Varying the Dimensionality of Cu(II)-Based Coordination Polymers Through Solvent Influence

 ,
, 
 , and
, and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
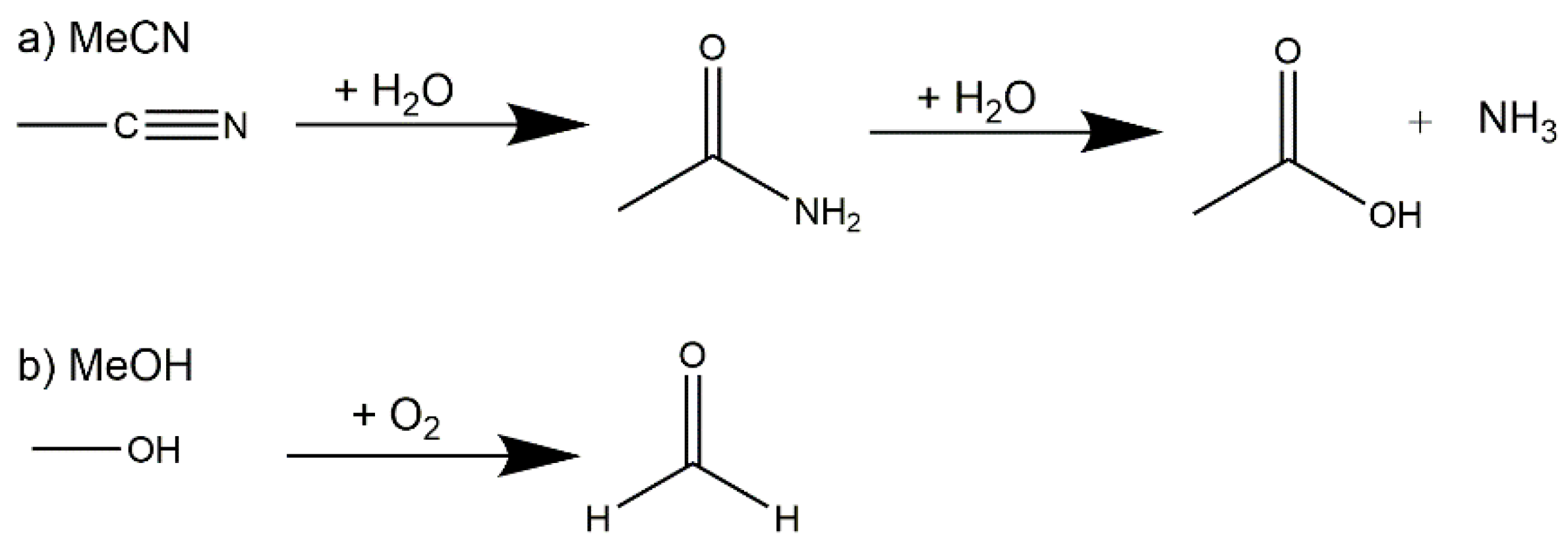
2. Results and Discussion
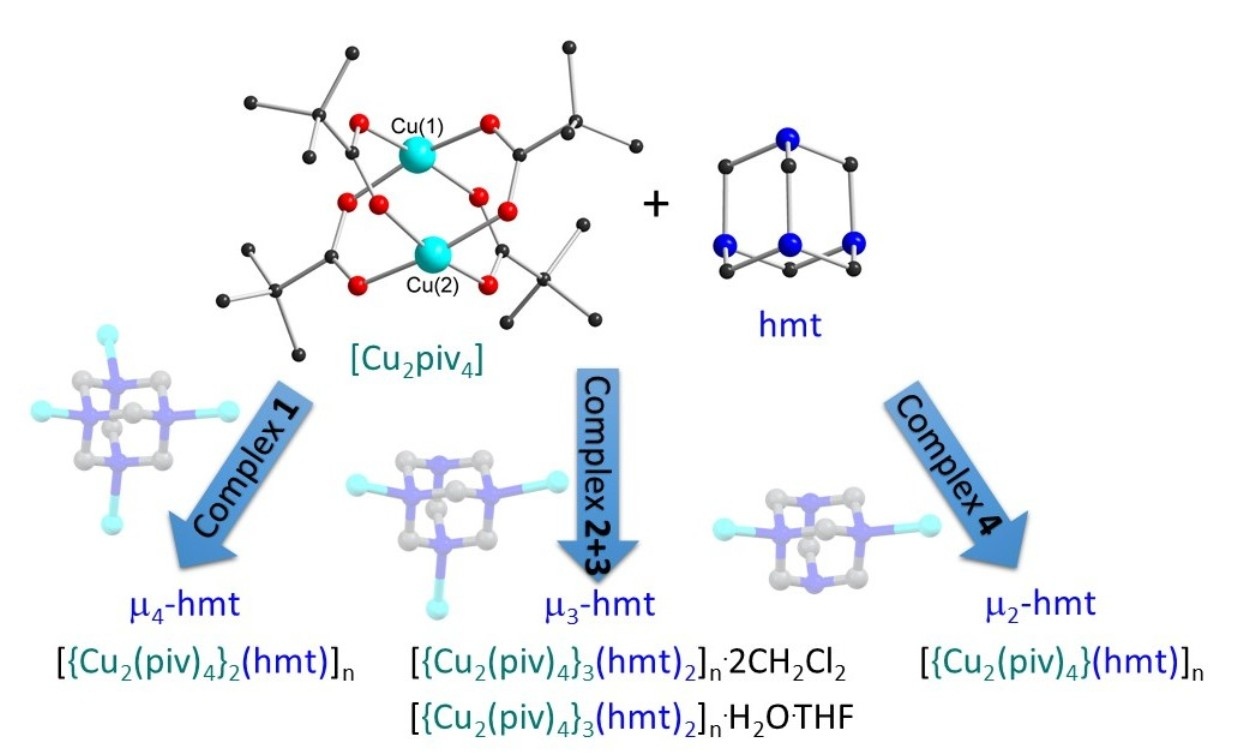
2.1. Synthetic Strategy
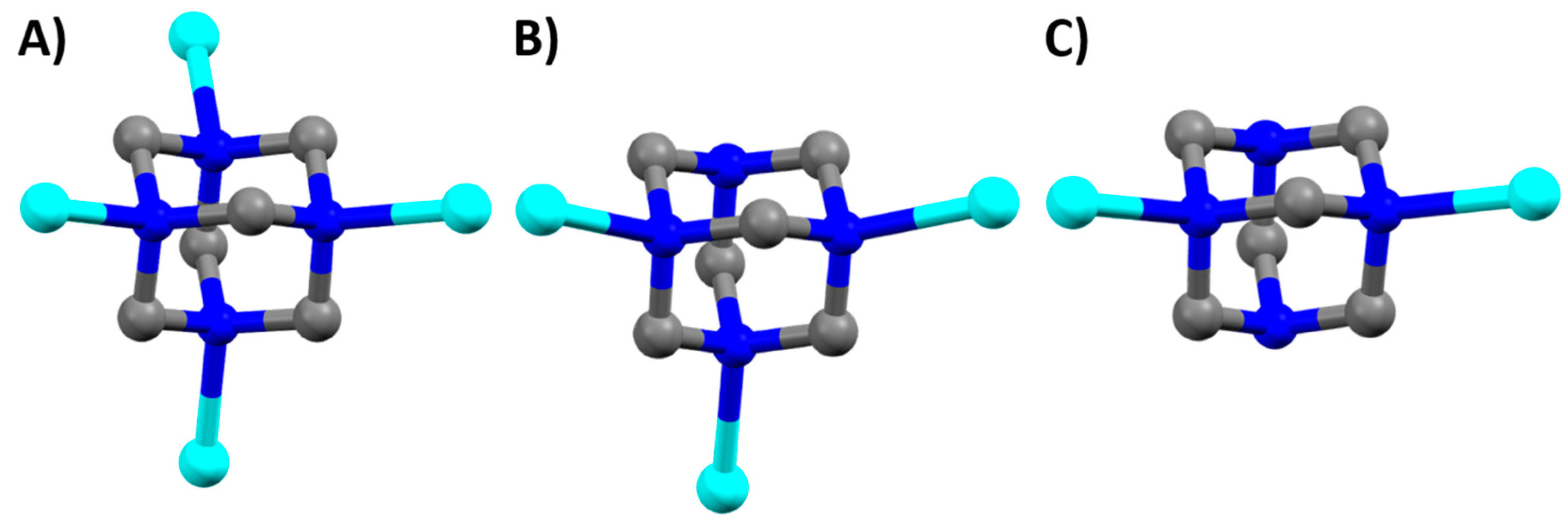
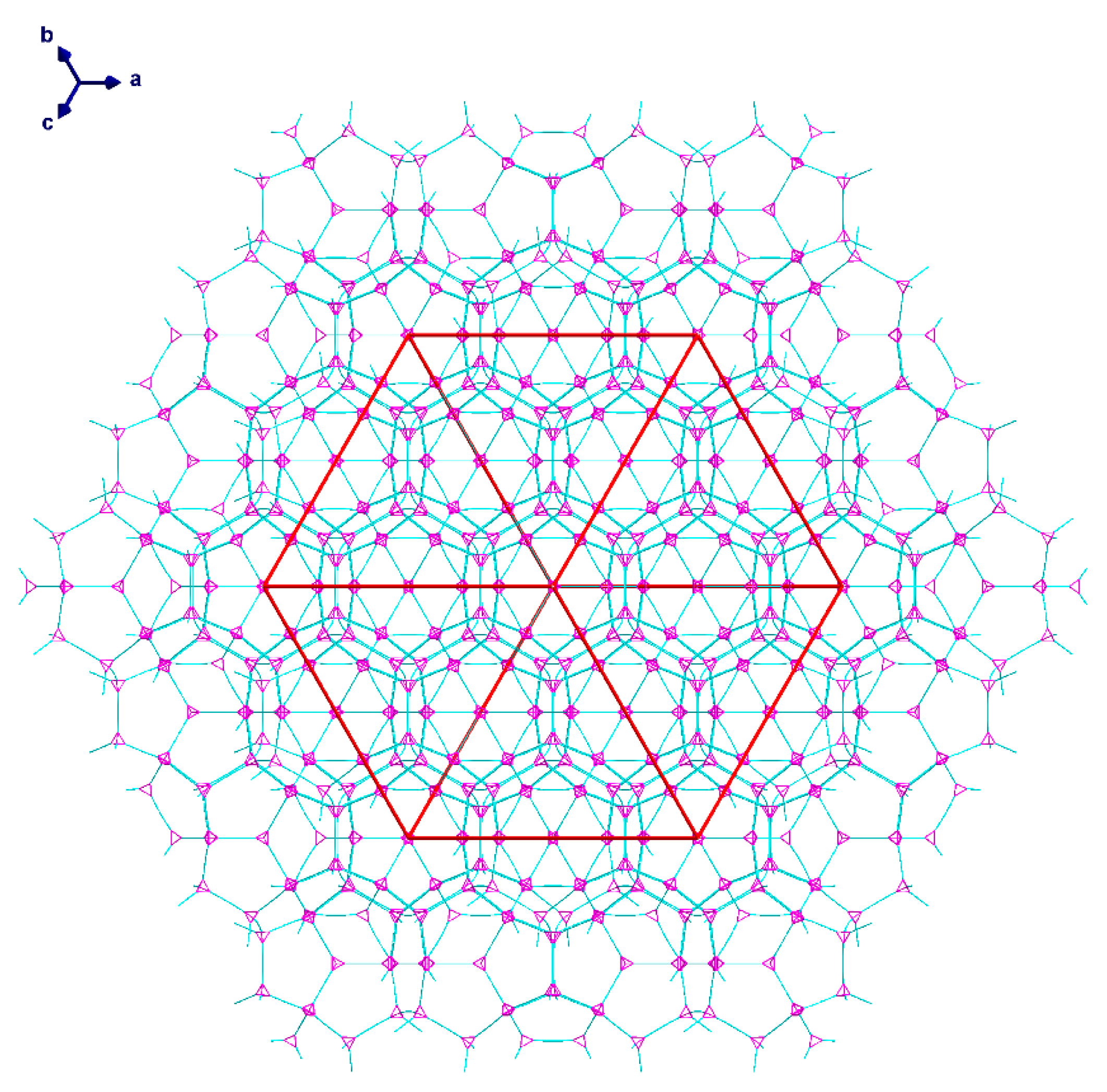
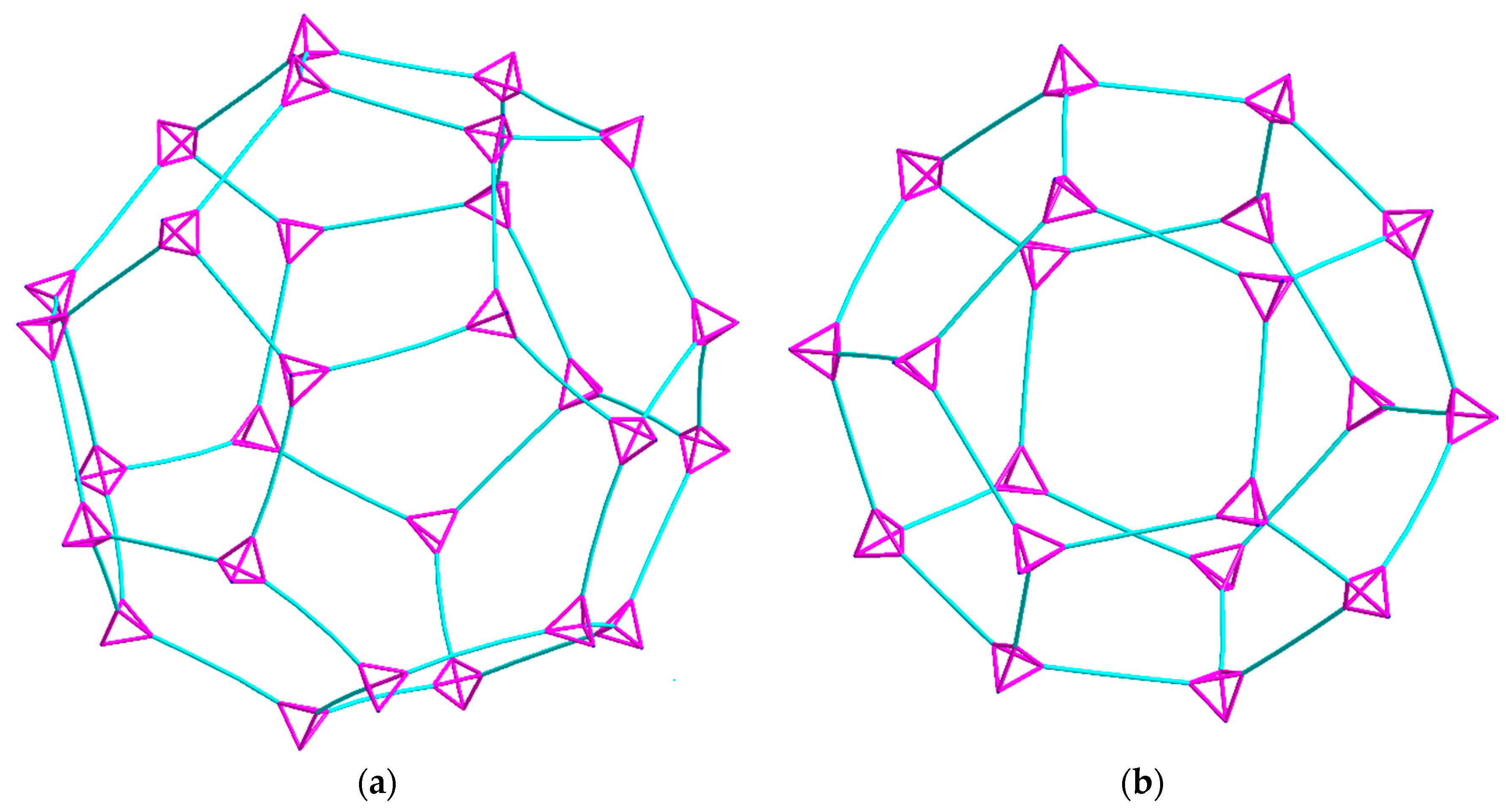
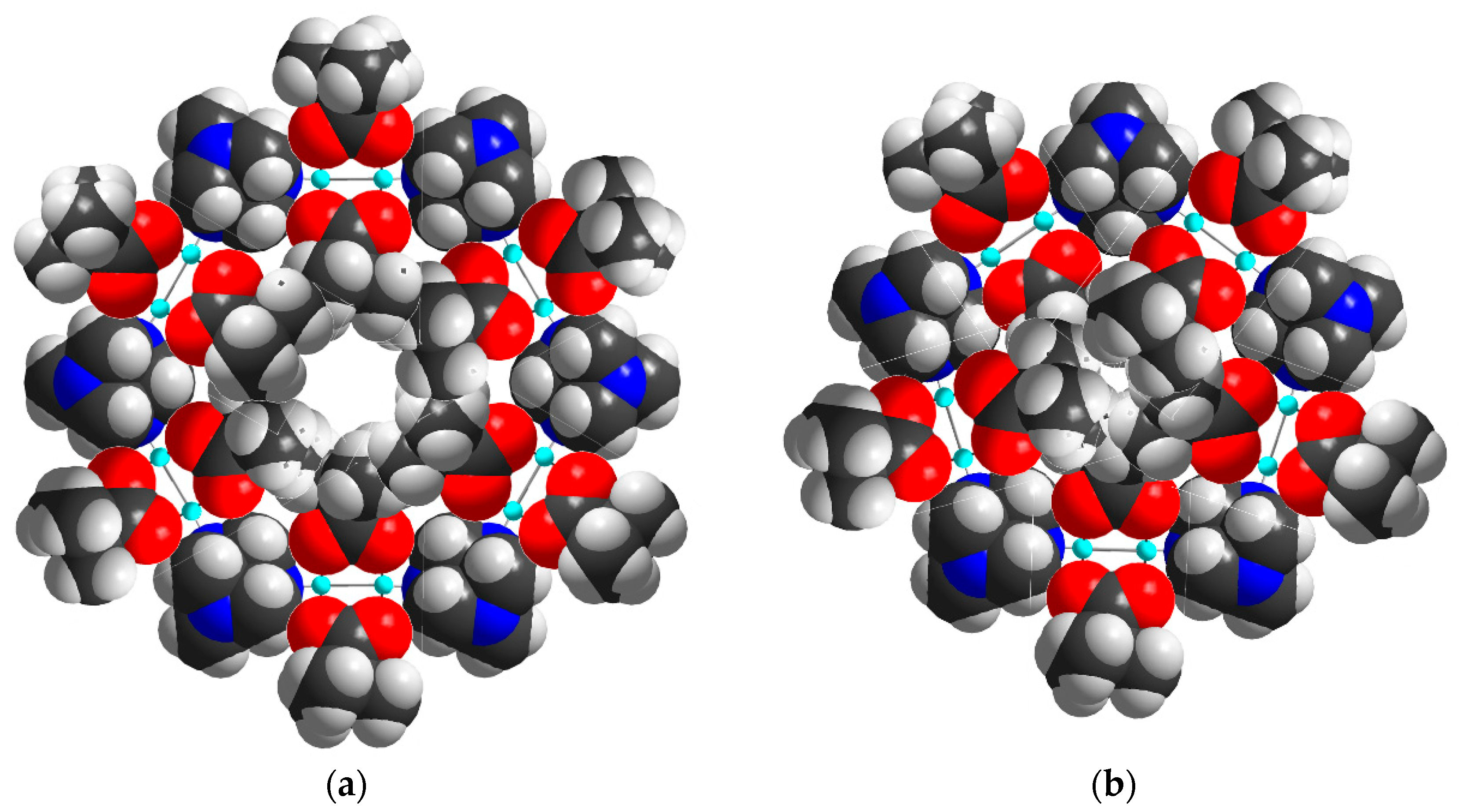
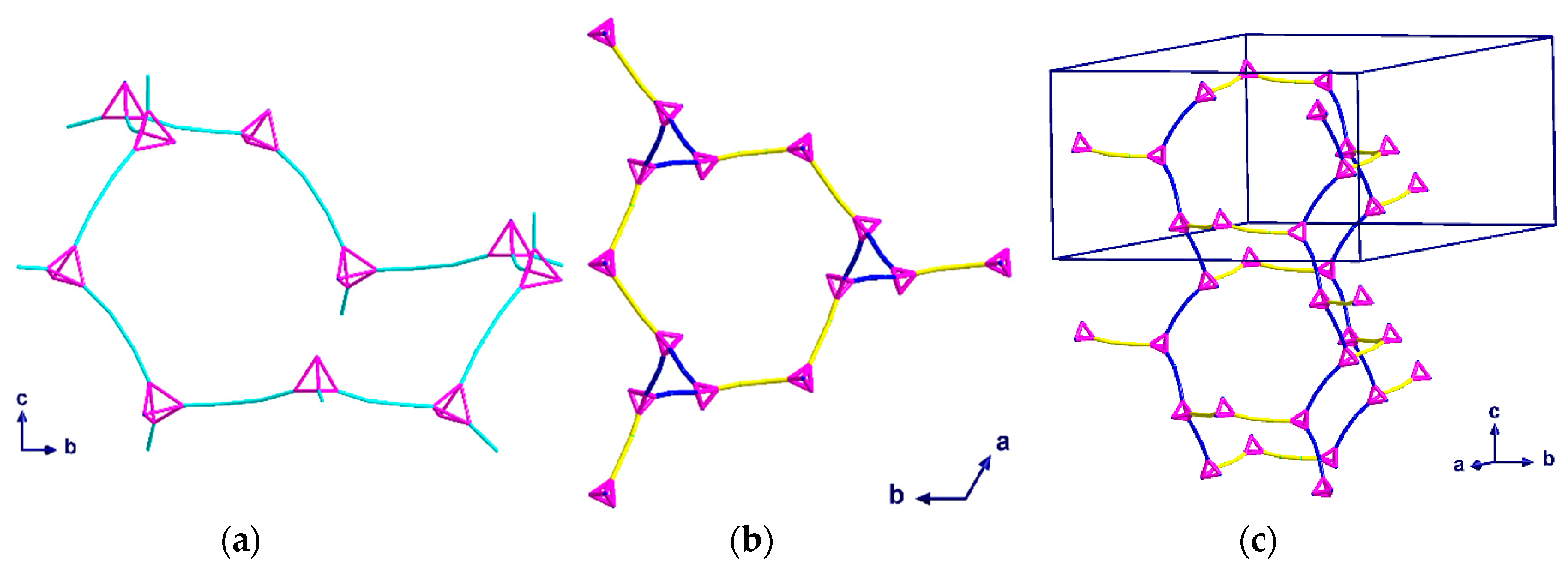
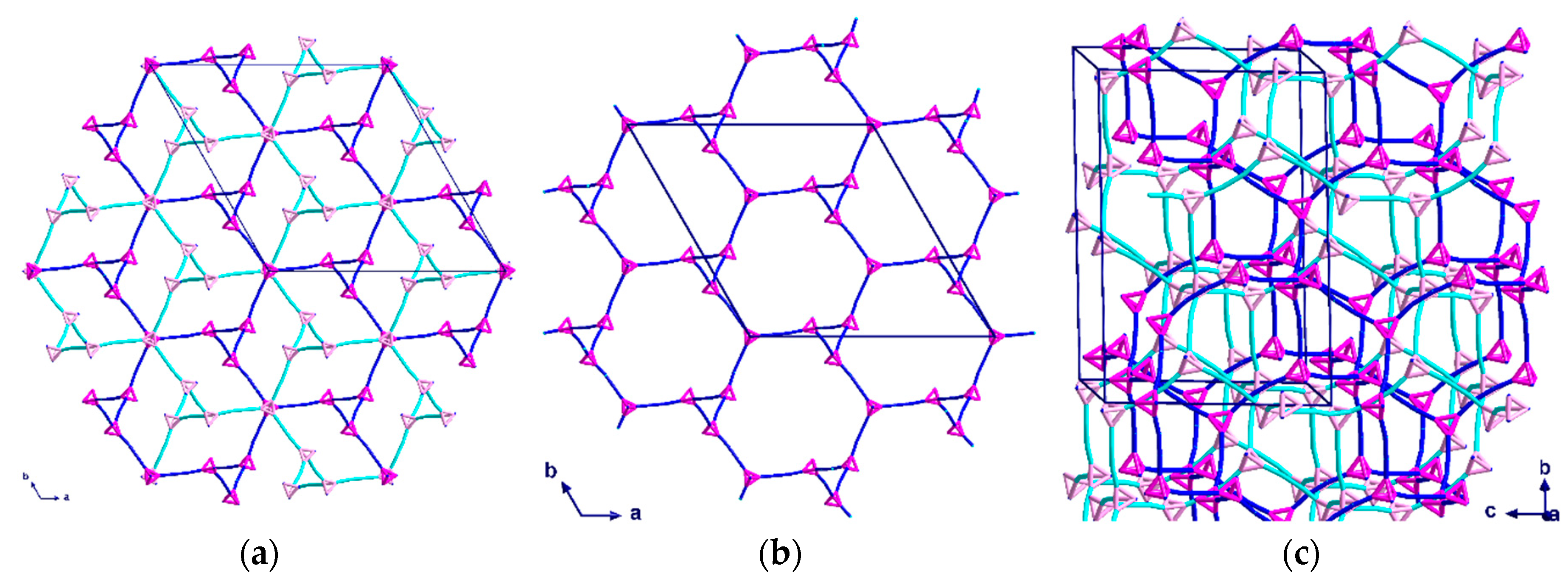
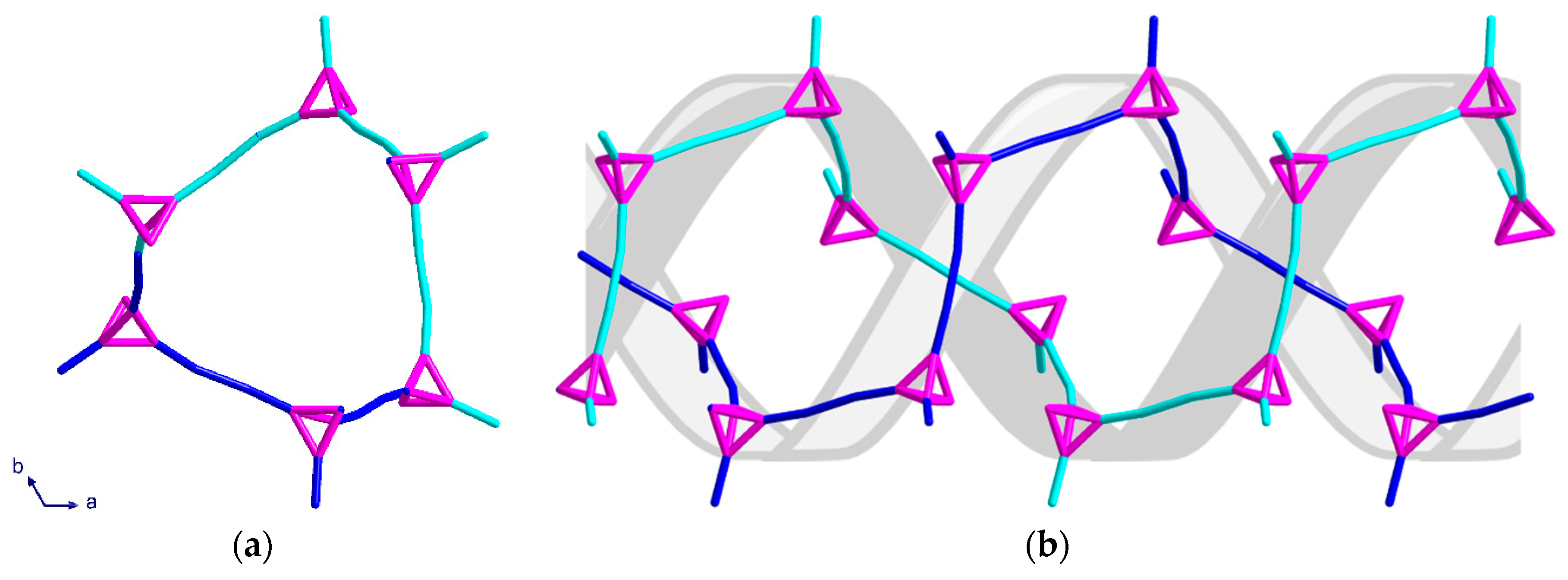
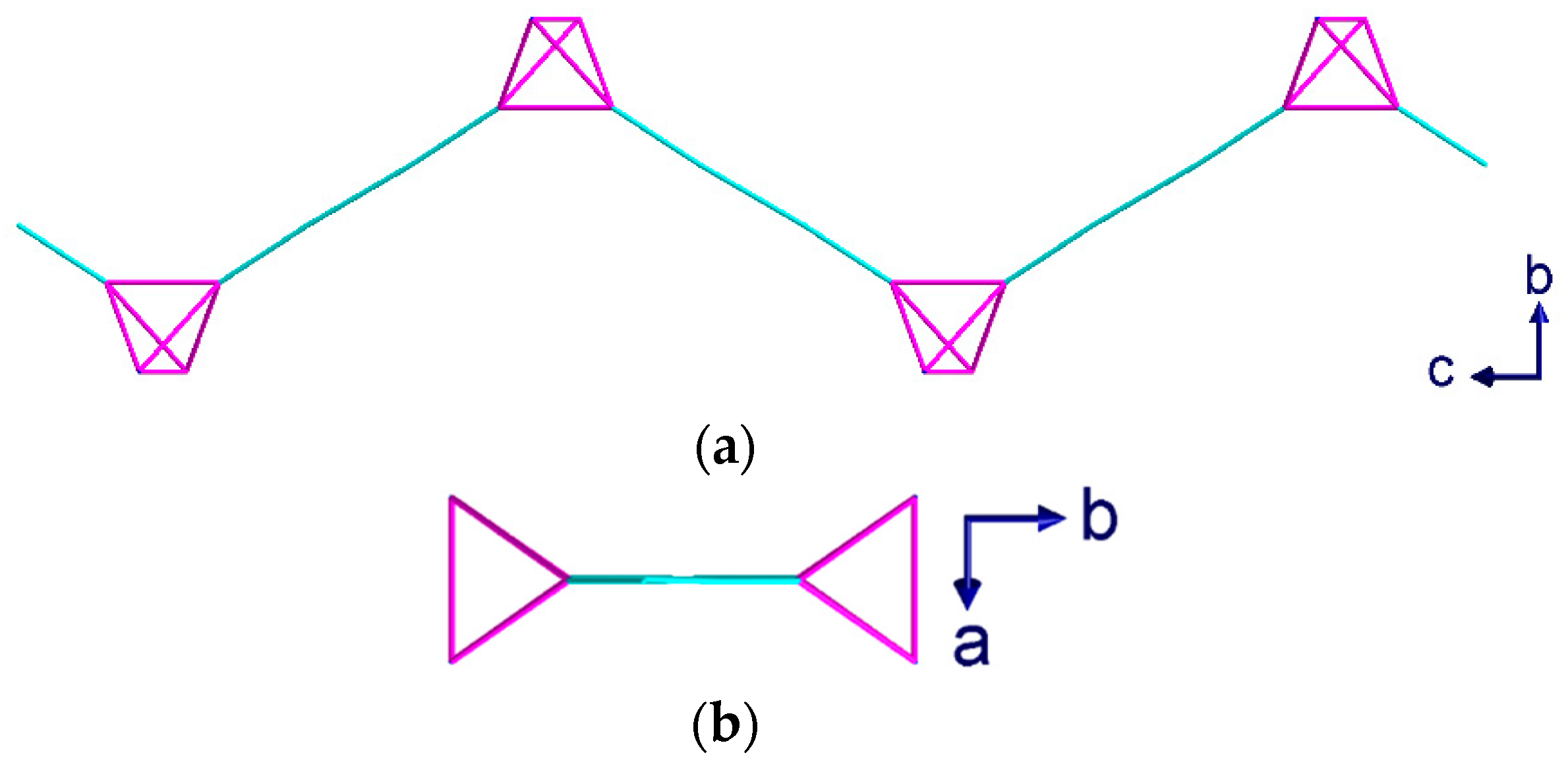
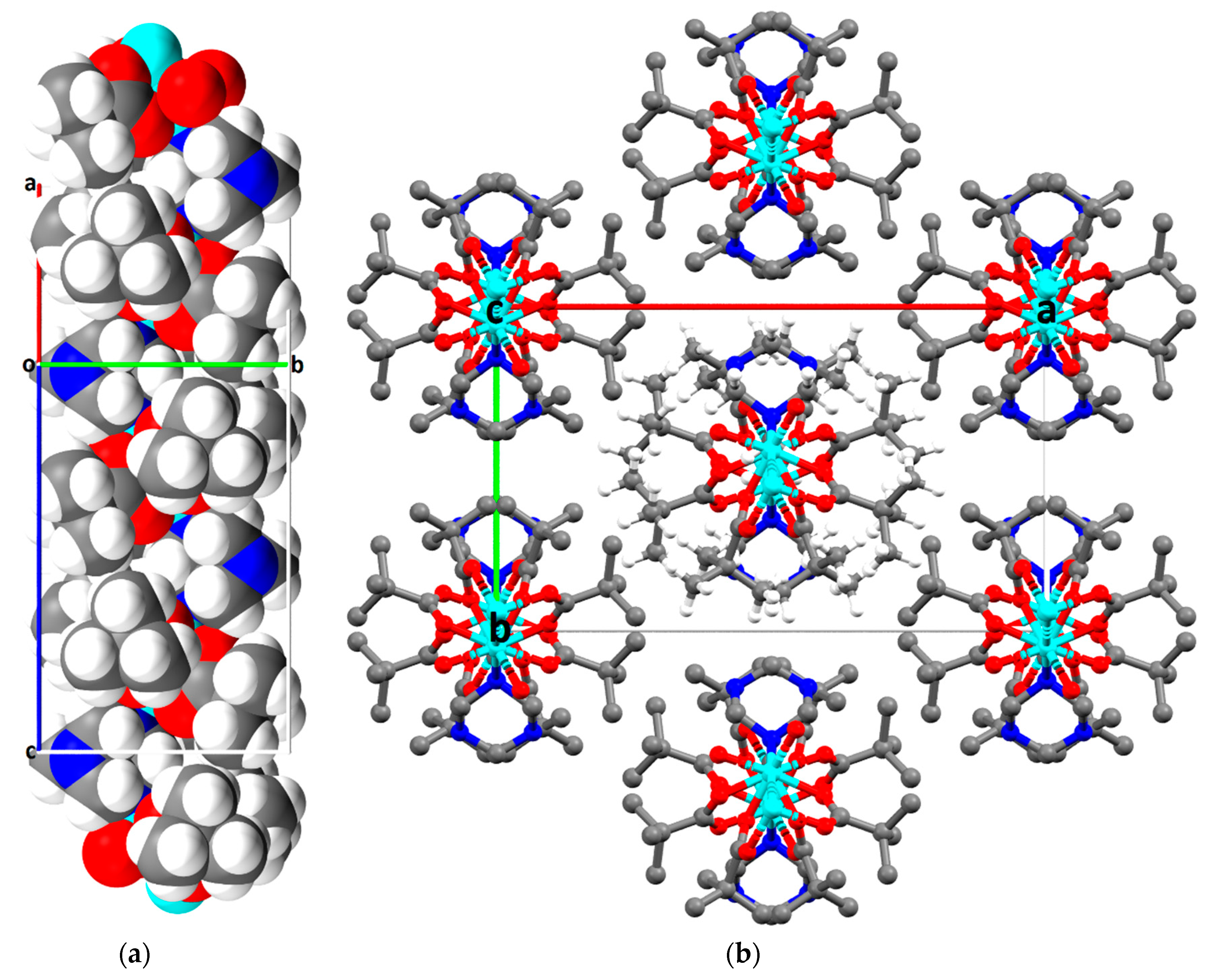
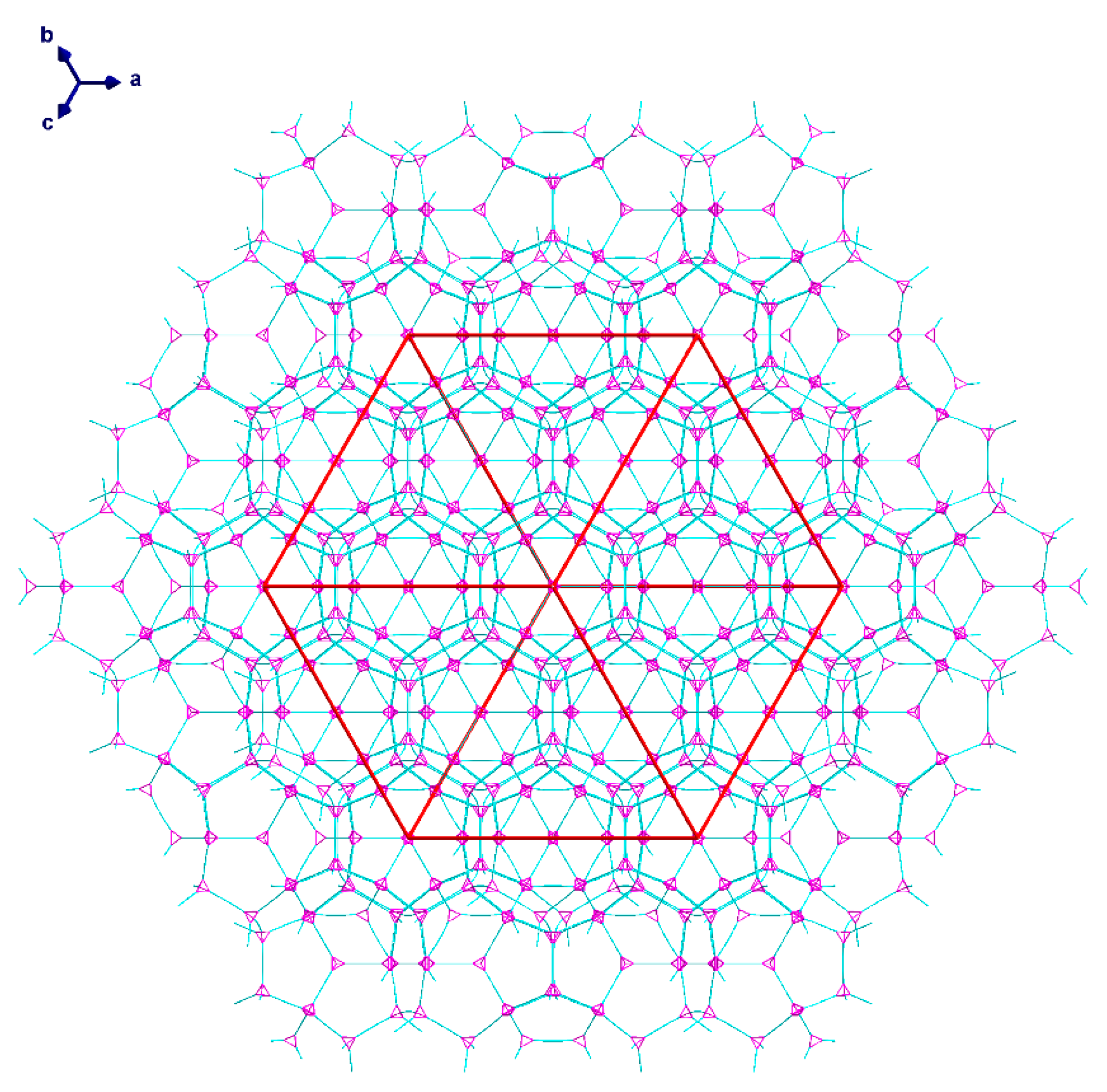
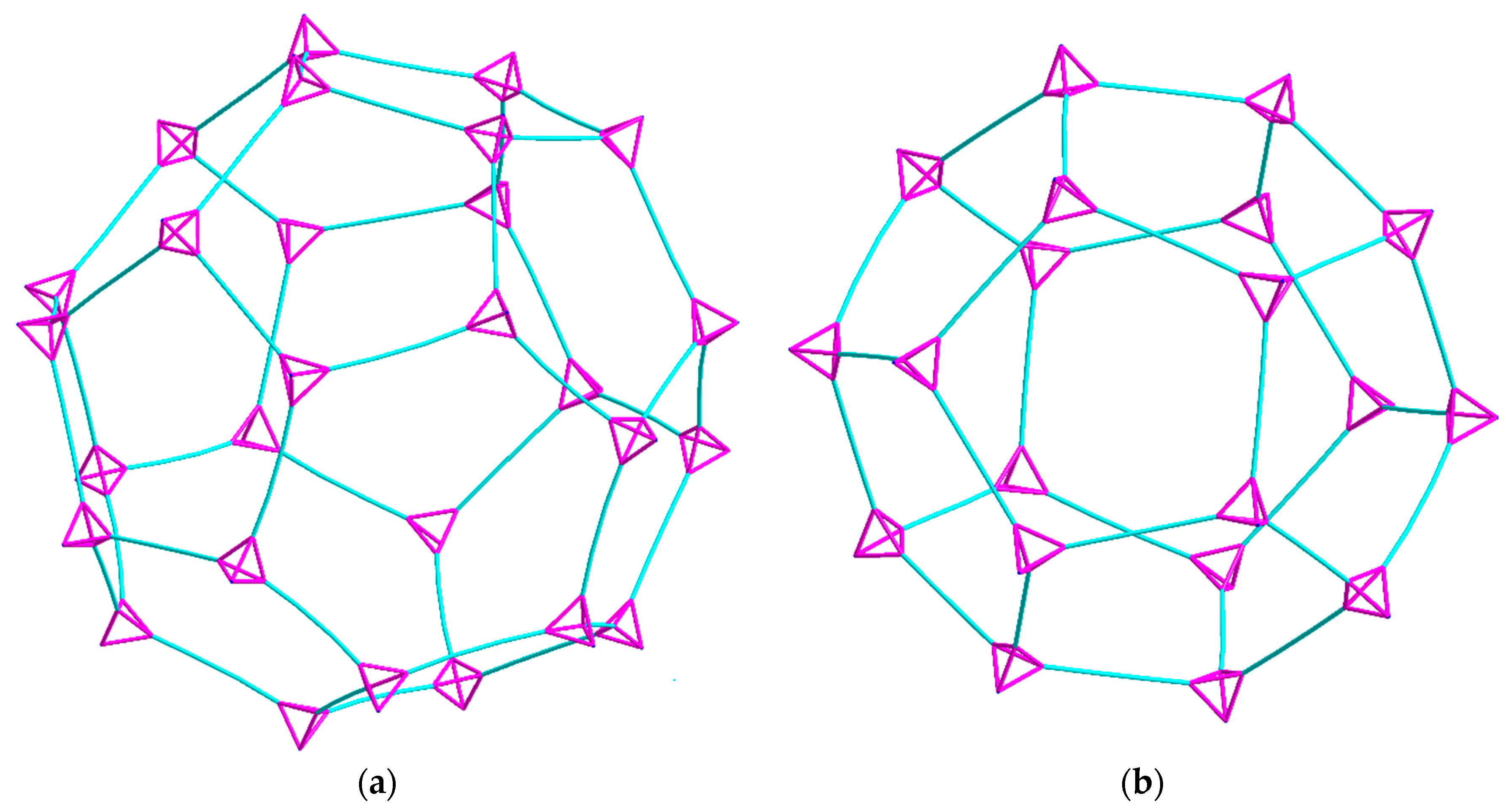
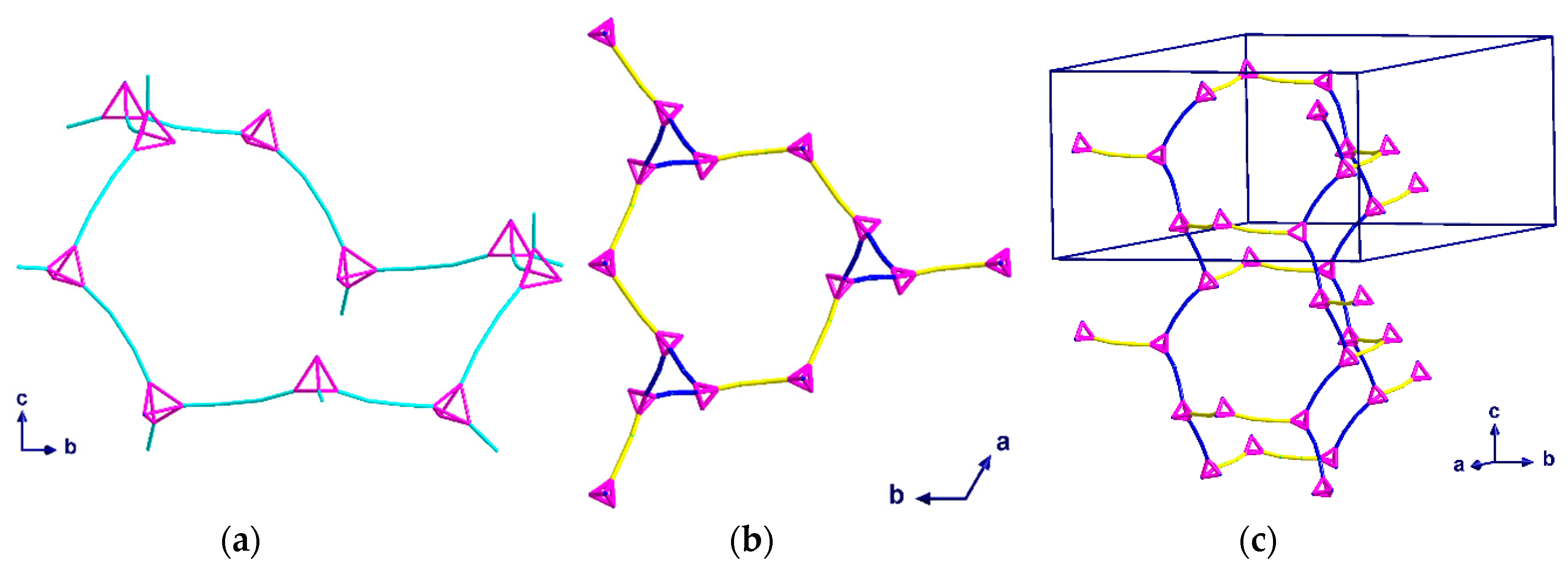
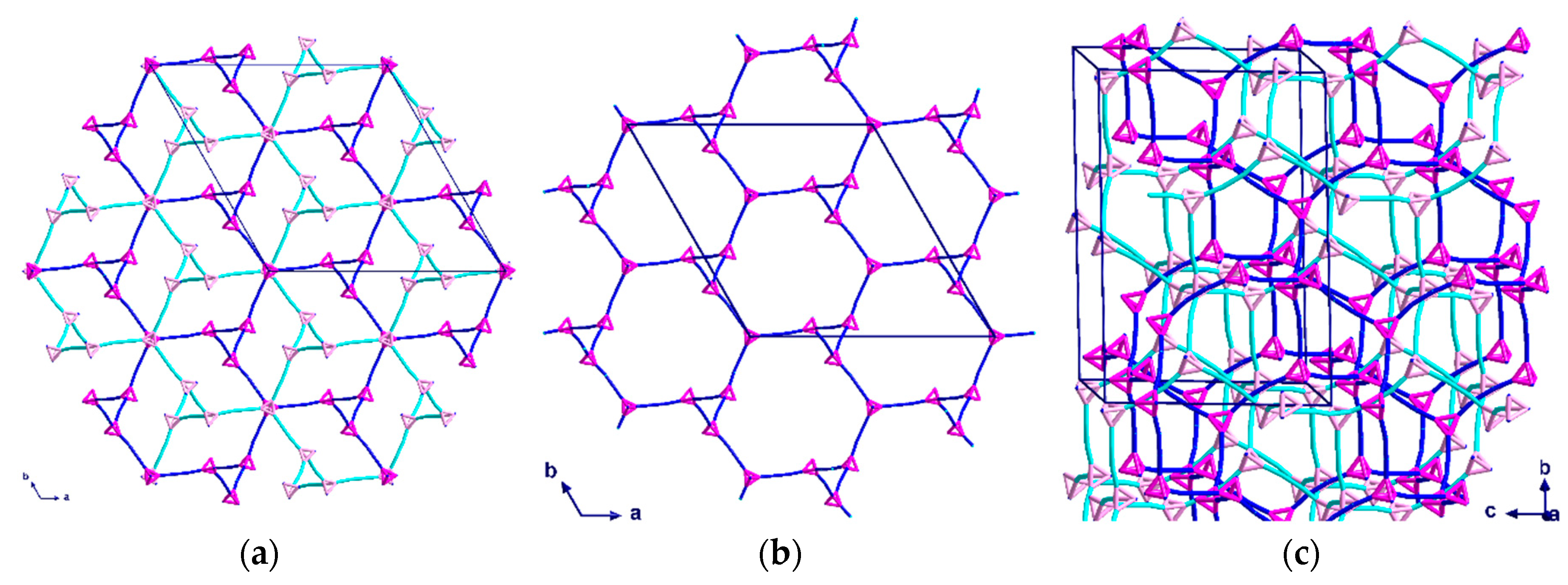
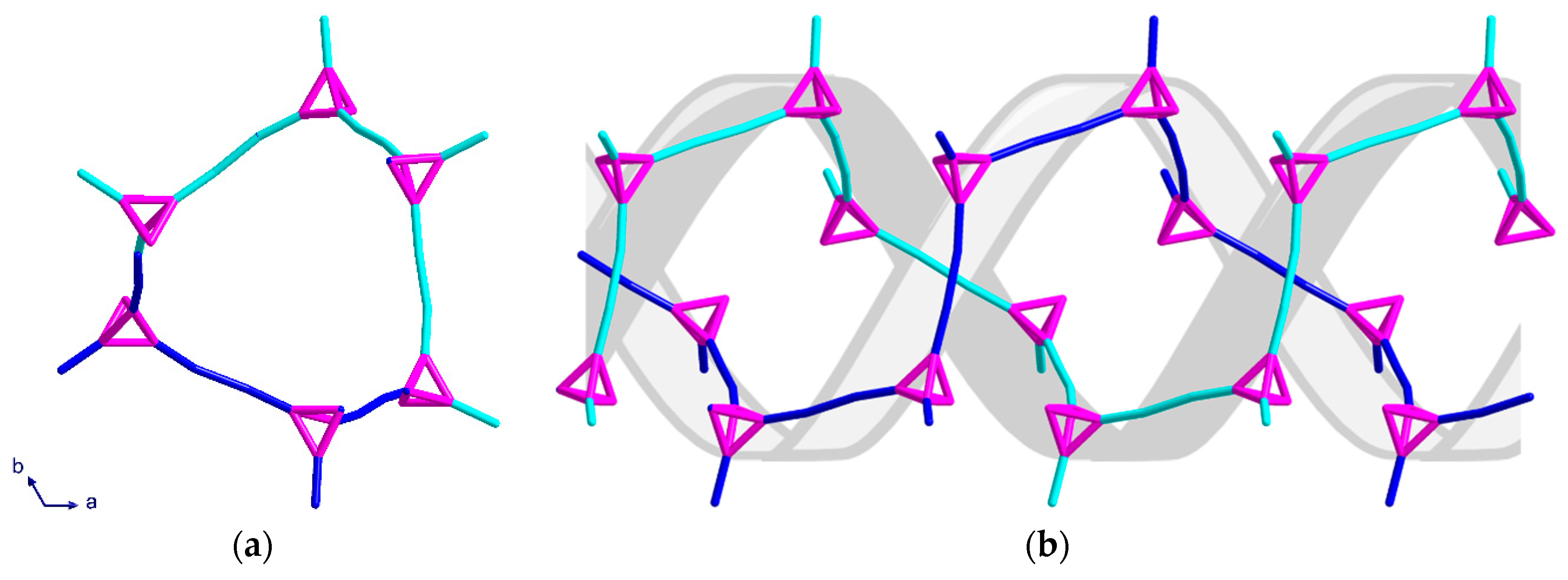
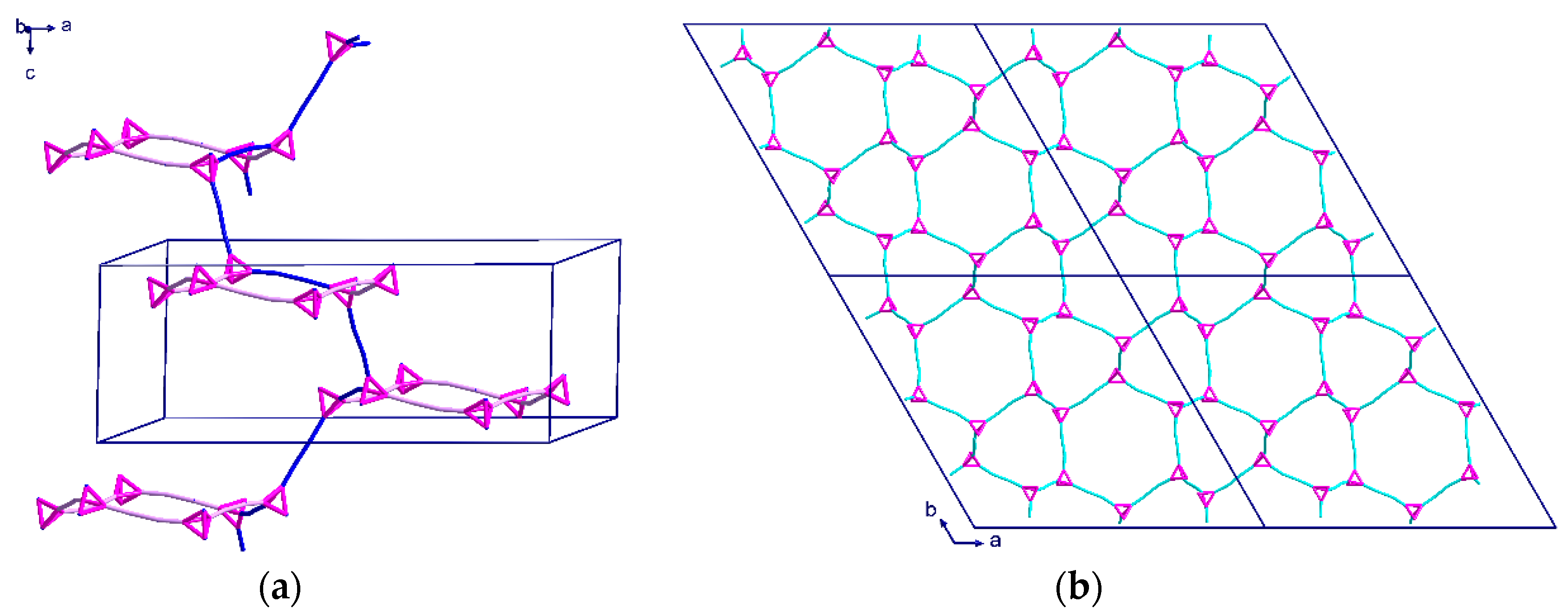
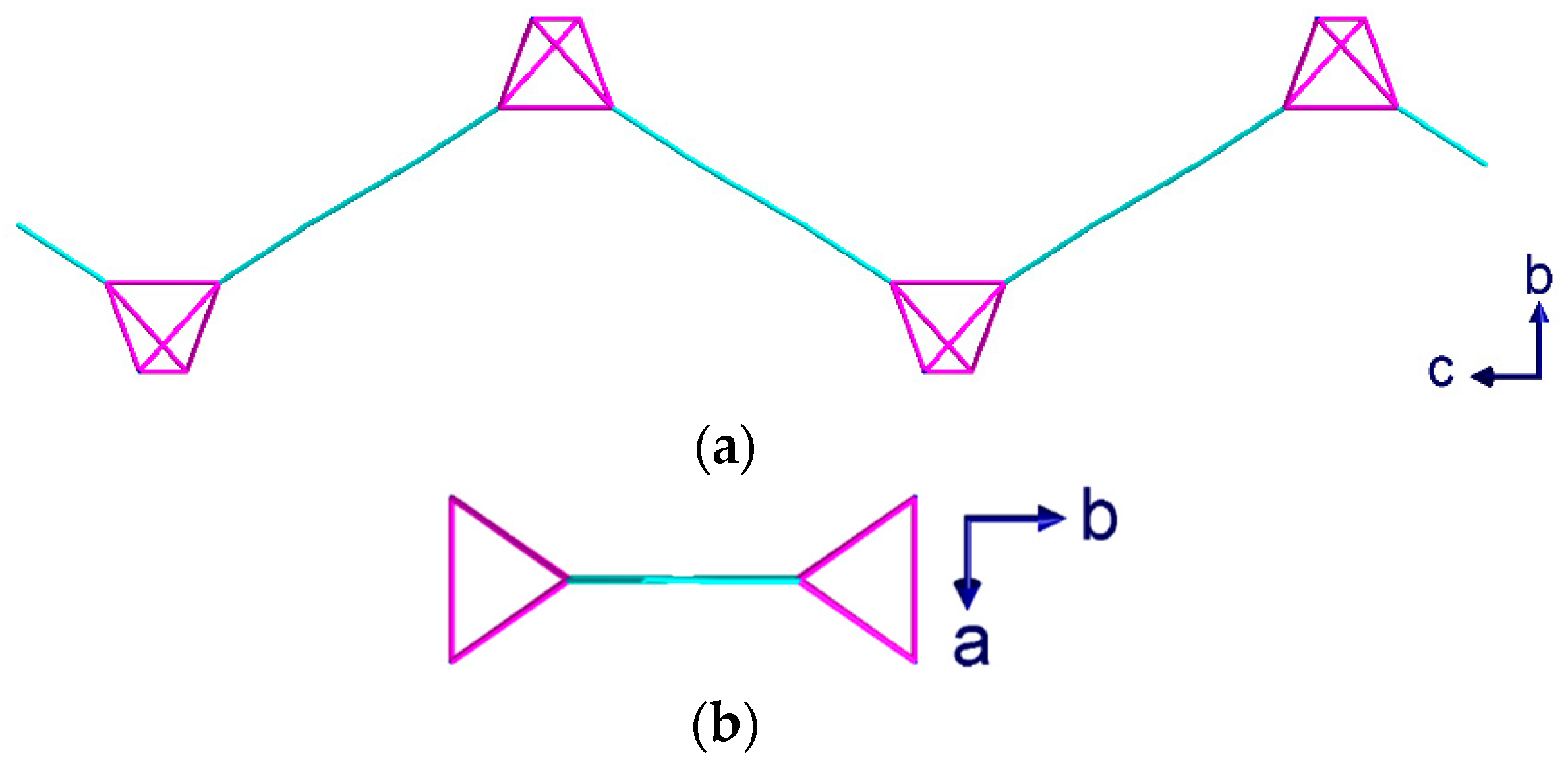
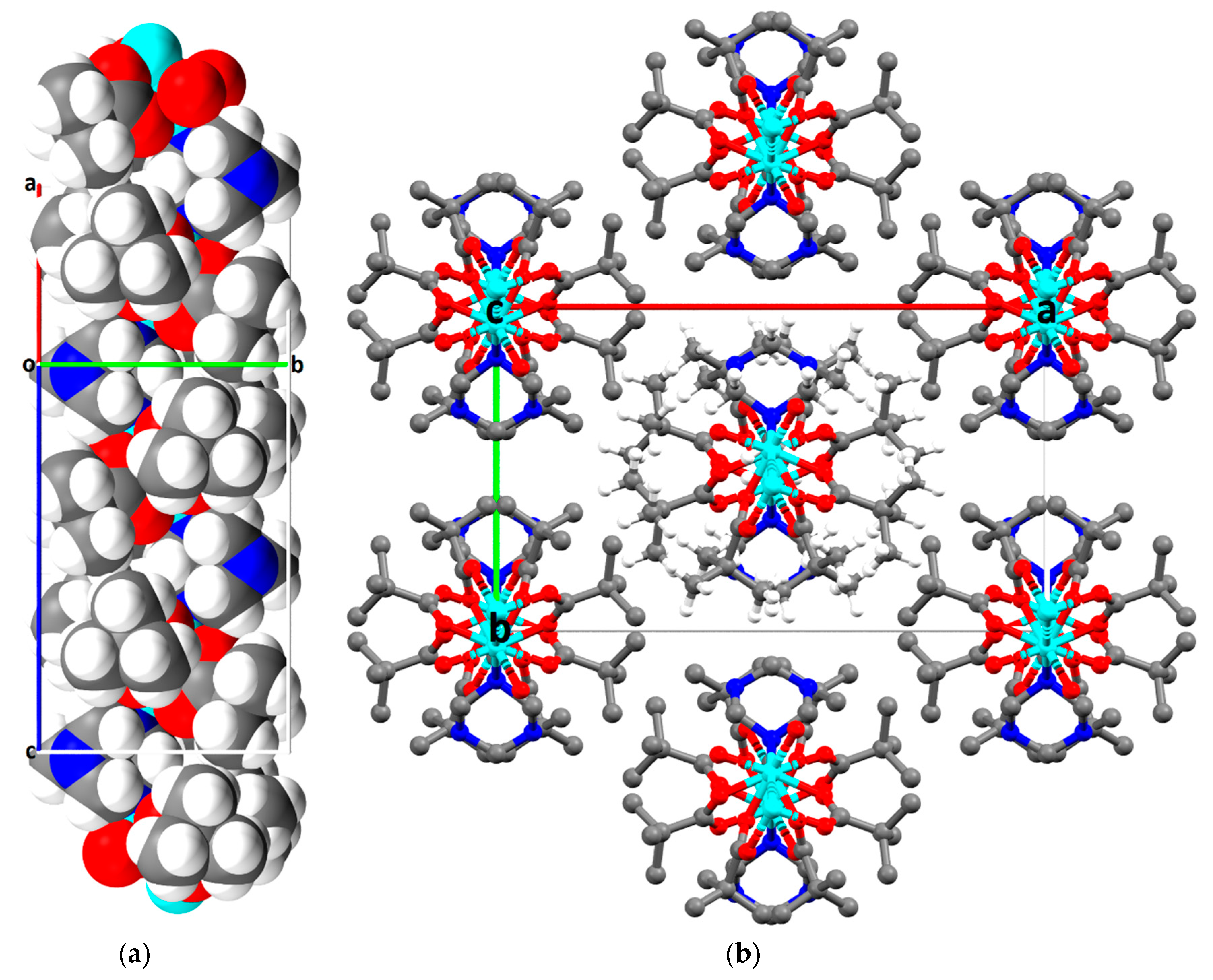
2.2. Description of Crystal Structures
3. Experimental Section
3.1. Materials and Instrumentation
3.2. Single-Crystal X-ray Structure Determinations
3.3. Synthesis of Complex (1)
3.4. General Procedure for the Synthesis of Complexes (2), (3), and (4)
3.4.1. Synthesis of Complex (2)
3.4.2. Synthesis of Complex (3)
3.4.3. Synthesis of Complex (4)
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bonneau, M.; Lavenn, C.; Ginet, P.; Otake, K.; Kitagawa, S. Upscale synthesis of a binary pillared layered MOF for hydrocarbon gas storage and separation. Green Chem. 2020, 22, 718–724. [Google Scholar] [CrossRef]
- Millward, A.R.; Yaghi, O.M. Metal organic frameworks with exceptionally high capacity for storage of carbon dioxide at room temperature. J. Am. Chem. Soc. 2005, 127, 17998–17999. [Google Scholar] [CrossRef] [PubMed]
- Ding, M.; Flaig, R.W.; Jiang, H.-L.; Yaghi, O.M. Carbon capture and conversion using metal–organic frameworks and MOF-based materials. Chem. Soc. Rev. 2019, 48, 2783–2828. [Google Scholar] [CrossRef] [PubMed]
- Gangu, K.K.; Maddila, S.; Mukkamala, S.B.; Jonnalagadda, S.B. Characteristics of MOF, MWCNT and graphene containing materials for hydrogen storage: A review. J. Energy Chem. 2019, 30, 132–144. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.-C.J.; Kitagawa, S. Metal-organic frameworks (MOFs). Chem. Soc. Rev. 2014, 43, 5415–5418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; An, B.; Lin, W. Metal–organic frameworks in solid–gas phase catalysis. ACS Catal. 2019, 9, 130–146. [Google Scholar] [CrossRef]
- Sheberla, D.; Sun, L.; Blood-Forsythe, M.A.; Er, S.; Wade, C.R.; Brozek, C.K.; Aspuru-Guzik, A.; Dincă, M. High electrical conductivity in Ni3(2,3,6,7,10,11-hexaiminotriphenylene)2, a semiconducting metal-organic graphene analogue. J. Am. Chem. Soc. 2014, 136, 8859–8862. [Google Scholar] [CrossRef] [PubMed]
- Talin, A.A.; Centrone, A.; Ford, A.C.; Foster, M.E.; Stavila, V.; Haney, P.; Kinney, R.A.; Szalai, V.; Gabaly, F.E.; Yoon, H.P.; et al. Tunable electrical conductivity in metal-organic framework thin-film devices. Science 2014, 343, 66–69. [Google Scholar] [CrossRef]
- He, X.; Nguyen, V.; Jiang, Z.; Wang, D.; Zhu, Z.; Wang, W.-N. Highly-oriented one-dimensional MOF-semiconductor nanoarrays for efficient photodegradation of antibiotics. Catal. Sci. Technol. 2018, 8, 2117–2123. [Google Scholar] [CrossRef]
- Baldoví, J.J.; Coronado, E.; Gaita-Ariño, A.; Gamer, C.; Giménez-Marqués, M.; Espallargas, G.M. A SIM-MOF: Three-dimensional organisation of single-ion magnets with anion-exchange capabilities. Chem. A Eur. J. 2014, 20, 10695–10702. [Google Scholar] [CrossRef]
- Wriedt, M.; Yakovenko, A.A.; Halder, G.J.; Prosvirin, A.V.; Dunbar, K.R.; Zhou, H.C. Reversible switching from antiferro- to ferromagnetic behavior by solvent-mediated, thermally-induced phase transitions in a trimorphic mof-based magnetic sponge system. J. Am. Chem. Soc. 2013, 135, 4040–4050. [Google Scholar] [CrossRef] [PubMed]
- Zeng, M.H.; Yin, Z.; Tan, Y.X.; Zhang, W.X.; He, Y.P.; Kurmoo, M. Nanoporous cobalt(II) MOF exhibiting four magnetic ground states and changes in gas sorption upon post-synthetic modification. J. Am. Chem. Soc. 2014, 136, 4680–4688. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Vieru, V.; Feng, X.; Liu, J.L.; Zhang, Z.; Na, B.; Shi, W.; Wang, B.W.; Powell, A.K.; Chibotaru, L.F.; et al. Influence of guest exchange on the magnetization dynamics of dilanthanide single-molecule-magnet nodes within a metal-organic framework. Angew. Chemie Int. Ed. 2015, 54, 9861–9865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castells-Gil, J.; Baldoví, J.J.; Martí-Gastaldo, C.; Espallargas, G.M. Implementation of slow magnetic relaxation in a SIM-MOF through a structural rearrangement. Dalton Trans. 2018, 47, 14734–14740. [Google Scholar] [CrossRef] [Green Version]
- Aulakh, D.; Liu, L.; Varghese, J.R.; Xie, H.; Islamoglu, T.; Duell, K.; Kung, C.-W.; Hsiung, C.-E.; Zhang, Y.; Drout, R.J.; et al. Direct imaging of isolated single-molecule magnets in metal–organic frameworks. J. Am. Chem. Soc. 2019, 141, 2997–3005. [Google Scholar] [CrossRef]
- Chen, Z.; Hanna, S.L.; Redfern, L.R.; Alezi, D.; Islamoglu, T.; Farha, O.K. Reticular chemistry in the rational synthesis of functional zirconium cluster-based MOFs. Coord. Chem. Rev. 2019, 386, 32–49. [Google Scholar] [CrossRef]
- Bertelli, M.; Carlucci, L.; Ciani, G.; Proserpio, D.M.; Sironi, A. Structural studies of molecular-based nanoporous materials. Novel networks of silver(I) cations assembled with the polydentate N-donor bases hexamethylenetetramine and 1,3,5-triazine. J. Mater. Chem. 1997, 7, 1271–1276. [Google Scholar] [CrossRef]
- Carlucci, L.; Ciani, G.; Proserpio, D.M.; Sironi, A. A three-dimensional, three-connected cubic network of the SrSi2 topological type in coordination polymer chemistry: [Ag(hmt)](PF6).cntdot.H2O (hmt = Hexamethylenetetraamine). J. Am. Chem. Soc. 1995, 117, 12861–12862. [Google Scholar] [CrossRef]
- Batten, S.R.; Hoskins, B.F.; Robson, R. Synthesis and rutilelike structure of [Cd(tcm)(hmt)(H2O)](tcm) (tcm- = Tricyanomethanide, C(CN)3-; hmt = Hexamethylenetetramine). Inorg. Chem. 1998, 37, 3432–3434. [Google Scholar] [CrossRef]
- Moulton, B.; Lu, J.; Zaworotko, M.J. Periodic tiling of pentagons: The first example of a two-dimensional -net. J. Am. Chem. Soc. 2001, 123, 9224–9225. [Google Scholar] [CrossRef]
- Zheng, G.; Zhang, H.-J.; Song, S.-Y.; Li, Y.-Y.; Guo, H.-D. Self-assembly ofp-sulfonatocalix[4]arene and a Ag–hmt coordination polymer into a porous structure. Eur. J. Inorg. Chem. 2008, 2008, 1756–1759. [Google Scholar] [CrossRef]
- Fang, S.-M.; Ma, S.-T.; Guo, L.-Q.; Zhang, Q.; Hu, M.; Zhou, L.-M.; Gao, L.-J.; Liu, C.-S. A photoluminescent 3D silver(I) polymer with mixed 2-naphthol-5-carboxylate and hexamethylenetetramine ligands, showing an unusual (3,4)-connected (6·7·8)2(4·6·7)2(4·7·8)2(72·8)2(62·7·83)(62·72·8·10) topology. Inorg. Chem. Commun. 2010, 13, 139–144. [Google Scholar] [CrossRef]
- Liu, C.-S.; Chang, Z.; Wang, J.-J.; Yan, L.-F.; Bu, X.-H.; Batten, S.R. A photoluminescent 3D silver(I) coordination polymer with mixed ligands anthracene-9,10-dicarboxylate and hexamethylenetetramine, showing binodal 4-connected (43·63)2(42·62·82)3 topology. Inorg. Chem. Commun. 2008, 11, 889–892. [Google Scholar] [CrossRef]
- Tong, M.-L.; Zheng, S.-L.; Chen, X.-M. Self-assembly of two- and three-dimensional coordination networks with hexamethylenetetramine and different silver(I) salts. Chem. Eur. J. 2000, 6, 3729–3738. [Google Scholar] [CrossRef]
- Köberl, M.; Cokoja, M.; Herrmann, W.A.; Kühn, F.E. From molecules to materials: Molecular paddle-wheel synthons of macromolecules, cage compounds and metal–organic frameworks. Dalton Trans. 2011, 40, 6834. [Google Scholar] [CrossRef]
- Yan, Y.; Juríček, M.; Coudert, F.-X.; Vermeulen, N.A.; Grunder, S.; Dailly, A.; Lewis, W.; Blake, A.J.; Stoddart, J.F.; Schröder, M. Non-interpenetrated metal–organic frameworks based on Copper(II) paddlewheel and oligoparaxylene-isophthalate linkers: Synthesis, structure, and gas adsorption. J. Am. Chem. Soc. 2016, 138, 3371–3381. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Wang, B.; Wang, X.; Xie, L.-H.; Li, J.; Xie, Y.; Li, J.-R. A copper(II)-paddlewheel metal–organic framework with exceptional hydrolytic stability and selective adsorption and detection ability of aniline in water. ACS Appl. Mater. Interfaces 2017, 9, 27027–27035. [Google Scholar] [CrossRef]
- Pickardt, J. Struktur von catena -μ-(Hexamethylentetramin-N, N’)-[tetra-μ-acetato-dikupfer(II)]. Acta Crystallogr. Sect. B Struct. Crystallogr. Cryst. Chem. 1981, 37, 1753–1756. [Google Scholar] [CrossRef]
- Liu, Q.; Li, Y.; Song, Y.; Liu, H.; Xu, Z. Three-dimensional five-connected coordination polymer [M2(C3H2O4)2(H2O)2(μ2-hmt)]n with 4466 topologies (M=Zn, Cu; hmt=hexamethylenetetramine). J. Solid State Chem. 2004, 177, 4701–4705. [Google Scholar] [CrossRef]
- Batten, S.R.; Hoskins, B.F.; Robson, R. Interdigitation, interpenetration and intercalation in layered cuprous tricyanomethanide derivatives. Chem. Eur. J. 2000, 6, 156–161. [Google Scholar] [CrossRef]
- Cao, J.; Huang, Z.; Cao, C.; Cheng, C.; Sun, C. catena -Poly[[tetra-μ-formato-κ 8 O : O ′-dicopper(II)]-μ-hexamethylenetetramine-κ 2 N 1 : N 5 ]. Acta Crystallogr. Sect. E Struct. Rep. Online 2013, 69, m690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.-J.; Chang, Z.; Zhang, A.-S.; Hu, T.-L.; Bu, X.-H. Copper(II) complexes with monocarboxylate ligands bearing different substituent groups: Synthesis and spectroscopic studies. Inorg. Chim. Acta 2010, 363, 1377–1385. [Google Scholar] [CrossRef]
- Konar, S.; Mukherjee, P.S.; Drew, M.G.B.; Ribas, J.; Chaudhuri, N.R. Syntheses of two new 1D and 3D networks of Cu(II) and Co(II) using malonate and urotropine as bridging ligands: Crystal structures and magnetic studies. Inorg. Chem. 2003, 42, 2545–2552. [Google Scholar] [CrossRef] [PubMed]
- Hazra, S.; Sarkar, B.; Naiya, S.; Drew, M.G.B.; Frontera, A.; Escudero, D.; Ghosh, A. Self-assembled molecular complexes and coordination polymers of Cd II, hexamine, and monocarboxylates: Structural analysis and theoretical studies of supramolecular interactions. Cryst. Growth Des. 2010, 10, 1677–1687. [Google Scholar] [CrossRef]
- Wei, X.; Aiqin, D.; Guang-Ming, B.; Xiong-Xin, P.; Ting-Ting, S.; Chun-Yan, H.; Hou-Qun, Y. Crystal structure of poly[dodekais(μ2-2,6-difluorobenzoato-κ2O:O′)-bis((μ3-hexamethylenetetramine-κ3N:N′:N′′)hexacopper(II)], C48H30N4O12F12Cu3. Z. Krist. New Cryst. Struct. 2018, 234, 25–27. [Google Scholar] [CrossRef]
- Hazra, S.; Sarkar, B.; Naiya, S.; Drew, M.G.B.; Ribas, J.; Diaz, C.; Ghosh, A. A self-assembled non-interpenetrating cubic diamondoid coordination polymer of hexamine with linear dicopper spacer: Structural and magnetic studies. Inorg. Chem. Commun. 2011, 14, 1860–1863. [Google Scholar] [CrossRef]
- Bhaskaran, B.; Trivedi, M.; Yadav, A.K.; Singh, G.; Kumar, A.; Kumar, G.; Husain, A.; Rath, N.P. Synthetic, spectral, structural and catalytic activity of infinite 3-D and 2-D copper(II) coordination polymers for substrate size-dependent catalysis for CO2 conversion. Dalton Trans. 2019, 48, 10078–10088. [Google Scholar] [CrossRef]
- Hu, L.-X.; Wang, F.; Kang, Y.; Zhang, J. Structural design of zeolitic cluster organic frameworks from hexamethylentetramine and copper-halide clusters. Cryst. Growth Des. 2016, 16, 7139–7144. [Google Scholar] [CrossRef]
- Kühne, I.A.; Griffiths, K.; Hutchings, A.-J.; Townrow, O.P.E.; Eichhöfer, A.; Anson, C.E.; Kostakis, G.E.; Powell, A.K. Stepwise investigation of the influences of steric groups versus counterions to target Cu/Dy complexes. Cryst. Growth Des. 2017, 17, 5178–5190. [Google Scholar] [CrossRef]
- Baur, E.; Rüetschi, W. Über bildung und zerfall von hexamethylentetramin. Helv. Chim. Acta 1941, 24, 754–767. [Google Scholar] [CrossRef]
- Vollhardt, K.P.C.; Schore, N.E. Organische Chemie, 5th ed.; Wiley-VCH: Weinheim, Germany, 2011. [Google Scholar]
- Asano, Y.; Tani, Y.; Yamada, H. A new enzyme “Nitrile hydratase” which degrades acetonitrile in combination with amidase. Agric. Biol. Chem. 1980, 44, 2251–2252. [Google Scholar] [CrossRef] [Green Version]
- Thimann, K.V.; Mahadevan, S. Nitrilase: I. Occurrence, preparation, and general properties of the enzyme. Arch. Biochem. Biophys. 1964, 105, 133–141. [Google Scholar] [CrossRef]
- Nagasawa, T.; Yamada, H. Microbial transformations of nitriles. Trends Biotechnol. 1989, 7, 153–158. [Google Scholar] [CrossRef]
- Martínková, L.; Vejvoda, V.; Křen, V. Selection and screening for enzymes of nitrile metabolism. J. Biotechnol. 2008, 133, 318–326. [Google Scholar] [CrossRef]
- Yamaguchi, K.; Matsushita, M.; Mizuno, N. Efficient hydration of nitriles to amides in water, catalyzed by ruthenium hydroxide supported on Alumina. Angew. Chem. Int. Ed. 2004, 43, 1576–1580. [Google Scholar] [CrossRef]
- Ramón, R.S.; Marion, N.; Nolan, S.P. Gold activation of nitriles: Catalytic hydration to amides. Chem. Eur. J. 2009, 15, 8695–8697. [Google Scholar] [CrossRef]
- Goto, A.; Endo, K.; Saito, S. RhI-catalyzed hydration of organonitriles under ambient conditions. Angew. Chemie Int. Ed. 2008, 47, 3607–3609. [Google Scholar] [CrossRef]
- Battilocchio, C.; Hawkins, J.M.; Ley, S.V. Mild and selective heterogeneous catalytic hydration of nitriles to amides by flowing through manganese dioxide. Org. Lett. 2014, 16, 1060–1063. [Google Scholar] [CrossRef]
- Marcé, P.; Lynch, J.; Blacker, A.J.; Williams, J.M.J. A mild hydration of nitriles catalysed by copper(II) acetate. Chem. Commun. 2016, 52, 1436–1438. [Google Scholar] [CrossRef]
- Kukushkin, V.Y.; Pombeiro, A.J.L. Additions to metal-activated organonitriles. Chem. Rev. 2002, 102, 1771–1802. [Google Scholar] [CrossRef]
- O’Connor, C. Acidic and basic amide hydrolysis. Q. Rev. Chem. Soc. 1970, 24, 553. [Google Scholar] [CrossRef]
- Jones, E.; Fowlie, G.G. Thermodynamics of formaldehyde manufacture from methanol. J. Appl. Chem. 2007, 3, 206–213. [Google Scholar] [CrossRef]
- Skovran, E.; Martinez-Gomez, N.C. Just add lanthanides. Science 2015, 348, 862–863. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, P.; Hess, M.; Müller, J.; Hildenbrand, K.; Bill, E.; Weyhermüller, T.; Wieghardt, K. Aerobic oxidation of primary alcohols (including methanol) by Copper(II)− and Zinc(II)−phenoxyl radical catalysts. J. Am. Chem. Soc. 1999, 121, 9599–9610. [Google Scholar] [CrossRef]
- Fang, Q.; Zhu, G.; Xue, M.; Sun, J.; Wei, Y.; Qiu, S.; Xu, R. A metal-organic framework with the Zeolite MTN topology containing large cages of volume 2.5 nm3. Angew. Chem. Int. Ed. 2005, 44, 3845–3848. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Hazra, S.; Naiya, S.; Sarkar, B.; Drew, M.G.B.; Ghosh, A. Structural variations in self-assembled coordination polymers constructed by some carboxylate bridged di-copper(II) nodes and hexamethylenetetramine spacers. Polyhedron 2013, 65, 193–199. [Google Scholar] [CrossRef]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kühne, I.A.; Carter, A.B.; Kostakis, G.E.; Anson, C.E.; Powell, A.K. Varying the Dimensionality of Cu(II)-Based Coordination Polymers Through Solvent Influence. Crystals 2020, 10, 893. https://doi.org/10.3390/cryst10100893
Kühne IA, Carter AB, Kostakis GE, Anson CE, Powell AK. Varying the Dimensionality of Cu(II)-Based Coordination Polymers Through Solvent Influence. Crystals. 2020; 10(10):893. https://doi.org/10.3390/cryst10100893
Chicago/Turabian StyleKühne, Irina A., Anthony B. Carter, George E. Kostakis, Christopher E. Anson, and Annie K. Powell. 2020. "Varying the Dimensionality of Cu(II)-Based Coordination Polymers Through Solvent Influence" Crystals 10, no. 10: 893. https://doi.org/10.3390/cryst10100893
APA StyleKühne, I. A., Carter, A. B., Kostakis, G. E., Anson, C. E., & Powell, A. K. (2020). Varying the Dimensionality of Cu(II)-Based Coordination Polymers Through Solvent Influence. Crystals, 10(10), 893. https://doi.org/10.3390/cryst10100893