for doxorubicin. J. Mater. Chem. B 2016, 4, 57–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mostaghaci, B.; Loretz, B.; Lehr, C.M. Calcium Phosphate System for Gene Delivery: Historical Background and Emerging Opportunities. Curr. Pharm. Des. 2016, 22, 1529–1533. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Mitchell, A.; Morcol, T.; Bell, S.J. Calcium phosphate nanoparticles induce mucosal immunity and protection against herpes simplex virus type 2. Clin. Diagn. Lab. Immunol. 2002, 9, 1021–1024. [Google Scholar] [CrossRef] [Green Version]
- Ali, I. New generation adsorbents for water treatment. Chem. Rev. 2012, 112, 5073–5091. [Google Scholar] [CrossRef]
- Mori, K.; Yamaguchi, K.; Hara, T.; Mizugaki, T.; Ebitani, K.; Kaneda, K. Controlled synthesis of hydroxyapatite-supported palladium complexes as highly efficient heterogeneous catalysts. J. Am. Chem. Soc. 2002, 124, 11572–11573. [Google Scholar] [CrossRef]
- Yoon, H.Y.; Lee, J.G.; Esposti, L.D.; Iafisco, M.; Kim, P.J.; Shin, S.G.; Jeon, J.-R.; Adamiano, A. Synergistic Release of Crop Nutrients and Stimulants from Hydroxyapatite Nanoparticles Functionalized with Humic Substances: Toward a Multifunctional Nanofertilizer. ACS Omega 2020, 5, 6598–6610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramírez-Rodríguez, G.B.; Dal Sasso, G.; Carmona, F.J.; Miguel-Rojas, C.; Pérez-de-Luque, A.; Masciocchi, N.; Guagliardi, A.; Delgado-López, J.M. Engineering Biomimetic Calcium Phosphate Nanoparticles: A Green Synthesis of Slow-Release Multinutrient (NPK) Nanofertilizers. ACS Appl. Bio Mater. 2020, 3, 1344–1353. [Google Scholar] [CrossRef]
- Rhee, S.-H. Synthesis of hydroxyapatite via mechanochemical treatment. Biomaterials 2002, 23, 1147–1152. [Google Scholar] [CrossRef]
- Kim, I.-S.; Kumta, P.N. Sol-gel synthesis and characterization of nanostructured hydroxyapatite powder. Mater. Sci. Eng. B 2004, 111, 232–236. [Google Scholar] [CrossRef]
- Drouet, C.; Bosc, F.; Banu, M.; Largeot, C.; Combes, C.; Dechambre, G.; Estournès, C.; Raimbeaux, G.; Rey, C. Nanocrystalline apatites: From powders to biomaterials. Powder Technol. 2009, 190, 118–122. [Google Scholar] [CrossRef] [Green Version]
- Jevtic, M.; Mitric, M.; Skapin, S.; Jancar, B.; Ignjatovic, N.; Uskokovic, D. Crystal structure of hydroxyapatite nanorods synthesized by sonochemical homogeneous precipitation. Cryst. Growth Des. 2008, 8, 2217–2222. [Google Scholar] [CrossRef]
- Di Chen, J.; Wang, Y.J.; Wei, K.; Zhang, S.H.; Shi, X.T. Self-organization of hydroxyapatite nanorods through oriented attachment. Biomaterials 2007, 28, 2275–2280. [Google Scholar] [CrossRef] [PubMed]
- Afshar, A.; Ghorbani, M.; Ehsani, N.; Saeri, M.; Sorrell, C. Some important factors in the wet precipitation process of hydroxyapatite. Mater. Des. 2003, 24, 197–202. [Google Scholar] [CrossRef]
- Kim, D.W.; Cho, I.-S.; Kim, J.Y.; Jang, H.L.; Han, G.S.; Ryu, H.-S.; Shin, H.; Jung, H.S.; Kim, H.; Hong, K.S. Simple large-scale synthesis of hydroxyapatite nanoparticles: In Situ observation of crystallization process. Langmuir 2010, 26, 384–388. [Google Scholar] [CrossRef]
- Chen, J.; Zheng, C.; Chen, G.A. Interaction of macro-and micromixing on particle size distribution in reactive precipitation. Chem. Eng. Sci. 1996, 51, 1957–1966. [Google Scholar] [CrossRef]
- Kazmierczak, T.; Schuttringer, E.; Tomažić, B.; Nancollas, G. Controlled composition studies of calcium carbonate and sulfate crystal growth. Croat. Chem. Acta 1981, 54, 277–287. [Google Scholar]
- Uskokovic, V. Challenges for the modern science in its descend towards nano scale. Curr. Nanosci. 2009, 5, 372–389. [Google Scholar] [CrossRef] [Green Version]
- Latocha, J.; Wojasiński, M.; Sobieszuk, P.; Ciach, T. Synthesis of hydroxyapatite in a continuous reactor: A review. Chem. Process Eng. 2018, 39, 281–293. [Google Scholar]
- Castro, F.; Ferreira, A.; Rocha, F.; Vicente, A.; Teixeira, J.A. Continuous-flow precipitation of hydroxyapatite at 37 C in a meso oscillatory flow reactor. Ind. Eng. Chem. Res. 2013, 52, 9816–9821. [Google Scholar] [CrossRef] [Green Version]
- Massart, D.L.; Vandeginste, B.G.; Buydens, L.; Lewi, P.; Smeyers-Verbeke, J.; Jong, S.D. Handbook of Chemometrics and Qualimetrics; Elsevier Science Inc.: Amsterdam, The Netherlands, 1998. [Google Scholar]
- Giardina, M.A.; Fanovich, M.A. Synthesis of nanocrystalline hydroxyapatite from Ca(OH)2 and H3PO4 assisted by ultrasonic irradiation. Ceram. Int. 2010, 36, 1961–1969. [Google Scholar] [CrossRef]
- Deng, S.; Lin, Z.T.; Tang, H.X.; Ullah, S.; Bi, Y.G. Rapid synthesis of hydroxyapatite nanoparticles via a novel approach in the dual-frequency ultrasonic system for specific biomedical application. J. Mater. Res. 2019, 34, 2796–2806. [Google Scholar] [CrossRef]
- Coelho, A. Topas Academic V5; Coelho Software: Brisbane, Australia, 2012. [Google Scholar]
- Hughes, J.M.; Cameron, M.; Crowley, K.D. Structural variations in natural F, OH, and Cl apatites. Am. Mineral. 1989, 74, 870–876. [Google Scholar]
- Walker, D.; Verma, P.K.; Cranswick, L.M.; Jones, R.L.; Clark, S.M.; Buhre, S. Halite-sylvite thermoelasticity. Am. Mineral. 2004, 89, 204–210. [Google Scholar] [CrossRef]
- Fischer, A.; Palladino, G. Trisodium citrate dihydrate. Acta Crystallogr. Sect. E Struct. Rep. Online 2003, 59, m1080–m1082. [Google Scholar] [CrossRef]
- Abràmoff, M.D.; Magalhães, P.J.; Ram, S.J. Image processing with ImageJ. Biophotonics Int. 2004, 11, 36–42. [Google Scholar]
- Martínez-Casado, F.J.; Iafisco, M.; Delgado-López, J.M.; Martínez-Benito, C.; Ruiz-Pérez, C.; Colangelo, D.; Oltolina, F.; Prat, M.; Gómez-Morales, J. Bioinspired Citrate–Apatite Nanocrystals Doped with Divalent Transition Metal Ions. Cryst. Growth Des. 2015, 16, 145–153. [Google Scholar] [CrossRef]
- Delgado-López, J.M.; Iafisco, M.; Rodríguez, I.; Tampieri, A.; Prat, M.; Gómez-Morales, J. Crystallization of bioinspired citrate-functionalized nanoapatite with tailored carbonate content. Acta Biomater. 2012, 8, 3491–3499. [Google Scholar] [CrossRef]
- Torrent-Burgués, J.; Gómez-Morales, J.; López-Macipe, A.; Rodríguez-Clemente, R. Continuous precipitation of hydroxyapatite from Ca/citrate/phosphate solutions using microwave heating. Cryst. Res. Technol. J. Exp. Ind. Crystallogr. 1999, 34, 757–762. [Google Scholar] [CrossRef]
- Dorozhkin, S.V. Calcium orthophosphates. J. Mater. Sci. 2007, 42, 1061–1095. [Google Scholar] [CrossRef]
- Iafisco, M.; Palazzo, B.; Martra, G.; Margiotta, N.; Piccinonna, S.; Natile, G.; Gandin, V.; Marzano, C.; Roveri, N. Nanocrystalline carbonate-apatites: Role of Ca/P ratio on the upload and release of anticancer platinum bisphosphonates. Nanoscale 2012, 4, 206–217. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Morales, J.; Iafisco, M.; Delgado-López, J.M.; Sarda, S.; Drouet, C. Progress on the preparation of nanocrystalline apatites and surface characterization: Overview of fundamental and applied aspects. Prog. Cryst. Growth Charact. Mater. 2013, 59, 1–46. [Google Scholar] [CrossRef] [Green Version]
- Kawase, M.; Suzuki, T.; Miura, K. Growth mechanism of lanthanum phosphate particles by continuous precipitation. Chem. Eng. Sci. 2007, 62, 4875–4879. [Google Scholar] [CrossRef]
- Pieper, M.; Aman, S.; Hintz, W.; Tomas, J. Optimization of a continuous precipitation process to produce nanoscale BaSO4. Chem. Eng. Technol. 2011, 34, 1567–1574. [Google Scholar] [CrossRef]
- Gecim, G.; Donmez, S.; Erkoc, E. Calcium deficient hydroxyapatite by precipitation: Continuous process by vortex reactor and semi-batch synthesis. Ceram. Int. 2020. [Google Scholar] [CrossRef]
- Calabrese, G.S.; Pissavini, S. From batch to continuous flow processing in chemicals manufacturing. AIChE J. 2011, 57, 828–834. [Google Scholar] [CrossRef]
- Koutsopoulos, S. Synthesis and characterization of hydroxyapatite crystals: A review study on the analytical methods. J. Biomed. Mater. Res. 2002, 62, 600–612. [Google Scholar] [CrossRef] [PubMed]
- Lakhmanan, B. Infrared Absorption spectrum of sodium citrate. J. Indian Inst. Sci. 2013, 39, 27. [Google Scholar]
- Termine, J.D.; Posner, A.S. Infra-red determination of the percentage of crystallinity in apatitic calcium phosphates. Nature 1966, 211, 268–270. [Google Scholar] [CrossRef]
- Kandori, K.; Kuroda, T.; Togashi, S.; Katayama, E. Preparation of calcium hydroxyapatite nanoparticles using microreactor and their characteristics of protein adsorption. J. Phys. Chem. B 2011, 115, 653–659. [Google Scholar] [CrossRef]
- Ivanchenko, P.; Delgado-López, J.M.; Iafisco, M.; Gómez-Morales, J.; Tampieri, A.; Martra, G.; Sakhno, Y. On the surface effects of citrates on nano-apatites: Evidence of a decreased hydrophilicity. Sci. Rep. 2017, 7, 8901. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Level | Sonication Amplitude (SA, %) | Flow Rate (FR, mL s−1) | Maturation Time (MT, h) |
|---|---|---|---|
| Level −1 | 20 | 0.7 | 0 |
| Level +1 | 100 | 2.8 | 24 |
| Sample Code | Design | ||
| Run 1 | +1 | +1 | +1 |
| Run 2 | +1 | +1 | −1 |
| Run 3 | +1 | −1 | +1 |
| Run 4 | +1 | −1 | −1 |
| Run 5 | −1 | +1 | +1 |
| Run 6 | −1 | +1 | −1 |
| Run 7 | −1 | −1 | +1 |
| Run 8 | −1 | −1 | −1 |
| Sample Code | Composition (wt. %) | D(002) (nm) | D(310) (nm) | D(002)/D(310) | Splitting Factor a | ||
|---|---|---|---|---|---|---|---|
| Hydroxyapatite (HA) | Na3(Cit) | Halite | |||||
| Run 1 | 64.8 ± 0.5 | 34.3 ± 0.5 | 0.9 ± 0.1 | 38.5 ± 1.9 | 7.2 ± 0.9 | 5.3 ± 0.4 | 4.2 ± 0.1 |
| Run 2 | 100 ± 0.1 | - | - | 22.6 ± 1.1 | 6.1 ± 0.7 | 3.7 ± 0.5 | 4.2 ± 0.1 |
| Run 3 | 87.6 ± 0.7 | 11.9 ± 0.7 | 0.5 ± 0.1 | 30.3 ± 1.5 | 6.1 ± 0.8 | 5.0 ± 0.6 | 4.3 ± 0.1 |
| Run 4 | 100 ± 0.1 | - | - | 24.2 ± 1.2 | 5.7 ± 0.7 | 4.2 ± 0.4 | 4.2 ± 0.1 |
| Run 5 | 69.9 ± 0.4 | 29.3 ± 0.4 | 0.9 ± 0.1 | 34.3 ± 1.7 | 6.1 ± 0.6 | 5.6 ± 0.6 | 4.6 ± 0.1 |
| Run 6 | 100 ± 0.1 | - | - | 22.6 ± 1.2 | 5.5 ± 0.5 | 4.1 ± 0.3 | 4.3 ± 0.1 |
| Run 7 | 90.2 ± 0.7 | 9.5 ± 0.7 | 0.4 ± 0.1 | 32.2 ± 1.6 | 5.0 ± 0.6 | 6.4 ± 0.5 | 4.2 ± 0.1 |
| Run 8 | 100 ± 0.1 | - | - | 23.8 ± 1.2 | 6.1 ± 0.7 | 3.9 ± 0.3 | 4.4 ± 0.1 |
| Batch | 100 ± 0.1 | - | - | 24.0 ± 1.1 | 7.1 ± 0.5 | 3.4 ± 0.4 | 3.5 ± 0.1 |
| Sample Code | Ca/P | Yield (g L−1) | Corrected Yield (g L−1) |
|---|---|---|---|
| Run 1 | 1.50 ± 0.01 | 3.8 ± 0.4 | 2.5 ± 0.3 |
| Run 2 | 1.53 ± 0.01 | 2.6 ± 0.3 | 2.6 ± 0.3 |
| Run 3 | 1.52 ± 0.01 | 3.4 ± 0.4 | 3.0 ± 0.3 |
| Run 4 | 1.52 ± 0.01 | 2.6 ± 0.3 | 2.6 ± 0.3 |
| Run 5 | 1.49 ± 0.01 | 3.5 ± 0.2 | 2.5 ± 0.2 |
| Run 6 | 1.54 ± 0.01 | 2.8 ± 0.3 | 2.8 ± 0.3 |
| Run 7 | 1.51 ± 0.01 | 3.4 ± 0.3 | 3.1 ± 0.3 |
| Run 8 | 1.59 ± 0.01 | 2.9 ± 0.3 | 2.9 ± 0.3 |
| Batch | 1.50 ± 0.01 | 1.6 ± 0.2 | 1.6 ± 0.2 |
| Sample Code | Z-Average (nm) | Dv(90) (nm) | PdI | ζ-Potential |
|---|---|---|---|---|
| Run 1 | 62 ± 2 | 61 ± 3 | 0.28 ± 0.04 | −23.3 ± 4.6 |
| Run 2 | 74 ± 4 | 70 ± 8 | 0.25 ± 0.01 | −28.6 ± 0.7 |
| Run 3 | 140 ± 12 | 398 ± 130 | 0.50 ± 0.02 | −33.6 ± 2.3 |
| Run 4 | 126 ± 5 | 336 ± 151 | 0.42 ± 0.01 | −24.5 ± 1.3 |
| Run 5 | 61 ± 4 | 62 ± 4 | 0.27 ± 0.05 | −31.3 ± 4.7 |
| Run 6 | 91 ± 8 | 97 ± 27 | 0.25 ± 0.01 | −31.3 ± 2.7 |
| Run 7 | 124 ± 12 | 175 ± 78 | 0.39 ± 0.02 | −33.6 ± 0.5 |
| Run 8 | 135 ± 36 | 151 ± 68 | 0.42 ± 0.01 | −32.6 ± 1.4 |
| Batch | 87 ± 1 | 90 ± 9 | 0.41 ± 0.01 | −19.1 ± 2.1 |
| Factors | Response Parameter | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| D(002) | D(310) | D(002)/D(310) | Ca/P | Yield | Splitting Factor | Z-Average | PdI | ζ-Potential | |
| SA | - | - | - | - | - | - | - | - | - |
| FR | - | - | - | - | - | - | (↓) | (↓) | - |
| MT | (↑) | - | (↑) | - | - | - | - | - | - |
| SA-FR | - | - | - | - | - | - | - | - | - |
| SA-MT | - | - | - | - | - | - | - | - | - |
| FR-MT | - | - | - | - | - | - | - | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Degli Esposti, L.; Dotti, A.; Adamiano, A.; Fabbi, C.; Quarta, E.; Colombo, P.; Catalucci, D.; De Luca, C.; Iafisco, M. Calcium Phosphate Nanoparticle Precipitation by a Continuous Flow Process: A Design of Experiment Approach. Crystals 2020, 10, 953. https://doi.org/10.3390/cryst10100953
Degli Esposti L, Dotti A, Adamiano A, Fabbi C, Quarta E, Colombo P, Catalucci D, De Luca C, Iafisco M. Calcium Phosphate Nanoparticle Precipitation by a Continuous Flow Process: A Design of Experiment Approach. Crystals. 2020; 10(10):953. https://doi.org/10.3390/cryst10100953
Chicago/Turabian StyleDegli Esposti, Lorenzo, Alessandro Dotti, Alessio Adamiano, Claudia Fabbi, Eride Quarta, Paolo Colombo, Daniele Catalucci, Claudio De Luca, and Michele Iafisco. 2020. "Calcium Phosphate Nanoparticle Precipitation by a Continuous Flow Process: A Design of Experiment Approach" Crystals 10, no. 10: 953. https://doi.org/10.3390/cryst10100953
APA StyleDegli Esposti, L., Dotti, A., Adamiano, A., Fabbi, C., Quarta, E., Colombo, P., Catalucci, D., De Luca, C., & Iafisco, M. (2020). Calcium Phosphate Nanoparticle Precipitation by a Continuous Flow Process: A Design of Experiment Approach. Crystals, 10(10), 953. https://doi.org/10.3390/cryst10100953